
Method Article
Hochauflösende Volume Imaging von Neuronen durch den Einsatz von Fluoreszenz-Ausschluss Methode und mikrofluidischen Geräten gewidmet
In diesem Artikel
Zusammenfassung
Volumen ist ein wichtiger Parameter zur physiologischen und pathologischen Eigenschaften der Zellen. Wir beschreiben eine fluoreszierende Ausschluss-Methode, flächenhafte Messung von in-vitro- neuronale Volumen mit Sub mikrometrische axiale Auflösung benötigt für die Analyse von Neuriten und dynamische Strukturen in neuronale Wachstum impliziert.
Zusammenfassung
Volumen ist ein wichtiger Parameter zur physiologischen und pathologischen Eigenschaften der Neuronen auf unterschiedlichen Zeitskalen. Neuronen sind ziemlich einzigartig Zellen über ihre erweiterte verzweigten Morphologien und erhöhen folglich mehrere methodische Herausforderungen für Volumen-Messung. Im besonderen Fall von in-vitro- neuronale Wachstum sollte die gewählte Methodik Sub mikrometrische axiale Auflösung, kombiniert mit Vollfeld Beobachtung auf Zeitskalen von Minuten, Stunden oder Tage umfassen. Im Gegensatz zu anderen Methoden wie Zelle Form Wiederaufbau mit konfokale Bildgebung, elektrisch-basierten Messungen oder Atomic Force Microscopy hat die neu entwickelte Fluoreszenz-Ausschluss-Methode (FXm) das Potenzial, diese Herausforderungen zu erfüllen. Obwohl es einfach in seinem Prinzip, erfordert jedoch Umsetzung einer hochauflösenden FXm für Neuronen, mehrere Anpassungen und eine spezielle Methodik. Wir stellen Ihnen hier eine Methode basiert auf der Kombination von Fluoreszenz Ausgrenzung, niedrig-Rauhigkeit Multi-Fächer mikrofluidischen Geräten und schließlich Micropatterning, in-vitro- Messung der lokalen neuronalen Volumen zu erreichen. Die hohe Auflösung zur Verfügung gestellt vom Gerät konnten wir messen das lokale Laufwerk von neuronalen Prozessen (Neuriten) und die Lautstärke etwas spezifischer Strukturen neuronale Wachstum, wie Wachstum Kegel (GCs) beteiligt.
Einleitung
Die genaue Kenntnis der zellulären Volumen hat erhöhte Aufmerksamkeit in den letzten Jahren angezogen, angetrieben von der Frage der Zelle Größe Homöostase in einzelnen-celled Mikroorganismen 1 und ganz allgemein in der mitotischen Zellen 2. Allerdings ist die Frage des Zellvolumens relevant auch für Post-mitotischen Zellen, für die Neuronen ein Paradebeispiel dar.
Volumen ist in der Tat eine wichtige Unterschrift der physiologischen und pathologischen Veranstaltungen auf verschiedenen Skalen und Zeitpunkten im neuronalen Leben von transienten axonalen Verformung elektrische Aktivität (Millisekunden Skala) 3 , die irreversible zugeordnet neuronale Schwellung auftreten während der asymptomatischen Phase der neurodegenerativen Erkrankungen (über Jahre beim Menschen) 4. Allerdings tritt die größte Volumenänderung in Zwischenzeit Maßstab von Tagen oder Wochen (abhängig von der betrachteten Organismus) während des neuronalen Wachstums. Die ausgedehnte und komplexe Morphologie der Neuronen wirft Fragen auf ein Vielfaches, unter denen die Regulierung der Zellengröße. Axonalen Länge und Durchmesser sind in der Tat streng regulierten in Vivo, mit Werten spezifisch für jedes neuronaler Typ 5,6.
Diese Fragen, komplex, um in Vivo, Adresse können auch in vereinfachter Form angesprochen werden in-vitro-. In diesem Ziel eine Methode gewidmet Volumenmessung schnell genug Wachstumsdynamik (d. h. in einer Zeitskala von Minuten) und kompatibel mit Beobachtung über Stunden oder Tage zu folgen ist erforderlich. Verschiedene Methoden wurden im Laufe der Jahre zu einer direkten oder indirekten Zugriff auf zelluläre Volumen in Vitroentwickelt. Zelle Rekonstruktion von confocal Imaging ist einer von ihnen, aber diese Methode impliziert Etikettierung und wiederholten Expositionen gegenüber Licht und zeigt eine begrenzte axiale Auflösung von ca. 500 nm 7. Beachten Sie, dass diese zwei letzten Nachteile durch eine anspruchsvolle und aktuelle Methode namens Gitter Licht-Blatt Mikroskopie 8teilweise überwunden werden. Atomic Force Microscopy hat gebrauchte 9 aber diese Scan-Methode ist durch Wesen langsam und mühsam. Darüber hinaus kann der Körperkontakt, die, den es mit der Zelle erfordert, die Messung, wenn man bedenkt die extreme Weichheit der Neuronen 10beeinträchtigen. Indirekte Methode mittels Impedanz oder Resonanz haben für verschiedene Zelltypen 11, aber sind nicht ausreichend für erweitert anhaftende Zellen wie Neuronen eingesetzt.
Eine der vielversprechendsten Methoden basiert auf das Maß der ausgeschlossenen Volumen von Zellen in einer engen Kammer gefüllt mit einem Fluoreszenzfarbstoff. Die Fluoreszenz-Ausschluss-Methode (FXm) ist einfach in seinem Prinzip, da es erfordert keine Kennzeichnung und eignet sich für schnelle, langfristige optische Bildgebung der Zell-Populationen mit einer potenziell Sub optische axiale Auflösung. Genauer gesagt, hängt die Auflösung in z die maximale Fluoreszenzintensität in der Kultur-Kammer (d. h. in Region frei von Zellen) geteilt durch den Dynamikbereich der Kamera, obwohl mehrere Schallquellen dieser ultimative Auflösung begrenzen. Diese Methode ist sehr mächtig, das Volumen der Migration von adhärenten Zellen 12 folgen oder Volumenänderung während der Mitose von Säugerzellen, wie gründlich in 13beschrieben zu studieren gewesen. Neuronen bilden jedoch eine methodische Herausforderung für FXm unter Berücksichtigung ihrer umfangreichen Verzweigung in Sub mikrometrische Prozesse.
Wir stellen Ihnen hier eine Methode führt zur Herstellung von glatten FXm Kammern zugänglich mit hoher Präzision die Lautstärke und Höhe des neuronalen Verzweigungen und dynamische Strukturen neuronale Wachstum wie Wachstum Kegel beteiligt.
Kammern müssen ähnliche Höhen als das Objekt zu messen, um axiale Auflösung optimieren. Daher haben wir verschiedene FXm-Geräte zeichnen sich durch zentrale Messung Kammern mit drei verschiedenen Höhen entwickelt. Die dünnste (3 µm in der Höhe) widmet sich der Neurit Messung: dieser niedrigen Höhe schließt Soma, die bleiben in der in der Nähe von 15 µm hohen Zwischenkammer. Dickere zentrale Kammern (10 und 12 µm) sind hoch genug, um die ganze Zelle Wachstum folgen. Das Gerät beinhaltet auch zwei Reservoirs befindet sich auf beiden Seiten der zentralen Kammer. Vier Löcher in Injektion (IH) sind so umgesetzt und werden wie folgt bezeichnet: den Einlass und Auslass einzuführen die zellulären Suspensionen in den Chip dienen, während die beiden anderen Stauseen ernähren.
Wir haben erste fabrizierten Kalibrierung Deckgläsern für Höhenmessungen mit Fotolack Strukturen der bekannter Geometrie. Wir haben dann frei wachsenden Neuronen, sondern auch morphologisch abhängigen Neuronen in Micropatterns der Adhäsion abgebildet.
Protokoll
Die Studie wurde durchgeführt, nach Richtlinien der Europäischen Gemeinschaft auf die Pflege und Verwendung von Labortieren: 86/609/EWG. Der Zweck der Forschung und das Protokoll sind im ethischen Anhang von ERCadg Projekt CellO, beschrieben, wurde genehmigt und wird regelmäßig von der ERCEA überprüft. Institut Curie Tierhaus erhielt Lizenz #C75-05-18, 24.04.2012, Berichterstattung an Comité d'Ethique En Matière d'Expérimentation Animale Paris Centre et Sud (nationale Registernummer: #59).
1. Herstellung der Form
Hinweis: Die Form umfasst Mittel- und fortgeschrittene Kammern angeschlossen an einen Einlass und eine Steckdose, plus zwei Stauseen (und Buchten) befindet sich auf beiden Seiten der zentralen Kammer.
- Flache Pinzette, 51 mm Durchmesser-Silizium-Wafer zu manipulieren und sie von einem Ort zum anderen zu übertragen.
- Zentralkammer Silikon Form
- Füllen Sie eine 2-mL-Kunststoff-Pipette mit positiven Photoresist. Positionieren Sie die Pipette in der Mitte einen Durchmesser von 51 mm-Silizium-Wafer und drücken Sie auf der Stausee bis auf etwa 75 % der Wafer-Oberfläche mit den Fotolack. Spin-Mantel bei 3000 u/min für 30 s.
- Übertragen den Wafer aus der Spin Coater auf eine Herdplatte mit einer Oberflächentemperatur von 100 ° C, für 50 s.
- Übertragen Sie den Wafer von der Herdplatte auf der Substrat-Halter (Chuck) der Mask Aligner. Durch die "DRIE Maske" aussetzen (siehe ergänzende Dateien "masks_neuron_volume_chips.tiff" und "masks_neuron_volume_chips.dxf").
- Entfernen Sie die Wafer aus dem Futter zu, dann tauchen Sie es in einem 100-mL-Glas kristallisieren Schale mit dem Entwickler (Verdünnung 1:4 in entionisiertem Wasser). Lassen Sie die Wafer und halten Sie es für 1 min Rühren sanft und kontinuierlich die kristallisierende Schale.
Hinweis: Um Reagenzien zu sparen, füllen Sie die kristallisierende Schale bis 1 cm in der Höhe maximal. - Zurücknehmen des Wafers vom Entwickler und Tauchen Sie es in einen 100-mL kristallisierenden Teller gefüllt mit entionisiertem Wasser für ca. 10 s.Then Ort der Wafer auf einem saugfähigen Papier und trocknen Sie ihn mit unter Druck stehenden Stickstoff mit einer Druckluftpistole Schlag.
- Legen Sie die Wafer für 50 s auf einer Herdplatte mit einer Oberflächentemperatur von 115 ° C.
- Führen Sie Deep Reactive Ion Etching (DRIE) mit den folgenden Parametern (weitere Informationen über die DRIE-Technik finden Sie in den Referenzen 14 und 15); Passivierung Schritt: 50 Sccm C4F8, er Unterstützung fließen 10 Sccm, CP 10 W, induktiv gekoppelten Plasma (ICP) 1500 W, Gesamt Druck 24 Mbar, Temperatur 18 ° C; Radierung Schritt: 100 Sccm SF6, er Unterstützung fließen 10 Sccm, CP 11 W, ICP 1500 W, Gesamt Druck 24 Mbar, Temperatur 18 ° C; Passivierung Zeit: 4 s; Ätzen-Zeit: 7 s; Gesamtdauer der Prozess: in der Regel 5 min für ca. 10 µm Tiefe geätzt.
- Lösen Sie der Fotolack durch Tauchen das Substrat in eine kristallisierende Schale gefüllt mit Aceton auf.
- Tauchen Sie die Wafer in einer Piranha-Lösung (2/3 H2O2 (30 %) und 1/3 H2SO4 (95 %)) für 5 Minuten.
Achtung: Immer zuerst H2O2 hinzufügen und dann H2SO4 und spülen mindestens 3 Mal entionisiertem Wasser nach der Reinigung.
- Fertigen Sie die Formen entsprechend Mittelstufe Kammern durch 1.2.1 zu 1.2.11 folgende Parameter, die in den Tabellen 1 bis 3aufgeführten Schritte ausführen.
Hinweis: Jede Tabelle entspricht einem bestimmten Gerät zeichnet sich durch eine bestimmte Höhe der zentrale Beobachtung Kammer.
Hinweis: Führen Sie den gesamten Prozess mit zunehmenden Photoresists Höhen (Masken 1 bis 3 für jedes Gerät siehe ergänzende Dateien "masks_neuron_volume_chips.tiff" und "masks_neuron_volume_chips.dxf").- Legen Sie die geätzten Silizium-Wafer auf dem Spin Coater-Substrat-Halter.
- Überprüfen Sie, dass der Spin Coater funktionsfähig ist, indem Sie überprüfen, dass das Streben des Substrats wirksam ist (das Substrat sollte spin und bleiben während der nominale Drehzahl).
- Mit der Höhe des Bereichs gezielt Dicke steigt die Viskosität von SU-8. Verwenden Sie immer 20-30 mL Flaschen zum Speichern von SU-8 Photoresists und gießen es auf dem Wafer vor Spin-Coating (SU-8 möglicherweise zu zähflüssig mit einer Kunststoff-Pipette bearbeitet werden soll).
- Gießen Sie negative Epoxy-Typ Photoresist SU-8 in der Mitte des Substrats bis die rund 75 % der Silizium-Wafer, dann Dreh-Mantel mit den Parametern in Zeile "Spincoating" der Tabellen angegeben.
- Ort der beschichteten Wafer auf eine Herdplatte für die Dauer und Temperatur in Zeile "weiche Backen" angegeben.
- Montieren Sie die geätzten Wafer und die angemessene "SU-8-Maske" auf die Mask Aligner.
- Richten Sie die geätzten Wafer mit der Maske mit der speziellen Ausrichtung Kreuze (typische Größe diese Kreuze: 50 µm × 150 µm) auf jede Maske entworfen.
Hinweis: Zwei Kreuze auf jeder Seite des Geräts sind ausreichend (unten links) und rechts oben. - Expose mit dem ich-Line der Mask Aligner (Wellenlänge 365 nm) mit der entsprechenden UV-Dosis, wie in Zeile "Exposition Energie" angegeben.
Hinweis: Die Belichtungszeit ist die Exposition Energie E spezifisch für jedes Fotolack durch die effektive Leistung der UV-Lampe dividiert, moduliert durch die Absorption der Maske: X (1 - AbsorptionMaske). Absorption ist ca. 20 % für flexible Masken und vernachlässigbar für Chrom schwer Maske.
X (1 - AbsorptionMaske). Absorption ist ca. 20 % für flexible Masken und vernachlässigbar für Chrom schwer Maske. - Ort der beschichteten Wafer auf eine Herdplatte für die Dauer und Temperatur in Zeile "Post Backen" angegeben.
- Bereiten Sie zwei 100 mL Glas kristallisieren Gerichte, mit der Entwickler, der andere leer. Tauchen Sie die Wafer in der Entwickler für die Dauer, die in der Zeile "Entwicklung" angegeben. Agitieren Sie sanft die kristallisierende Schale entlang Entwicklung
- Streuen Sie die Wafer mit Isopropanol über die leere kristallisierenden Schale für ca. 5 s. Schließlich legen Sie die Wafer auf ein saugfähiges Papier und trocknen Sie ihn mit unter Druck stehenden Stickstoff mit einer Druckluftpistole Schlag.
- Die beschichtete Wafer auf eine Herdplatte für die Dauer und Temperatur in Zeile "harten Backen" angegebenen Ort (optional).
Hinweis: Dieser Schritt ist nützlich, um zu vermeiden, Risse in den Fotolack und bieten eine homogene Fläche für die nächsten Schritte. - Wiederholen Sie die Schritte 1.3.1 - 1.3.10 für die in Tabelle 1 bis 3aufgeführten Produktionsprozesse benötigt.
- Nach dem letzten Schritt der Photolithographie, Silanize der letzte Meister Schimmel besteht aus 3 Schichten von negativen Photoresists durch die Entsendung von zwei 100 µL Tröpfchen von Trichloro(1H,1H,2H,2H-perfluoro-octyl) Silan auf jeder Seite des Meisters in einer 100-mm-Petrischale. Versiegeln Sie die Petri-Scheibe mit einem Kunststoff Paraffin-Film zu und inkubieren Sie 20 min bei Raumtemperatur (RT).
Hinweis: Der master-Form ist fertig und kann mehrfach verwendet werden.
- Legen Sie die geätzten Silizium-Wafer auf dem Spin Coater-Substrat-Halter.
2. Herstellung von PDMS-chip
- Gießen Sie 90 g Silizium-basierten organisches Polymer (Polydimethylsiloxan: PDMS) in einen 100-mL-Plastikbecher. Fügen Sie 10 g seine Härtemittel (01:10 in Gewicht). Rühren Sie die Mischung mit einer Kunststoff-Pipette für 2-3 min.
Hinweis: Mischen Sie 90 g von PDMS mit 10 g härter für die Herstellung von 6 Chips. - Legen Sie die Mischung in ein Vakuum Exsikkator und Pumpe für ca. 30 min in der PDMS eingeschlossenen Luftbläschen zu entfernen.
- Legen Sie die master-Form in einer Petrischale P100 und gießen Sie 15 mL des Gemisches auf dem Schimmel mit einer Spritze.
Hinweis: 15 mL führt zu einer gesamten Chip-Höhe von 1,5 mm. - Legen Sie die PDMS-Form-Struktur in einem Vakuum Exsikkator und Pumpe bis gibt es keine Luftblasen mehr an der Oberfläche der PDMS platzen.
Hinweis: Dieser Schritt dauert etwa 30 Minuten. - Drücken Sie die Wafer am unteren Rand der Petrischale mit einem Kegel Spitze PDMS Totvolumen unterhalb der Wafer zu vermeiden. Vorgeheizten Sie das PDMS-Schimmel-Gerät im Backofen bei 70 ° C für mindestens 2 h eingestellt.
- Entformt PDMS-Block unter einer chemischen Haube mit einem flachen Spatel aus rostfreiem Stahl und durch Gießen Isopropanol auf dem Chip. Der Edelstahl-Bank der Haube aufsetzen.
- Rund um die Silizium-Wafer geschnitten und entformt das PDMS-Replikat mit einem Skalpell.
- Rund (lassen Sie eine 2-mm-Marge) geschnitten Sie den Chip mit einem Skalpell oder einer Rasierklinge.
- Punch Buchten durch Drücken der 1, 5 mm Durchmesser Puncher fest und betätigen sie zu schneiden und schnitzen das Loch des Einlasses. Machen Sie dasselbe auf vier speziellen Zonen des Chips wo Flüssigkeit injiziert wird.
- Reinigen Sie den Chip kleben und peeling Klebeband auf der mikrostrukturierten Seite. Streuen Sie Isopropanol auf beiden Seiten. Trocknen Sie dann den Chip mit unter Druck stehenden Stickstoff mit einer Druckluftpistole Schlag.
3. Herstellung von gemusterten Deckgläsern (24 × 24 mm2)
Hinweis: Manipulieren Sie Deckgläsern mit gebogenen Pinzette.
- Poly-Ornithin (PLO) Muster
- Wenden Sie ein O2 Reinigung Plasma auf Glasdeckgläser an. Plasmaparameter: Druck Abpumpen: 0,25 Mbar; O2 Angebot Dauer: 3 min; Gas-Durchfluss: 10 Sccm; maximale Abweichung: ±5 Sccm; Plasma-Dauer: 3 min; Ansprechdruck: 0,36 Mbar; maximale Abweichung: ±0.10 Mbar; Leistung: 50 W; maximale Abweichung: 5 %; Entlüftung Dauer: 45 s.
- Mix-484 µL Essigsäure und 56 µL 3-Methacryloxypropyl-Trimethoxysilane, komplett mit absoluten Ethanol zu einem Gesamtvolumen von 15 mL zu erhalten.
- Mit einer 1-mL-Spitzenkonus, setzte einen Tropfen von 500 µL dieser Lösung auf jeder Deckglas, warten ca. 2-3 min. mit einem Reinraum-Mikrofaser-Wischer trocken.
Hinweis: Silanisiert Glasdeckgläser können bis zu 1 Monat bei Raumtemperatur innerhalb von Kunststoffboxen versiegelt mit einem Kunststoff Paraffin-Film gespeichert werden. - Ein Spin Coater befindet sich in einer Reinraumumgebung legen Sie jedes Deckgläschen auf. Ein Tropfen von einem positiven Photoresist, die rund 75 % des Deckglases (ca. 500 µL) und Spin-Mantel bei 4000 u/min für 30 s, Enddicke von 0,45 µm zu erreichen.
- Legen Sie die Deckgläsern für 1 min auf eine Kochplatte mit einer Oberflächentemperatur von 115 ° C.
- Mit einem Mask Aligner aussetzen jedes Deckglas bei einer Wellenlänge von 435 nm (G-Linie) durch die spezielle Maske entsprechend den Parametern der Fabrikant (UV-Dosis über 50-60 mJ.cm–1)
- 2 Glasschalen kristallisierenden bereiten, mit dem Entwickler (keine Verdünnung), die andere mit entionisiertem Wasser.
- Tauchen Sie eins nach dem anderen jedes Deckglas im Entwickler für 1 min kontinuierlich und sanft unter Rühren das kristallisierende Gericht. Dann tauchen Sie die Wafer in entionisiertem Wasser für ca. 5 S. Ort der Wafer auf einem saugfähigen Papier und trocknen Sie es mit unter Druck stehenden Stickstoff mit einer Druckluftpistole Schlag.
- Gelten Sie eine Aktivierung O2 Plasma mit den gleichen Parametern wie in 3.1.1.
- Unter der Haube abzusetzen vier 170 µL Tropfen einer Lösung von 100 µg/mL PLO pro P100 Petrischale. Setzen Sie die gemusterte Gesicht von den Deckgläsern auf jedem der diese Tropfen. Versiegeln der Petrischale mit einem Kunststoff Paraffin-Film um Austrocknen zu vermeiden. Über Nacht bei RT inkubieren
Hinweis: Die PLO-Lösung sollte an den Deckgläsern durch Kapillarität bleiben. - Bereiten Sie vier Empfänger (in der Regel P60 Petrischalen), füllen Sie drei davon mit PBS und das vierte mit entionisiertem Wasser. Bereiten Sie zwei Glasschalen kristallisieren von reinem Ethanol.
- Nehmen Sie jede Deckglas aus der Petrischalen, Tauchen Sie es in das erste PBS-Bad für 10-15 s, evakuieren Sie die Flüssigkeit auf eine absorbierende Gewebe zu, indem das Deckglas vertikal auf der Seite, fügen Sie es mit z.B. der gemusterten aufgedeckte innerhalb der Ethanol-Bad.
Hinweis: Sobald die Auflösung der Fotolack abgeschlossen ist, wird es schwierig, die Dekorseite das Deckglas zu finden. Daher ist es wichtig, in diesem Stadium, seine Lage zu verfolgen. - Legen Sie das Gericht in einem Ultraschallbad Sonikator kristallisieren Ethanol (120 W / 35 kHz) und lassen Sie den Fotolack für 3 min aufgelöst werden.
Hinweis: Ändern Sie die Ethanol-Bad alle 4 Deckgläsern zur Verdünnung von PBS zu begrenzen, die die Auflösung der Fotolack beeinträchtigen könnten. - Nehmen Sie das Deckglas aus dem Ethanol-Bad, dann tauchen Sie es mehrere Male in der zweiten PBS-Bad. Überprüfen Sie die Oberfläche Aspekt wiederholt bis die fettig-ähnliche Oberfläche, die Ergebnisse aus den verbleibenden Flüssigkeitsfilm Ethanol verschwindet.
- Für 5-10 s das Deckglas in das dritte PBS Bad eintauchen. Dann übertragen sie sofort das deionisiertes Wasserbad. Legen Sie das Deckglas auf ein saugfähiges Papier und trocknen Sie es mit unter Druck stehenden Stickstoff mit einer Druckluftpistole Schlag.
Hinweis: Die letzten Spülgang in entionisiertem Wasser wird verwendet, um die Bildung von PBS Kristalle während der Trocknung zu vermeiden.
- Photoresist Strukturen zur Höhenkalibrierung
- Führen Sie nur Schritte 3.1.4. zu 3.1.8. Verwendung der speziellen Maske (Maske "Fotolack Streifen", siehe zusätzliche Datei "Mask_Photoresist-stripes.dxf").
(4) chip-Montage und endgültige Umsetzung
- Chip-Montage am unteren Glasschalen
- Der Plasmakammer für Oberflächenaktivierung der PDMS-Chip und die Glasschale auf dem verklebenden umgesetzt. Parameter: Abpumpen Druck: 0,25 Mbar; O2 Angebot Dauer: 3 min; Gas-Durchfluss: 10 Sccm; maximale Abweichung: ±5 Sccm; Plasma-Dauer: 30 s; Ansprechdruck: 0,40 Mbar; maximale Abweichung: ±0.10 Mbar; Leistung: 50 W; maximale Abweichung: 5 %; Entlüftung Dauer: 45 s.
- Legte sie sanft den aktivierten PDMS-Chip in Kontakt mit Glas-Deckglas und zart üben Druck auf die Kanten des Chips, den Chip auf dem Deckglas zu verbinden. Um die Haftfestigkeit zu erhöhen, stellen Sie das Gerät in den Ofen bei 70 ° C für 5 bis 10 Minuten.
Hinweis: Drücken Sie nicht auf Teile, die mit Säulen, sie zu sehr unter Druck zusammenbrechen könnte. - Unter der Haube (d. h. bei RT) und innerhalb von 30 min nach Verklebung, platzieren Sie eine 10-µL Spitzenkonus gefüllt mit einer Lösung von 100 µg/mL PLO am IHs, dann die Flüssigkeit zu injizieren. Die Lautstärke um einen Tropfen an der Spitze der einzelnen IHs zu bilden. Dann fügen Sie mithilfe einer 1-mL-Spitzenkonus PBS in der Petrischale rund um den Chip.
- Lassen Sie den Chip bei RT mit einem Minimum von 2 h Inkubationszeit. Für Übernachtung Inkubation Versiegeln der Petrischale mit einem Kunststoff Paraffin-Film um Austrocknen zu vermeiden.
Hinweis: Die Flüssigkeit sollte nicht außerhalb des Chips sonst auslaufen, die der Chip verworfen werden muss. - Drücken Sie ein 10-µL Spitzenkonus leicht in jede IH und die überschüssige Flüssigkeit aufzusaugen. Dann bleiben Sie vollständig des Spitze Kegels in die Steckdose und die restliche Flüssigkeit erarbeiten.
- Ersetzen Sie PLO durch Laminin folgt den Anweisungen in den Schritten 4.1.5 (Entleerung) und 4.1.3 (Füllung). 1 h bei RT inkubieren.
- Ersetzen Sie Laminin Kulturmedium folgende Anweisungen in den Schritten 4.1.5 (Entleerung) und 4.1.3 (Füllung). Zusammensetzung des Kulturmediums: MEM 81,8 %; Natrium-Pyruvat 100 mM 1 %; Glutamax 200 mM 1 %; Pferd Serum 5 %; B27 ergänzen 2 %, N2 Ergänzung 1 %, Gentamicin 0,2 %; Filtern Sie die Lösung mit einem 220-nm-Filter. Mit einer 1-mL-Spitzenkonus, ersetzen Sie auch die PBS rund um den Chip durch dieses Medium.
- Setzen Sie den Chip in den Brutschrank bei 37 ° C und 5 % CO2 für mindestens 5 Stunden (oder über Nacht) vor dem Neuron Aussaat geregelt.
- Chip-Montage mit gemusterten Deckgläsern
Hinweis: Wenn die gemusterten Deckgläsern positiven Photoresist Referenzobjekte enthalten, führen Sie Schritte 4.2.1 zu 4.2.9. Andernfalls führen Sie nur Schritt 4.2.3, Stick legte das PDMS-Gerät auf die PLO gemustert Deckglas gemäß 4.1.2, Kulturmedium in und um den Chip dann gehen Sie zu Schritt 4.2.10.- Hinterlegen Sie einen Tropfen Wasser auf einem rechteckigen dicken Mikroskop-Objektträger und kleben Sie das Deckglas auf den Objektträger durch Kapillarität (nicht gemustert zugewandten Seite der Objektträger). Unter dem Mikroskop der Fotolack Streifen im hinteren Teil der Objektträger mit einem Filzstift markieren.
- Die Maske-Inhaber der Mask Aligner Ornamentglas Deckgläschen auflegen. Setzen Sie auf die Zeichen mit dem Filzstift Photoresist Referenzobjekte zu zentrieren.
- Führen Sie den Plasma-Schritt, wie unter 4.1.1 auf dem PDMS-Chip.
- Platzieren Sie den PDMS-Chip auf dem mobilen Substrat-Halter (Chuck) der Mask Aligner.
Hinweis: Um die optischen Kontrast zu erhöhen, legen Sie einen Silizium-Wafer unter dem PDMS-Chip. Die Silizium-Wafer sollte fest auf dem Futter während der Alignment-Prozess (verwenden Sie ein durchsichtiges Klebeband festhalten an das Futter) wegziehen lassen. - Heben Sie das Futter an der Grenze der mechanischen Kontakt, den Chip mit dem Array von Fotolack Streifen befindet sich auf dem Deckglas auszurichten.
- Erreichen Sie mechanischen Kontakt zwischen dem Chip und dem Deckglas durch heben dem Spannfutter bis die Oberfläche der PDMS Touch Säulen der Glas-Deckglas Abschluss.
- Senken Sie das Futter. Entfernen Sie das Deckglas jetzt auf den Chip aus der Maske Halterung verklebt. Dann stellen Sie das Gerät in einer 35-mm-Petrischale und übertragen alles in den Ofen (Temperatur: 70 ° C) für 5 bis 10 min zur Erhöhung der Festigkeit der Verklebung.
- Führen Sie wie in Schritt 4.1.3.
Hinweis: Wenn mit PLO Deckgläsern gemustert, gehen Sie direkt von Schritt 4.2.7 4.2.9 verstärken. - Ersetzen Sie PLO Beschichtung mittlere folgende Verfahren in den Schritten 4.1.5 (Entleerung) und 4.1.3 (füllen) beschrieben. Mit einer 1-mL-Spitzenkonus, ersetzen Sie auch die PBS rund um den Chip durch dieses Medium.
- Setzen Sie den Chip in den Brutschrank bei 37 ° C und 5 % CO2 bis Neuron Aussaat, mit einem Minimum von 5 h Inkubationszeit geregelt.
(5) Neuron Kultur
- Bereiten Sie 100 mL Dissektion Medium (HH-Mittel) durch Mischen 10 mL HBSS 10 X 2 mL Hepes 1 M und 88 mL steriles Wasser in einen 200-mL-Kunststoff-Kolben.
Hinweis: Die HH-Medium kann am Vortag der Kultur vorbereitet werden. - Sezieren Sie Hippocampus von E18 Mäuse Embryo aus einer Mutter eingeschläfert durch zervikale Dislokation (C57BL/6J Mäuse von Charles Rivers) extrahiert. Schritte der Präparation sind z.B. detaillierte in 16.
- Legen Sie Hippocampus in ein Kunststoffrohr mit Trypsin (0,3 mL Trypsin 2,5 %, w/o EDTA in 2,7 mL HH Medium) für 10 min bei 37 ° C um chemische Dissoziation zu initiieren.
- Verwerfen Sie fast alle Flüssigkeit und ersetzen Sie es mit 5 bis 10 mL HH mit Einweg Plastik Pipetten. Tun Sie es 3 Mal. Verwenden Sie für die letzte Füllung 1 mL Medium statt HH Plattieren.
- Gewebe mit einer 1 mL Spitzenkonus durch aufstrebende und Auswerfen das volle Volumen mehrmals, Vermeidung von Luftblasen Herstellung und Verwendung von nicht mehr als 15-20 Passagen mechanisch zu trennen.
- Bereiten Sie in einen separaten Behälter 500 µL eine Lösung mit 5 µL Zellsuspension in 45 µL PBS verdünnt. Nehmen Sie 1 µL dieser Lösung mit einer 10 µL Pipette und legen Sie die verdünnte Suspension in einen Malassez-Zelle-Zähler. Verwenden Sie die Angaben in 17 zur Schätzung der Anzahl der Zellen.
Hinweis: Ein einzelnes Hippocampus bietet in der Regel rund 0,5 Millionen von Neuronen. - Zentrifuge bei 100 X g für 6 min bei RT.
- Verwerfen Sie den Überstand und ersetzen Sie es durch das Volumen der Beschichtung Medium erforderlich, um eine Konzentration von 10 Millionen Zellen/mL zu erreichen. Aufschwemmen Zellen durch sukzessive streben und Auswerfen die Zellsuspension mit einer 1-mL-Spitzenkegel.
- Ausarbeiten der Beschichtung Medium in den Chip vorhanden (siehe Schritt 4.1.5). Sammeln Sie 2-3 µL der frisch resuspendierte Lösung mit einer 10 µL Pipette und injizieren sie am Einlass (siehe das Injektionsverfahren in Schritt 4.1.3 beschriebenen). Wiederholen Sie den gleichen Vorgang an der Steckdose sofort.
- Über die gleiche Menge an Beschichtung Medium in jedem Behälter Spritzen (siehe Schritt 4.1.3).
Hinweis: Beachten Sie schnell den Chip unter die Lupe genommen, die Dichte der Zellen zu überprüfen. Die Größenordnung der optimale Zelle Dichte entspricht etwa 5-10 Zellen innerhalb der quadratische Fläche von 4 Säulen begrenzt (ca. 0,3 mm2, d. h. über 15-20 Zellen pro mm2). - Schließlich wiederholen Sie Schritt 5.10 verwenden stattdessen 0,5-1 µL Zellsuspension, die gezielte Zelldichte zu erreichen.
- Platzieren Sie den kernigen Chip in einem Inkubator bei 37 ° C 5 % CO2geregelt.
(6) Fluoreszenz Ausgrenzung Beobachtung
- Ersatz des Kulturmediums durch bildgebendes Medium.
- Bereiten Sie bildgebende Medium wie 4.1.7 zu, sondern verwenden Sie stattdessen MEM frei von Phenol rot und fluoreszierende Dextran. In diesem Ziel verdünnen Dextran (Molekulargewicht 10.000 g/Mol, konzentrierte sich auf 10 mg/mL in PBS-Stammlösung) zur Erreichung eine Endkonzentration von 0,5-1 mg/mL im bildgebendes Medium.
Hinweis: Verwendung Dextran konjugiert mit Absorption/Emission Maxima 496/524 oder 650/668. Bevorzugen Sie die erste, 0,45 µm hohen positiven Photoresist Strukturen (get rid of ihre Auto-Fluoreszenz in der roten Bandbreite) und das zweite Bild Neuronen (weniger toxisch) Bild. - Alle Buchten mit einer 10 µL Pipette zu leeren und neu füllen sie vollständig mit dem bildgebendes Medium (siehe Schritte 4.1.3 und 4.1.5 für die präzise Methodik der mittlere Ersatz).
- Bereiten Sie bildgebende Medium wie 4.1.7 zu, sondern verwenden Sie stattdessen MEM frei von Phenol rot und fluoreszierende Dextran. In diesem Ziel verdünnen Dextran (Molekulargewicht 10.000 g/Mol, konzentrierte sich auf 10 mg/mL in PBS-Stammlösung) zur Erreichung eine Endkonzentration von 0,5-1 mg/mL im bildgebendes Medium.
- Bildgebung
- Platzieren Sie den Chip unter einem Epifluoreszenz-Mikroskop verfügt über eine Klimakammer auf 37 ° C und 5 % CO2geregelt. Verwendung einer 40 X, numerische Apertur (NA) 0,8 trocknen Objektive, 30 % der vollen Ladung (volle Leistung: 3 W) und 30 ms Belichtungszeit. Bilder von Zellen im Fokus (von einzelnen auf mehrere aufeinander folgende Bilder bei Zeitraffer Experimente) zu erwerben.
- Bildanalyse
- Bilder mit dem Hintergrund Reduktion Routine in dedizierte Software implementiert zu einem homogenen Hintergrund zu normalisieren. Siehe 13 für die Details der Bildverarbeitungs-Schritte in dieser Software enthalten. Das Ausgabebild hat ein. MAT-Format.
- Zu konvertieren. MAT-Datei in. TIFF-8-Bit-Bilder mit der Routine setzen ergänzendes Material (conversion_mattotiff.m, fordert ein importfilevol.m).
- Führen Sie Import > Bildsequenz auf ImageJ, bauen ein Video von der. TIFF-Bilder.
- Berechnen Sie die mittlere Intensität P von einer quadratischen Fläche in der Mitte eine Säule (Bezugsobjekt) und die mittlere Intensität B von einer quadratischen Fläche der Kammer frei von Zellen (Hintergrund, d. h. Höhe Null) lokalisiert. Siehe 18 ein Beispiel für detaillierte Methodik der Bildverarbeitung.
Hinweis: Die seitlichen Dimension der quadratischen Bereiche verwendet, da Intensität Referenzen müssen etwa die Hälfte der Säule Durchmesser, eine ausreichende Anzahl von Pixeln zu Vermeidung von Lichtverschmutzung aus der Säule Kanten. - Verwenden Sie die Werte für P und B, um die lineare Umwandlung von Intensität ich bis Höhe hRechtswissenschaften zu etablieren:

mit hc der bekannten Höhe der Kammer und
und
Hinweis: PDMS zeigt keine nachweisbaren Autofluoreszenz. - Wählen Sie einen Bereich um die Zone von Interesse, mit ImageJ Intensität zu integrieren (siehe 19 für weitere Details) und der Umbau Recht erwarb 6.3.5, Messen Sie das Volumen von einem Zellkompartiment anwenden.
Hinweis: Die Zone von Interesse möglicherweise ausgewählt anhand der Fluoreszenz der subzellulären Elemente wie Aktin, z. B.für die Auswahl des Wachstums in den GFP-LifeAct Neuronen, wählen Sie die Zone des Interesses an der GFP Emission Kanal speichern die Kontur dieser Kegel Zone mit einem ROI-Manager-Tool, dann Maßnahme das Zellvolumen eingeschlossen innerhalb derselben Zone in der Emission-Kanal von Dextran (in rot).
Ergebnisse
Das Ergebnis des Prozesses der Herstellung in den Abschnitten 1 und 2 beschrieben wird anhand der Bilder der Figur 1A-1 b und die Kurve der Abbildung 1. Die Tabelle der Abb. 1 zeigt die Rauheitskennwerte von zwei verschiedenen repräsentativen Bereichen der PDMS-Chip, d. h. in den Mittel-und 20 µm hoch nächste Zwischenkammer. Eine Abnahme der Rauheit um einen Faktor von etwa 7 eingeholt wurde mit geätzten Si-Wafer statt SU-8 Fotolack. FXm wurde dann zuerst auf ein Photoresist Streifen aus bekannter Geometrie (Abbildung 2A) innerhalb einer 10 µm hohe Kammer angewendet. Nach der Bildverarbeitung und Intensität zur Höhe Umkehr (siehe das Diagramm der Abbildung 2 b), FXm Profile durchgeführt auf Querschnitte entlang dieser Streifen (Abbildung 2) bieten die gewünschte Höhenprofile (Abb. 2D). Abb. 2D zeigt den Vergleich zwischen den Profilen mit mechanischen Profilometry und FXm Methoden erzielt. Diese Profile, einschließlich Rand und Plateau, sind sehr ähnlich, Validierung der Methode. Beachten Sie, dass die Streuung der FXm Daten nicht repräsentativ für die ultimative Auflösung der Methode, wie in Abbildung 3 und Abbildung 4, beurteilt, sondern ergibt sich aus der geringen Intensität eingesetzt, um eine mögliche Wirkung von dem sehr schwachen zu vermeiden Auto-Fluoreszenz von Photolack in der GFP-Kanal.
Dann beobachteten wir Neuriten in 3 µm und 10 µm hohen Kammern (Abbildung 3). Die Standardabweichung der Hintergrundgeräusche ist etwa 18 nm nach Intensität, Höhe-Konvertierung und Hintergrund-Korrektur. Dieser Wert ist etwas höher als der physische Rauheit des PDMS Flächen gegossen auf Silizium-Oberflächen (12 nm, siehe Abbildung 1) aber viel niedriger als die Rauheit auf PDMS gemessen von SU-8 Formen erhalten. Diese Ergebnisse zeigen den Mehrwert der Bohren von Brunnen in Silizium-Wafer, anstatt Löcher in SU-8 Photoresist Säulen werfen zu öffnen. Ein niedriger Wert erlaubt ein high-Signal zu Rauschverhältnis und sehr klare Bilder in Volumen wie die in Abbildung 3Aangezeigt. Als ein Beispiel für die Daten, die aus solchen Bildern abgerufen werden können, berechnet das Volumen von 1,6 µm (d.h. 10 Pixel) breite Neuriten (siehe das Diagramm der Abbildung 3 b) in Scheiben schneiden. Verwendung in erster Näherung eine lineare Anpassung dieser Daten bietet einen mittlere Neuriten Höhenwert von etwa 400 nm, verglichen mit z.B. der axonale Durchmesser 500 nm in 10 Tage alten Welpen innerhalb der Corpus Callosum- 5 gefunden. Wir haben auch FXm mit Micropatterns Adhäsion bestehend aus seriell verschweißten 2 µm und 6 µm breite Streifen von 30 µm Länge kombiniert. Unser Ziel war die Untersuchung des Einflusses der Neurit Breite auf die 3D-Form. Abbildung 3 zeigt eine 3D Darstellung in der falschen Farbe eine ganze Neuron Bild in einer großen Kammer 10 µm. Neuriten verbreiten auf 2 µm und 6 µm breite Streifen, während die Soma auf die Extremität der größte Streifen befindet. Höhenprofile wurden in drei verschiedenen Querschnitten gezogen. In Übereinstimmung mit der Grafik in Abbildung 3Aangezeigt die Oberfläche über die Erhöhung der Querschnitte mit der Neurit Breite (Abbildung 3D) integriert.
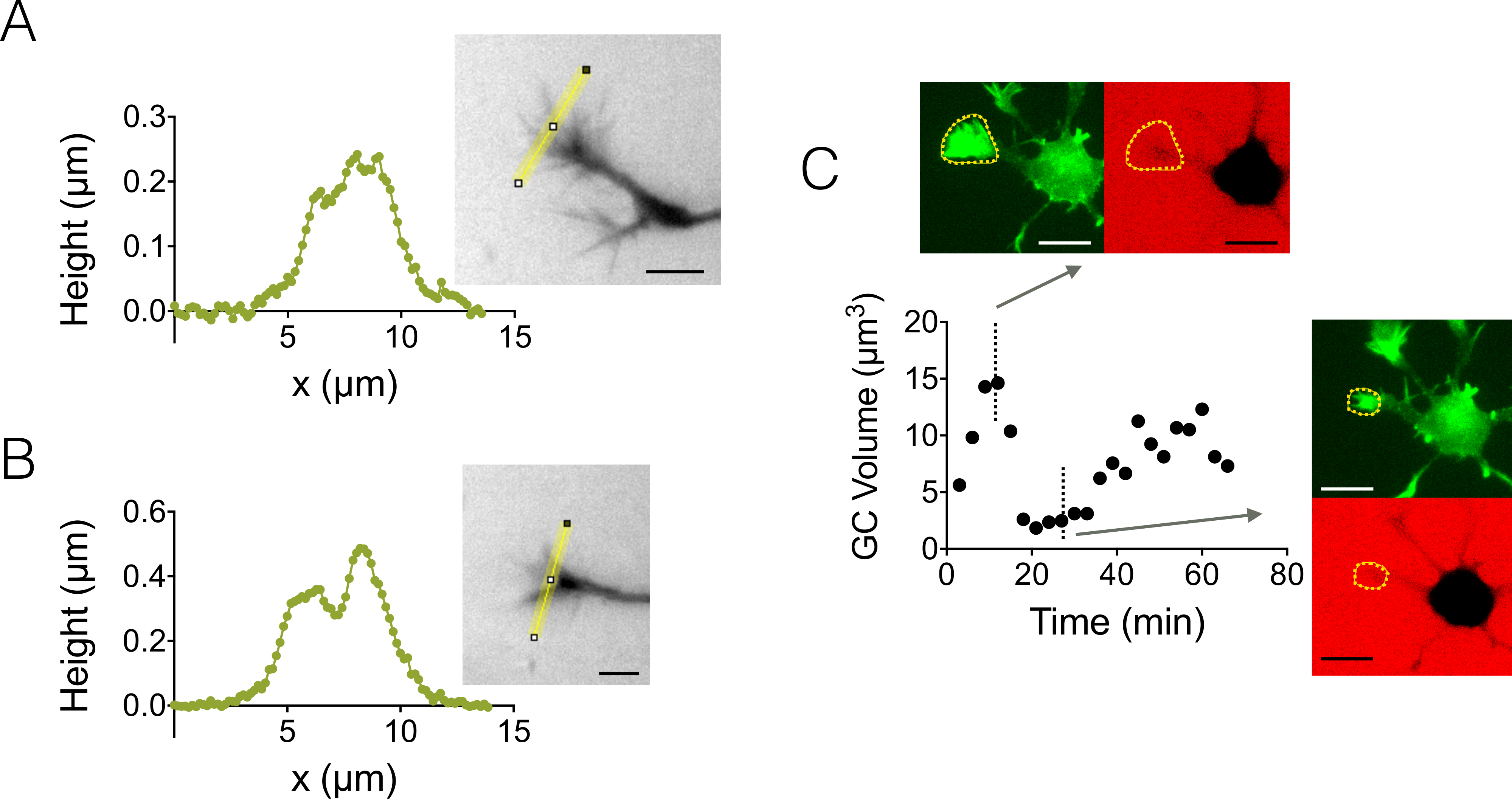
Wir weiters Wachstum Kegel (GC) 3D-Strukturen. Abbildung 4 A-B zeigt zwei verschiedene GC Profile erhalten in einer 3 µm hohe Kammer, die ihre verzweigten Unterkonstruktion hervorheben. Darüber hinaus führten wir Zeitraffer Experimente durchführen, um die Dynamik des Volumens der GCs in einer 12 µm hohe Kammer zu folgen. Abbildung 4 zeigt einen Zyklus der schrumpfenden und Reaktivierung einer bestimmten GC innerhalb einer Zeitskala von ein paar Dutzend Minuten. Durch den Einsatz von GFP-Lifeact Mäusen waren Wachstum Kegel in der GFP Emissionswellenlänge lokalisiert (510 nm) von ihrer hohen Aktin-Konzentration. Die Oberfläche bei der Wellenlänge identifiziert wurde verwendet, um über die Dextran Emissionswellenlänge bei 647 integrieren nm um GC Volumen zu berechnen. Abbildung 4 zeigt die Verteilung der GC Volumen an Ort zu verschiedenen Zeitpunkten schließlich auf drei verschiedene Neuronen, zentriert auf einen Wert von ca. 6 µm3.

Abbildung 1: FXm PDMS Kammern. (A) Regelungen der vier verschiedenen wichtigsten Schritte der Mikrofabrikation führt in die endgültige Form. Die Lage der Einlass, Auslass und Stauseen sind angegeben. Skalieren Sie Bars: 1 mm. (B) Bild der PDMS FXm Kammer unter Verwendung einer optische Profilometer. Dieses Bild zeigt die zentrale Kammer mit 3 Reihen von 10 µm hohen Säulen und die Mittelstufen Kammern von 20, 50 und 90 µm in der Höhe. Maßstabsleiste: 500 µm. (C) Querschnittsansicht des Chips an die beiden gestrichelten Linien gezeichnet (B). Gelb/Gold: Querschnitt entlang Säulen, blau: Querschnitt zwischen den Säulen. (D) bedeuten die PDMS Rauheit Messwerte auf 50 × 50 µm-2 -Gebieten auf Silizium und auf 20 µm hohen SU-8 Zwischenkammer geformt (siehe Pfeile für den Standort dieser Bereiche). Mittelwerte wurden aus den Messungen von drei verschiedenen Bereichen erhalten. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 2: Kalibrierung der FXm-Methode mit einem Fotolack Streifen als das Objekt des Interesses. (A) GLP-Fluoreszenz-Aufnahme in einer 10 µm hohe Kammer gefüllt mit 10.000 MW Dextran absorbierende bei 488 nm bei 1 mg/mL. (B: Hintergrund, P: Säule). Beobachtung mit einem trockenen 40 X NA 0,8 Ziel. Maßstabsleiste: 50 µm. (B) lineare Kalibrierung Recht entnommen die mittlere Intensität der beiden farbigen Rechtecke dargestellt in A. (C) Fluoreszenz Intensität Profil bezogen auf der Ebene der blau gestrichelten Linie dargestellt (A), überqueren den Fotolack Streifen (0,45 µm hohen positiven Photoresist). (D) Vergleich der gewonnenen mechanischen Profilometer (schwarze Punkte) und FXm nach Intensität, Höhe Konvertierung der Daten (b) (blaue Punkte) Profile. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 3: Neurit Volume Imaging. (A) Neurit erstreckt sich in die zentrale 3 µm hohe Kammer von Soma befindet sich in den nächsten 15 µm Zwischenkammer. Bildgebung durchgeführt mit 10.000 MW Dextran absorbierende bei 488 nm und 40 X, NA 0,8 trocknen Ziel. Der Inset erhalten nach dem Gebrauch den Hintergrund Reduktion Routine Highlights die beiden Neuriten und gewählt, um das Diagramm auf der rechten Seite zu plotten. Skalieren Sie Bars: 30 µm. (B) Neurit Slice Volumen als Funktion der Neurit breite gewonnenen 22 Profile (Durchschnitt auf 10 Pixel, d. h. auf einem 1,6 µm "Neurit Slice") in (A) gezeigt. Die durchgezogene Linie stellt eine lineare Anpassung der Neigung 0,4 µm durch den Ursprung. (C) Falschfarbenbild eines gemusterten Neurons auf einen selbstklebenden Streifen hergestellt aus aufeinanderfolgenden 2 µm und 6 µm breite Stümpfe (in weiß dargestellt). Messungen wurden in einer 10 µm hohe Kammer gefüllt mit 10.000 MW Dextran absorbierende bei 647 nm und mit einer 40 X NA 0,8 trocknen Ziel. (D) Höhenprofilen entsprechend farbige Striche angezeigt (c), halten die gleiche Farbcode. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 4: statische und dynamische Wachstum Kegel Bildgebung. (A-B) Wachstum Kegel Höhenprofile erhalten in einer großen Kammer 3 µm nach Intensität zur Umkehr Höhe entlang der gelben Linien im zugehörigen Bilder angezeigt. Beobachtung durchgeführt, mit einer Füllung mit 10.000 MW Dextran absorbierende bei 488 nm und 40 X, NA 0,8 trocknen Ziel. (C) ganze Neuron Bildgebung in einer 12 µm hohe Kammer gefüllt mit 10.000 MW Dextran absorbierende bei 647 nm. Beobachtungen wurden in zwei fluoreszierende Kanäle: GFP für Wachstum Kegel Lokalisierung (gelb gestrichelt) und CY5 um GC Volumen von Fluoreszenz Ausgrenzung zu berechnen. Die Oberfläche enthalten gelb gestrichelt wurde zur GC Volumen zu berechnen. Die Grafik zeigt die Variation des GC Volumen über Zeit und damit verbundenen Morphologien in GLP und CY5 Kanäle zu zwei repräsentative unterschiedlichen Zeitpunkten. Alle Daten wurden übernommen, mit einer 40 X NA 0,8 trocknen Ziel alle 3 min. Maßstabsleisten: 10 µm. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
| Schritt | 1:8 µm Schicht Maske | Maske 02:30 µm Schicht | Maske 03:40 µm Schicht |
| SU-8 Typ | 2007 | 2025 | 2050 |
| Spincoating | 30 s @ 2000 u/min | 30 s @ 3050 u/min | 30 s bei 3250 u/min |
| Weiche Backen | 3 min @ 95 ° C | 2 min @ 65 ° C + 6 min. @ 95 ° C | 3 min @ 65 ° C + 7 min. @ 95 ° C |
| Exposition-Energie | 110 mJ/cm2 | 155 mJ/cm2 | 170 mJ/cm2 |
| Nach Exposition Backen | 4 min @ 95 ° C | 1 min @ 65 ° C + 6 min. @ 95 ° C | 2 min @ 65 ° C + 7 min. @ 95 ° C |
| Entwicklung | 2 min 30 s | 5 min | 6 min |
| Harte Backen (optional) | 3-5 min. @ 200 ° C | 3-5 min. @ 200 ° C | 3-5 min. @ 200 ° C |
Tabelle 1: Photolithographie Schritte durchgeführt, ein Gerät mit einer zentralen Kammer 12 μm Höhe bauen. Höhen der Mittelstufen Kammern: 20, 50 und 90 µm.
| Schritt | Maske 01:10 µm Schicht | Maske 02:30 µm Schicht | Maske 03:40 µm Schicht |
| SU-8 Typ | 2007 | 2025 | 2050 |
| Spincoating | 30 s @ 1500 u/min | 30 s @ 3050 u/min | 30 s bei 3250 u/min |
| Weiche Backen | 3 min @ 95 ° C | 2 min @ 65 ° C + 6 min. @ 95 ° C | 3 min @ 65 ° C + 7 min. @ 95 ° C |
| Exposition-Energie | 125 mJ/cm2 | 155 mJ/cm2 | 170 mJ/cm2 |
| Nach Exposition Backen | 4 min @ 95 ° C | 1 min @ 65 ° C + 6 min. @ 95 ° C | 2 min @ 65 ° C + 7 min. @ 95 ° C |
| Entwicklung | 2 min 30 s | 5 min | 6 min |
| Harte Backen (optional) | 3-5 min. @ 200 ° C | 3-5 min. @ 200 ° C | 3-5 min. @ 200 ° C |
Tabelle 2: Photolithographie Schritte durchgeführt, ein Gerät mit einer zentralen Kammer von 10 μm Höhe bauen. Höhen der Mittelstufen Kammern: 20, 50 und 90 µm.
| Schritt | Maske 01:12 µm Schicht | Maske 02:32 µm Schicht | Maske 03:40 µm Schicht |
| SU-8 Typ | 2015 | 2025 | 2050 |
| Spincoating | 30 s bei 3250 u/min | 30 s @ 2500 u/min | 30 s bei 3250 u/min |
| Weiche Backen | 3 min @ 95 ° C | 2 min @ 65 ° C + 5 min. @ 95 ° C | 3 min @ 65 ° C + 7 min. @ 95 ° C |
| Belichtungszeit | 140 mJ/cm2 | 157 mJ/cm2 | 170 mJ/cm2 |
| Nach Exposition Backen | 4 min @ 95 ° C | 1 min @ 65 ° C + 5 min. @ 95 ° C | 2 min @ 65 ° C + 7 min. @ 95 ° C |
| Entwicklung | 3 min | 5 min | 6 min |
| Harte Backen (optional) | 3-5 min. @ 200 ° C | 3-5 min. @ 200 ° C | 3-5 min. @ 200 ° C |
Tabelle 3: Photolithographie Schritte durchgeführt, ein Gerät mit einer zentralen Kammer 3 μm Höhe bauen. Höhen der Mittelstufen Kammern: 18, 50 und 90 µm.
Ergänzende Daten 1: masks_neuron_volume_chips.tiff. Schematische Darstellung der Masken verwendet, um das PDMS-Gerät (DRIE Maske und Masken 1-3) zu fabrizieren. Bitte klicken Sie hier, um diese Datei herunterladen.
Ergänzende Daten 2: Datei "masks_neuron_volume_chips.dxf". Elektronische Dateien ermöglicht, um die DRIE Maske und Masken 1-3 zu fabrizieren. Bitte klicken Sie hier, um diese Datei herunterladen.
Ergänzende Daten 3: "Mask_Photoresist-stripes.dxf". Elektronische Dateien ermöglicht, um die Maske für die Fotolithografie Photoresist Streifen zu fabrizieren. Bitte klicken Sie hier, um diese Datei herunterladen.
Ergänzende Daten 4: conversion_mattotiff.m-Datei Klicken Sie bitte hier, um diese Datei herunterzuladen.
Ergänzende Daten 5: importfilevol.m-Datei Klicken Sie bitte hier, um diese Datei herunterzuladen.
Diskussion
Volumen-Bildgebung von Neuronen stellt eine Herausforderung für die FXm-Technik durch die langen und dünnen Erweiterungen dieser Zellen. Dieses Protokoll beschreibt Varianten des gleichen Typs von mikrofluidischen Gerät Neuron Bildgebung gewidmet.
Neben den Aspekten von mikrofluidischen Design die Wahl des Ziels ist von grundlegender Bedeutung für die Fluoreszenz Ausgrenzung Bildgebung und impliziert einen Kompromiss zwischen Ortsauflösung und die Bildschärfe. Es hat 13 gezeigt, dass eine hohe NA führt zu einer Tiefenschärfe kleiner als die Höhe der Kammer war nicht nachteilig für die Präzision der Volumenmessung, wenn Bildgebung mit Schwerpunkt durchgeführt wurde und wenn ein ausreichender Spielraum zwischen die Kontur des Objekts der ich gelassen wird Geschäftsgelegenheiten und die Grenzen der Integration Oberfläche. Allerdings beeinträchtigt die Verwendung einer Kammer deutlich höher als die Schärfentiefe der Bildschärfe durch Photon Diffusion, welche glättet die Kanten der Objekte von Interesse. Die Herstellung einer hohen Kammer mit 3 µm reduziert das seitliche Weichzeichnen und lieferte außergewöhnlich gut definierten fluoreszierende Ausgrenzung Bilder auch mit hohen NA (0,8) 40 X Ziele neuronale Verzweigungen mit hoher Ortsauflösung zu visualisieren.
Chip-Montage ist ein entscheidender Schritt, insbesondere bei 3 µm hohen Kammern, aber vorsichtig Manipulation wie unter 4.1.2 vermeidet den Zusammenbruch des Daches. Die hohe Fläche zum Volumenverhältnis, diese dünnen Kammern zugeordnet Frage auch die der Stabilität der Dextran Konzentration im Laufe der Zeit. Wir haben überprüft, ob die Oberflächenabsorption der Dextran nach einer Nacht der Inkubation unbedeutend war: nach Dextran durch PBS ersetzt, der Unterschied der Intensität zwischen der Säule und der Hintergrund war etwa 1 pro 1000 der Anfangsintensität Kontrast zwischen Diese beiden Regionen in Anwesenheit von Dextran. Beachten Sie, dass Neuronen auf der Unterseite Deckglas und auf dem Dach des PDMS halten können. Dieser Effekt verschwindet, wenn mit gemusterten Deckgläsern (d. h. wenn wir nicht anhaftende Moleküle innerhalb der PDMS-Kammer inkubieren), wie die Beschichtung auf der Unterseite der Kammer daher streng lokalisiert.
Abgesehen von ihrer anspruchsvollen Morphologie Neuronen eignen sich eher für FXm aufgrund der Tatsache, dass eine erhebliche Einschränkung der Methode, d. h. Dextran Endozytose, sehr begrenzt ist in diesen Zellen. Wir wählen eine 10 kDa Formulierung langfristig unterdrücken reichen (in Stunden) sichtbaren Endozytose Phänomene.
Zusammenfassend ist die konzeptionelle Einfachheit der FXm durch eine Reihe von experimentellen Probleme ausgeglichen, die durch dieses Protokoll, z. B. nanometrischen PDMS Rauheit und mikrometrische Kammer Höhe oder Untergrundkorrektur für die Unebenheiten korrigieren gelöst wurden die PDMS Decke zwischen Säulen. Die Verwendung einer engen mikrofluidischen Kammer zu beschränken, das fluoreszierende Medium führt jedoch ein paar spezifische Einschränkungen wie die Notwendigkeit der Unterstützung Säulen, die die wirksame Oberfläche für Zelladhäsion oder die Notwendigkeit der zentralen Soma ausschließen senkt Kammer, neuronale Erweiterungen mit der höchsten Klarheit zu beobachten, die Regionen der Zelle für hochauflösende Beobachtung zugänglich einschränkt. Eine mögliche Weiterentwicklung dieser Methode wäre get rid of dieser physischen Entbindung durch eine optische ersetzt werden. Die Neuentwicklung des Lichtblattmikroskopie könnte in Zukunft vorteilhaft mit FXm kombiniert werden.
Offenlegungen
Die Autoren erklären keinen Interessenskonflikt.
Danksagungen
Die Autoren ChiLab, Materialien und Microsystems Labor - Politecnico di Torino - DISAT, in der Person von Prof. C F Pirri, Dr. M Cocuzza und Dr. S L Marasso, bestätigen möchten für ihre wertvolle Unterstützung bei der Prozessentwicklung und Gerät Herstellung. Wir danken Victor Racine von Quantacell für Diskussion und Unterstützung in der Bildverarbeitung. Wir sind dankbar, Isabelle Grandjean und Manon Chartier von Animal Facility des Institut Curie für ihre Unterstützung für Mäuse, und Pablo Vargas und Ana-Maria Lennon (Institut Curie) für die Bereitstellung von uns mit der GFP LifeAct Mäusen. Wir sind dankbar, Olivier Thouvenin vom Institut Langevin und Clotilde Cadart, Larisa Venkova und Matthieu Piel vom Institut Curie - UMR 144, für ihre Hilfe für das Verständnis der Fluoreszenz unter Ausschluss Methode. Zu guter Letzt danken wir die technologische Plattform der Institut Pierre-Gilles de Gennes (CNRS UMS 3750) für ihre Unterstützung in Microfabrication. Diese Arbeit wurde teilweise unterstützt von der europäischen Forschung Rat Advanced Grant Nr. 321107 "CellO" PSL Université (SwithNeuroTrails Projekt), ANR Investissement Avenir und IPGG Labex und Equipex.
Materialien
| Name | Company | Catalog Number | Comments |
| Equipments | |||
| Plasmalab System 100 | Oxford Instruments | To perform DRIE | |
| MJB4 mask aligner | SUSS MicroTec | SU-8 photolithography | |
| NXQ 4006 Mask aligner | Neutronix Quintel | Photolithography associated to DRIE | |
| Plasma cleaner | Diener Electronic | Pico PCCE | |
| Cell culture hood | ADS Laminaire | Optimale 12 | |
| Centrifuge | Thermo Fisher Scientific | Heraeus Multifuge X1R | |
| Incubator 37 °C 5% CO2 | Panasonic | MCO-170AICUVH-PE IncuSafe CO2 Incubator | |
| Epifluorescence microscope | Leica | DMi8 | |
| PDMS Oven between 65 and 80 °C | Memmert | ||
| Mechanical profilometer | Veeco | Dektak 6M Stylus Profiler | |
| Optical profilometer | Veeco | Wyko NT9100 | |
| Vacuum desiccator | Verrerie Villeurbanaise (Kartel Labware) | 230KAR | Diameter 200 mm |
| Ultrasonic bath sonicator | Labo Moderne | SHE1000 | Volume of the bath: 0.8 liter |
| Hotplate | Stuart SD162 | SD162 | |
| Masks | |||
| DRIE | Supplementary data | ||
| Su8 Mask 1 | Supplementary data | ||
| Su8 Mask 2 | Supplementary data | ||
| Su8 Mask 3 | Supplementary data | ||
| S1805 calibration stripes | Supplementary data | ||
| Small laboratory equipment | |||
| 1.5 mm hole puncher | Sigma-Aldrich | 29002519 (US reference) | |
| Scalpel or razor blade | |||
| 9" Stainless Steel Flat Spatula with Spoon | VWR International | 82027-532 | To demold PDMS |
| Top Lip Wafer Handling | VWR International | 63042-096 | |
| Curved tweezer | FST | Dumont #7 Forceps - Standard / Dumoxel | To manipulate glass coverslips |
| Substrates | |||
| Silicon wafer | Prolog Semicor Ltd | ||
| 24x24 mm glass coverslips | VWR | 631-0127 | |
| Photoresists and developpers | |||
| AZ 1518 positive photoresist | Microchemicals GmbH | Before DRIE process, thicknes 1.8 µm | |
| AZ 351B developer | Microchemicals GmbH | To develop positive AZ 1518 photoresist | |
| SU-8 2007 | MicroChem | ||
| SU-8 2025 | MicroChem | ||
| SU-8 2050 | MicroChem | ||
| PGMA developer | Technic | To develop SU-8 negative photoresist | |
| Microposit S1805 resist | Chimie Tech Services | Positive photoresist used to obtain 0.5µm high structures | |
| MF 26A developer | Chimie Tech Services | To develop positive S1805 photoresist | |
| Laboratory consumables | |||
| Disposable plastic pipette 3 mL | LifeTechnologies - ThermoFisher | ||
| P100 Petri dishes | TPP | 93100 | |
| 20 mL syringe | Terumo | SS+20ES1 | |
| Transparent scotch tape | |||
| Square wipes | VWR | 115-2148 | |
| Parafilm | DUTSCHER | 90260 | Plastic paraffin film |
| Chemicals | |||
| (3-Methacryloxypropyl)trichlorosilane | abcr | AB 109004 | |
| PDMS and curing agent Sylgard 184 | Sigma-Aldrich | 761036 | |
| isopropanol | W292907 | ||
| poly-ornithine | Sigma-Aldrich | P4957 - 50 mL | |
| Ethanol absolute | Sigma-aldrich | 02865 | 99.8% |
| 3-methacryloxypropyl-trimethoxysilane | Sigma-aldrich | M6514-25ML | (C4H5O2)-(CH2)3- Si(OCH3)3 |
| acetic acid | Sigma-aldrich | 71251-5ML-F | |
| Dextran 10kW conjugated with Alexa488 | LifeTechnologies - ThermoFisher | D22910 | Absoprtion at 488 nm |
| Dextran 10kW conjugated with Alexa647 | LifeTechnologies - ThermoFisher | D22914 | Absoprtion at 647 nm |
| Culture medium | |||
| MEM | LifeTechnologies - ThermoFisher | 21090-022 | |
| Horse Serum | LifeTechnologies - ThermoFisher | 26050088 | |
| B27 | LifeTechnologies - ThermoFisher | 12587-010 | |
| Glutamax 200 mM | LifeTechnologies - ThermoFisher | 35050-061 | |
| Sodium Pyruvate GIBCO 100 mM | LifeTechnologies - ThermoFisher | 11360-070 | |
| Gentamicin | LifeTechnologies - ThermoFisher | 15710-049 | |
| PBS | Sigma-Aldrich | D8537-500ML | |
| HBSS 10x | LifeTechnologies - ThermoFisher | 14180-046 | |
| Hepes 1M | LifeTechnologies - ThermoFisher | 15630-056 | |
| trypsin-EDTA | Sigma-Aldrich | 59418C-100ML | |
| Neurobasal | LifeTechnologies - ThermoFisher | 21103-049 | |
| Neurobasal without phenol red | LifeTechnologies - ThermoFisher | 12348-017 | |
| Softwares | |||
| Routine in Matlab for background normalization | Quantacell | Contact Victor Racine: victor.racine@quantacell.com | |
| ImageJ | To select specific ROI for image analysis | ||
| Routine | ImageJ | Supplementary data | |
| Routine | Matlab | Supplementary data |
Referenzen
- Jun, S., Taheri-Araghi, S. Cell-size maintenance: universal strategy revealed. Trends Microbiol. 23 (1), 4-6 (2015).
- Zlotek-Zlotkiewicz, E., Monnier, S., Cappello, G., Le Berre, M., Piel, M. Optical volume and mass measurements show that mammalian cells swell during mitosis. J. Cell Biol. 211 (4), 765-774 (2015).
- Kim, G. H., Kosterin, P., Obaid, A. L., Salzberg, B. M. A mechanical spike accompanies the action potential in Mammalian nerve terminals. Biophys J. 92 (9), 3122-3129 (2007).
- Iacono, D., et al. Neuronal Hypertrophy in Asymptomatic Alzheimer Disease. J Neuropathol Exp Neurol. 67 (6), 578-589 (2008).
- Cheli, V. T., et al. Conditional Deletion of the L-Type Calcium Channel Cav1. 2 in Oligodendrocyte Progenitor Cells Affects Postnatal Myelination in Mice. J Neurosci. 36 (42), 10853-10869 (2016).
- Kneynsberg, A., Collier, T. J., Manfredsson, F. P., Kanaan, N. M. Quantitative and semi-quantitative measurements of axonal degeneration in tissue and primary neuron cultures. J Neurosci Methods. 266, 32-41 (2016).
- Fouquet, C., et al. Improving axial resolution in confocal microscopy with new high refractive index mounting media. PloS one. 10 (3), (2015).
- Aguet, F., et al. Membrane dynamics of dividing cells imaged by lattice light-sheet microscopy. Mol Biol Cell. 27 (22), 3418-3435 (2016).
- Roland, A. B., et al. Cannabinoid-induced actomyosin contractility shapes neuronal morphology and growth. eLife. 3, (2014).
- Lu, Y. B., et al. Viscoelastic properties of individual glial cells and neurons in the CNS. Proc Natl Acad Sci USA. 103 (47), 17759-17764 (2006).
- Chen, J., Xue, C., Zhao, Y., Chen, D., Wu, M. H., Wang, J. Microfluidic impedance flow cytometry enabling high-throughput single-cell electrical property characterization. Int J Mol Sci. 16 (5), 9804-9830 (2015).
- Bottier, C., et al. Dynamic measurement of the height and volume of migrating cells by a novel fluorescence microscopy technique. Lab Chip. 11, 3855-3863 (2011).
- Cadart, C., et al. Fluorescence eXclusion Measurement of volume in live cells. Methods Cell Biol. 139, 103-120 (2017).
- Marasso, S. L., et al. A novel graphene based nanocomposite for application in 3D flexible micro-supercapacitors. Materials Research Express. 3 (6), 065001(2016).
- Welch, C. C., Goodyear, A. L., Wahlbrink, T., Lemme, M. C., Mollenhauer, T. Silicon etch process options for micro-and nanotechnology using inductively coupled plasmas. Microelectronic Engineering. 83 (4), 1170-1173 (2006).
- Fath, T., Ke, Y. D., Gunning, P., Götz, J., Ittner, L. M. Primary support cultures of hippocampal and substantia nigra neurons. Nat. Protoc. 4 (1), 78-85 (2008).
- abcam. Counting cells using a hemocytometer. , Available from: http://www.abcam.com/protocols/counting-cells-using-a-haemocytometer (2017).
- ImageJ. Getting intensity values from single ROI. Image Intensity Processing. , Available from: https://imagej.net/Image_Intensity_Processing (2017).
- ImageJ. 19. Tools. ImageJ User Guide. , Available from: https://imagej.nih.gov/ij/docs/guide/146-19.html (2017).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten