
Method Article
Kombination von Analyse von DNA in eine grobe Virion-Extraktion mit der Analyse der RNA aus den infizierten Blättern, entdecken neue Virus-Genom
In diesem Artikel
Zusammenfassung
Hier präsentieren wir einen neuen Ansatz zur Identifizierung von Pflanzenviren mit Doppel-Strang DNA Genom. Wir verwenden standard-Methoden zu extrahieren von DNA und RNA aus den infizierten Blättern und Next Generation Sequencing durchzuführen. Bioinformatische Werkzeuge montieren Sequenzen in Contigs, Contigs Vertretung Virus Genome zu identifizieren und Genome taxonomischen Gruppen zuweisen.
Zusammenfassung
Dieser Ansatz Metagenom wird verwendet, um Pflanzenviren mit kreisförmigen DNA Genome und ihre Zeugnisse zu identifizieren. Oft Pflanze DNA-Viren, die in niedrige Titer in ihrem Wirt auftreten oder können nicht mechanisch geimpft werden, auf einen anderen Host sind schwer zu propagieren, um eine höhere Titer von infektiösem Material zu erreichen. Infizierte Blätter sind Boden in einem milden Puffer mit optimalen pH-Wert und Ionischen Zusammensetzung empfohlen für die meisten Bacilliform Para Retroviren zu reinigen. Harnstoff wird verwendet, Einschlusskörperchen aufzubrechen, die Virionen Falle und zelluläre Komponenten auflösen. Differentielle Zentrifugation liefert weitere Trennung der Virionen von pflanzlichen Verunreinigungen. Proteinase K Behandlung entfernt dann die Capsids. Dann wird die virale DNA konzentriert und für Next Generation Sequencing (NGS) verwendet. Die NGS-Daten dienen zum montieren Contigs die eingereicht werden, NCBI-BLASTn, eine Teilmenge der Virus-Sequenzen in das generierte Dataset zu identifizieren. In einer parallelen Pipeline ist RNA isoliert aus den infizierten Blättern mit einer standard spaltenbasierten RNA Extraktionsmethode. Dann wird Ribosom Erschöpfung durchgeführt, um für eine Teilmenge von mRNA und Virus Transkripte zu bereichern. Montierte Sequenzen von RNA Sequenzierung (RNA-Seq) abgeleitet abgegeben NCBI-BLASTn, eine Teilmenge der Virus-Sequenzen in diesem Dataset zu identifizieren. In unserer Studie haben wir zwei verwandte in voller Länge Badnavirus Genome in den beiden Datensätzen identifiziert. Diese Methode wird bevorzugt, eine andere Möglichkeit die Gesamtbevölkerung von kleinen RNA-Sequenzen zur Pflanze Virus Genomsequenzen rekonstruieren extrahiert. Dieses letztere metagenomische Pipeline erholt sich Virus im Zusammenhang mit Sequenzen, die sind Retro-Transkription Elemente eingefügt in das pflanzliche Genom. Damit verbunden ist, Biochemische und molekulare Tests weiter aktiv Krankheitserreger zu erkennen. Der Ansatz in der vorliegenden Studie dokumentiert erholt sich Sequenzen Vertreter der Replikation von Viren, die wahrscheinlich aktive Virus-Infektion hinweisen.
Einleitung
Aufstrebenden Pflanzenkrankheiten fahren Forscher entwickeln neue Werkzeuge, um die richtige kausale Agents zu identifizieren. Erste Berichte über neue oder wiederkehrende Virosen basieren auf häufig auftretende Symptome wie Mosaik und Fehlbildungen des Blatt, Vene, clearing, Kleinwuchs, welken, Läsionen, Nekrose oder andere Symptome. Der Standard für die Meldung eines neuen Virus, wie der kausale Agent für eine Krankheit ist, es von anderen kontaminierende Erreger zu trennen in geeigneten Host zu verbreiten und vermehren sich die Krankheit durch impfen in gesunde Pflanzen von der ursprünglichen Wirtsarten. Die Einschränkung bei diesem Ansatz ist, dass ein Insekt oder andere Vektoren für die Übertragung zu einem geeigneten Wirt oder zurück zu den ursprünglichen Wirtsarten viele Gattungen von Pflanzenviren abhängen. In diesem Fall die Suche nach der geeigneten Vektor kann verlängert werden, möglicherweise gibt es Schwierigkeiten, Labor Kolonien des Vektors zu etablieren und weitere Anstrengungen sind erforderlich, um ein Protokoll für die experimentelle Übertragung zu entwickeln. Wenn die Voraussetzungen für die erfolgreiche Übertragung Laborstudien nicht erreicht werden können, fällt die Arbeit hinter den Standard für die Meldung einer neuen Viruskrankheit. Für Viren, die in ihrer natürlichen Wirte auf sehr niedrige Titer auftreten, müssen Forscher alternative Gastgeber für die Vermehrung um ausreichende infektiöse Vorräte für die Durchführung von Recherchen erhalten identifizieren. Dies kann auch ein Hindernis für den Anbau von Stammkulturen1Virus-Arten, die nur ein paar Pflanzen infizieren.
In den letzten Jahren beschäftigen Wissenschaftler immer häufiger Hochdurchsatz-NGS und metagenomische Ansätze Virus Sequenzen aufzudecken, die in der Umgebung vorhanden, die möglicherweise nicht im Zusammenhang mit einer bekannten Krankheit vorhanden sind, aber taxonomische Arten und Gattungen zugeordnet werden kann 2 , 3 , 4. solche Ansätze zur Entdeckung und Kategorisierung des genetischen Materials in einer deutlichen Umgebung bieten die Möglichkeit, Virus-Vielfalt in der Natur oder ihre Präsenz in einem bestimmten Ökosystem beschreiben bestätigt jedoch nicht unbedingt zu einem Rahmen für die Festlegung kausale Agenten für eine offensichtliche Krankheit.
Gattung Badnavirus gehört zur Familie Caulimoviridae von Pararetroviruses. Diese Viren sind Bacilliform in Form mit kreisförmigen Doppelstrang DNA Genome von etwa 7 bis 9 kb. Alle Pararetroviruses werden durch ein RNA-Zwischenprodukt repliziert. Pararetroviruses gibt es als Episomes und unabhängig von der Pflanze chromosomale DNA-5,6zu replizieren. Feldstudien über Virus Populationen zeigen, dass diese Virus-Populationen genetisch komplexer sind. Darüber hinaus Informationen über eine Reihe von pflanzengenomen von Hochdurchsatz-Sequenzierung haben aufgedeckt, zahlreiche Beispiele von Badnavirus Genom Fragmente pflanzengenomen durch illegitime Integrationsveranstaltungen eingefügt. Diese endogene Badnavirus-Sequenzen sind nicht unbedingt in Zusammenhang mit Infektionen7,8,9,10,11. Anschließend wird die Verwendung von NGS, neue Badnaviruses als kausale Agent der Krankheit zu identifizieren durch die Teilgesamtheit Vielfalt episomal Genome sowie das Auftreten von endogenen Sequenzen12,13erschwert.
Zwar gibt es nicht eine optimale Pipeline für die Entdeckung von Roman Pararetrovirus Genomen, gibt es zwei Möglichkeiten, diese Viren als kausale Agenten für die Krankheit zu identifizieren. Eine Methode ist die Bereicherung für kleine RNA-Sequenzen aus den infizierten Blättern und dann montieren Sie diese Sequenzen um das Virus Genome(s)14,15,16,17wieder zusammenzusetzen. Ein weiterer Ansatz ist die rollenden Kreis Verstärkung (RCA), kreisförmige DNA-Virus Genome18zu verstärken. Der Erfolg von RCA hängt das Alter des Blattes und der Virustiter im ausgewählten Gewebe. Die RCA-Produkte sind Beschränkungsverdauung unterzogen und in Plasmide für direkte Sequenzierung19,20,21kloniert.
Canna gelbe Mottle virus (CaYMV) ist ein Badnavirus und wird beschrieben als die ätiologische Ursache der gelben Mottle Krankheit in Canna, obwohl nur ein 565 bp Fragment des Genoms von infizierten Cannas22zuvor isoliert wurde. Eine zeitgenössische Studie identifiziert CaYMV in Alpinia Purpurata (Blüte Ingwer; CaYMV-Ap)23. Das Ziel dieser Studie war, komplette Badnavirus Genomsequenzen von infizierten Canna Lilien zu erholen. Wir beschreiben ein Protokoll zum Virus von pflanzlichen Verunreinigungen zu reinigen, und dann isolieren virale DNA aus dieses Präparat und bereiten eine DNA-Bibliothek für den Einsatz in NGS. Dadurch entfällt die Notwendigkeit zwischen-Molekulare Verstärkung Schritte. Wir isolieren auch mRNA von infizierten Pflanzen für RNA-FF NGS, die RNA-Seq umfasst mit jeden Nukleinsäure-Vorbereitung durchgeführt wurde. Montierten Contigs befanden sich auf dem Badnavirus Taxon in beiden Datensätzen über das nationale Zentrum für Biotechnologie und Information (NCBI) grundlegende lokale Suche Ausrichtwerkzeug für Nukleinsäuren (BLASTn) beziehen. Wir ermittelten die Genome von zwei Badnavirus Arten24.
Protokoll
1. allgemeine Virus Reinigung durch Differentielle Zentrifugation mit Standard-Methode von Covey Et al. 25
- Zuerst, 80-100 g Blätter von Kranken Pflanzen schneiden und Schleifen in einem waring Blender bei 4 ° C mit 200 mL Puffer Schleifen (0,5 M NaH2PO4, 0,5 M Na2HPO4 (pH 7,2). und 0,5 % (w/V) Na2SO3). Tragen Sie einen Laborkittel und Handschuhen für alle Schritte dieses Verfahrens.
- Übertragen Sie anschließend die Homogenat (300 mL) auf einen 1,0 L-Becher. Das Homogenat in einem chemischen Kapuze 18 g Harnstoff und 25 mL 10 % nichtionische Waschmittel (t-Okt-C6H4-(OCH2CH2)9OH) hinzufügen.
Hinweis: Für diesen Schritt ist es am besten, Schutzbrille und eine einfache Atemmaske für persönliche Schutzausrüstung zu tragen. - Rühren mit Magnetrührer kurz in der Kapuze und den Becher mit der Folie bedecken. Dann die Folie abgedeckt-Becher zu einem kalten Raum übertragen und rühren mit einem Magnetrührer über Nacht bei 4 ° c
- Übertragen Sie das Homogenat um Rotor Flaschen (250 mL Behälter) und Zentrifuge in einem festen Winkel-Rotor bei 4.000 x g für 10 min bei 4 ° c zentrifugiert Zurückzugewinnen Sie in einem chemischen Abzug überstand und Filter durch 4 Schichten von Gaze.
- Teilen Sie das Homogenat unter 38,5 mL Polypropylen Zentrifuge Röhren und Zentrifuge für 2,5 h bei 40.000 x g bei 4 ° C. In der Regel Überprüfen Sie das Vorhandensein eines grünen Pellets an der Unterseite des Rohres und einer weißen Pellet entlang der Länge des Rohres. Der Überstand Gießen Sie ab und behalten Sie beide Pellets; Legen Sie Proben auf Eis.
Hinweis: Die grüne Tablette enthält Chloroplasten, Stärke und anderen Organellen. - Arbeiten in einer chemischen Kapuze, verwenden Sie ein Kautschuk-Polizist um die Pellets zu trennen. Aufschwemmen der weißen Pellet in jeder Rotor-Flasche in 1 mL DdH2O im Laufe von 1-2 h unter Beibehaltung der Suspensionen über Nacht bei 4 ° C erlauben die Materialien, die vollständig in Lösung zu lösen. Zentrifugieren Sie die Federung bei 6.000 x g und bei 4 ° C für 10 min um die restlichen Schmutz entfernen.
- Zentrifugieren Sie die konzentrierte Suspension bei 136.000 x g für 2 h bei 4 ° C zu Virionen Pellets. Die Pellets in 1 mL des Puffers (50 mM Tris-HCl, pH 7.5, 5 mM MgCl2) aufzuwirbeln.
Hinweis: Ein optionaler Schritt ist zur Behandlung von Virionen mit DNAse I (10 µg/mL) für 10 min bei 37 ° C, nicht Encapsidated DNA, d. h. verunreinigen Chloroplasten und mitochondriale DNA zu entfernen. Dann inaktivieren die DNAse I durch 1 mM EDTA hinzufügen. - Unterbrochen Sie Virionen mit 40 µL 2 µg/µL Proteinase K bei 37 ° C für 15 Minuten werden.
- Arbeiten Sie in einem chemischen Kapuze Virion DNA durch organische Extraktion zu erholen. Tragen Sie einen Gesichtsschutz, Handschuhe und einen Laborkittel während der Extraktion zum Schutz gegen mögliche akute gesundheitliche Auswirkungen. Die Probe 1 Volumen von Phenol-Chloroform-Isoamyl Alkohol (49:50:1) hinzu und schütteln von hand für 20 S. Zentrifuge bei Raumtemperatur für 5 min bei 16.000 x g. Entfernen der oberen wässrigen Phase und Übertragung auf einen neuen Schlauch. Wiederholen Sie diese Extraktion zweimal oder öfter. Die organische Phase indem man sie in einer Glasflasche für angemessene institutionelle Chemikalienentsorgung26Abfall entsorgen.
- Die DNA mit Ethanol Niederschlag zu konzentrieren. Verwenden von 0,3 M Endkonzentration von Natriumacetat (pH 5,2) und 2,5 Volumina von 95 % Ethanol. Proben bei-20 ° C für 30-60 min und Zentrifuge bei 13.000 x g für 10-20 min zu den DNA-26Pellets zu platzieren.
- Arbeiten bei einem Labortisch, Aufschwemmen Sie das DNA-Pellet in 1 mL der 0,1 mM TE-Puffer (pH 8,0). Filer die Aussetzung durch eine kommerzielle Gel Filtration Spalte (normalerweise verwendet für Polymerase-Kettenreaktion (PCR) Aufräumen), Salze und niedermolekularen Material, das NGS behindern könnten zu beseitigen.
- Analysieren Sie Proben von 1 % Agarose-Gelelektrophorese mit Interkalation Bromid Färbung um die Qualität der Präparate anzuzeigen. Bewerten Sie die Qualität der DNA mit einem Spektralphotometer Nanodrop.
Hinweis: Ein Verhältnis von Probe Extinktion bei 260 λ und 280 λ zwischen 1,85 und 2.0 gibt in der Regel, dass die Vorbereitung "sauber" von Verunreinigungen ist und der gewünschten Qualität. - Qualitätsanalyse von DNA (verwenden Sie 5 Pg 10 ng) mit einem Chip-basierte Kapillarelektrophorese Instrument.
Hinweis: Ausgabequalität zeigt saubere Gipfeln, DNA-Fragmente verteilt nach Größe auf einer X-Achse darstellt. Peakhöhe zeigt Fülle des Fragments. Zerklüftete Gipfel zeigen teilweise degradierte Fragmente oder chemischen Verunreinigungen. Molligen Rundungen repräsentieren einen Abstrich von DNA, die schlechte Qualität
(2) Bibliothek Vorbereitung mit DNA und Emulsion basierenden klonalen Verstärkung (EmPCR Verstärkung)
Hinweis: Die Bibliothek ist in der Regel durch eine NGS-Anlage bereit, kundenorientierte Arbeit durchführt.
- Eine Lösung der DNA (> 200 ng) mit einem Vernebler der DNA Fragmente umwandelt zu scheren. Verbinden Sie die kommerzielle Adapter nach dem Handbuch Anweisungen27.
- Führen Sie EmPCR Amplifikation von DNA-Probe nach der Hersteller Anweisungen28,29,30. Der Waschschritt dreimal wiederholen, und nach jedem Waschen, pellet-Perlen in ein Minicentrifuge 10 S. verwerfen des Überstands nach jedem Waschen.
Hinweis: Das Verfahren beginnt mit der Vorbereitung der Aufnahme Perlen durch Waschen in der kommerziellen Waschpuffer mit dem Kit zur Verfügung gestellt. EmPCR wird häufig für Vorlage Verstärkung für NGS verwendet. - Hitze denaturieren die DNA oder RNA bei 95 ° C für 2 min und dann 4 ° C bis zum Gebrauch. Einsatz 200 Millionen Moleküle der DNA/RNA, 5 Millionen Erfassung Perlen in einem Endvolumen von 30 µL. vorbereiten eine mock Probe neben die DNA/RNA-Probe und führen Sie die folgenden Schritte mit der Nukleinsäure Probe sowie die mock Probe.
- Führen Sie Emulgierung durch aufschütteln der Röhre Emulsion Öl für 10 s mit maximaler Geschwindigkeit, dann Gießen Sie den gesamten Inhalt (4 mL) in einem Kunststoff rühren Rohr, das mit einem Plattform-Homogenisator kompatibel ist. Legen Sie die mitreißende Röhre auf der Plattform, die Emulsion bei 2.000 u/min, 5 min zu mischen.
- 100 µL-Aliquots der Emulsion zu verzichten, in 8 Streifen Cap Röhren oder in eine 96-Well-Platte. Kappe die Schläuche oder die Dichtplatte und EmPCR unter Verwendung des Herstellers Empfohlene Programm28durchführen.
Hinweis: Nach die PCR abgeschlossen ist, überprüfen Sie die Brunnen zu sehen, ob die Emulsion intakt ist und fahren dann. Den gesamte Brunnen zu verwerfen, wenn die Emulsion gebrochen ist. - Einen Laborkittel zu tragen und arbeiten in einer chemischen Kapuze, die amplifizierten DNA-Perlen (ADB) zu sammeln. Vakuum Aspirieren Sie die Emulsion aus dem Brunnen und sammeln Sie die Perlen in einer 50 mL-Tube. Spülen Sie die Brunnen zweimal mit 100 µL Isopropanol und Aspirieren Sie abspülen, um die gleichen 50 mL-Tube.
- Wirbel der gesammelten Emulsionen und Aufschwemmen die ADB mit Isopropanol zu einem Endvolumen von 35 mL. Pellet-ADB bei 930 X g für 5 min. Entfernen des Überstands und 10 mL Puffer zu verbessern. Wirbel der ADB und dann waschen durch Zugabe von Isopropanol zu 40 mL Endvolumen Zentrifuge und entsorgen der Überstand nach jedem Waschen und Waschschritt zweimal wiederholen.
- Führen Sie eine endgültige waschen mit Ethanol anstelle von Isopropanol. Fügen Sie Verbesserung Puffer 35 mL Endvolumen, Wirbel und Pellet Perlen bei 930 X g für 5 min. Überstands entfernen, aber lassen 2 mL Puffer zu verbessern.
- Die Aussetzung zu einem Microcentrifuge Schlauch übertragen und kurz um die ADB pellet Zentrifugieren. Spülen Sie nach der Überstand verwerfen, die ADB-Pellet zweimal mit 1 mL Verbesserung Puffer. Zentrifugieren und den überstand nach jedem Waschen zu verwerfen.
- Zur Vorbereitung auf die DNA Bibliothek Wulst Bereicherung fügen Sie 1 mL 1 N NaOH der Perlen hinzu. Wirbel der ADB und dann für 2 min bei Raumtemperatur inkubieren. Zentrifugieren und den überstand verwerfen. Wiederholen Sie waschen einmal.
- Fügen Sie 1 mL glühen Puffer, dann Wirbel der ADB und 2 min bei Raumtemperatur inkubieren. Kurz zentrifugiert und den überstand entsorgen. Wiederholen Sie diesen Schritt noch einmal mit 100 µL glühen Puffer.
- Um Sequenzierung Primer an die DNA zu tempern, fügen Sie 15 µL Seq Primer a und 15 µL Seq Primer B im Kit enthalten. Kurz mischen durch aufschütteln und Microcentrifuge Schlauch in einem Heizblock bei 65 ° C für 5 min. Transfer für 2 min Eis.
- Waschen Sie dreimal mit 1,0 mL Puffer glühen. Vortex für 5 s und entsorgen des Überstands jedes Mal.
- Messen Sie vor der Sequenzierung die Anzahl der Perlen, die kommerzielle Wulst Zähler. Es sollte mindestens 500.000 angereicherte Perlen.
Hinweis: Der Wulst-Counter ist ein spezielles Gerät, das die Perlen in einem mitgelieferten Microcentrifuge Schlauch misst.
3. allgemeine mRNA Isolierung und Synthese ausgehend von infiziert Canna verlässt, die Probe mittels RT-PCR für CaYMV mittels berichtet diagnostische Primer DsDNA
- Tragen Sie einen Laborkittel und Latex-Handschuhe für den Personenschutz in alle weiteren Schritte. Arbeiten bei einem Labortisch, sammeln Sie 12 Proben aus den Blättern und Tauchen Sie ein die Proben in flüssigem Stickstoff. Verwenden Sie eine Rührwerksmühle für Homogenisierung. Verwenden Sie eine kommerzielle Kit, die eine Spalte basierenden Standardmethode für die Gesamtanlage RNA Isolierung bietet. Die Guanidin-Herstellung Lyse Puffer zur Verfügung gestellt durch das Kit das zerkleinerte Mahlgut und schütteln für 20 s.
- Ethanol und mischen Sie gründlich, nach Kit Anweisungen. Eine Spin-Spalte, die RNA an die Membran bindet fügen Sie jeder Homogenat hinzu. Waschen Sie dreimal und eluieren Sie RNA in eine Erholung Rohr24.
- Quantifizieren Sie die RNA mit einem Spektralphotometer, um das Verhältnis der Extinktion bei 260 λ und überprüfen die RNA Integrität 280 λ. mit 1 % Agarose-Gelelektrophorese gebeizt mit Interkalation Bromid zu messen.
Hinweis: Eine Extinktion Verhältnis zwischen 1,85 und 2.0 zeigt, dass die Vorbereitung der gewünschten Qualität. Behandlung von RNA mit DNase I (10 µg/mL) für 10 min bei 37 ° C. Verwenden Sie eine kommerzielle Spin-Spalte, um RNA in RNase-freies Wasser31konzentrieren. Pool RNA-Proben, bevor Sie fortfahren. - Mithilfe einer rRNA Entfernung Kit um Pflanze ribosomaler RNA zu entfernen. Aliquoten magnetische Beads Microcentrifuge Schlauch und waschen zweimal Wasser mit RNase-freie. Wirbel der Röhre aliquoten zu Aufschwemmen, legen Sie das Rohr auf einem Magnetstativ und warten auf Flüssigkeit zu löschen. Den überstand entsorgen und ersetzen durch die magnetische Wulst Wiederfreisetzung Lösung. Vortex aufschwemmen und fügen Sie 1 µL RNase-Inhibitor.
Hinweis: Diese Kits verwenden Oligo-dT an magnetischen Perlen, die an mRNA hybridisieren gebunden. Die Methode verwendet standard Magnet-Perlen-Trenntechnik, Transkripte24wiederherzustellen. - Kombinieren Sie 500 ng zu 1,25 µg RNA, RNase-freies Wasser und Reaktion Puffer zur Verfügung gestellt vom Kit. Legen Sie die Mischung für 10 min bei 50 ° C. Vom Herd nehmen und gewaschenen magnetische Beads in RNAse freies Wasser hinzufügen. Vortex kurz und auf 5 min bei Raumtemperatur.
- Auf einem Magnetstativ legen Sie und warten Sie, bis die Flüssigkeit klar. Übertragen Sie den überstand zu einem frischen Microcentrifuge Schlauch. Auf Eis legen.
- Verwenden Sie eine Projektmappe-basierte Capture-Methode für die Anreicherung von exosomen und 200 ng RNA, die cDNA-Bibliothek vorzubereiten.
Hinweis: Die Doppelstrang-cDNA-Bibliothek ist in der Regel durch eine NGS-Anlage bereit, kundenorientierte Arbeit durchführt. - Fragment der RNS mit einem kommerziellen RNA-Fragmentierung-Lösung (0,136 g ZnCl2 und 100 mM Tris-HCl pH 7,0). 18 µL RNA-Lösung 2 µL hinzufügen (200 ng insgesamt). Röhren kurz in den Microcentrifuge Spin, die Proben bei 70 ° C für 30 s und Übertragung auf Eis legen. Stoppen Sie die Reaktion mit 2 µL 0,5 M EDTA pH 8.0 und 28 µL 10 mM Tris-HCl pH 7,5.
- Binden Sie RNA an magnetischen Kügelchen durch Mischen bei Raumtemperatur für 10 min. Einsatz einen magnetischen Konzentrator, die Perlen zu sammeln und entsorgen Sie den überstand. Waschen Sie die Perlen dreimal mit 200 µL 70 % Ethanol. Verwerfen jedes waschen und dann an der Luft trocknen Aufschwemmen der gebeizte Perlen bei Raumtemperatur für 3 min. in 19 µL 10 mM Tris-HCl pH 7,5.
- Tempern von zufälligen Primern, fragmentierte RNA durch Erhitzen auf 70 ° C für 10 min und dann das Rohr auf Eis für 2 min. Vorbereitung der erste Strang und zweiten Strang cDNA mit einem standard kommerzielle cDNA Synthese-Kit.
- Die Doppel-Strang cDNA mit einem magnetischen Wulst-Konzentrator zu reinigen. Waschen Sie mit 800 µL 70 % Ethanol dreimal. Verwerfen jedes waschen und Lufttrocknen Aufschwemmen der Pellets bei Raumtemperatur für 3 min. in 16 µL 10 mM Tris-HCl pH 7,5. Verwenden des magnetischen Wulst-Konzentrators, um die Perlen von der doppelsträngige cDNA zu trennen ist nun auf die Lösung. Entfernen Sie die cDNA durch in eine neue 200 µL PCR-Röhrchen pipettieren.
- Führen Sie Fragment Ende Reparatur mit Taq Polymerase und eine Mischung aus Deoxyribonucleotides, die von einem kommerziellen Bibliothek vorbereitungssatz zur Verfügung gestellt. Das kommerzielle Kit bietet vorverdünnt Adapter an jedem Ende der Doppel-Strang cDNA mit kommerziellen Ligase bei 25 ° C für 10 min hinzufügen.
4. NGS der DNA Bibliothek vorbereitet von Crude Virus Vorbereitung und DsDNA Bibliothek aus mRNA vorbereitet
- Verwenden Sie einen standard Hochdurchsatz pyrosequenzierung und befolgen Sie alle empfohlenen Hersteller-Protokolle, um direkte Ablesung der DNA-Sequenzen zu erzeugen. Verwenden Sie kommerzielle Sequenzierung Reagenzien, einschließlich eindringmittel markierte Nukleotide.
Hinweis: Siehe Angaben des Herstellers mit dem Instrument zur Verfügung gestellt. - Durchführung von Post-Sequenzierung Analyse mit Genom-Montage-Software, die automatisch liest um den ersten Satz von Contigs mit einer durchschnittlichen Länge von < 700 BP. produzieren montiert verwenden die FastQC-Software auf der Website von iPlant/CyVerse die Qualitätskontrolle durchführt Kontrollen auf den rohen Sequenz Daten32. Wählen Sie Sequenzen mit Phred Scores ≥ 30 weiterhin längere Sequenzen aus kleineren Sequenz liest24 mit Zuordnung und Amplifikate Software rekonstruieren.
Hinweis: Siehe Anleitungen des Herstellers. - Reichen diese montierten Contigs NCBI-BLASTn Analyse, MEGABLAST-Standard-Modul als auch Viridplantae (USt: 33090) und Viren (USt: 10239) wie die Begrenzung organismal33nennt. Sammeln Sie die Subpopulation von Contigs, die hohe Ähnlichkeit mit gemeldeten Badnavirus Genomen in einen Bericht zeigen.
- Überprüfen Sie, ob die verknüpften Gerüste, die einen oder mehrere Kandidaten in voller Länge Virus Genome, darstellen richtig produzieren in-Frame Sequenzen, die derselben Organisation wie der standard Badnavirus Genom. Um dies zu tun, geben Sie das Virusgenom Kandidat in voller Länge in ein Plasmid Zeichnung Software. Bestätigen Sie dann die ersten 15 Nukleotide besteht aus einer tRNAerfüllt (TGGTATCAGAGCGAG) der unter Badnaviruses hoch konserviert ist. Suchen Sie die potenziellen Polyadenylation Signal am 3'-Ende des Genoms. Beschriften Sie das komplette Genom um das Vorhandensein von zwei kleinen ORFs und einem großen ORF Codierung ein funkionalen identifizieren. Verwenden Sie dann die ExPASy Portal übersetzen Werkzeug, um die Badnavirus ORF1 und ORF2 ORF3 Übersetzung Produkte34zu identifizieren.
Hinweis: Diese wissenschaftliche Software ist kostenlos und wird erzeugen kreisförmige DNA, identifizieren Sie alle offenen Lesung umrahmt, und bietet einen unmittelbaren Ausgang um sicherzustellen, dass die Folge in voller Länge kreisförmige DNA Genom darstellt. - Verwenden Sie open-Source Sequenz Vergleich Mehrfachwerkzeuge, Muskel- und CLUSTALW, um das Virus-Genom gewonnenen DNA und RNA Analysen35,36vergleichen.
- Suchen Sie die NCBI Nukleotid-Datenbank, um die vollständige Genomsequenzen 30 Badnavirus Arten zu erhalten und als Dokument im .fasta-Format zu exportieren. Laden Sie Sequenzen auf eine Software, die evolutionäre genetische Analyse der Folgen zusammen mit den Virus Genomsequenzen erzielten NGS führt. Generieren Sie mehrere Reihenfolge Ausrichtungen und maximale Wahrscheinlichkeit Bäume mit Muskel-37.
(5) Qualitätsbeurteilung von De Novo Sequenzierung durch PCR-Amplifikation des Virus-Genom von infizierten Pflanzen
- Geben Sie die neu identifizierten in voller Länge Badnavirus Genomsequenzen (.fasta-Format) in das kostenlose Onlinetool Primer3 PCR Primer38ableiten. Primer-Sets, die 1.000-1.500 bp über die gesamte Länge des Virus Genome(s) versetzte Produkte werden zu identifizieren. Senden Sie die Sequenzen an eine Service-Einrichtung, die zu synthetisieren und liefern PCR-Primer.
Hinweis: Die Ausgabe identifiziert akzeptabel Grundierung Paare mit gemeinsamen und akzeptable Schmelztemperaturen und präzise Grundierung Orten entlang der eingeführten Sequenzen. - Arbeiten bei einem Labortisch und tragen einen Laborkittel und Handschuhen, isolieren 5 µg DNA aus den Virus infizierte und gesunde Kontrolle Blättern mit einem automatisierten Verfahren, das standard paramagnetischen Zellulose Partikel zur Isolierung von DNA aus pflanzlichen Material39 beinhaltet. Blattmaterial (20-40 mg) in flüssigem Stickstoff in einem Microcentrifuge Schlauch Einfrieren und Schleifen mit einem Rührwerksmühle. Kombinieren Sie die Probe mit Lyse Puffer in einem Microcentrifuge Schlauch und jede Probe fügen Sie RNase A hinzu. Die Probe kurz und für 10-20 s Wirbel spin die Probe, feste Partikel zu entfernen.
Hinweis: Zellulose PARAMAGNETISCHE Teilchen haben hohe DNA-bindende Kapazität und hohe Erträge von reine DNA zu isolieren. Die standard kommerzielle Kieselsäure Spalte Methoden zur DNA-Isolierung extrahieren nicht effizient DNA aus einer Vielzahl von Pflanzenarten. Infolgedessen gibt es Dutzende von Methoden, die Änderungen dieser Verfahren zur Verbesserung der Effizienz für die einzelnen Pflanzenarten sind. Die automatisierte paramagnetischen Zellulose Partikel Methode wurde gewählt, weil es mehr und hochwertigere DNA von mehr als 25 krautige Angiospermen Arten40ergibt. - Verwenden Sie kommerzielle Reagenz Patronen für automatisierte paramagnetischen DNA-Isolierung. Jede kommerzielle Reagenz Patrone und Transfer Pflanze lysate, die gleiche Patrone 300 µL Nuklease freies Wasser hinzufügen. Setzen Sie die Patrone in die Patrone Rack, legen Sie einen Kolben in den Brunnen am nächsten zur Elution Röhre und platzieren Sie Elution Puffer in der Elution Rohr zu. Laden Sie Patronen in die automatisierte Nukleinsäure-Isolierung Maschine und führen Sie die Pflanze DNA-Isolierung Protokoll41,42.
- Führen Sie PCR eine Reihe von überlappenden PCR-Produkte ableiten. Verwenden Sie 5 µM jeder vorwärts- und Grundierung mit 35 Zyklen der PCR Verstärkung. Verwenden Sie die folgenden Radsport Bedingungen: Denaturierung bei 95 ° C für 60 s, Glühen bei 50 ° C für 45 s und Erweiterung bei 72 ° C für 1-2 min mit der letzten Erweiterung bei 72 ° C für ca. 7-10 min. Verwendung einer vorgefertigten Gel Filtration Spalte zur Beseitigung Salze und niedermolekularen Material als in Schritt 1.231.
- Berechnen einer 3:1 molare Verhältnis von PCR-Produkt, Vektor, bestimmen die Menge des PCR-Produktes zu verbinden bis 50 ng der linearisierten pGEM Plasmid43. Verwenden Sie ein Steuerelement einfügen DNA zu bestimmen, ob die Verbindlichkeiten effizient zu arbeiten. Durchführen der Ligatur über Nacht mit T4 DNA-Ligase (3 U/µL) bei 4 ° C. Dann transformieren kommerziell vorbereitet JM109 kompetenten Escherichia coli Zellen. Verwendung steuern 100 Pg ungeschnitten Plasmid DNA als Positivkontrolle für effiziente Umwandlung. Platte 100 µL der transformierten Zellen auf LB-Agar-Platten mit antibiotischen und blau/weißen Auswahl aufgespaltenen Plasmiden26wiederherstellen. Inkubieren Sie Platten für 16-24 h bei 37 ° c
Hinweis: Die pGEM-Vektor hat eine LacZ-Gen, das β-Galaktosidase kodiert. Transformierte Bakterien auf einem Teller angebaut wird, enthält 100 µg/mL Ampicillin, 0,5 mM IPTG, 80 µg/mL 5-bromo-4-chloro-3-indoyl-β-D-galactopyranosidase (X-gal) aufgrund der Aktivität der β-Galaktosidase blau. Das pGEM-Plasmid ist in einer Weise, die das LacZ gen stört linearisiert. Kolonien, die die PCR-Produkt-Einsätze enthalten stören das LacZ-Gen und X-gal nicht verstoffwechseln zu tun. Diese Kolonien sind weiß. Somit können Kolonien mit einer Einlage von denen ohne einen Einsatz durch die Farbe der Kolonie (weiß und blau)26unterschieden werden. - DNA aus drei Kolonien mit einem standard spaltenbasierten Plasmid Isolierung Kit39zu isolieren. Sequenz drei Plasmide Transformation Produkt. Vergleichen Sie jede DNA-Sequenz mit der de Novo zusammengebaut Virus Genome von NGS produziert. Verwenden Sie CLUSTALW, um die Sequenzen auszurichten und um sicherzustellen, dass sie entsprechend bestellt werden.
Ergebnisse
Diese veränderten Virus Reinigung Methode bereitgestellt, eine Bereicherung des Virus DNA nützlich zur Identifikation von zwei Virus-Arten von NGS und Bioinformatik. Nachdem das Homogenat bei 40.000 x g für 2,5 h zentrifugiert wurde, gab es einen grünen Pellet am Boden des Röhrchens und einem weißen Pellet entlang der Länge. Das grüne Pellet wurde in einem Microcentrifuge Schlauch Nukleinsäuretablette und das weißen Pellet wurde in zwei Mikrozentrifugenröhrchen Nukleinsäuretablette. PCR wurde mit standard CaYMV PCR-Diagnostik-Primern durchgeführt, und Produkte wurden in die solubilized weißen Pellet und nicht die grüne Kugel (Abb. 1A) nachgewiesen. Ein Beispiel für die groben Vorbereitung war von Transmissions-Elektronenmikroskopie untersucht und wir beobachteten Bacilliform Partikel Messen 124-133 nm Länge (Abbildung 1B). Dies ist innerhalb der prognostizierten modal Länge von den meisten Badnaviruses. DNA wurde extrahiert aus der weißen und grünen Pellets und Nukleinsäuretablette getrennt. In Abbildung 1Cluden wir 5 µL DNA extrahiert von den grünen und weißen Pellet Probe (1,6 µg DNA für die grüne Fraktion) und 3,1 µg DNA für die weißen Bruchteil um 0,8 % Agarose gel Elektrophorese und analysierten die DNA nach Interkalation Bromid Färbung. Die grüne Fraktion enthaltenen niedermolekularen DNA, während der weiße Anteil zwei Bänder aus höherem Molekulargewicht DNA sowie das geringere Molekulargewicht DNA (Abbildung 1C) produziert. Das Gel, dargestellt in Abbildung 1C für 40 min. bei 100 V ausgeführt wurde und der Abstrich auf Spur 3 legt nahe, dass die Gel Spannung gesenkt werden sollte, um klarere Bands produzieren. Diese Daten deuten darauf hin, dass die weiße Kugel für Virionen angereichert wurde. Die DNA (0,6 µg/mL) Konzentration der weißen Probe entnommen war gering, aber ausreichend für NGS, die erfordert ein Minimum von 10 ng DNA um fortzufahren. Fragmentierte DNA wurden verwendet, um eine Bibliothek auf NGS vorzubereiten.
Parallel wurde RNA aus infizierten Canna Pflanzen (Abbildung 1D) für Hochdurchsatz-RNA-FF gewonnen. Ein standard Workflow erfolgte für Bibliothek Vorbereitung, NGS, Erstellen von Contigs und virale Genomsequenzen (Abbildung 1E) zu identifizieren. Die Ausgangsresultate hindern, DNA und RNA als Ausgangsstoffe wurden verglichen.
Wir erhalten 188.626 rohe DNA liest von NGS mit DNA isoliert von der rohen Virus Vorbereitung. Liest in 13.269 Contigs versammelt waren und BLASTn wurde verwendet, um das NCBI-Dataset von Nukleotidsequenzen suchen (mit Viridplantae TaxID: 33090 und Virus USt: 10239 als begrenzenden Organismen) (Abbildung 1E). Die NCBI-BLASTn-Ergebnisse zeigten, dass 93 % der de Novo montiert Contigs zellulären Sequenzen waren, 22 % unbekannt waren, und 0,3 % Virus Contigs (Abb. 2A). Die Mehrheit der Contigs kategorisiert, wie zelluläre Sequenzen wie Mitochondrien identifiziert wurden oder Chloroplast DNA. Innerhalb des Datasets der Virus Contigs, 32 % der Virus Contigs wurden im Zusammenhang mit Mitgliedern des Caulimoviridae (die nicht Badnavirus Sequenzen waren) und 58 % davon wurden im Zusammenhang mit Badnavirus. Von dem Virus Contigs 29 % waren sehr ähnlich (e < 1 x 10-30), CaYMV isolieren V17 ORF3-gen (EF189148.1), Zuckerrohr-Bacilliform-Virus zu isolieren Batavia D, vollständige Genom (FJ439817.1), und Banane Streifen CA Virus komplette Genom ( KJ013511). Innerhalb dieser Population gab es lange Contigs, die zwei volle Länge Genome ähnelte.
Hochdurchsatz-RNA-Seq produziert 153.488 gereinigte einzelne Sequenz liest mit einer durchschnittlichen Länge von < 500 BP. Contig Montage lesen reduziert dies auf 8.243 Contigs. Diese wurden eingereicht, NCBI-BLASTn (mit Viridplantae TaxID: 33090 und Virus USt: 10239 als begrenzenden Organismen) und die Ausgänge platziert 76 % der Contigs in einer Kategorie Pflanze zellulären Sequenzen, 23 % waren unbekannt, und 0,1 % wurden als Virus Contigs (kategorisiert Abbildung 2 ( B). näherer Betrachtung der Bevölkerung die 0,1 % der Bevölkerung des Virus Contigs festgestellt, dass 68 % davon Caulimoviridae (Abbildung 2B) zugeordnet wurden. Drei große Contigs innerhalb dieser Population wurden mit hohen Ähnlichkeit identifiziert (e < 1 X 10-30), CaYMV isolieren V17 ORF3-gen (EF189148.1) Zuckerrohr Bacilliform-Virus zu isolieren, Batavia D, vollständige Genom (FJ439817.1) und Banane-Streifen CA-Virus vollständige Genom (KJ013511). Prüfung der drei Contigs, traten wir manuell zwei davon, eine Full-Length Virusgenom zu produzieren.
Wir verglichen die Virus-Genom Länge Contigs produziert von DNA und RNA Sequenzierung als gegenseitige Gerüst, das Vorhandensein von zwei Full-Length Virus Genome zu bestätigen. Eine Full-Length Virusgenom von 6.966 bp wurde vorläufig benannt Canna gelbe Mottle verbundenen Virus 1 (CaYMAV-1)(Abbildung 3). Das zweite Genom wurde 7.385 bp und eine Variante des CaYMV infizieren Alpinia Purpurata (CaYMV-Ap01)(Abbildung 3).
Zu guter Letzt PCR Zündkapseln, die entworfen wurden, um Klon ~ 1.000 bp Fragment von jedem Virus dienten differentiell beide Genome in einer Population von 227 Canna-Pflanzen für neun kommerziellen Sorten erkennen. In vielen Fällen wurden einzelne Pflanzen mit beiden Viren infiziert. Wir bieten ein Beispiel für RT-PCR-Nachweis von CaYMAV-1 und CaYMV-Ap01 in 12 Werken. Drei davon waren positiv nur für CaYMV-Ap01 und neun positiv für beide Viren (Abbildung 3B).

Abbildung 1 : Virus-Nukleinsäure-Vorbereitungen und NGS Workflow. (A) Agarose (1,0 %) gel-Elektrophorese von 565 bp-PCR-Fragmente von CaYMV Genomen. Zwei PCR-Produkte wurden in Proben aus der weißen Pellet (Bahnen 1, 2) vorbereitet, aber nicht in die grüne Pellet-Probe (Bahn 3) nachgewiesen. Positivkontrolle (+) stellt ein PCR-Produkt von infizierten Pflanzen-DNA, die mit einem automatisierten Verfahren mit standard paramagnetischen Zellulose Partikel isoliert wurde verstärkt. Lane L enthält die DNA-Leiter als Standard für die Messung der Größe des linearen DNA-Bänder in Probe Bahnen verwendet. (B) Beispiel der Viruspartikel von Transmissions-Elektronenmikroskopie im weißen Pellet erholt von Rohöl Fraktionierung von infizierten Canna Blätter angezeigt. (C) Agarose (0,8 %) gel-Elektrophorese von DNA erholte sich von den grünen (Spur 1) und weiß (Bahn 2) pellets, positiv getestet durch PCR im Bedienfeld "A." Die rote und gelbe Punkte neben Bahn 2 identifizieren zwei hochmolekularen DNA-Bänder, die in den weißen Bruch auftreten. (D) Agarose (1 %) gel-Elektrophorese von Gesamt-RNS durch spaltenbasierten RNA Reinigung wiederhergestellt. Lane L enthält die DNA-Leiter als Standard für die Messung der Größe des linearen Bändern in Probe Bahnen verwendet. Spur 1-6 enthält RNA isoliert von infizierten Canna Blätter, die zusammengefasst wurden auf eine einzelne Probe für Ribo-Erschöpfung und RNA-f (E) schematische Pipeline von Nukleinsäure-Isolationen, Bibliothek Vorbereitung, Sequenzierung, Contig Montage und Virusgenom Entdeckung. Bitte klicken Sie hier für eine größere Version dieser Figur.
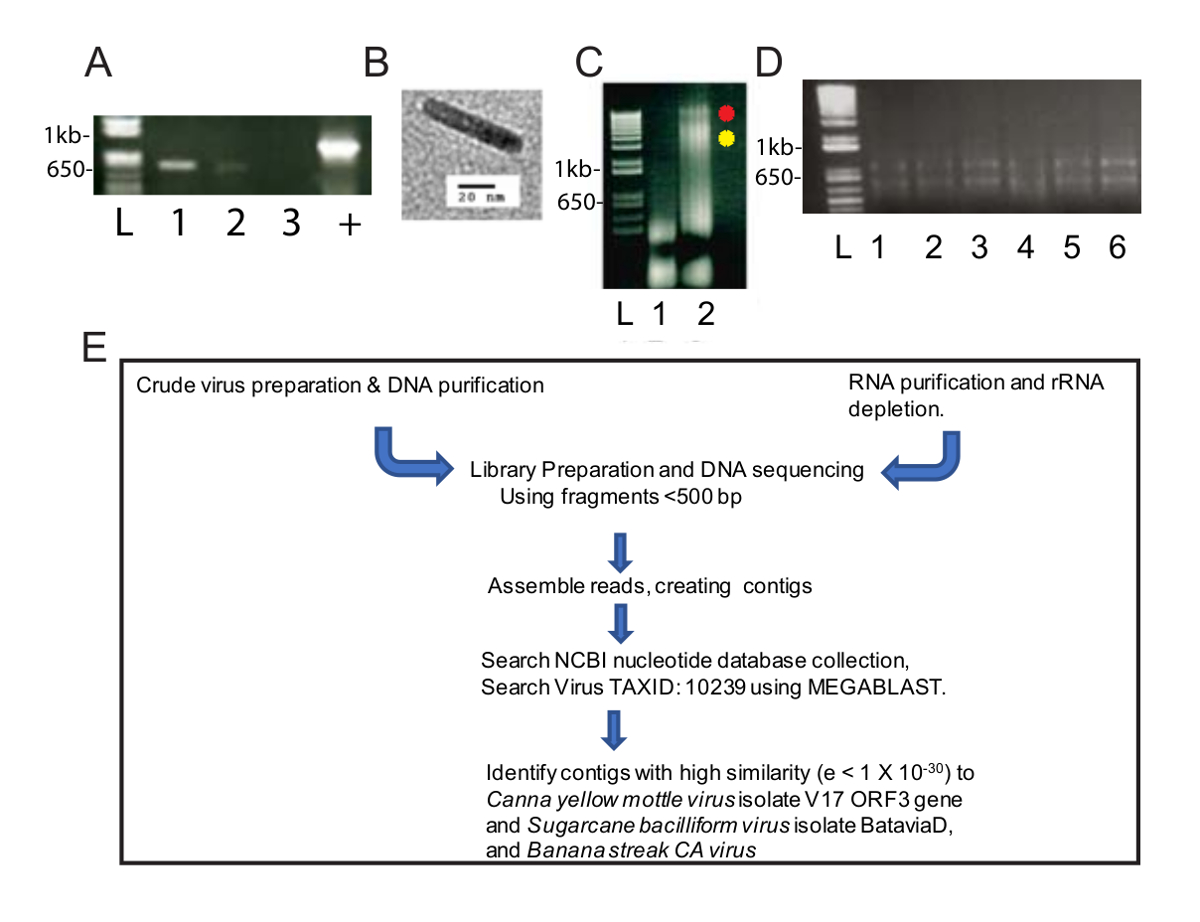{kind=link}

Abbildung 2 : Krone Diagramme visualisieren die taxonomischen Kategorien von Contigs. (A) das Diagramm auf der linken Seite zeigt die Fülle und taxonomische Verteilung von Contigs aus Rohöl Virus Vorbereitung zusammengestellt. Das rechte Diagramm zeigt die Proportionen des Virus Contigs der Familie Caulimoviridae , Badnavirus Gattung und drei nah verwandte Arten zugeordnet. (B) zeigt das Panel auf der linken Seite die Fülle von Contigs RNA-Seq abgeleitet basierend auf deren taxonomische Verteilung. Auf der rechten Seite ist der Graph zeigt die Fülle von Contigs innerhalb der Bevölkerung des Virus Contigs der Familie Caulimoviridae , Badnavirus Gattung und drei nah verwandte Arten zugeordnet. Bitte klicken Sie hier für eine größere Version dieser Figur.
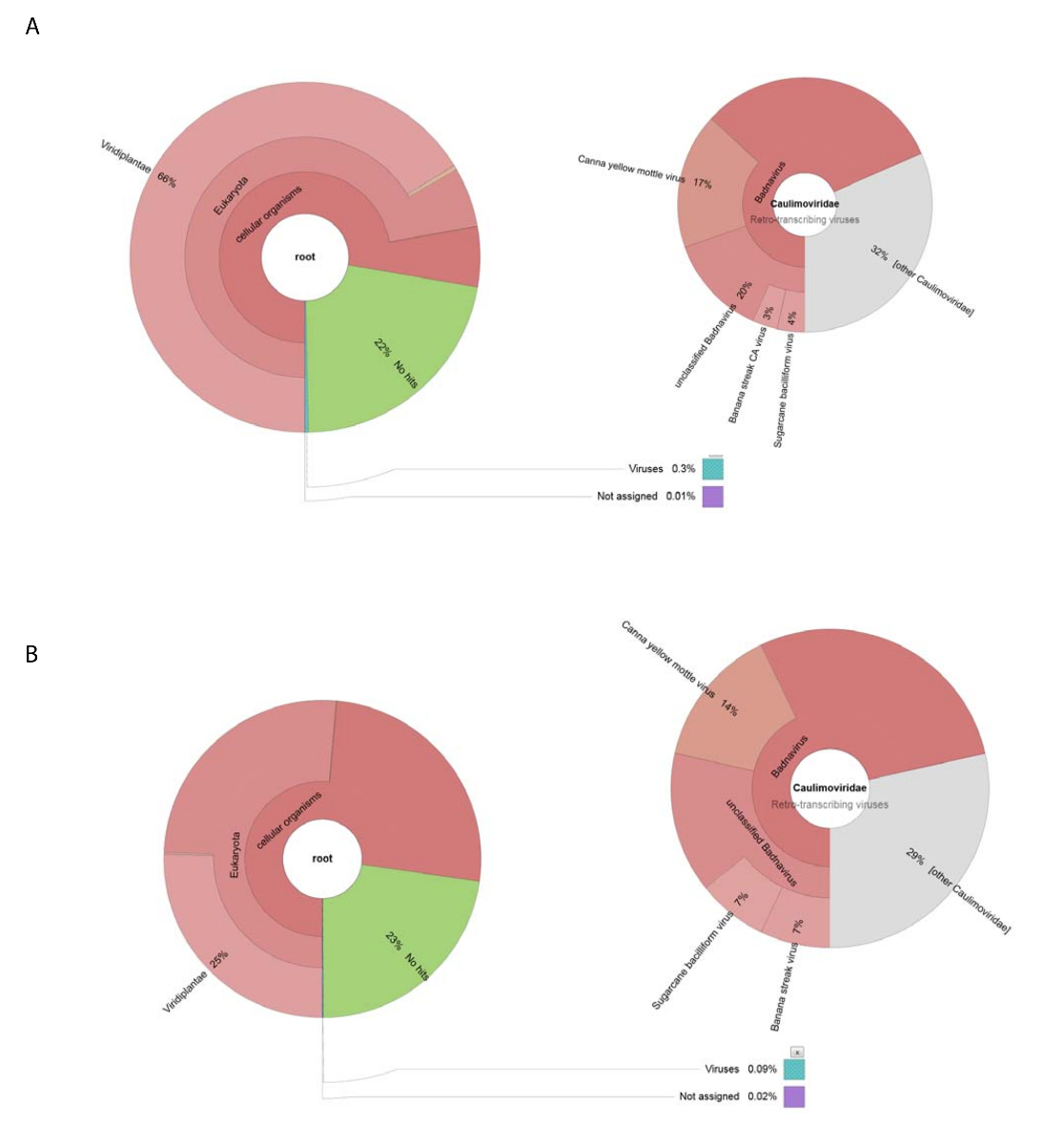{kind=link}

Abbildung 3 . Charakterisierung von CaYMAV-1 und CaYMV-Ap01 Genomen. (A) schematische Darstellung der Canna gelbe Mottle Virus 1 zuordnen (CaYMAV) und Canna gelbe Mottle Virus ähnlich wie das Genom isoliert von Alpinia Purpurata (CaYMV-Ap01). Nukleotid-Positionen 1-10 wird als Beginn des Genoms identifiziert und enthält eine tRNAtraf Anticodon Website typisch für die meisten Badnavirus Genomen. Stop und Start Positionen für die Übersetzung der offenen Leserahmen (ORF) 1 und 2 liegen. Diese Proteine haben unbekannte Funktionen. ORF3 ist ein funkionalen mit Zinkfinger (ZnF), Protease (Pro), Reverse Transkriptase (RT) und RNAse H-Domänen. Eine 3' poly(A) Signalsequenz ist für beide Virus-Genom erhalten. (B) RT-PCR-Analyse wurde durchgeführt mit RNA isoliert vom Virus infizierte Blätter und Grundierungen, die CaYMAV und CaYMV-Ap01 erkennen. In der gleichen Bevölkerung von 12 Pflanzen waren drei mit CaYMV-Ap01 nur infiziert während der verbleibenden mit CaYMAV und CaYMV-Ap01 infiziert waren. (+) zeigt positive Kontrolle und (-) zeigt negative Kontrolle. Diese Zahl ist von Wijayasekara Et Al. reproduziert/geändert 24 mit Erlaubnis. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
Diskussion
In den letzten Jahren wurde eine Vielzahl von Methoden eingesetzt, um pflanzliche Virus Biodiversität in natürlichen Umgebungen zu studieren, darunter bereichernd für virusähnliche Partikel (VLP) oder Virus spezifische RNA oder DNA-2,3,44, 45,46 . Diese Methoden werden von NGS und bioinformatische Analyse gefolgt. Das Ziel dieser Studie war den kausalen Agent eine häufige Erkrankung in eine kultivierte Pflanze zu finden. Die Krankheit wurde berichtet, das Ergebnis von einem unbekannten Virus, die hat unbehüllte Bacilliform Teilchen, und nur ein 565 bp Fragment wurde für die geklonte47. Diese Information war ausreichend für vorherige Forscher hypothetisch das Virus der Gattung Badnavirus innerhalb der Familie Caulimoviridaezuweisen. Während vorherige Berichte vermutet, dass Canna Mottle Krankheit in Canna Lilien das Ergebnis einer einzigen Badnavirus war, mit dem Metagenomik Ansatz in dieser Studie haben wir festgestellt, dass die Krankheit durch zwei vorläufige Badnavirus Arten24verursacht wurde. So ist die Stärke mit einem Metagenom Ansatz, um den kausalen Agent einer Krankheit zu entdecken, dass wir jetzt Situationen zu erkennen wo es mehr als eine Ursache sein kann.
Unser Ansatz, die Kombination von DNA und RNA Sequenzierungsdaten ist gründlich und zeigt auch, dass die Ergebnisse mit zwei Ansätzen konsistente Ergebnisse gezeitigt und das Vorhandensein von zwei verwandten Viren bestätigte. Wir beschäftigt eine modifizierte Verfahren zur Isolierung von Caulimoviruses und produziert eine Stichprobe, wurde associated Virus-Nukleinsäuren bereichert und, innerhalb der Virus-Kapsid geschützt waren. Ein Servicelabor erhielt den Auftrag zur Durchführung der DNA-Sequenzierung. Das wesentliche Konzept für die de Novo Sequenzierung ist, dass die DNA-Polymerase die fluoreszierenden beschriftet Nukleotide in einem DNA-Strang Vorlage beim sequenziellen Zyklen der DNA-Synthese enthält. Die Contigs montiert gefolgt von NGS wurden in einer bioinformatischen Workflow produzieren ein paar Contigs, die als Virus Contigs identifiziert wurden eingereicht. Eine weitere Bestätigung von zwei Viren Genome10,24,48,49,50 wurde durch bioinformatische Analyse der RNA-Seq Daten aus Ribo-abgereicherte RNA Vorbereitungen erhalten. Ein interessantes Ergebnis war zu erfahren, dass die Bevölkerungen der Sequenzen von DNA und RNA Sequenzierung wiederhergestellt sofern ähnliche Verteilungen von nicht-viralen und viralen Nukleinsäuren. Für die Sequenzierung von DNA und RNA, < 0,5 % der Sequenzen waren Virus Ursprungs. Innerhalb der Bevölkerung des Virus Sequenzen 78-82 % der Familie Caulimoviridaegehörte. Durch den Vergleich der montierten Virus Contigs von DNA und RNA Sequenzierung, haben wir bestätigt, dass die zwei montierten Genome in beiden Datensätzen aufgetreten.
Ein Anliegen der Verwendung nur DNA-Sequenzierung, um das neue Virus-Genom zu identifizieren ist, dass das Badnavirus Genom einer offenen kreisförmige DNA. Wir vermutet, dass überlappende Diskontinuitäten im Genom Sequenzen Hindernisse für die Genom-Montage von Contigs darstellen könnte. Erstuntersuchung der DNA-Sequenzierung Ergebnisse ergab zwei ähnliche Viren Genome. Wir die Hypothese auf, dass diese Genome entweder genetische Vielfalt einer Spezies vertreten, die nicht untersucht worden ist, oder vertretene zwei Arten Co infizieren die gleiche Pflanze24. Daher aktiviert die kollektive bioinformatische Analyse von Datensätzen durch die NGS DNA und RNA Sequenzierung, die Bestätigung der Anwesenheit von zwei Genome in voller Länge.
Gibt es einen weiteren Bericht, der eine alternative Methode für die Extraktion VLP und Nukleinsäuren aus Anlage Homogenates für metagenomischen Untersuchungen, basierend auf Verfahren, die DNA von Blumenkohl-Mosaik-Virus (CaMV; eine Caulimovirus)3erholen entwickelt. Dieser Ansatz identifiziert Roman RNA und DNA-Virus Sequenzen in nicht kultivierten Pflanzen. Die Schritte abgeleitet aus dem Caulimovirus-Isolierung-Verfahren in dieser Studie verwendet, um den kausalen Agent auf eine Erkrankung der Kulturpflanzen zu entdecken sind im Gegensatz zu den Schritten zum Extrahieren VLP natürlich infizierten Pflanzen24abgeleitet. Der Erfolg der beiden veränderte Methoden legt nahe, dass das Rahmen-Verfahren zur Caulimovirus Isolierung ein wertvoller Ausgangspunkt für metagenomischen Untersuchungen von Pflanzenviren im Allgemeinen sein kann.
Offenlegungen
Die Autoren haben nichts preisgeben.
Danksagungen
Forschung wurde durch Oklahoma Zentrum für die Förderung von Wissenschaft und Technik angewandte Forschung Programm Phase II AR 132-053-2 finanziert; und von der Oklahoma Abteilung der Landwirtschaft Sonderkulturen Research Grant Program. Wir danken Dr. HongJin Hwang und der OSU Bioinformatik Core Facility, die durch Zuschüsse von NSF (EOS-0132534) und NIH (2P20RR016478-04, 1P20RR16478-02 und 5P20RR15564-03) unterstützt wurde.
Materialien
| Name | Company | Catalog Number | Comments |
| NaH2PO4 | Sigma-Aldrich St. Louis MO | S5976 | Grinding buffer for virus purification |
| Na2HPO4 | Sigma-Aldrich | S0751 | Grinding buffer for virus purification |
| Na2SO3 | Thermo-Fisher Waltham, MA | 28790 | Grinding buffer for virus purification |
| urea | Thermo-Fisher | PB169-212 | Homogenate extraction |
| Triton X-100 | Sigma-Aldrich | X-100 | Homogenate extraction |
| Cheesecloth | VWR Radnor, PA | 21910-107 | Filter homogenate |
| Tris | Thermo-Fisher | BP152-5 | Pellet resuspension& DNA resuspension buffers |
| MgCl2 | Spectrum, Gardena, CA | M1035 | Pellet resuspension buffer |
| EDTA | Spectrum | E1045 | Stops enzyme reactions |
| Proteinase K | Thermo-Fisher | 25530 | DNA resuspension buffer |
| phenol:chloroform:isoamylalcohol | Sigma-Aldrich | P2069 | Dissolve virion proteins |
| DNAse I | Promega | M6101 | Degrade cellular DNA from extracts |
| 95% ethanol | Sigma-Aldrich | 6B-100 | Virus DNA precipitation |
| Laboratory blender | VWR | 58984-030 | Grind leaf samples |
| Floor model ultracentrifuge &Ti70 rotor | Beckman Coulter, Irving TX | A94471 | Separation of cellular extracts |
| Floor model centrifuge and JA-14 rotor | Beckman Coulter | 369001 | Separation of cellular extracts |
| Magnetic stir plate | VWR | 75876-022 | Mixing urea into samples overnight |
| Rubber policeman | VWR | 470104-462 | Dissolve virus pellet |
| 2100 bioanalyzer Instrument | Agilent Genomics, Santa Clare, CA | G2939BA | Sensitive detection of DNA and RNA quality and quantity |
| 2100 Bioanalyzer RNA-Picochip | 5067-1513 | Microfluidics chip used to move, stain and measure RNA quality in a 2100 Bioanalyzer | |
| 2100 Bioanalyzer DNA-High Sensitive chip | 5067-4626 | Microfluidics chip used to move, stain and measure DNA quality in a 2100 Bioanalyzer | |
| Nanodrop spectrophotometer | Thermo-Fisher | ND-2000 | Analysis of DNA/RNA quality at intermediate steps of procedures |
| Plant total RNA isolation kit | Sigma-Aldrich | STRN50-1KT | Isolate RNA for RNA-seq |
| RNase-free water | VWR | 10128-514 | Resuspension of DNA and RNA for NGS |
| RNA concentrator spin column | Zymo Research, Irvine, CA | R1013 | Prepare RNA for RNA-seq |
| rRNA removal kit | Illumina, San Diego, CA | MRZPL116 | Prepare RNA for RNA-seq |
| DynaMag-2 Magnet | ThermoFisher | 12321D | Prepare RNA for RNA-seq |
| RNA enrichment system | Roche | 7277300001 | Prepare RNA for RNA-seq |
| Agarose | Thermo-Fisher | 16500100 | Gel analysis of DNA/RNA quality at intermediate steps of procedures |
| Ethidium bromide | Thermo-Fisher | 15585011 | Agarose gel staining |
| pGEM-T +JM109 competent cells | Promega, Madison, WI | A3610 | Clone genome fragments |
| pFU Taq polymerase | Promega | M7741 | PCR amplify virus genome |
| dNTPs | Promega | U1511 | PCR amplify virus genome |
| PCR oligonucleotides | IDT, Coralvill, IA | Custom order | PCR amplify virus genome |
| Miniprep DNA purification kit | Promega | A1330 | Plasmid DNA purification prior to sequencing |
| PCR clean-up kit | Promega | A9281 | Prepare PCR products for cloning |
| pDRAW32 software | ACAClone | Computer analysis of circular DNA and motifs | |
| MEGA6.0 software | MEGA | Molecular evolutionary genetics analysis | |
| Primer 3.0 | Simgene.com | ||
| Quant-iT™ RiboGreen™ RNA Assay Kit | Thermo-Fisher | R11490 | Fluorometric determination of RNA quantity |
| GS Junior™ pyrosequencing System | Roche | 5526337001 | Sequencing platform |
| GS Junior Titanium EmPCR Kit (Lib-A) | Roche | 5996520001 | Reagents for emulsion PCR |
| GS Jr EmPCR Bead Recovery Reagents | Roche | 5996490001 | Reagents for emulsion PCR |
| GS Junior EmPCR Reagents (Lib-A) | Roche | 5996538001 | Reagents for emulsion PCR |
| GS Jr EmPCR Oil & Breaking Kit | Roche | 5996511001 | Reagents for emulsion PCR |
| GS Jr Titanium Sequenicing kit* | Roche | 5996554001 | Includes sequencing reagents, enzymes, buffers, and packing beads |
| GS Jr. Titanium Picotiter Plate Kit | Roche | 5996619001 | Sequencing plate with associated reagents and gaskets |
| IKA Turrax mixer | 3646000 | Special mixer used with Turrax Tubes | |
| IKA Turrax Tube (specialized mixer) | 20003213 | Specialized mixing tubes with internal rotor for creating emulsions | |
| GS Nebulizers Kit | Roche | 5160570001 | Nucleic acid size fractionator for use during library preparations |
| GS Junior emPCR Bead Counter | Roche | 05 996 635 001 | Library bead counter |
| GS Junior Bead Deposition Device | Roche | 05 996 473 001 | Holder for Picotiter plate during centrifugation |
| Counterweight & Adaptor for the Bead Deposition Devices | Roche | 05 889 103 001 | Used to balance deposition device with picotiter plate centrifugation |
| GS Junior Software | Roche | 05 996 643 001 | Software suite for controlling the instrument, collecting and analyzing data |
| GS Junior Sequencer Control v. 3.0 | Roche | (Included in item 05 996 643 001 above) | |
| GS Run Processor v. 3.0 | Roche | (Included in item 05 996 643 001 above) | |
| GS De Novo Assembler v. 3.0 | Roche | (Included in item 05 996 643 001 above) | |
| GS Reference Mapper v. 3.0 | Roche | (Included in item 05 996 643 001 above) | |
| GS Amplicon Variant Analyzer v. 3.0 | Roche | (Included in item 05 996 643 001 above) |
Referenzen
- Dijkstra, J., Jager, C. P. Practical Plant Virology : Protocols and Exercises. , Springer-Verlag. Berlin Heidelberg. 1 edn (1998).
- Roossinck, M. J. Plant virus metagenomics: biodiversity and ecology. Annu Rev Genet. 46, 359-369 (2012).
- Melcher, U., et al. Evidence for novel viruses by analysis of nucleic acids in virus-like particle fractions from Ambrosia psilostachya. J Virol Methods. 152 (1-2), 49-55 (2008).
- Stobbe, A. H., Schneider, W. L., Hoyt, P. R., Melcher, U. Screening metagenomic data for viruses using the e-probe diagnostic nucleic Acid assay. Phytopathology. 104 (10), 1125-1129 (2014).
- Borah, B. K., et al. Bacilliform DNA-containing plant viruses in the tropics: commonalities within a genetically diverse group. Mol Plant Pathol. 14 (8), 759-771 (2013).
- Bousalem, M., Douzery, E. J., Seal, S. E. Taxonomy, molecular phylogeny and evolution of plant reverse transcribing viruses (family Caulimoviridae) inferred from full-length genome and reverse transcriptase sequences. Arch Virol. 153 (6), 1085-1102 (2008).
- Geering, A. D., et al. Banana contains a diverse array of endogenous badnaviruses. J Gen Virol. 86, Pt 2 511-520 (2005).
- Kunii, M., et al. Reconstruction of putative DNA virus from endogenous rice tungro bacilliform virus-like sequences in the rice genome: implications for integration and evolution. BMC Genomics. 5, 80(2004).
- Laney, A. G., Hassan, M., Tzanetakis, I. E. An integrated badnavirus is prevalent in Figure germplasm. Phytopathology. 102 (12), 1182-1189 (2012).
- Gambley, C. F., Geering, A. D., Steele, V., Thomas, J. E. Identification of viral and non-viral reverse transcribing elements in pineapple (Ananas comosus), including members of two new badnavirus species. Arch Virol. 153 (8), 1599-1604 (2008).
- Gayral, P., et al. A single Banana streak virus integration event in the banana genome as the origin of infectious endogenous pararetrovirus. J Virol. 82 (13), 6697-6710 (2008).
- Lyttle, D. J., Orlovich, D. A., Guy, P. L. Detection and analysis of endogenous badnaviruses in the New Zealand flora. AoB Plants. 2011, 008(2011).
- Le Provost, G., Iskra-Caruana, M. L., Acina, I., Teycheney, P. Y. Improved detection of episomal Banana streak viruses by multiplex immunocapture PCR. J Virol Methods. 137 (1), 7-13 (2006).
- Singh, K., Talla, A., Qiu, W. Small RNA profiling of virus-infected grapevines: evidences for virus infection-associated and variety-specific miRNAs. Funct Integr Genomics. 12 (4), 659-669 (2012).
- Alfson, K. J., Beadles, M. W., Griffiths, A. A new approach to determining whole viral genomic sequences including termini using a single deep sequencing run. J Virol Methods. 208, 1-5 (2014).
- Kreuze, J. F., et al. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology. 388 (1), 1-7 (2009).
- Zheng, Y., et al. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology. 500, 130-138 (2017).
- James, A. P., Geijskes, R. J., Dale, J. L., Harding, R. M. Molecular characterisation of six badnavirus species associated with leaf streak disease of banana in East Africa. Annals of Applied Biology. 158 (3), 346-353 (2011).
- Baranwal, V. K., Sharma, S. K., Khurana, D., Verma, R. Sequence analysis of shorter than genome length episomal Banana streak OL virus like sequences isolated from banana in India. Virus Genes. 48 (1), 120-127 (2014).
- Sukal, A., Kidanemariam, D., Dale, J., James, A., Harding, R. Characterization of badnaviruses infecting Dioscorea spp. in the Pacific reveals two putative novel species and the first report of dioscorea bacilliform RT virus 2. Virus Res. 238, 29-34 (2017).
- BÖmer, M., Turaki, A. A., Silva, G., Kumar, P. L., Seal, S. E. A sequence-independent strategy for amplification and characterisation of episomal badnavirus sequences reveals three previously uncharacterised yam badnaviruses. Viruses. 8 (7), (2016).
- Momol, M. T., Lockhart, B. E. L., Dankers, H., Adkins, S. Canna yellow mottle virus detected in Canna in Florida. Plant Health Progress. , August 2-4 (2004).
- Zhang, J., et al. Characterization of Canna yellow mottle virus in a new host, Alpinia purpurata, in Hawaii. Phytopathology. 107 (6), 791-799 (2017).
- Wijayasekara, D., et al. Molecular characterization of two badnavirus genomes associated with Canna yellow mottle disease. Virus Res. 243, 19-24 (2018).
- Covey, S. N., Noad, R. J., al-Kaff, N. S., Turner, D. S. Caulimovirus isolation and DNA extraction. Methods Mol Biol. 81, 53-63 (1998).
- Sambrook, J., Fritsch, E. F., Maniatis, T. Molecular cloning: A laboratory manual. 2nd edn. , Cold Spring Harbor Press. (1989).
- Radford, A. D., et al. Application of next-generation sequencing technologies in virology. J Gen Virol. 93, Pt 9 1853-1868 (2012).
- Kanagal-Shamanna, R. Emulsion PCR: Techniques and Applications. Methods Mol Biol. 1392, 33-42 (2016).
- Getts, D. R., et al. Targeted blockade in lethal West Nile virus encephalitis indicates a crucial role for very late antigen (VLA)-4-dependent recruitment of nitric oxide-producing macrophages. J Neuroinflammation. 9, 246(2012).
- van Dijk, E. L., Jaszczyszyn, Y., Thermes, C. Library preparation methods for next-generation sequencing: tone down the bias. Exp Cell Res. 322 (1), 12-20 (2014).
- Gel filtration principles and methods. GE Healthcare. , (2010).
- Goff, S., et al. The iPlant Collaborative: Cyberinfrastructure for Plant Biology. Frontiers in Plant Science. 2, (2011).
- Lin, Z., et al. Next-generation sequencing and bioinformatic approaches to detect and analyze influenza virus in ferrets. J Infect Dev Ctries. 8 (4), 498-509 (2014).
- Artimo, P., et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 40, Web Server issue 597-603 (2012).
- Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 5, 113(2004).
- Hung, J. H., Weng, Z. Sequence Alignment and Homology Search with BLAST and ClustalW. Cold Spring Harb Protoc. 2016 (11), (2016).
- Sohpal, V. K., Dey, A., Singh, A. MEGA biocentric software for sequence and phylogenetic analysis: a review. Int J Bioinform Res Appl. 6 (3), 230-240 (2010).
- Untergasser, A., et al. Primer3--new capabilities and interfaces. Nucleic Acids Res. 40 (15), 115(2012).
- Dhaliwa, A. DNA extraction and purification. Mater Methods. 3, 191(2013).
- Moeller, J. R., Moehn, N. R., Waller, D. M., Givnish, T. J. Paramagnetic cellulose DNA isolation improves DNA yield and quality among diverse plant taxa. Appl. Plant Sci. 2 (10), (2014).
- Moeller, J. R., et al. Paramagnetic cellulose DNA isolation improves DNA yield and quality among diverse plant taxa. Appl. Plant Sci. 2 (10), (2014).
- Grooms, K. Review: Improved DNA Yield and Quality from Diverse Plant Taxa. , (2015).
- Nishimori, A., et al. In vitro and in vivo antivirus activity of an anti-programmed death-ligand 1 (PD-L1) rat-bovine chimeric antibody against bovine leukemia virus infection. PLoS One. 12 (4), 0174916(2017).
- Rojas, M. R., Gilbertson, R. L. Plant Virus Evolution. Roossinck, M. J. 1, Springer-Verlag. 27-51 (2008).
- Roossinck, M. J. The big unknown: plant virus biodiversity. Curr Opin Virol. 1 (1), 63-67 (2011).
- Roossinck, M. J., Martin, D. P., Roumagnac, P. Plant Virus Metagenomics: Advances in Virus Discovery. Phytopathology. 105 (6), 716-727 (2015).
- Momol, M. T., Lockhart, B. E. L., Dankers, H., Adkins, S. Plant Health Progress. , Online (2004).
- Eni, A., Hughes, J. D., Asiedu, R., Rey, M. Sequence diversity among badnavirus isolates infecting yam (Dioscorea spp.). Archives of Virology. 153 (12), Ghana, Togo, Benin and Nigeria. 2263-2272 (2008).
- Harper, G., et al. The diversity of Banana streak virus isolates in Uganda. Arch Virol. 150 (12), 2407-2420 (2005).
- Muller, E., Sackey, S. Molecular variability analysis of five new complete cacao swollen shoot virus genomic sequences. Arch Virol. 150 (1), 53-66 (2005).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten