
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Primer für die Immunhistochemie auf gefriergeschnitten Rat Brain Tissue: Beispiel Färbung für Mikroglia und Neuronen
In diesem Artikel
Zusammenfassung
This introductory level protocol describes the reagents, equipment, and techniques required to complete immunohistochemical staining of rodent brains, using markers for microglia and neuronal elements as an example.
Zusammenfassung
Immunhistochemie ist eine weit verbreitete Technik zur Erfassung der Anwesenheit, den Ort und die relative Häufigkeit von Antigenen in situ. Dieser Einführungs Level-Protokoll beschreibt die Reagenzien, Geräte und Techniken erforderlich, um die immunhistochemische Färbung von Hirngewebe von Nagern zu vervollständigen, mit Markern für Mikrogliazellen und neuronale Elemente als Beispiel. Genauer gesagt, dieses Papier ist ein Schritt-für-Schritt-Protokoll für fluoreszierende Visualisierung von Mikroglia und Neuronen über die Immunhistochemie für Iba1 und Pan-neuronalen sind. Fluoreszenzdoppelmarkierung ist besonders nützlich für die Lokalisierung mehrerer Proteine innerhalb der gleichen Probe, die Bereitstellung der Möglichkeit, genau zu beobachten Wechselwirkungen zwischen Zelltypen, Rezeptoren, Liganden und / oder der extrazellulären Matrix in Relation zueinander sowie Protein Co- Lokalisierung innerhalb einer einzelnen Zelle. Im Gegensatz zu anderen Visualisierungstechniken kann Fluoreszenz Immunhistochemie Färbeintensität Abnahmedie Wochen bis Monate nach Färbung, es sei denn, entsprechende Vorkehrungen getroffen werden. Trotz dieser Einschränkung in vielen Anwendungen Fluoreszenzdoppelmarkierung über Alternativen wie 3,3'-Diaminobenzidintetrahydrochlorid (DAB) oder alkalische Phosphatase (AP) bevorzugt, da die Fluoreszenz zeiteffizienter und ermöglicht genauere Unterscheidung zwischen zwei oder mehr Marker. Die Diskussion enthält Tipps zur Fehlerbehebung und Beratung zum Erfolg zu fördern.
Einleitung
Immunohistochemie ist ein Verfahren zum Nachweis von Antigenen (dh Proteine) in Gewebeschnitten unter Verwendung von primären Antikörpern, die spezifisch gegen die Antigene von Interesse binden. Immunhistochemie wurde von JR Marrack 1934 Pionierarbeit geleistet, als er festgestellt, dass Antikörper könnten Antigene mit großer Spezifität 1 zu lokalisieren. Ab 1942 einige der ersten in-vitro-Studien unter Verwendung von fluoreszierenden Antikörpern immunhistochemisch visualisieren veröffentlicht wurden 2,3, wonach die erste in vivo histochemische Studie veröffentlicht 4. Während der 1960er drei Jahrzehnte nach dem Beginn des immunhistochemische Verfahren begann, Enzym-konjugierten Antikörper als sekundäre Reagentien verwendet werden. Diese Methoden wurden gleichzeitig und unabhängig voneinander in Frankreich und in den USA 5,6 entwickelt. Heute, ein breites Spektrum von Antikörpern bietet unendlich viele Möglichkeiten für die Immunhistochemie Studien 7.
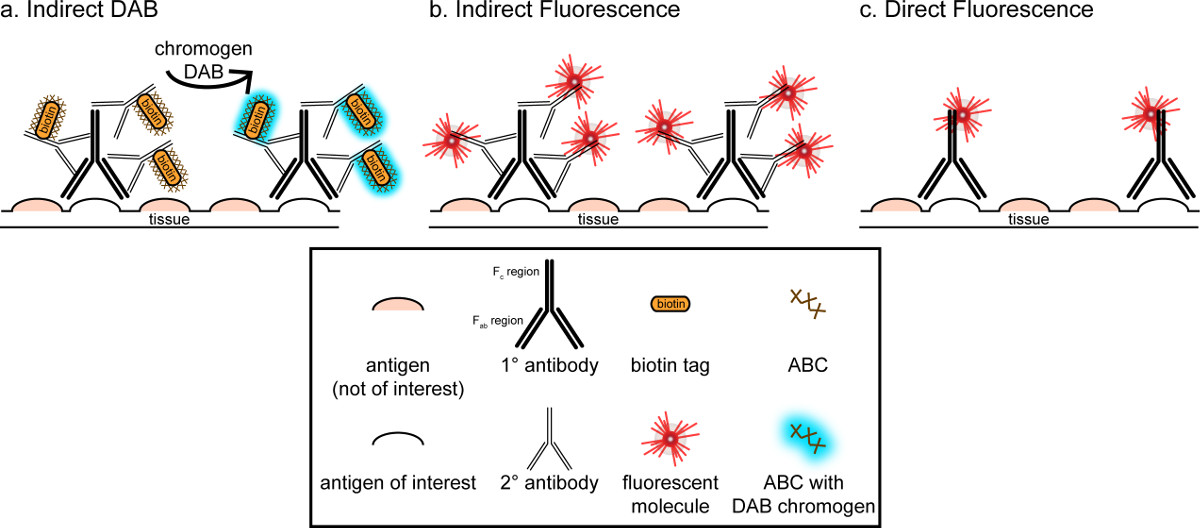
"> Das übergeordnete Ziel dieser Korrespondenz ist es, eine kurze Einführung in immunhistochemischen Färbung bereitzustellen; es ist nicht als eine umfassende und abschließende Bewertung dieser Technik werden in der Methode beschrieben werden immunhistochemischen Techniken für zwei Antigene präsentiert (Marker für die Mikroglia und. Neuronen) für die Färbung von Paraformaldehyd perfundiert beginnt Saccharose kryogeschützt, gefriergeschnitten Rattenhirn. Die immunhistochemische Färbung mit Blockierungs unspezifische Antigen-Bindung an die Hintergrundfärbung zu reduzieren. Als nächstes wird die Inkubation mit dem primären Antikörper können zur Bindung an ein spezifisches Antigen im Gewebe. Im Anschluss an die primären Antikörper, ein anderer Antikörper, bezeichnet als sekundärer Antikörper, aufgebracht wird, um den primären Antikörper auf einen konjugierten Visualisierungssignal 8 zu verknüpfen. Der sekundäre Antikörper zielt auf das Immunglobulin G (IgG) Domäne spezifisch für die Spezies, in denen das primäre Antikörper gezüchtet wurde. Der sekundäre Antikörper verstärkt das Signal, des primären Antikörpers, da die Fab Regionen ter sekundären Antikörper binden sich an mehreren Stellen auf dem IgG-Domäne des primären Antikörpers. Entweder Enzymen oder fluoreszierenden Moleküle an die Fc-Bereiche der sekundären Antikörper konjugiert ermöglichen Visualisierung. Beispielsweise ist ein Kaninchen-Anti-Iba1 Primärantikörper ein Kaninchen-IgG-Molekül spezifisch für Iba1. Wenn Esel-Anti-Kaninchen-IgG als sekundärer Antikörper verwendet, wird es zu erkennen und zu mehreren Regionen des Kaninchen-Anti-Iba1 IgG (siehe 1), zu binden. Der Esel-Antikörper kann durch verschiedene Verfahren sichtbar gemacht werden. Diese Entsprechung konzentriert sich auf Erfassung eines Fluorophors an den sekundären Antikörper, der den primären Antikörper erkennt, zur Sichtbarmachung durch Fluoreszenzmikroskopie konjugiert. In fluoreszierenden Immunhistochemie kann eine Kernfärbung wie Hoechst oder DAPI verwendet, um alle Kerne zu visualisieren.
Abbildung 1: SchEmatic Darstellung direkte vs. indirekte Antikörper-Markierungstechniken. Antikörper binden an das Antigen von Interesse und kann durch sekundäre Antikörper, die gegen die Spezies des primären Antikörper erzeugt amplifiziert werden. Diese Technik kann durchgeführt werden unter Verwendung von Avidin-Biotin-Komplex (ABC) zur Verstärkung und DAB zur Visualisierung (A) oder einem direkt konjugierten fluoreszierenden Sekundärantikörper (B). Alternativ können primäre Antikörper direkt mit vielen verschiedenen Tags, einschließlich Biotin oder ein Fluorophor (C) konjugiert werden. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
Ein alternatives Verfahren zur Visualisierung von immunhistochemischen Färbung verwendet 3,3'-Diaminobenzidintetrahydrochlorid (DAB; siehe 1 und 2). Dies unterscheidet sich von Fluoreszenz, die durch Verwendung eines biotinylierten oderMeerrettich-Peroxidase (HRP) konjugierten Sekundärantikörper, die ein Enzym, um DAB zu einem Niederschlag, der unter Hellfeldmikroskopie sichtbar zu konvertieren bietet. In Fällen, in denen eine einzige Antigen von Interesse ist oder Färbung ist erforderlich, langlebig zu sein, kann DAB besser geeignet als Fluoreszenzfärbung ist. Allerdings DAB-Färbung nicht zur Differenzierung zwischen mehreren Markern gut geeignet, vor allem, wenn zwei nukleäre Antigene von Interesse. Informationen zur DAB Materialien und Protokoll Änderungen, wenden Tabelle 1. Alternativ Nitroblautetrazoliumchlorid / 5-Brom-4-chlor-3-indolyl-phosphat (NBT / BCIP) kann verwendet werden, um eine alkalische Phosphatase (AP) konjugierten sekundären visualisieren Antikörper.

Abb. 2: Repräsentative Bilder von Nickel-verstärkten DAB einfach markierten Rattenhirngewebeschnitten Rattenhirn-sections, die mit Nickel-verstärkten DAB für Iba1 (A) und Pan-neuronalen (B) markiert sind, ermöglichen eine lang anhaltende Analyse von Mikrogliazellen oder Neuronen allein. Maßstabsleiste 20 um. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
Man muß bedenken die geschätzte Häufigkeit des Antigens von Interesse innerhalb des Gewebes analysiert. Indirekten Methoden (wie oben beschrieben) sind nützlich für Ziele mit geringer Häufigkeit. Wenn das Antigen von Interesse in besonders hoher Zahl, können direkte Verfahren aufgebracht werden. Direkte Verfahren umfassen einen primären Antikörper, der direkt an ein Visualisierungssignal konjugiert ist, und damit keine sekundären Antikörpers erforderlich ist. Diese Methode vereinfacht die Färbung Prozess, aber beseitigt die durch indirekte Methoden erreicht Verstärkung. Mit Hilfe eines direkt konjugierten primären Antikörper beseitigt auch Kreuzreaktivität Sekundärantikörperals Doppelmarkierung.
Diese Mitteilung beschreibt die Protokoll für Doppelmarkierung mit Iba1 und Pan-neuronalen (Details in Tabelle 1). Iba1 Flecken Mikroglia-Aktivierung in vielen Staaten, darunter verzweigte, hyperverzweigte, aktiviert, amoeboid und Stange. Pan-neuronalen Flecken neuronalen Axonen, Dendriten, und Soma. Seit Iba1 Flecken meisten Mikroglia und Pan-neuronalen Ziele das Neuron ist diese Kombination von Flecken nützlich gewinnen ein umfassendes Verständnis von Mikroglia-Neuron-Interaktionen.
Zusammengefasst beruht die immunhistochemische Färbung auf die sorgfältige Auswahl von Antikörpern. Da die Fragestellung wird präziser können Antikörper, Antigene, um alternative angehoben wünschen übrig. Um einen bestimmten Mikrogliaaktivierung Staaten abzielen, kann man sich dafür entscheiden, CD45 oder CD68-Antikörper verwenden, anstatt Iba1. Ferner wird bei der Arbeit mit Mäusen, F4 / 80 können die erforderlichen Ergebnisse zu liefern. In ähnlicher Weise kann die neuronale Elemente spezifisch mit Antikörpern ra gezieltsierten gegen den Zellkern, synapse (Pre- oder Post), Axon, und Wachstumskegel. Darüber hinaus gibt es andere Markierungen, die das Alter des Neurons (Doppel-Cortin, NeuN) unterscheiden und neuronalen Regeneration (GAP-43).
Access restricted. Please log in or start a trial to view this content.
Protokoll
HINWEIS: Alle Verfahren wurden in Einklang mit den institutionellen Animal Care und Verwenden Committee (IACUC) von der Universität von Arizona durchgeführt. Eine Liste der empfohlenen Materialien und Geräte können in Tabelle 1 gefunden werden.
1. Gewebepräparation
- Perfusion
- Euthanize Nager mit einer Überdosis von Natriumpentobarbital (25 mg / kg, IP), und transkardial perfundiert mit phosphatgepufferter Salzlösung (PBS), bis sie vollständig ausgeblutet (3-5 min) bei einer Flussrate von 8 ml / min. Für umfassende Perfusion Anweisungen finden Sie Gage et al 2012 9.
- Unmittelbar im Anschluss an PBS spülen, fixieren Gewebe durch Perfusion mit 4% Paraformaldehyd in PBS für 15-20 min bei einer Flussrate von 8 ml / min.
- Entfernen Gehirn und in 4% Paraformaldehyd für 24 h, gefolgt von einer abgestuften Saccharoselösungen (15%, 30%, 30%, in der Reihenfolge, in Tris gepufferter Kochsalzlösung) auf 4 ° C. Übertragen Sie das Gehirn, um die nachfolgende Saccharoselösung only Nachdem sich das Gehirn in jeder Lösung versenkt. Anmerkung: Normalerweise, 5 Tage in jeder Lösung ist ausreichend Zeit für Gewebe zu sinken.
- Tissue Freezing und Kryoschneiden
- Platzieren des Gehirns in Einbettungsmedium, wie OCT-Verbindung und tauchen in Isopentan bei einer Temperatur von -35 ° C. Ermöglichen das Gehirn für ein Minimum von 10 min Einfrieren und dann bei -80 ° C lagern. Probleme können entstehen, wenn Fleiß Temperatur wird nicht übernommen; finden Sie Informationen zur Fehlerbehebung Diskussion.
- Geschnitten serielle Kranzschnitte mit einer Dicke von 20 & mgr; m und einer Temperatur von -20 ° C. Sammeln Gewebe auf positiv geladene Objektträger. Hirnschnitte können in einer Dia-Box in der Folie bei -80 ° C eingewickelt in einer Zip-Top-Tasche und dort zentral langfristig platziert werden. Diese Art der Lagerung verursacht einen doppelten Begrenzungs die Exposition gegenüber Luft und Frost zu verhindern.
2. Gewebeverarbeitung
HINWEIS: Beispiel Ausrüstung und Materialien rzur Färbung equired sind in Abbildung 3 dargestellt. Alternativen zur Verfügung stehen, jedoch werden diese Bilder zu unterstützen diejenigen, die mit immunhistochemischen Färbung geeignete Produkte vor dem Kauf zu visualisieren.

Abbildung 3: Beispiel Produkte zur immunhistochemischen Färbung erforderlich Die in (A) gezeigt, Black Box ist ein idealer Feuchtigkeitskammer für Immunfluoreszenz, wie Objektträger werden vor Licht ohne die Notwendigkeit, auf Folie wickeln Sie das Feld geschützt.. Nach Schnitte können Folien in einer Box, wie die in (B) gezeigt, gelben Kasten gelagert werden. Wickeln Sie das Feld dicht in der Folie und Platzierung in einem zip-Top-Tasche vor dem Gefrier hilft den Gewebeproben von Gefrierbrand zu schützen. Ein Beispiel für Folien ist in (C), wobei in (D) dargestellt verschiedenen färbekasten und da (E ). Deckgläser können in Größe und Dicke (F) variieren jedoch 1,2 dicken Deckgläser bieten schöne Imaging-Ergebnisse auf den meisten aufrecht und konfokale Mikroskope. Ein Bleistift, wie die in (G) gezeigt ist, kann verwendet werden, um Folien zu markieren. Filzstiften sollte vermieden werden, da die Tinte ausgeführt werden kann, die sowohl die Färbung und die Fähigkeit zu bestimmen, welche die Probe. Ein Mini-pap pen, wie sie in (H) gezeigt, ermöglicht eine abstoßende Grenze auf Objektträger gezogen werden.
- Slide Vorbereitung
- Folien aus dem Gefrierschrank und Tauwetter entfernen bei Raumtemperatur.
- Optional: Wenn Abschnitte wurden zuvor von Dias schwebte, legen Sie aufgetaut Dias in einem Ofen bei 60 ° C für nicht mehr als 4 Stunden, um zu verhindern, dass Gewebeschnitte von schwimmenden off Folien.
- Die Objektträger in einem Objektträgergestell und entsprechenden Gericht.
- Wasch gleitet dreimal in PBS für jeweils 5 Minuten, Ändern Lösung zwischen Wäschen. Von diesem Schritt nach vorn, zu vermeiden, dass Abschnitons ohne Flüssigkeit über längere Zeiträume. Hinweis: Sofern Teile trocknen, wird die Hintergrundfärbung erhöht und aussagekräftige Daten nicht zuverlässig erreicht werden.
- Folien aus dem Gefrierschrank und Tauwetter entfernen bei Raumtemperatur.
- Gewebefärbung
- In einem lichtdichten Box Färbung, erstellen Sie eine "feuchte Kammer" mit fusselfreien Gewebe mit VE-Wasser eingeweicht.
- Trocknen Sie die Ränder der Objektträger mit einem fusselfreien Tuch, verwenden Sie einen Mini pap Stift, um eine flüssigkeitsabweisende Grenz ganz am Rand des Objektträgers zu machen, weg von Gewebeschnitten. Diese Grenze sollte ausreichend Raum zwischen dem Meniskus der Flüssigkeit und dem Rand des Gewebes, so dass die Oberflächenspannung nicht beeinflusst Markierung sicherzustellen.
HINWEIS: Der Pap pen abweisende Grenz kann vor 2.1.3 angelegt, wenn die Antikörper von Interesse nicht Mikrowelle Antigen-Retrieval verlangen. Wenn der PAP-Stift wurde vor dem Waschen in PBS aufzuerlegen, ist die Integrität der flüssigkeitsabweisenden Grenze bei diesem Schritt geprüft. Verwenden Sie ein Mini-pap Stift, um in Öffnungen in der Grenze zu füllen. - Mit Dias horizontal gelegt, Block nicht-spezifische Antigen-Bindung durch Inkubation in 4% v / v Serum in PBS (Blocklösung). Pipette 300 ul Block Lösung pro Objektträger für 1 h bei Raumtemperatur. Sicherzustellen, dass der Block-Lösung erstreckt sich zu dem pap Stift an der Kante der Folie und das Gewebe vollständig abdeckt ungleichmäßige Anfärbung durch Oberflächenspannung in der Nähe des Gewebes zu vermeiden.
- Verwenden Serum von der gleichen Spezies in dem sekundären Antikörper durchgeführt wird. Anmerkung: In diesem Verfahren werden die sekundären Antikörper in Esel gemacht und damit Eselsserum verwendet. Wenn Sekundärantikörper aus zwei oder mehr verschiedenen Arten verwendet werden, umfassen Serum von jeder Spezies.
- Pipette primären Antikörper auf Objektträger. Hinweis: Antikörperkonzentrationen für diese Färbung haben bei 1 optimiert: 5000 und 1: 500 für Iba1 und Pan-neuronalen sind. Diese Konzentrationen haben sich als sinn Färbung mit einer Abwesenheit von Hintergrundfärbung zu zeigen.
- Verdünnen Block Lösung1% Serum in PBS und fügen primären Antikörper. Pipette 300 ul primären Antikörperlösung in 1% Serum pro Folie. Wieder zu beachten, die Flüssigkeit an der Kante des pap Stift. Inkubieren über Nacht bei 4 ° C.
- Fügen Sie drei Kontrollobjektträger: eine, die weder Iba1 noch Pan-neuronalen Antikörper enthält, eine mit Iba1 ohne Pan-neuronalen Antikörpers und eines mit Pan-neuronalen Antikörper ohne Iba1. Fleck diese Folien in der gleichen Flucht mit den gleichen Lösungen, aber die primären Antikörper weggelassen werden, um die nicht-spezifische Bindung der sekundären Antikörper zu testen.
- Am nächsten Morgen, Wasch gleitet dreimal in PBS für jeweils 5 Minuten, Ändern Lösung zwischen den Waschgängen.
- Fluoreszierende Antikörper sind lichtempfindlich, daher von dieser Schritt nach vorn, Lichtexposition zu minimieren, indem sichergestellt Waschbehälter werden in Folie eingewickelt und Hybridisierung Kästen sind entweder schwarz oder im Dunkeln inkubiert. Pipettieren Sie die entsprechenden Sekundärantikörper auf allen Folien und Inkubation für60 min bei Raumtemperatur in einer Konzentration von 1: 250 in Blocklösung (siehe Schritt 2.2.3) in einer lichtdichten "feuchte Kammer" (siehe Schritt 2.2.1).
- Verwenden Sekundärantikörper unterschiedlicher Wellenlängen. Hier für den primären Antikörper Kaninchen-anti-Iba1 Verwenden Esel anti- Kaninchen 594 als geeigneter sekundärer Antikörper. Für den primären Antikörper Maus-Anti-Pan-neuronalen Verwenden Esel-anti-Maus 488 als geeigneter sekundärer Antikörper. Alternativ können Sie Anti-Kaninchen-488 und Anti-Maus-594.
- Wasch gleitet dreimal in PBS für jeweils 5 min.
- Optional: durchführen Kernfärbung.
- Platz in Hoechst (oder andere Kernfärbung) in einer Konzentration von 0,03 ug / ml in doppelt destilliertem H 2 0 für genau 60 sec.
- Wasch gleitet dreimal in PBS für jeweils 5 min.
- In ddH 2 0 waschen.
- Eindecken
- Deckglasobjektträger mit einer wässrigen Einschlussmittel, wie beispielsweise Fluoromount-G oder ProlongGold. Achten Sie darauf, um alle Blasen mit einem Wattestäbchen entfernen.
Hinweis: Andere Befestigungsmittel verwendet werden könnte, hat sich jedoch hohe Durchschlag zwischen den Farbstoffen, die durch einige wenige Tage nach Eindecken festgestellt worden. - Verwenden klarem Nagellack, um die Kanten zu versiegeln, verhindern, dass die Abschnitte vor dem Austrocknen durch Verdunstung. Lassen Sie Nagellack auf eine lichtdichte Behälter trocken, während in flachen Folien und bei Raumtemperatur bleiben, und dann in einem lichtdichten Behälter in Folie bei 4 ° C eingewickelt zu speichern.
- Deckglasobjektträger mit einer wässrigen Einschlussmittel, wie beispielsweise Fluoromount-G oder ProlongGold. Achten Sie darauf, um alle Blasen mit einem Wattestäbchen entfernen.
3. Abbilden der gefärbten Gewebe
- Mikroskopie
- Lassen Sie den Nagellack, um für mindestens eine Stunde trocknen vor Beginn der Mikroskopie, welche in einem abgedunkelten Raum stattfinden sollte.
- Erwerben Mikrophotographien mit einem konfokalen oder Forschungsmikroskop mit einer fluoreszierenden Lichtquelle und einer digitalen Kamera Anhaftung. Mit Hilfe der beiliegenden Software, stellen Sie die Belichtung für jede Wellenlänge - 405, 488, und 594 - getrennt. Hinweis: Tiefenabbildungsanweisungen sollten Online vom Mikroskop-Hersteller zur Verfügung.
- Erwerben Mikroaufnahmen in jedem Kanal, ohne die Teile oder Einstellung des Fokus. Nehmen Sie Bilder in Farbe, in Graustufen oder alternativ und konvertieren, um danach zu färben.
Hinweis: Farb- oder Graustufenbilder von jedem Kanal können in der Nachbearbeitung zusammengetragen werden. - Sicherzustellen, dass die Gewebeabschnitte nicht auf Umgebungslicht oder Licht mikroskopisch für lange Zeit ausgesetzt, als Photobleich der Abschnitte auftritt. Um dies zu vermeiden, erhöhen Sie die Belichtungszeit, anstatt die Licht- / Laserintensität.
- Schalten Sie nicht die Fluoreszenzlichtquelle innerhalb von 30 Minuten eingeschaltet.
Hinweis: Schalten Sie die Quelle an und aus kurz hintereinander kann die Lebensdauer der Leuchtstofflampe zu verringern.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Diese Färbung Protokoll führt Rattenhirngewebeschnitten, die Mikroglia fluoreszenz im Kanal 594 (rot) markiert sind und Neuronen in dem Kanal 488 markiert (grün; siehe Abbildung 4). Wenn eine Kernfärbung durchgeführt wurde, wird es in der 405-Kanal (blau) zeigen. Bilder können in verschiedenen Kanälen zwischen zwei beliebigen Kanälen aufgenommen werden und überlagert zum direkten Vergleich der drei Kanäle oder. Viele digitale Erfassungssoftware Suiten verfügen diese Funktionalität. Doppelmar...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Das übergeordnete Ziel dieser Mitteilung war die Immunhistochemie Verfahren für den Leser einzuführen. Hierzu wird das Beispiel der Doppelmarkierung mit Iba1 und Pan-neuronale Antigene an Mikroglia und Neuronen in Paraformaldehyd perfundiert beobachten, wurde Saccharose kryogeschützt, gefriergeschnitten Rattenhirn verwendet.
Diese Technik lässt sich an endlosen Zwecken dienen werden. Ein Array von verschiedenen Antigenen in einer Vielzahl von Gewebetypen, wie beispielsweise, jedoch nich...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
The authors have nothing to disclose.
Danksagungen
The authors would like to thank Mr. Ryan Hart and Mr. Arriz Lucas for their invaluable feedback on this communication. This work was supported by NIH NINDS R01 NS065052 and Phoenix Children’s Hospital Mission Support Funds.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| Fisherbrand Superfrost Plus Glass Slides | Fisher Scientific | 22-034-979 | Used for tissue mounting (1.2.2) |
| Oven | Thermo Scientific | 51028112 | Used for tissue drying (2.1.1) |
| Mini Pap pen | Life Technologies | 00-8877 | Used in step 2.2.2 |
| Andwin Scientific Tissue-tek Slide Staining Dish | Fisher Scientific | 22-149-429 | Used for all washes during staining (2.2), as well as the Hoechst step (2.2.8) |
| Kimwipes | Fisher Scientific | 06-666-A | Used for drying slides (2.2) |
| Black Staining Box | Ted Pella | 21050 | Used for blocking and staining steps (2.2) |
| Normal Donkey Serum | Fisher Scientific | 50-413-253 | Used for block and antibody incubation (2.2) |
| Mouse α-Pan-neuronal | Millipore | MAB2300 | Used for primary antibody (2.2.4) |
| Rabbit α-Iba1 | Wako Chemical | 019-19741 | Used for primary antibody (2.2.4) |
| Donkey α-rabbit 594 | Jackson ImmunoResearch | 711-585-152 | Used for secondary antibody (2.2.6) |
| Donkey α-mouse 488 | Jackson ImmunoResearch | 715-545-150 | Used for secondary antibody (2.2.6) |
| Caterer's foil | Any | N/A | Used in steps 1.2.2 and 2.3.2 |
| Fluoromount-G | Southern Biotech | 0100-01 | Used for coverslipping (2.2.8) |
| Coverslips | Fisher Scientific | 12544E | Used for coverslipping (2.2.8) |
| Clear Nail Polish | Any | N/A | Used for coverslipping (2.2.8) |
| Axio Observer.Z1 and LSM 710 (laser scanning, confocal) | Carl Zeiss | N/A | Used for imaging (3) |
| Axioskop A2 | Carl Zeiss | N/A | Used for imaging (3) |
| CitriSolv | FisherScientific | For DAB protocol | |
| ABC | Vector Laboratories | PK-6100 | For DAB protocol |
| DAB Peroxidase kit | Vector Laboratories | SK-4100 | For DAB protocol |
| Biotinylated horse α-rabbit IgG | Vector Laboratories | BA-1100 | For DAB protocol |
| Biotinylated horse α-mouse IgG | Vector Laboratories | BA-2001 | For DAB protocol |
| 30% Hydrogen Peroxide | FisherScientific | H325-500 | For DAB protocol |
| Wheaton slide racks and staining dishes | TedPella | 21043 | For DAB protocol |
| Masterflex perfusion pump and tubing | Cole-Parmer | Used for perfusion (1.1.1 and 1.1.2) | |
| Andwin scientific tissue-tek CRYO-OCT compound (case of 12) | Fisher Scientific | 14-373-65 | Used for tissue freezing (1.2.1) |
| Thermometer (-50 to 50 C) | Fisher Scientific | 15-059-228 | Used for tissue freezing (1.2.1) |
| Cryostat | Leica | CM3500S | Used for tissue sectioning (1.2.2) |
| Staining Dish, Plastic with 2 Lids | Grale Scientific | 353 | For antigen retrival |
| 20 Place Staining Rack, Slides Horizontal | Grale Scientific | 354 | For antigen retrival |
| Microwave | Any | N/A | For antigen retrival |
Referenzen
- Marrack, J. R. Chemistry of antigens and antibodies. Nature. 134, 468-469 (1934).
- Coons, A. H., Creech, H. J., Jones, R. N., Berliner, E. The demonstration of pneumococcal antigen in tissues by the use of fluorescent antibody. J Immunol. 45, 159-170 (1942).
- Marshall, J. M. Localization of adrenocorticotropic hormone by histochemical and immunochemical methods. The Journal of experimental medicine. 94, 21-30 (1951).
- Mellors, R. C. Histochemical demonstration of the in vivo localization of antibodies: antigenic components of the kidney and the pathogenesis of glomerulonephritis. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 3, 284-289 (1955).
- Nakane, P. K., Pierce, G. B. Enzyme-labeled antibodies: preparation and application for the localization of antigens. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 14, 929-931 (1966).
- Avrameas, S., Uriel, J. Method of antigen and antibody labelling with enzymes and its immunodiffusion application. Comptes rendus hebdomadaires des seances de l'Academie des sciences. Serie D: Sciences naturelles. 262, 2543-2545 (1966).
- Cuello, A. C. Immunohistochemistry. , Wiley. (1983).
- Junqueira, L. C. U., Mescher, A. L. Junqueira's basic histology : text and atlas. , Thirteenth edition / edn, (2013).
- Gage, G. J., Kipke, D. R., Shain, W. Whole animal perfusion fixation for rodents. Journal of visualized experiments : JoVE. , (2012).
- Christensen, N. K., Winther, L. Education Guide: Immunohistochemical (IHC) staining methods. Kumar, G. L., Rudbeck, L. , DAKO North America. Carpinteria, California. 103-108 (2009).
- Wang, G., Achim, C. L., Hamilton, R. L., Wiley, C. A., Soontornniyomkij, V. Tyramide signal amplification method in multiple-label immunofluorescence confocal microscopy). Methods. 18, 459-464 (1999).
- Feldengut, S., Del Tredici, K., Braak, H. Paraffin sections of 70-100 mum: a novel technique and its benefits for studying the nervous system. Journal of neuroscience methods. 215, 241-244 (2013).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten