
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Umsetzung einer kohärenten Anti-Stokes-Raman-Streuung (CARS)-System auf einem Ti: Saphir und OPO-Laser-basierte Standard-Laser-Scanning-Mikroskop
In diesem Artikel
Zusammenfassung
Coherent Anti-Stokes-Raman-Streuung (CARS) Mikroskopie auf Basis von Eigenschwingung von Molekül Bindungen erlaubt die markierungsfreie chemisch selektive Live Cell Imaging. Diese Arbeit stellt die Implementierung einer komplementären Mikroskopietechnik auf einem Standard-Multiphotonen-Laserrastermikroskop auf Basis eines Femtosekunden- Ti: Saphir-Laser und einem OPO-Laser.
Zusammenfassung
Laser-Scanning-Mikroskope ein Femtosekunden Ti kombiniert: Saphir-Laser und ein optischer parametrischer Oszillator (OPO), um die Laserlinie zu duplizieren sind für Biologen zur Verfügung stehen. Diese Systeme werden hauptsächlich für Mehrkanal-Zwei-Photonen-Fluoreszenzmikroskopie. Jedoch ohne jegliche Modifikation, komplementäre nichtlineare optische Mikroskopie wie Erzeugung der zweiten Harmonischen (SHG) oder Erzeugung der dritten Harmonischen (THG) kann auch mit diesem Set-up durchgeführt werden, so dass die markierungsfreie Abbildung von strukturierten Molekülen oder wässrigen mittel- Lipid-Grenzflächen. Diese Techniken sind gut geeignet für die in-vivo - Beobachtung, sondern sind in der chemischen Spezifität beschränkt. Chemisch selektive Bildgebung kann aus inhärente Schwingungssignale auf Basis von Raman-Streuung erhalten werden. Die konfokale Raman-Mikroskopie liefert räumliche 3D-Auflösung, aber es erfordert eine hohe mittlere Leistung und lange Aufnahmezeit. Zur Überwindung dieser Schwierigkeiten, die jüngsten Fortschritte in der Lasertechnik die Entwick erlaubtwicklung von nichtlinearen optischen Schwingungsmikroskopie, insbesondere kohärente Anti-Stokes-Raman-Streuung (CARS). daher CARS-Mikroskopie hat sich als ein mächtiges Werkzeug für die biologische und Live Cell Imaging, durch chemisch-Mapping Lipide (über CH-Streckschwingung), Wasser (über OH-Streckschwingungen), Proteine oder DNA. In dieser Arbeit beschreiben wir die Umsetzung der CARS-Technik auf einem Standard-OPO-gekoppelten Multi Laser-Scanning-Mikroskop. Es basiert auf dem in Zeitsynchronisation der beiden Laserlinien, die durch die Länge von einer der Laserstrahlpfad eingestellt wird. Wir stellen eine Schritt-für-Schritt-Implementierung dieser Technik auf einem bestehenden Multisystem. Eine grundlegende Hintergrund in der experimentellen Optik ist hilfreich, und das vorgestellte System keine teuren Zusatzausstattung erforderlich. Wir zeigen auch, CARS Abbilden auf Myelinscheiden der Ischiasnerv von Nagetier erhalten, und wir zeigen, dass diese Abbildungs gleichzeitig mit anderen nichtlinearen optischen Abbildungsdurchgeführt werden kann, wie beispielsweise Standard-two-Photonen-Fluoreszenz-Technik und Erzeugung der zweiten Harmonischen.
Einleitung
Optische Mikroskopie hat in lebenden biologischen Systemen mit subzellulärer Auflösung eine wichtige Technik zur zerstörungsfreien Visualisierung dynamischer Prozesse geworden. Die Fluoreszenzmikroskopie ist derzeit der beliebteste Bildgebungskontrast in lebenden Zellen verwendet aufgrund seiner hohen Spezifität und Sensitivität 1. Eine große Palette von Fluoreszenzsonden entstanden (exogene Farbstoffe, genetisch kodierten Proteine, Halbleiter-Nanopartikel). Verschiedene Probenbeleuchtung Fluoreszenzgestützte Techniken blühten haben (wie konfokaler oder Zweiphotonenmikroskopie) 3D - Bildgebung durchzuführen und einen Hauptnachteil dieser Technik zu verringern , die 2 Photobleaching. Weitere Einschränkungen sind das Erfordernis der Markierungsfluorophor, weil die meisten molekularen Spezies sind nicht eigen fluoreszierende und daher sind diese Fluorophore müssen künstlich in der abgebildeten Probe eingeführt werden. Diese künstliche Manipulation kann störend sein, vor allem für kleine Moleküle oder Topf induziertzielle Phototoxizität. Diese Gründe machen Fluoreszenzmikroskopie nicht gut geeignet für die In-vivo - Beobachtungen. Daher ist die Verwendung von optischen Abbildungstechniken mit hoher Empfindlichkeit und spezifische molekulare Kontraste ohne die Verwendung von Fluoreszenzmolekülen äußerst wünschenswert, in der biomedizinischen Wissenschaft.
Mehrere nichtlineare optische Bildgebungsverfahren ohne Kennzeichnung oder Färbung entstanden, einschließlich Erzeugung der zweiten Harmonischen (SHG) 3,4 und der dritten Harmonischen (THG) 5. SHG - Mikroskopie wurde an Bild strukturelle Anordnungen auf supramolekularer Ebene wie Mikrotubuli oder Kollagen 6 verwendet. THG erzeugt aus optischen Heterogenitäten wie Schnittstelle zwischen einem wässrigen Medium und Lipiden 7. THG wurde auch auf Bild Myelin 8,9 demonstriert. Beide Techniken können auf einem Zwei-Photonen-Fluoreszenzmikroskop und erfordern nur einen Laserstrahl durchgeführt werden. Allerdings erfordern sie hohe Leistung Laserintensität (in der Regel 50mW bei 860 nm für SHG 10, 25 - 50 mW bei 1,180 nm für THG 9), die in lebenden Proben schädlich ist, und zwar um die chemische Spezifität nicht liefern, die eindeutig Bild spezifische biologische Strukturen erforderlich ist.
Chemisch selektive Bildgebung kann von inhärenten molekularen Schwingungssignale auf Basis von Raman-Streuung erhalten werden. Wenn ein Lichtstrahl Materie auftrifft, können Photonen, die von Atomen oder Molekülen absorbiert und gestreut werden. Die meisten der gestreuten Photonen die gleiche Energie haben, das heißt, die Frequenz, wie die einfallenden Photonen. Dieser Vorgang wird als Rayleigh-Streuung genannt. Bezeichnet die Raman - Streuung eine geringe Anzahl von Photonen wird jedoch mit einem unelastischen Streuprozeß unterscheidet sich von der Frequenz der einfallenden Photonen, dh bei einer optischen Frequenz gestreut. Der Unterschied in der Energie stammt aus Anregung von Schwingungsmoden auf molekularen Struktur und Umgebung abhängt. Daher spontane Raman-Streuung provchemisch selektive Iden Bildgebung wie verschiedene Moleküle haben spezifische Schwingungsfrequenzen. Jedoch ist es wegen der extrem schwachen Signals begrenzt. Die konfokale Raman - Mikroskopie wurde entwickelt und bietet räumliche 3D - Auflösung, aber es erfordert eine hohe mittlere Leistung und lange Aufnahmezeit 11. Zur Überwindung dieser Schwierigkeiten, die jüngsten Fortschritte in der Lasertechnologie der Anstieg der nicht - linearen optischen Schwingungs Mikroskopie erlaubt, insbesondere kohärente Anti-Stokes - Raman - Streuung (CARS) 11,12,13.
CARS ist ein dritter Ordnung nichtlinearen optischen Prozess. Drei Laserstrahlen, bestehend aus einem Pumpstrahl bei der Frequenz ω P, ein Stokes - Strahl bei der Frequenz ω S und einen Sondenstrahl (meistens ist die Pumpe) in einer Probe fokussiert und erzeugen eine Anti-Stokes - Strahl bei der Frequenz ω AS = ( 2ω P - ω S) 14. Der Anti-Stokes-Signal kann wesentlich verbessert werden, wenn die Frequenzdifferenzzwischen der Pumpe und der Stokes - Strahlen auf eine Raman molekulare Schwingung Ω R = abgestimmt ist (ω P - ω S). CARS-Signal wird auf mehreren Photon-Wechselwirkung basiert. Es erzeugt daher ein kohärentes Signal Größenordnungen stärker als spontane Raman-Streuung.
CARS - Mikroskopie wurde erstmals experimentell durch Duncan et al. 15. Zumbusch et al. Verbessert dann die Technik, durch Verwendung von zwei fokussierten Nahinfrarot-Femtosekundenlaserstrahlung mit einer Objektivlinse mit hoher numerischer Apertur, so dass die Phasenanpassungsbedingung von PKW und vermeidet die Zwei-Photonen nicht-resonanten Hintergrund 16. Daher CARS - Mikroskopie hat sich als ein mächtiges Werkzeug für die lebenden Zellen und Gewebe - Bildgebung, die durch chemische Moleküle wie Lipide (über CH - Streckschwingung) 17,18, Wasser (über OH - Streckschwingungen) Nachweis, Proteine, DNA in lebenden Zellen 19,20 sondern auch chemische Verbindung deuteriertems für pharmazeutische und kosmetische Anwendungen 21 22.
Die Hauptbeschränkung der nichtlinearen Mikroskopie stammt von der Kompliziertheit und die Kosten der optischen Quellen. Ein CARS System erfordert zwei Wellenlänge durchstimmbare Laser mit kurzen Pulsdauern und mit zeitlich und räumlich synchronisierte Impulsfolgen. Frühe CARS Mikroskope wurden auf Basis von zwei synchronisierten Pikosekunden - Ti: Saphir - Laser 20. Saphir - Laser zum Erzeugen eines Superkontinuum- Lichtquelle 23: CARS Bildgebung wurde ebenfalls aus einem einzigen Femtosekunden Ti erhalten. Vor kurzem zusammengesetzt Laserquellen eines einzelnen Femtosekunden Ti: Saphir-Laser eine abstimmbare optische parametrische Oszillatoren (OPO) Pumpen wurden für die CARS-Mikroskopie verwendet. Dieser Aufbau ermöglicht eigen zeitlich synchronisierten Strahlen mit einer Differenz von Frequenz zwischen der Pumpe und dem Stokes - Strahl die das gesamte molekulare Schwingungsspektrum 24. Darüber hinaus Laser-Scanning-Mikroskope auf der Basis eines Dreh-Schlüssel fs-Laser und ein OPO, in erster Linie für die Zwei-Photonen-Fluoreszenz (TPF) verwendet werden, sind jetzt nicht die Physiker zur Verfügung. Das Potential solcher Aufbauten können erheblich, ohne Ergänzungsanlage durch den Einbau von anderen nichtlinearen optischen Abbildungs verbessert werden, da jede nichtlineare (NLO) Bildgebungsmodalität zu spezifischen Strukturen oder Molekülen empfindlich ist. Multimodale NLO - Bildgebung nutzt daher das Potenzial von NLO - Mikroskopie für komplexe biologische Proben 25. Die Kopplung dieser Techniken hat die Untersuchung vieler biologischer Fragestellungen erlaubt, insbesondere auf Fettstoffwechsels, Haut oder Krebsentwicklung 26, Entwicklung Skelettmuskel 27, atherosklerotische Läsionen 28. Darüber hinaus gibt die Implementierung von Laserstrahlabtastung mit CARS die Fähigkeit , mit hoher Rate Bildgebung, dh ein ansprechendes Werkzeug dynamische Prozesse in vivo zu untersuchen.
Das Ziel dieser Arbeit ist es, jeden Schritt zu zeigen, t zu implementierener CARS Technik auf einem Standard-Multi Laser-Scanning-Mikroskop. Das Mikroskop wird basierend auf einem fsec Ti: Saphir-Laser und ein OPO (vom Ti gepumpt: Laser Saphir) für Biologen durch eine Software betrieben. Die Integration wurde durch Einstellen der Länge von einer der Laserstrahlpfad, um in der Zeit, die zwei Strahlen zu synchronisieren ausgeführt. Wir beschreiben die Schritt-für-Schritt Durchführung dieser Technik, die nur grundlegende Hintergrund in experimentellen Optik erfordert. Wir zeigen auch CARS auf Myelinscheiden von Ischiasnerv von Nagetieren gewonnen Bildgebung, und wir zeigen diese Bildgebung können mit anderen nichtlinearen optischen Bildgebung, wie Standard-Zwei-Photonen-Fluoreszenz-Technik und Erzeugung der zweiten Harmonischen gleichzeitig ausgeführt werden.
Access restricted. Please log in or start a trial to view this content.
Protokoll

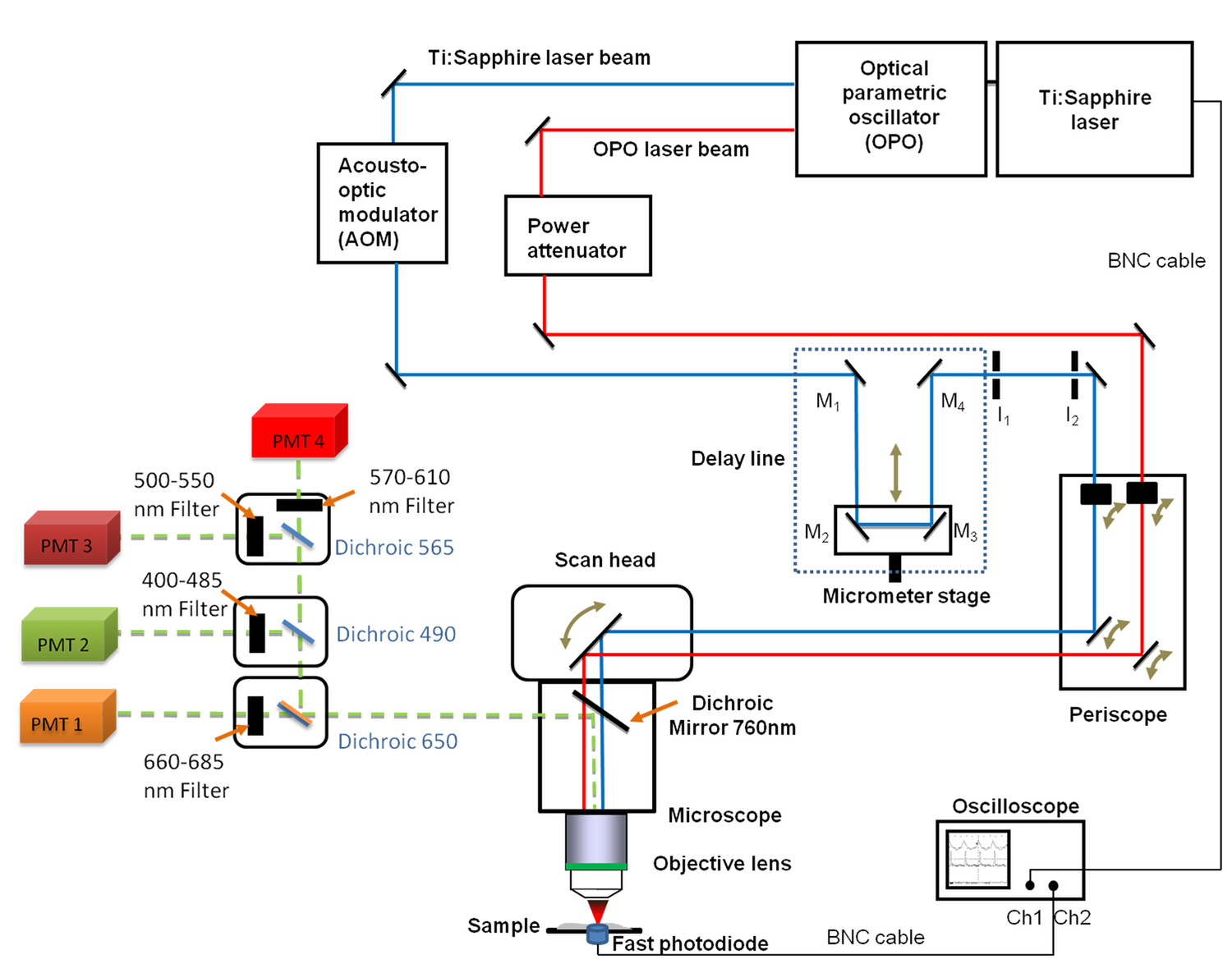
Abbildung 1. Schematische Darstellung des allgemeinen Aufbaus Es umfasst die Ti:. Saphir (680 - 1.080 nm) und die OPO (1050 - 1300 nm) Laser, die Verzögerungsleitung mit den vier Spiegeln (M 1 bis M 4), das schnelle Oszilloskop, die Photodiode und zwei feste Irisblenden I 1 und I 2. Spiegel M 2 und M 3 werden auf einer linearen Translationsstufe fixiert ermöglicht die Verzögerungsleitungslänge mit einer Mikrometer - Auflösung zu ändern. A 660 -. 685 nm Bandpassfilter wurde vor der Photovervielfacherröhre (PMT) für CARS Bildgebung positioniert Bitte klicken Sie hier , um eine größere Version dieser Figur zu sehen.
{kind=link}
1. Inbetriebnahme des Lasersystems
- Stellen Sie sicher, dass der Ti: Saphir-Wellenlänge auf 800 nm eingestellt ist, oder definierendiese Wellenlänge auf dem Ti: Saphir-Netzteil-Controller. Drehen Sie den Schlüssel aus dem Standby auf Ein zum Einschalten des Ti: Saphir-Laser.
- Schalten Sie den OPO-Laser an der Rückseite des OPO-Controller und öffnen Sie den Ti: Saphir-Verschluss auf dem Ti: Saphir-Netzteil-Controller.
- Schalten Sie den Tablet-Computer die OPO zu pumpen. Klicken Sie auf die OPO Verbunden und Remote-Connected-Symbole auf dem Tablett. Warten Sie 30 - 40 min zum Aufwärmen.
- Schalten Sie das Mikroskop Computer und schalten Sie die "Mikroskop-Komponenten" Schalter. Starten Sie die Software durch einen Doppelklick auf das Symbol auf dem Desktop.
- In die Software Acquisition Registerkarte öffnen Sie das Laserwerkzeug im Setup - Manager beider Laser von der Software zu arbeiten. Wählen Ti: Saphir - Laser On und OPO - Laser auf. Überprüfen Sie den Wert der optischen Laserleistung (typische Werte von 3.700 mW bei 800 nm und 700 mW bei 1000 nm).
- Zum Strahlengang und Lasern zu konfigurieren,öffnen Sie das Lichtpfad - Werkzeug im Setup - Manager Werkzeuggruppe und überprüfen Sie die erste Photomultiplier (PMT) boxen.
- Um zu überprüfen , die Ti: Saphir - Laser öffnen Fleck am Ausgang des Ziels, die Kanäle Werkzeug in der Werkzeuggruppe Acquisition Parameter. Wählen Sie den Ti: Saphir - Leistung bei niedrigen Wert (ca. 1%), reduzieren Sie die Verstärkung auf 0 (kein Bild zu diesem Zeitpunkt erforderlich) und klicken Sie auf die kontinuierliche , um den Suchvorgang zu starten , den Laserstrahl durch das Mikroskopobjektiv zu starten. Überprüfen die Anwesenheit eines roten Fleck durch direkte Beobachtung durch Positionierung des IR Laser-Sichtkarte am Ausgang des Luftmikroskopobjektiv (10x).
- Um zu überprüfen , die OPO Laserpunkt, stoppen Sie den Scan des Ti: Saphir - Laser , indem Sie auf die Schaltfläche Stop klicken. Wählen Sie die OPO Kraft bei niedrigen Wert in der Kanäle - Fenster und klicken Sie auf den Continuierliche Taste.
2. Mikroskopeinstellungen
- Stellen Sie manuell die dichroitischen Spiegel mit einem Cut-off-Wellenlänge bei 760 nm in dem Sideport-Regler in der Unendlichkeit Raum oberhalb des Objektivrevolvers das Licht bis zu 760 nm von der Probe in die PMTs in nichtdescannten Detektion (NDD) Modus zu starten.
- Stellen Sie den Schmalbandpassfilter bei 660-685 nm im NDD Reflektorwürfel vor PMT1 nur die CARS bei 670 nm-Signal aufzeichnen, die Ergebnisse in dieser Arbeit vorgestellten zu reproduzieren.
- Legen Sie ein Schmalbandfilter im Bereich von 500 bis 550 nm im NDD Reflektorwürfel vor PMT3 für die Fluoreszenzbeobachtung des Myelin. Legen Sie ein Schmalbandfilter im Bereich von 565 bis 610 nm im Reflektorwürfel vor PMT4 für SHG Beobachtung.
- So wählen Sie in der Software die Aufzeichnung des Signals auf dem Detektor mit dem Ad - hoc - Bandpassfilter, öffnen Sie das Lichtpfad - Werkzeug im Setup - Manager - Menü in der Registerkarte Acquisition. Aktivieren Sie die gewünschte PMT (Kontrollkästchen) und eine Farbe für diesen Kanal auswählen. In dieser Arbeit wurde das Grün für CARS, rot für Fluoreszenz und Magenta für SHG gewählt.
3. zeitliche Synchronisation
Hinweis: Die beiden Laserstrahlen stammen aus demselben Ti: Saphir-Laser, aber der OPO Strahl verzögert wird, wenn es erzeugt wird, um die beiden Strahlen nicht zeitlich synchronisiert werden, wenn sie das Mikroskop erreichen. Das Ziel hier ist eines der beiden Strahlen zu verzögern sie in der Zeit neu zu synchronisieren, bevor sie das Mikroskop erreichen.
- Verbinden Sie sich mit BNC-Kabel den Eingangskanal CH1 des Oszilloskops mit dem elektrischen BNC-Laser-Ausgang (Sync. Out). Schließen Sie den Eingangskanal CH2 des Oszilloskops an die Photodiode und wählen Sie den CH1 Kanal als Triggerkanal durch TRIGGER MENU drücken Sie dann die Hauptmenütaste Quelle und anschließend die Seiten-Menü - Taste, die dem Kanal entspricht ausgewählt CH1.
- Position und fixieren mit optischem Befestigungspfosten die Photodiode in der Brennebene eines Luftmikroskopobjektiv (10x) oder im Strahlengang des Mikroskopes nach dem Ziel zu entfernen. Hinweis: Falls erforderlich, entfernen Sie den Kondensator und dessen Träger.
- In der Kanäle - Werkzeug (Acquisition Parameter Werkzeuggruppe), definieren die Ti: Saphir - Laser - Wellenlänge bei 830 nm bei geringer Leistung (dh weniger als 1% der vollen Leistung). In der Werkzeugerfassungsmodus, reduzieren den Scanbereich auf einen Punkt, um die Photodiode mit dem kleinsten Strahl zu beleuchten. Schalten Sie den Laser - Scan auf dem Continuous - Taste anklicken.
- Drücken Sie AUTOSET auf dem Oszilloskop Frontplatte und manuell die Position der Photodiode bewegen , um die Pulszüge auf dem Bildschirm zu bekommen. Drücken Sie RUN / STOP - Taste , um die Anzeige einzufrieren.
- Um eine Kopie des Oszilloskops Anzeige speichern, legen Sie eine 3,5die GPIB-Anschluss auf der Rückseite des Geräts an einen Computer Zoll-Diskette in das Diskettenlaufwerk oder verbinden. Dann drücken Sie SHIFT HARDCOPY MENU, drücken Sie FORMAT (Haupt-) TIFF - Bildformat zu wählen und im Menü Port den Ausgangskanal angeben. Drücken Sie HARDCOPY Taste , um die Oszilloskop - Darstellung der Impulsfolgen des Ti aufnehmen: Laser Saphir.
- Schalten Sie den Ti: Saphir - Laser - Scan , indem Sie auf die Schaltfläche Stop klicken. Durch Kanäle Werkzeug Anklicken definieren die OPO - Signal bei 1107 nm und geringer Leistung. Schalten Sie die OPO-Laser-Scan und notieren Sie die Pulszüge des OPO-Laser auf dem Oszilloskop. Schalten Sie die OPO-Laser-Scan.
- Vergleichen Sie die zeitliche Verschiebung zwischen dem Ti: Saphir und die OPO-Signale.
HINWEIS: Die zeitliche Verschiebung t Verschiebung gibt die Länge der L Delayline - Verzögerungsleitung , die nach der Gleichung umgesetzt werden muss: L Verzögerungsleitung = c5; t verschieben , wobei c die Lichtgeschwindigkeit ist. - Wählen Sie eines der Laserlinien.
HINWEIS: In dieser Arbeit wird die Ti: Saphir-Laser-Linie wurde gewählt, weil Freiraum in der Nähe von dieser Laserlinie zur Verfügung stand. Darüber hinaus ermöglicht diese Wahl die erneute Ausrichtung der Laserlinie mit sichtbarem Laserlicht zu erreichen. - Öffnen Sie die Laserlinie durch die Schutzrohre an der Stelle entfernt, wo die Verzögerungsleitung umgesetzt werden.
Vorsicht! Tragen Sie geeignete Schutzbrille und entfernen Kette Armbänder oder Uhr aus Handgelenke. - Wählen Sie eine Wellenlänge im sichtbaren Bereich um in der Lage zu sein , einfach um den Laserstrahl zu beobachten (700 nm zum Beispiel bei niedriger Leistung in der Kanäle - Werkzeug der Software). Schalten Sie den Laser-Scan.
- Platzieren und Set mit optischen Montagepfosten zwei Irisblenden entlang der offenen Laserlinie. Positionieren Sie eine Blende am Ausgang der Verzögerungsleitung und legen Sie die anderen Iris am Eingang des Periskop.
HINWEIS: Die periscope Kontrollen durch zwei von der Software den Winkel des Eintritts des Laserstrahls in den Scankopf des Laser-Scanning-Mikroskop gesteuerte Spiegel motorisiert. - Verringern Sie die Irisblendenöffnung und passen Sie die Membran positioniert den Laserstrahl Weg zu passen. Fix sie auf dem optischen Tisch. Stellen Sie die vertikale Position einer dritten mobilen Irisblende, die Höhe des Laserstrahls zu überprüfen, während nacheinander die vier Spiegel der Verzögerungsleitung zu positionieren.
HINWEIS: Diese Irisblenden als Steuerung für die Wiederausrichtungsverfahren durch mit dem Pfad zu folgen dienen wird. - Platzieren der Spiegel M1 auf einem kompakten kinematischen Spiegel am Eingang der Verzögerungsleitung Halterung befestigt (wie in 1 gezeigt) und stellen seine Position und seine Ausrichtung der Strahlhöhe bei der Verwendung des mobilen Irisblende zu halten. Platz Spiegel M2 und M3 (auch auf kompakten kinematischen Spiegelhalterungen montiert) bei 90 ° auf die Translationsstufe, die bei Midco positioniert werdenurse. Stellen Sie sie die Verzögerungsleitungslänge zu passen, wie zuvor berechnet.
- Stellen Sie die Orientierung von M2 und M3 mit der Verwendung des mobilen Irisblende. Set M4 (auch auf einem kompakten Halterung befestigt) am Ausgang der Verzögerungsleitung (kurz vor der Iris I 1 , wie in 1 gezeigt) und sorgfältig einzustellen seine Position und den Winkel des Laserstrahls Pfad durch die zwei feststehenden Irisblenden zu passen.
- Positionieren Sie den Laser - Sichtkarte am Ausgang des Mikroskopobjektivs und überprüfen Sie das Laserstrahlprofil , indem Sie auf Continuous auf der Laser - Scan einzuschalten. Beachten Sie eine einheitliche helle Scheibe. Bei Bedarf leicht die Orientierung der M4 einzustellen.
- Position wieder die schnelle Photodiode unter dem Laserstrahl in der Probe Fokusebene des Mikroskops. Beachten Sie die zeitliche Verschiebung zwischen dem Ti: Saphir-Laserstrahl und den OPO Strahl auf dem Oszilloskop.
Hinweis: Falls erforderlich, um die Länge Verzögerungsleitung ändern, indem das gesamte System zu bewegen M2, M3 montiert auf derÜbersetzung Stufe (ohne Übersetzung Bühne Einstellung zu ändern) beide Impulse zu synchronisieren. Änderungen von wenigen Zentimetern kann erforderlich sein.
4. räumliche Überlappung der Strahlen
Hinweis: Um eine CARS-Signal, die räumliche Überlappung der beiden Laserstrahlen erzeugen erforderlich. Die alternative Beleuchtung beider Strahlen auf den gleichen Perlen mit zwei unterschiedlichen Fluoreszenzfarbstoffen gefärbt überall können die räumliche Verschiebung, um anzuzeigen, verwendet werden. Die Feineinstellung der Spiegelpositionen können dann die Verschiebung zu minimieren.
- Verwenden vormontiert fluoreszierende Mikrokugeln. Oder montieren Mikrokügelchen in Suspension auf sauberen Mikroskop-Objektträger, wie unten beschrieben:
- Vor Probenahme-Mix (auf einem Cortex-Mischer oder durch Beschallen) die Perlen Lösung sicher sein, dass die Kügelchen gleichmäßig suspendiert sind.
- Bewerben 5 ul der Beadsuspension auf die Oberfläche einer Folie und verbreiten mit der Pipettenspitze. Warten, bis die Tröpfchen trocknen und dann anwenden 5 ul Halterunging Medium, wie Glycerin, Wasser oder Sionsöl über die trockene Probe von Kügelchen. Decken Sie die Probe mit einem Deckglas und Dichtung das Deckglas mit schnell trocknenden Klebstoff oder geschmolzenem Paraffin.
- Legen Sie die fluoreszierenden Polystyrolkügelchen fixiert auf einem Objektträger unter dem 20X Wasser Ziel. Fügen Sie einige Tropfen Wasser, um das Ziel einzutauchen.
- Den Fokus auf die Perlen zu erreichen, die Reiter in der Software von der Laser - Scanning - Modus auf die direkte Beobachtung der Probe mit dem Auge zu wechseln Suchen öffnen, indem Sie die Online - Taste drücken. Öffnen Sie das Ocular Werkzeug , um die Ad - hoc - Filter und schalten Sie die Halogenlampe zu wählen , indem Sie auf die Symbole klicken.
- Entfernen Sie manuell in den Sideport Schieber im Unendlichen Raum und verwenden Sie den Fokustrieb des Mikroskops den dichroitischen Spiegel der Probe Ebene zu fokussieren, indem Sie die Perlen mit den Okularen zu beobachten. Ersetzen Sie den dichroitischen Spiegel.
- In demSuchen Sie Registerkarte, wechseln Sie in den Laser - Scanning - Modus , indem Sie die Taste Offline - Taste. Gehen Sie auf die Registerkarte Acquisition die Parameter für das Scannen zu definieren: Wählen Sie die Bildgröße auf 512 Pixel, eine Scangeschwindigkeit von 9, eine Mittelung von 1, die eine Bittiefe von 8 Bit und erhöhen den Scanbereich auf das Maximum.
- Im Kanäle Werkzeug der Akquisition Registerkarte, fügen Sie einen Track (Spur 1) , wenn nicht bereits erstellt. Wählen Sie die Wellenlänge bei 830 nm und niedriger Leistung für den Ti:. Saphir Laserstrahl Kreuzen Sie die Farbe Grün in der Spur 1 Box aus der Kanäle - Fenster und in der PMT3 oder der PMT4 Box aus dem Lichtweg - Fenster.
- Im Kanäle Werkzeug der Akquisition Registerkarte, fügen Sie eine zweite Spur (Track 2). Wählen Sie die Wellenlänge bei 1107 nm und niedriger Leistung für den OPO Laserstrahl. Markieren Sie die Farbe Rot in der Spur 2 Box aus der Kanäle - Fenster und in der PMT3 Box aus der Light Path - Fenster.
- Stellen Sie die Verstärkung der beiden Spuren bis 600. Dann nacheinander anzuwenden den Scan der beiden Strahlen auf die Probe auf Continuous durch einen Klick.
- Beobachten Sie das Bild im Bildbereich in der 2D-Ansicht. In der Anzeigeansicht Option Steuerblock, stellen Sie die Helligkeit der Anzeige.
Hinweis: Falls erforderlich, bewegen etwas Fokussiertrieb die Fokusebene der Perlen zu finden. Stellen Sie die Ernte und zoomen Sie das Bild in einem einzelnen Kügelchen oder in einer Gruppe von benachbarten Perlen. - Verwenden Sie das Periskop-Controller die Strahlen in xy-Ebene zu überlappen. In der Software, öffnen Sie die Registerkarte pflegen. Klicken Sie auf die Systemoptionen und zeigt das Motorisierte Periscope - Tool - Fenster. Verwenden Sie Grob- und Feineinstellungen der Periskop Spiegel des Ti: Saphir-Laserstrahl, um im Raum beide Bilder zu synchronisieren.
- Für die Periskop Manipulation, verwenden Sie die erste Einstellleisten für vertikale und dem second eine für horizontale Bewegungen des Laserstrahls. Bewegen des Strahls mit dem Eingangsspiegel, bis das Bild leicht sichtbar ist, und dann für die Laserintensität mit dem Ausgangsspiegel des Periskops kompensieren durch Klicken auf "Eingang" und "Ausgang".
- Um vertikal die Strahlen überlappen, in der Pflege Registerkarte öffnen Sie das Collimator Werkzeug und stellen Sie den Wert der Brennweite des Ti: Saphir - Laserstrahl.
- Bewegen Sie das Ziel, vertikale Position, um die Differenz der Fokus auf beiden Bildern zu überprüfen. Oder nehmen Sie einen Z-Stapel der Probe durch Öffnung in der Akquisition auf das Register Z-Stapel-Tool und wählen Sie die verschiedenen Parameter (Bereich, die Anzahl der Scheiben). Drücken Sie Ortho in der Bildschirmbereich, um die Strahlen in axialer Querschnitt zu sehen. Maximieren Sie die z-Überlappung durch die gleiche Prozedur mehrmals zu tun.
5. Schluss Anpassungen und Coherent Anti-Stokes-Raman-Streuung (CARS) Signal Beobachtung von Olivenöl Droplets
- Setzen Sie einen Tropfen Olivenöl auf einer Glasplatte und decken Sie es mit einem Deckglas. Fügen Sie einige Tropfen Wasser ein 20X Wassertauch Ziel einzutauchen. Konzentrieren Sie am Rand des Deckglases durch die Okulare mit (wie zuvor erläutert in 4.2).
- In der Kanäle - Werkzeug der Registerkarte Acquisition, wählen Sie im Track 1 die Wellenlänge bei 830 nm für den Ti: Saphir - Laserstrahl und bei 1107 nm für die OPO. Markieren Sie beide Laser in Spur 1 eine simultane Abtastung beider Laser zu bekommen. Set Kräfte bei niedrigen Wert für einen Start.
- Im Light Path - Fenster wählen Sie PMT1. Schalten Sie den Laserscans durch auf dem Continuous - Schaltfläche klicken. Bewegen Sie leicht den Fokus des Laserlichts in die Öl dünne Schicht zu liefern.
- Falls erforderlich, erhöhen die optische Leistung beider Laser. Stellen Sie die Helligkeit der Anzeige im Display-Ansicht Option Steuerblock. Langsam bewegen Übersetzung Stufe der Verzögerungsleitung, bis das Signal sig wirdsentlich verbessert.
- Nachdem die feinen Ausrichtungen abgeschlossen sind, prüfen Sie, ob es wirklich ein CARS-Signal ist: Bewegen Sie die Übersetzung der Bühne leicht; die Intensität des Signals muß schwächer. Und / oder schalten einen der Laserstrahl aus, entweder Ti: Saphir-Laser oder OPO. Auch hier muss es mit dem CARS-Signal im Vergleich eine starke Verfall der Intensität sein.
- Um das zu erreichen Signal die maximale CARS, wählen Sie die Option auf der Software einen Wert der mittleren Intensität des gesamten Bildes zur Verfügung zu stellen (in der Histo Ansicht des Bildschirmbereichs Registerkarte). Stellen Sie die Wellenlänge (einige nm), dann gibt die x, y, z-Positionen des Fokusstrahls zu maximieren den Intensitätswert bedeuten.
6. Gehäuse des Lichtweges der Verzögerungsleitung
- Da das endgültige System für Nicht-Physiker gewidmet ist, schließen Sie den Lichtweg der Verzögerungsleitung mit Rohren oder einem Gehäuse Box, direkten Zugang zu schädlichen nicht sichtbare hohe Spitzenleistung Laserstrahl zu vermeiden. Achten Sie darauf, einen Zugriff auf die Übersetzungsstufe zu liefernKnopf.
7. Wavelength Tuning für CARS
- Verwenden Sie die Gleichung
 die Laserwellenlängen auf die gewünschte Raman Schwingung abzustimmen. Um die Ergebnisse zu reproduzieren in dieser Arbeit Bild CARS präsentiert Signal von CH - Bindungen mit Streckschwingung von 3015 cm -1, wählen λ Ti: Saphir = 830 nm und λ OPO = 1095 nm.
die Laserwellenlängen auf die gewünschte Raman Schwingung abzustimmen. Um die Ergebnisse zu reproduzieren in dieser Arbeit Bild CARS präsentiert Signal von CH - Bindungen mit Streckschwingung von 3015 cm -1, wählen λ Ti: Saphir = 830 nm und λ OPO = 1095 nm.
HINWEIS: Raman charakteristischen Schwingungsfrequenzen in biologischen Proben beobachtet, wie Wasser, CH Bindung kann in Evans et al gefunden werden 13 oder in Ellis et al 29... - Verwenden Sie die Gleichung
 die Emissionswellenlänge von CARS-Signal zu bestimmen. Für CH - Bindung Bildgebung von Autos, ein Schmalbandfilter bei 670 nm , da λ CARS wählen = 670 nm mit Laserwellenlängen in 7.1 dargestellt.
die Emissionswellenlänge von CARS-Signal zu bestimmen. Für CH - Bindung Bildgebung von Autos, ein Schmalbandfilter bei 670 nm , da λ CARS wählen = 670 nm mit Laserwellenlängen in 7.1 dargestellt.
ANMERKUNG: Eine Handy-Anwendung ist available λ CARS zu berechnen aus λ P und λ S - Werte (siehe Referenz 30).
8. Die Beobachtung der CARS-Signal und Stained Myelin von Ischiasnerv Cuts
Hinweis: Alle Tierversuche wurden in Übereinstimmung mit institutionellen Regelungen durchgeführt.
- Bereiten Sie die Axial- und Längsschnitte Ischiasnervs auf einem Objektträger wie in Özçelik präsentiert et al. 31.
- Bereiten Sie die fluoromyelin rote Farblösung durch die Stammlösung 300-fach in PBS verdünnt. Fluten die Nerven Schnitte mit der Färbungslösung für 20 min bei RT. Entfernen Sie die Lösung und waschen 3 Mal für 10 min mit PBS.
- Positionieren Sie die Schnitte unter dem 20X Wassertauchziel. Legen Sie ein Deckglas. Fügen Sie einige Tropfen PBS um das Ziel zu tauchen und stellen Sie den Fokus des Objektivs, ein klares Bild der Schnitte durch die Okulare zu erhalten (wie zuvor in 4.2 beschrieben).
- Im Track 1, wählen Sie den Ti: Saphir und die OPO - Laser und definieren ihre Wellenlängen 830 nm und 1095 nm. Im Light Path - Fenster wählen Sie PMT1 und grüne Farbe.
- In Track 2, wählen Sie die OPO - Laser nur (Wellenlänge bei 1095 nm). Im Light Path - Fenster wählen Sie PMT4 und rote Farbe.
- Für beide Laser, wählen Sie Low-Power und die Verstärkung zu 600 für einen Start gesetzt. Schalten Sie den Laserscans und stellen Sie die folgenden Parameter zu verbessern CARS und Fluoreszenz-Signal Kontrast: Leistungswerte, Übersetzung Stufe Knopf (sehr leicht), Wellenlängen (wenige nm), Display-Intensität.
- Um die endgültigen Bilder mit hoher Auflösung aufzunehmen, wählen Sie in der Werkzeugerfassungsmodus die folgenden Parameter: Bildgröße von 1024 Pixel, Scangeschwindigkeit von 7, Mittelung von 4. Klicken Sie auf die Schaltfläche Schnapp für ein einzelnes Bild aufzunehmen. Speichern Sie das Bild in der geschützten Formauf das Bild und die volle Aufnahmeparameter aufzuzeichnen.
9. Beobachtung von CARS und SHG Signale von Ischiasnerv Cuts
- Bereiten Sie den Ischiasnerv wie in Özçelik präsentiert et al. 31.
- Folgen Sie dem Verfahren als 8 teilweise erklärt, ein Bild durch die Okulare zu bekommen und CARS-Signal Parameter zu wählen (Track 1).
- In Track 2, wählen Sie die OPO - Laser nur (Wellenlänge bei 1095 nm). Im Light Path - Fenster wählen Sie PMT3 und Farbe Magenta.
- Folgen Sie dem Verfahren, wie erklärt in Teil 8 auf den Laserscans zu wechseln und Bilder mit hoher Auflösung zu speichern.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Die Pulsfolgefrequenz von Standard-Ti: Saphir-Laser ist in der Regel etwa 80 MHz. Der OPO hat die gleiche Frequenz, da es durch die Ti gepumpt wird: Saphir-Laser. Ein schnelles Oszilloskop von mindestens 200 MHz ist daher erforderlich. Eine schnelle Photodiode im Bereich von 600 bis 1.100 nm ist ebenfalls erforderlich. Die maximale zeitliche Verschiebung tritt auf, wenn die Ti: Saphir und die OPO - Signale von 1 / (2 × 80 × 10 6) = 6,2 ns verschoben sind. Es entspricht...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Der schwierigste Teil der Arbeit ist die zeitliche Synchronisation der Laserstrahlen. Es erfordert eine schnelle Photodiode mit einem schnellen Oszilloskop kombiniert, aber nur eine grobe zeitlich überlappend auf den ersten durchgeführt werden. Dann wird eine weitere Einstellung von wenigen cm erforderlich. Schließlich Mikrometer bewegt sich durch eine lineare Translationsstufe ermöglicht die Durchführung der endgültigen Feineinstellung der Verzögerungsleitungslänge, um die CARS-Signal auszulösen. Dieses Signal...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
The authors declare that they have no competing financial interests.
Danksagungen
The authors want to thank Dr. Philippe Combette (IES, UM, Montpellier, France) for the loan of the fast oscilloscope and acknowledge financial supports from Montpellier RIO Imaging (MRI). HR acknowledges ANR grants France Bio Imaging (ANR-10-INSB-04-01) and France Life Imaging (ANR-11-INSB-0006) infrastructure networks for coherent Raman imaging developments. This work was mainly supported by an European Research Council grant (FP7-IDEAS-ERC 311610) and an INSERM - AVENIR grant to NT.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| Oscilloscope | Tektronix | TDS 520D | 500 MHz |
| Photodetector | Thorlabs | DET08C/M, T4290 | 5 GHz InGaAs, 800 - 1,700 nm |
| Ti:Sapphire laser Chameleon Ultra Family II | Coherent | ||
| Optical parametric oscillator OPO Compact Family | APE Berlin | ||
| Axio Examiner microscope LSM 7 MP | Carl Zeiss | ||
| Motorized periscope | Newport | ||
| Objective W Plan-Apochromat 20X/1.0 | Carl Zeiss | ||
| Beam combiner | Carl Zeiss | ||
| Acousto-optic modulator | Carl Zeiss | ||
| OPO power attenuator | Carl Zeiss | ||
| Photomultiplier tube | Carl Zeiss | ||
| ZEN software | Carl Zeiss | ||
| Bandpass filters | Carl Zeiss | LSM BiG 1935-176 | 400 - 480 nm; 500 - 550 nm; 465 - 610 nm |
| Dichroic mirror | Carl Zeiss | Cutoff wavelength 760 nm | |
| Silver mirrors | Newport | 10D20ER.2 | λ/10, 480 - 20,000 nm, Quantity 4 |
| Single-axis translation stage with standard micrometer | Thorlabs | PT1/M | Quantity 1 |
| Aluminium breadboard | Thorlabs | MB1015/M | Quantity 1 |
| Mirror mount | Thorlabs | KMSS/M | Quantity 4 |
| Mirror holder for Ø1" Optics | Thorlabs | MH25 | Quantity 4 |
| Iris diaphragms | Thorlabs | ID8/M | Quantity 3 |
| Protective box | Thorlabs | TB4, XE25L900/M, T205-1.0, RM1S | Quantity 1 |
| Optical posts | Thorlabs | TR40/M, PH50/M, PH75/M, BA2/M | Quantity 8 (lengths depending on the set-up) |
| 661 - 690 nm bandpass filter | Semrock | 676/29 nm BrightLine® single-band bandpass filter | Quantity 1 |
| Fluorescent beads | ThermoFisher | TetraSpeck™ Fluorescent Microspheres Size Kit | |
| Laser viewing card | Thorlabs | IR laser viewing card | |
| Laser safety glass | Newport | LV-F22.P5L07 | |
| FluoroMyelin™ Red Fluorescent Myelin Stain | ThermoFisher | F34652 |
Referenzen
- Valeur, B., Berberan-Santos, M. N. Molecular Fluorescence: Principles and Applications. , 2nd Edition, Wiley-VCH Verlag GmbH. (2012).
- Denk, W., Strickler, J. H., Webb, W. W. Two-photon laser scanning fluorescence microscopy. Science. 248 (4951), 73-76 (1990).
- Moreaux, L., Sandre, O., Mertz, J. Membrane imaging by second-harmonic generation microscopy. JOSA B. 17 (10), 1685-1694 (2000).
- Zoumi, A., Yeh, A., Tromberg, B. J. Imaging cells and extracellular matrix in vivo by using second-harmonic generation and two-photon excited fluorescence. Proc. Natl. Acad. Sci. USA. 99 (17), 11014-11019 (2002).
- Yelin, D., Silberberg, Y. Laser scanning third-harmonic-generation microscopy in biology. Opt. Express. 5 (8), 169-175 (1999).
- Campagnola, P. J., Millard, A. C., Terasaki, M., Hoppe, P. E., Malone, C. J., Mohler, W. A. Three-dimensional high-resolution Second-Harmonic Generation imaging of endogenous structural proteins in biological tissues. Biophys. J. 81 (1), 493-508 (2002).
- Olivier, N., et al. Cell lineage reconstruction of early zebrafish embryos using label-free nonlinear microscopy. Science. 329 (5994), 967-971 (2010).
- Farrar, M. J., Wise, F. W., Fetcho, J. R., Schaffer, C. B. In vivo imaging of myelin in the vertebrate central nervous system using third harmonic generation microscopy. Biophys. J. 100 (5), 1362-1371 (2011).
- Lim, H., Sharoukhov, D., Kassim, L., Zhang, Y., Salzer, J. L., Melendez-Vasquez, C. V. Label-free imaging of Schwann cell myelination by third harmonic generation microscopy. Proc. Natl. Acad. Sci. U.S.A. 111 (50), 18025-18030 (2014).
- Strupler, M., Pena, A. M., Hernest, M., Tharaux, P. L., Martin, J. L., Beaurepaire, E., Schanne-Klein, M. C. Second harmonic imaging and scoring of collagen in fibrotic tissues. Opt. Express. 15 (7), 4054-4065 (2007).
- Cheng, J. X., Xie, X. S. Coherent anti-Stokes Raman scattering microscopy: Instrumentation, theory, and applications. J. Phys. Chem. B. 108 (3), 827-840 (2004).
- Volkmer, A. Vibrational imaging and microspectroscopies based on coherent anti-Stokes scattering microscopy. J. Phys. D: Appl. Phys. 38, R59-R81 (2005).
- Evans, C. L., Xie, X. S. Coherent anti-Stokes Raman scattering microscopy: chemical imaging for biology and medicine. Annu. Rev. Anal. Chem. 1, 883-909 (2008).
- Mukamel, S. Principles of nonlinear optical spectroscopy. , Oxford University Press. New York. (1995).
- Duncan, M. D., Reintjes, J., Manuccia, T. J. Scanning coherent anti-Stokes Raman microscope. Opt. Lett. 7 (8), 350-352 (1982).
- Zumbusch, A., Holtom, G. R., Xie, X. S. Three-dimensional vibrational imaging by coherent anti-Stokes Raman scattering. Phys. Rev. Lett. 82 (20), 4142-4145 (1999).
- Folick, A., Min, W., Wang, M. C. Label-free imaging of lipid dynamics using Coherent Anti-Stokes Raman Scattering (CARS) and Stimulated Raman Scattering (SRS) microscopy. Curr. Opin. Genet. Dev. 21 (5), 585-590 (2011).
- Wang, P., Liu, B., Zhang, D., Belew, M. Y., Tissenbaum, H. A., Cheng, J. X. Imaging lipid metabolism in live Caenorhabditis elegans using fingerprint vibrations. Angew. Chem. Int. Ed. Engl. 53 (44), 11787-11792 (2014).
- Min, W., Freudiger, C. W., Lu, S., Xie, X. S. Coherent nonlinear optical imaging: beyond fluorescence microscopy. Annu. Rev. Phys. Chem. 62, 507-530 (2011).
- Cheng, J. X., Jia, Y. K., Zheng, G., Xie, X. S. Laser-scanning coherent anti-Stokes Raman scattering microscopy and applications to cell biology. Biophys J. 83 (1), 502-509 (2002).
- Chiu, W. S., Belsey, N. A. N., Garrett, L., Moger, J., Delgado-Charro, M. B., Guy, R. H. Molecular diffusion in the human nail measured by stimulated Raman scattering microscopy. Proc Natl. Acad. Sci. U.S.A. 112, 7725-7730 (2015).
- Chen, X., Grégoire, S., Formanek, F., Galey, J. -B., Rigneault, H. Quantitative 3D molecular cutaneous absorption in human skin using label free nonlinear microscopy. J. of Control. Release. 200, 78-86 (2015).
- Kano, H., Hamaguchi, H. In vivo multi-nonlinear optical imaging of a living cell using a supercontinuum light source generated from a photonic crystal fiber. Opt. Express. 14 (7), 2798-2804 (2006).
- Brustlein, S., Ferrand, P., Walther, N., Brasselet, S., Billaudeau, C., Marguet, D., Rigneault, H. Optical parametric oscillator-based light source for coherent Raman scattering microscopy: practical overview. J. Biomed. Opt. 16 (2), 021106(2011).
- Chen, H., et al. A multimodal platform for nonlinear optical microscopy and microspectroscopy. Opt. Express. 17 (3), 1282-1290 (2009).
- Yue, S., Slipchenko, M. N., Cheng, J. X. Multimodal nonlinear optical microscopy. Laser Photonics Rev. 5 (4), 496-512 (2011).
- Sun, Q., Li, Y., He, S., Situ, C., Wu, Z., Qu, J. Y. Label-free multimodal nonlinear optical microscopy reveals fundamental insights of skeletal muscle development. Biomed Opt Express. 5 (1), 158-166 (2013).
- Le, T. T., Langohr, I. M., Locker, M. J., Sturek, M., Cheng, J. X. Label-free molecular imaging of atherosclerotic lesions using multimodal nonlinear optical microscopy. J. Biomed. Opt. 12 (5), 054007(2007).
- Ellis, D. I., Cowcher, D. P., Ashton, L., O'Hagana, S., Goodacre, R. Illuminating disease and enlightening biomedicine: Raman spectroscopy as a diagnostic tool. Analyst. 138, 3871-3884 (2013).
- A•P•E Angewandte Physik & Elektronik GmbH. , Germany. Available from: http://www.ape-berlin.de/en/page/calculator (2015).
- Ozçelik, M., et al. Pals1 is a major regulator of the epithelial-like polarization and the extension of the myelin sheath in peripheral nerves. J Neurosci. 30 (11), 4120-4131 (2010).
- Heinrich, C., Hofer, A., Ritsch, A., Ciardi, C., Bernet, S., Ritsch-Marte, M. Selective imaging of saturated and unsaturated lipids by wide-field CARS-microscopy. Opt. Express. 16 (4), 2699-2708 (2008).
- Kyriakidis, N. B., Skarkalis, P. Fluorescence spectra measurement of olive oil and other vegetable oils. J. AOAC Int. 83 (6), 1435-1439 (2000).
- King, R. Microscopic anatomy: normal structure. Handb. Clin. Neurol. 115, 7-27 (2013).
- Monsma, P. C., Brown, A. FluoroMyelin Red is a bright, photostable and non-toxic fluorescent stain for live imaging of myelin. J. Neurosci. Methods. 209 (2), 344-350 (2012).
- Wang, H., Fu, Y., Zickmund, P., Shi, R., Cheng, J. X. Coherent anti-stokes Raman scattering imaging of axonal myelin in live spinal tissues. Biophys. J. 89 (1), 581-591 (2005).
- Wang, H. W., Fu, Y., Huff, T. B., Le, T. T., Wang, H., Cheng, J. X. Chasing lipids in health and diseases by coherent anti-Stokes Raman scattering microscopy. Vib. Spectrosc. 50 (1), 160-167 (2009).
- Jung, Y., Tam, J., Jalian, H. R., Anderson, R. R., Evans, C. L. Longitudinal, 3D in vivo imaging of sebaceous glands by coherent anti-stokes Raman scattering microscopy: normal function and response to cryotherapy. J. Invest. Dermatol. 135 (1), 39-44 (2015).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten