
Method Article
Strukturelle Studien von Makromolekülen in Lösung mit kleinen Winkel Röntgenstreuung
In diesem Artikel
Zusammenfassung
Hier zeigen wir, wie kleine Winkel x-ray Scattering (SAXS) genutzt werden können, um Informationen auf niedriger Auflösung Umschläge repräsentieren die makromolekularen Strukturen zu erhalten. Bei Verwendung in Verbindung mit hochauflösenden strukturelle Techniken wie Röntgen-Kristallographie und magnetischen Kernresonanz bieten SAXS detaillierte Einblicke in multidomain Proteine und makromolekulare komplexe in Lösung.
Zusammenfassung
Protein-Protein-Interaktionen mit Proteinen mit mehreren globulären Domänen präsentieren technische Herausforderungen für die Bestimmung, wie eine solche komplexe Form und wie die Domänen orientierte/positioniert sind. Hier wird ein Protokoll mit dem Potenzial für die Aufklärung welche bestimmte Domänen vermitteln Wechselwirkungen im Mehrkomponenten-System durch ab-initio Modellierung beschrieben. Eine Methode für die Berechnung der Lösung Strukturen von Makromolekülen und ihre Versammlungen ist vorgesehen, die Integration von Daten aus kleinen Winkel Röntgenstreuung (SAXS), Chromatographie und atomarer Auflösung Strukturen zusammen in ein Hybridansatz beinhaltet. Ein konkretes Beispiel ist der Komplex der Full-length Nidogen-1, die extrazelluläre Matrixproteine montiert und bildet eine verlängerte, gekrümmte Nanostruktur. Eines seiner globulären Domänen misst Laminin γ-1, die Strukturen der Basalmembran. Dies bildet die Grundlage für die Bestimmung der genauerer Strukturen flexibel multidomain Proteinkomplexe und Synchrotron Quellen gepaart mit Robotik und Größe Ausgrenzung Chromatographie Automatisierungssysteme aktiviert ist. Diese Kombination ermöglicht schnelle Analyse in mehrere Oligomere Staaten kurz vor SAXS Datenerfassung getrennt sind. Die Analyse liefert Informationen über den Radius der Gyration, Partikel Dimension, molekulare Form und domänenübergreifendes Paarung. Das Protokoll zur Erzeugung von 3D-Modellen der komplexe durch den Einbau hochauflösende Strukturen der Komponente Proteine ist auch gegeben.
Einleitung
Zellen enthalten komplexe Netzwerke von Proteinen, die als molekulare Maschinen zur Durchführung Zellfunktionen wie Signalisierung Kaskaden und strukturelle Integrität zu handeln. Die Art und Weise, in der diese verschiedenen Komponenten bewegen und interagieren im dreidimensionalen Raum, ergeben sich die spezifischen Funktionen der Makromoleküle. Die Bedeutung von Protein-Struktur, Dynamik und Wechselwirkungen bei der Bestimmung der Funktion hat die Notwendigkeit ständig weiterentwickelnde, komplexe Techniken, um diese Eigenschaften zu messen zur Verfügung gestellt. Von diesen Kernspinresonanz (NMR) und Röntgen-Kristallographie (XRC) vor kurzem Cryo-Elektron Mikroskopie (CEM) bieten hochauflösende strukturelle Informationen. Allerdings XRC und CEM Ausbeute Strukturen eines viele biomolekularen Staaten und fehlen Informationen über die Dynamik der Proteinstruktur, während 3D Strukturaufklärung durch NMR auf kleinere kugelförmige Proteine in der Regel begrenzt ist. Eine Möglichkeit, diese Einschränkungen zu überwinden ist, kleine Winkel x-ray Scattering (SAXS) um molekulare Umschläge groß, multidomain oder komplexiert Systeme zu erzeugen, und kombinieren Sie hochauflösende starre makromolekularen Strukturen um die globale erhellen zu nutzen Architektur und dynamischen Eigenschaften.
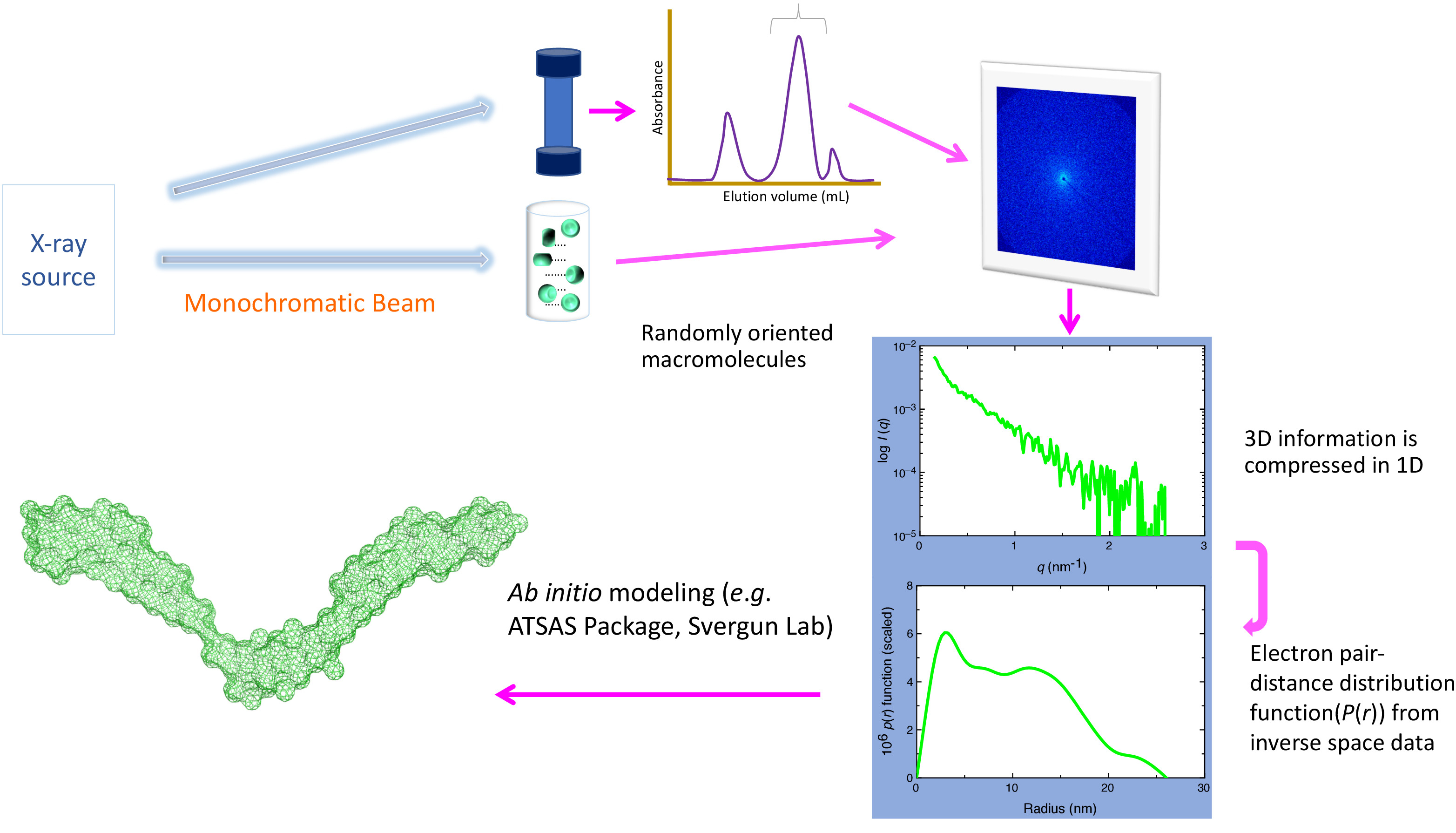
SAXS produziert mit niedriger Auflösung Umschläge der makromolekularen komplexe mit einer Auflösung von ca. 10-20 Å 1Einblick nicht nur in der Struktur, sondern auch die dynamischen Eigenschaften, die die Anlage anzeigt. Obwohl SAXS Röntgenstrahlen nutzt, um molekulare Struktur zu entdecken, ist es im Gegensatz zu XRC, isotrope Wirrlage der Partikel in Lösung nicht führt, Beugung, sondern Streuung, die atomaren Auflösung Ausbeute kann nicht. Stattdessen wird ein Elektron "Umschlag" von dem Makromolekül generiert, die durchschnittlich die Konformationen darstellt, die das Makromolekül anzeigt. Diese Informationen können in direkte Montage der zuvor gelösten atomarer Auflösung Strukturen verwendet werden, Regionen Flexibilität in ein einziges Protein oder Untereinheit Organisation oder Dynamik in einem größeren, Multi-Protein-Komplex ableiten. SAXS Daten werden bei Synchrotrone mit hochenergetischen monochromatischen Röntgenstrahlen oder aus internen Quellen, die eine schwächere Röntgenstrahlen Quelle erfordern Stunden anstatt Sekunden Belichtungszeit Probe (Abbildung 1) zu bieten. SAXS Daten werden oft von mehreren Proben mit einem einzigen Versuchsaufbau und Puffer, erfordern einen längeren Zeitraum eine Runde nützliche Daten auf einem System zu sammeln. Proben, deshalb sollte stabil und nicht aggregiert für mindestens ein paar Stunden basierend auf überprüfbare Qualitätskontrollmethoden wie dynamische Lichtstreuung (DLS) und/oder analytische Ultrazentrifuge (AUC) Analyse, qualitativ hochwertige SAXS Daten2 zu erhalten sein , 3. hier bieten wir eine praktische Beschreibung des SAXS, die Prinzipien hinter seine Verwendung, Vorteile, Einschränkungen und Probenvorbereitung und Fokus stark auf die Datensammlung und Analyse, zusammen mit berühren kurz auf- ab-initio Modellierung mit Hilfe der extrazelluläre Matrix Proteine Nidogen-1 und Laminin γ-1 als experimentelles Beispiel.
Prinzipien, Vorteile und Grenzen der SAXS:
Der leitenden Instandsetzungsprinzipien hinter SAXS ist relativ einfach: eine Lösung der Prozess-Vorbereitung des Macromolecule(s) von Interesse befindet sich innerhalb einer Kapillare und eine hochenergetische einfarbig Röntgenstrahl ausgesetzt ist. Die Photonen dazu führen, dass Elektronen in der Atomhülle oszillierende, beginnen in einer sphärischen Welle wird von der gleichen Energie und Wellenlänge emittiert. Da jedes Elektron schwingen wird, ein konstanter Hintergrund erzielt werden, und der daraus resultierenden Elektronendichte von dem Makromolekül kontrastiert in den Hintergrund. Die daraus resultierende Streuung Intensität wird als Funktion der Streuwinkel, 2Θ gesammelt (Abbildung 1).
Während andere Techniken wie XRC, NMR und CEM Strukturinformationen auf atomarer Ebene bieten, gibt es mehrere Vorteile für SAXS, die anderen Techniken zur Verfügung stellen können. SAXS kann in fast jedem Puffer durchgeführt werden und erfordert besondere Probenvorbereitung. Dies ist besonders wichtig bei der Untersuchung das Verhalten und die Struktur der Makromoleküle unter verschiedenen Bedingungen, wie z. B. das Vorhandensein oder Fehlen von Mono- oder zweiwertigen kationen oder Änderungen im pH4,5. SAXS hat die Fähigkeit, flexibel Regionen ein Makromolekül6, etwas Informationen über die anderen aufgeführten Techniken mit kämpfen können. Daher können SAXS verwendet werden, da eine starke kostenlose Technik mit den stabilen teilen ein Makromolekül, studierte mit XRC, NRM oder CEM und die gesamte Makromolekül oder komplexe in geringer Auflösung mit SAXS analysiert und kombiniert mit verschiedenen Analysetools als FoXSDock7 oder CRYSOL8. Da SAXS eine Lösungstechnik ist, wird es oft verwendet, um zu bestätigen, wenn statische Strukturen wie denen, die von XRC konsequent Lösung6. SAXS hat auch den Vorteil des Seins eine Technik, die eine relativ kleine Menge der Probe Investitionen (in der Regel 50-100 µL) und eine relativ kleine Menge von Experiment Zeit (30 min - 1 h) erfordert.
Die größte Einschränkung SAXS ist die Anfälligkeit für Probe-Aggregation und/oder Abbau, falsche strukturelle Vorhersagen führen kann. Eine Aggregation, sogar so niedrig wie 5 %, kann das Licht in sehr hohen Mengen, führt zu einer Überschätzung der maximalen Partikel Dimension (D-max) und Radius of Gyration (Rg) streuen. Auf der anderen Seite kann Probe Abbau zu einer Unterschätzung der molekularen Eigenschaften führen. Diese Sicherheitsanfälligkeit ergibt sich aus SAXS wird eine Mittelung Technik, was bedeutet, dass die Probe Homogenität ist entscheidend für die zuverlässige und reproduzierbare Ergebnisse zu erzielen. Jede Probe, die von SAXS analysiert werden sollten, daher mehrere Methoden der Reinigung und Homogenität überprüft, wie Geruchsstoffen und gebürtige Gelelektrophorese, Größe Ausgrenzung Chromatographie, dynamische Lichtstreuung unterziehen und analytische Ultrazentrifugation. Oft werden SAXS Beamlines Proben durch Hochleistungs-Flüssigkeitschromatographie als eine abschließende Qualitätskontrolle vor SAXS (S-SAXS)3,9Schritt laufen. SAXS Daten sollten in mehreren Konzentrationen und Rg von jeder Datensatz sollte verglichen werden, gewährleisten eine große Ähnlichkeit zur Vermeidung von interpartikuläre Interaktionen und Aggregation, führt zu einer Überschätzung des Teilchens Abmessungen, was zu ungenauen Datenanalyse und Modellierung. Da die Streuung auf Konzentration und Größe abhängt, erfordern kleinere Makromoleküle eine spezifischere Optimierung der Konzentrationsbereich. Dies liegt an der Gegenseitigkeit Theorem, wo große Größen zu kleinen Winkeln und kleinen Größen gegenüber großen Winkeln streuen. Dies manifestiert sich bei der Datenerhebung, wo ichO ist proportional zu R-6, wo R ist der Radius der Partikel. Eine letzte Einschränkung von SAXS ist das Potenzial für Strahlenschäden der Probe während der Belichtung, die Verzerrung der Daten führen kann. Es empfiehlt sich, Sample-Qualität zu vergleichen, vor und nach SAXS Probe um sicherzustellen, dass dies nicht geschieht.
Protokoll
(1) SAXS probieren, Vorbereitung und Datenerfassung
- Probenvorbereitung:
- SAXS Experimente erfordern homogenen, stabilen und nicht aggregieren Proteinproben; beobachten Sie Stabilität und Oligomere Zustand mit Größe Ausgrenzung Chromatographie (S), DLS und/oder AUC vor der Datenerhebung.
- Proben zu unterwerfen (Nidogen-1 und Laminin γ-1 in diesem Fall) zu DLS Analyse und Tricine SDS-PAGE, Probe Reinheit10zu visualisieren.
Hinweis: Beispiele können eine Reihe von Konzentrationen (1-4 mg/mL) je nach ihrer Größe, ihrem Lösungsverhalten wie Selbstassoziation und Aggregation sowie Stabilität decken; Hier wurden fünf Konzentrationen der Nidogen-1, (139 kDa), drei Laminin γ-1 (109 kDa) und vier von der S-gereinigte äquimolaren Komplex als zuvor beschriebenen10vorbereitet.
- Datensammlung:
- Sammeln Sie SAXS Daten mit einem Inhouse-System oder Synchrotron, nach den Richtlinien des Herstellers oder Anlage.
Hinweis: In dieser Arbeit verwendete Daten wurden mit einem Inhouse-System (siehe Tabelle der Materialien), enthält eine 3-Pinhole-Kamera ausgestattet mit + 002 Mikrofokus versiegelt Rohr (Cu Kα Strahlung bei 1,54 Å) und konfokale Max-Flux (CMF) Optik mit 40 w. Das System ist ausgestattet mit einem 200 nm mehradrigen 2D Detektor für die Datenerfassung. Bieten jedoch mit der Verfügbarkeit von modernen Synchrotrone in Frankreich, Deutschland, UK, USA und anderen Ländern, die Zugang zu einem S-SAXS Set-up, das erleichtert die Trennung einer Prozess-Zubereitung vor möglichen Aggregation/Abbau, wir jetzt routinemäßig Datenerhebung im Synchrotron-Einrichtungen. Ein kürzlich erschienenen Artikel auf eine DNA-G-Quadruplex-11 ist ein Beispiel für eine S-SAXS Data Collection-Strategie. In diesem Fall die SAXS Daten im Bereich von 0,08 ≤ f ≤ 0,26 Å für 3 Stunden für Nidogen-1 (2.0, 2.5, 3.0, 3,5 und 4,0 mg/mL); die Laminin γ-1 (1.5, 2.0 und 2.5 mg/mL) und ihre Anlage (0,8, 1,0, 1,25 und 1,5 mg/mL). - Reduzieren Sie Daten für Puffer und Proben mit Processing-Software speziell für das System. Subtrahieren Sie den Puffer-Beitrag von Protein-Daten mit einem Programm wie PRIMUS/qt12 (Abb. 2A).
- Sammeln Sie SAXS Daten mit einem Inhouse-System oder Synchrotron, nach den Richtlinien des Herstellers oder Anlage.
(2) Datenanalyse
Hinweis: Derzeit gibt es einige Software-Pakete, die nützlich für SAXS Datenanalyse: ScÅtter43 (Download verfügbar auf www.bioisis.net), BioXtas RAW44und die ATSAS Suite13. Dieser Abschnitt enthält eine Übersicht über allgemeine Maßnahmen ergriffen werden, wenn roh SAXS analysieren Daten mithilfe der ATSAS Suite Programm und konkrete Schritte, von der ATSAS 2.8.1 unternommen herunterladen. Andere Programme können verwendet werden und werden später kurz besprochen.
- Puffer-Subtraktion
Hinweis: Diese Schritte sind für statische SAXS Proben nur relevant.- Wählen Sie die Option "Öffnen" Menü in PRIMUS/qt und die Datendateien von Interesse. Beachten Sie, dass Datendateien müssen im ASCII-Format, in denen die erste Spalte der s-Vektor-Achse ist und die zweite Spalte die Intensität ist. Wiederholen Sie diesen Schritt für Daten, die für den Puffer selbst durch Einfügen dieser Daten in dem gleichen Menü.
- Wählen Sie "Subtrahieren" in der Datenverarbeitung-Fenster, das eine subtrahierte Streuung Kurve darstellt, nur Streuung von Makromolekül von Interesse zu erzeugen, wird. Wiederholen Sie diesen Schritt für jede Konzentration.
- Guinier Analyse
- Guinier Analyse durchführen, einen Puffer zu laden subtrahiert Scatter Kurve in PRIMUS/qt in Schritt 2.1.1 wie zuvor beschrieben.
- Klicken Sie auf "Radius of Gyration" die hervorgehen wird, bei der Eröffnung der Primus Guinier Assistent; ein Grundstück von ln(I) Vs Q2 wird angezeigt.
- Um zu erhalten verwenden eine vorläufige Rg die "AUTORG"-Funktion, die ist, dass ein externes Modul eingebaut PRIMUS/Qt finden die "Autorg"-Schaltfläche, und klicken Sie darauf.
- Geben Sie mehrere Dateien auf einmal, indem sie alle markieren und Einfügen in das Menü auf der rechten Seite, ähnlich wie 2.1.1.
- Verwenden Sie die Guinier-Plot zur Bewertung der Datenqualität zuvor erstellte; die grüne Linie unter die Guinier-Plot zeigt die Residuen Handlung repräsentieren eine Linearität der Passung. Beachten Sie, dass nicht-Linearität in der Guinier-Analyse könnte ein Zeichen der Probe Aggregation und weitere Analysen in diesem Fall nicht durchgeführt werden sollten.
Hinweis: Eine lineare Guinier Passform verleiht ein Rg mit einem kleinen Fehler (< 5 %) und schlägt vor, eine qualitativ hochwertige Probe.
- Kratky-Analyse
- Laden Sie die Daten in einer ähnlichen Weise visualisiert werden, wie oben beschrieben.
- Klicken Sie auf "Feld auswählen" neben den Namen der Datei. Dies wird die Daten in einem separaten Fenster zu zeichnen.
- Klicken Sie auf "PLOT" gefolgt von "Kartky Handlung" Auswahl im Dropdown-Menü.
- Dies wird die Daten als "Q2 x L(q) Vs Q" zu zeichnen. Beachten Sie, dass kugelförmige Proteine einen Gaußschen Spitzenwert, anzeigen, während entfaltet Proteine ein Plateau statt einer Spitze zeigt und ähneln einer hyperbolischen Grundstück17.
- Die Datenzusammenführung
- Last-Puffer subtrahiert Daten für jede Konzentration in PRIMUS/qt erneut, wie in Schritt 2.1.1.
- Um die Daten zusammenführen, klicken Sie einfach auf den Button "MERGE" im Fenster "Verarbeitung".
- Überprüfen Sie jede Kurve und ich Waage-Nummer, die auf die Verdünnungen hergestellt aus der ursprünglichen Probe korreliert.
Hinweis: Proben in höherer Konzentration Anzeigen weniger Lärm in der Tail-Region der Kurven.
- Fallt Verteilung
- Um die P(r) Darstellung zu erstellen, laden Sie die zusammengeführten Datenkurven in PRIMUS/qt wie zuvor beschrieben.
- Klicken Sie auf die "Entfernung Distribution" zum Öffnen eines neuen Fensters Präsentation der zusammengeführten Daten der Intensität des gestreuten Licht vs. q und paar-Entfernung Verteilung Funktion Plot auf der rechten Seite.
Hinweis: Die Informationen auf der rechten Seite zeigt die Gesamtqualität der paar-Verteilung Funktion Entfernungsberechnungen. - Passen Sie den Datenbereich der zusammengeführten Daten erheblichen Lärm Ende der raw-Daten zu vermeiden.
- Weglassen von Datenpunkten in der Nähe der Strahl-Haltestelle in der Low-Q-Region.
- Dmax, Beginn mit einem Bereich von ~ 5 Mal die Rg aus der Guinier-Analyse zu bestimmen. Verringern Sie diesen Wert schrittweise, bis das fallt Grundstück nicht abrupt auf Null auf der Y-Achse fällt und verfügt nicht über ein Longtail vor nähert sich Null.
- Überprüfen Sie, dass der experimentellen Rg/ich0 (abgeleitet von Guinier Annäherung) und P(R) Rg/ich0Zahlen sind ähnlich.
Hinweis: In einigen Fällen ist ein weiterer Manipulation auf der Range von Daten, Datenpunkte und ALPHA (Regularisierung Parameter die dem Programm, das Verhältnis sagt von wie viel Aufmerksamkeit die Glätte der Verteilung im Vergleich zur Anpassung der experimentellen Daten) auch erforderliche21 , ein guter Qualität P(R) Grundstück zu erhalten.
(3) einb Initio Wulst Modellierung und im Durchschnitt
- Wenn bei mehreren Konzentrationen erhobenen Daten zusammengeführt werden, oder die Daten mit S-SAXS minimiert und Kratky Plot, P(R) Grundstück und Guinier Analyse überprüft worden, berechnen mit niedriger Auflösung Strukturen von Makromolekülen und Ihre komplexe. Diese Pipeline kann zur Lösung Strukturen und Wechselwirkungen von Nukleinsäuren, Proteinen und Nukleinsäuren-Eiweiß oder Protein-Protein-komplexe10,11,21,22,23 studieren ,24,25,26,27,28,29,30,31,32 ,33,34. Eines der beliebtesten Programme ist DAMMIN, entwickelt von Svergun35 und ein Teil der ATSAS Paket13, die simulierte Glühen Protokolle mit Eingabe Vorausinformation Rg und Dmax beschäftigt . Die ab-initio Modellierungsansätze und Prinzipien beschrieben ausführlich an anderer Stelle18,36.
Ergebnisse
Die oben beschriebene Daten Analyse-Ansatz wurde eingesetzt, um die Rg und Dmax für Nidogen-1, Laminin γ-1 und ihrer Anlage mit der Funktion P(R) zu berechnen. Wir erhalten Rg Werte von 7,20 (±0.10) nm, 8.10 (±0.20) nm und 10,9 (±0.4) nm für Nidogen-1, Laminin γ-1 und ihrer Anlage bzw. (Abbildung 2A-B). Darüber hinaus ist D-max Werte von 24 nm, 26 nm und 35 nm für Nidogen-1, Laminin γ-1 und ihrer Anlage bzw. wurden (Abbildung 2)10 erhalten. Das DAMMIF-Programm wurde verwendet, um mit niedriger Auflösung Strukturen Nidogen-1 und Laminin erhalten γ-1, die vorschlugen, dass beide Proteine in Lösung eine erweiterte Form annehmen. Die Werte für Nidogen-1 (~ 1 und 0,8) und Laminin γ-1 X und NSD (~0.9 und 0,8 bzw.) waren auch im akzeptablen Bereich. Die Ausrichtung der hochauflösende Strukturen erlaubt zwei Domänen Nidogen-1 und zwei Laminin γ-1, auf ihre mit niedriger Auflösung Strukturen unter Verwendung SAXS Identifikation ihrer N - und C-terminalen Regionen10.
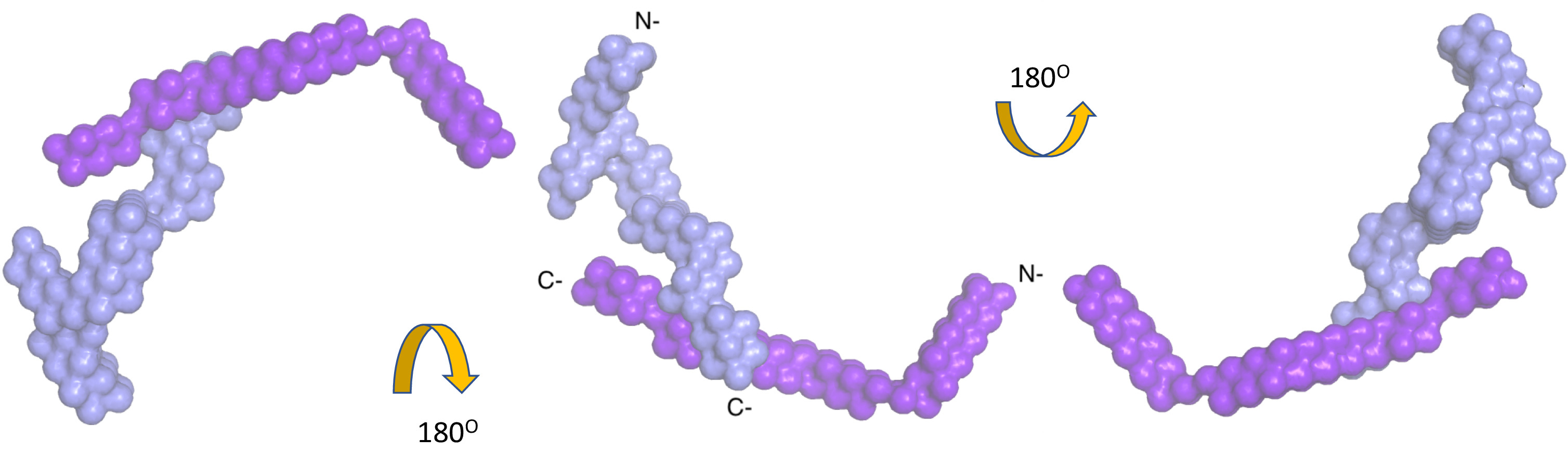
Nidogen-1 wurde als interagierenden Partner von Laminin γ-139,40 identifiziert und die Interaktion Website wurde mithilfe von Röntgen-Kristallographie an der C-terminalen Domänen41zugeordnet. Hochauflösende Strukturen beteiligt jedoch nur interagierenden Domänen und nicht das Full-length Nidogen-1 oder die gesamte Laminin γ-1 Arm. Daher gereinigt wir komplexe enthaltenden Nidogen-1 (volle Länge) und Laminin γ-1 Arm Ermittlung der interagierenden Regionen sowie hinsichtlich um die relative Orientierung der N-terminalen Domänen der beiden Proteine zu studieren. Die SAXS-Daten für den Komplex ergab eine Rg von 10,9 (±0.4) nm und eine DMax. 35 nm. Wir nutzten MONSA um die niedrig aufgelöste Struktur des gesamten Komplexes zu erhalten, schlug vor, dass in der Tat nur die C-terminalen Region der beiden Proteine teilnehmen bei der Vermittlung von Interaktionen, während der Rest der Domains sind weit voneinander entfernt ( Abbildung 3, 1 Video).

Abbildung 1. Schaltpläne SAXS Set-up. Eine Prozess-Zubereitung von Biomolekülen oder ihre komplexe ist bereit, gefolgt von Exposition mit hochenergetischen Röntgenstrahlen. Abhängig von der Quelle (z.B. Inhouse vs. Synchrotron) kann die Energie der Röntgenstrahlen und der Probe Quelle Abstand variieren. Die Röntgenstrahlen Streumuster (das hängt von der Größe und Form von Biomolekülen) aufgezeichnet und radial gemittelt, um eine 1-dimensionale Plot (1D) zu erhalten, der Informationen über die Intensität des gestreuten Lichts in Bezug auf den Streuwinkel enthält. Als Puffer Moleküle auch das Licht streuen, werden die Beiträge aus diesen Molekülen subtrahiert, um eine Lichtstreuung von Biomolekülen von Interesse zu erhalten. Am Synchrotron, vor die SAXS Datenerhebung erfolgt in der Regel auch eine zusätzliche Reinigungsstufe mit Inline-Größe Ausgrenzung/Hochleistungs-Chromatographie (Ansicht von oben). Dieser Schritt ist entscheidend für die aggregierte und/oder geschädigter Produkte sowie alle ungebundenen Biomoleküle aus der Anlage entfernen entfernen. 1D Streuung Plot wird in Elektronenpaar-Entfernung Verteilung Grundstück (ParzelleP(R)), bietet der Radius der Drehung und maximale Partikelgröße Dimension der Biomoleküle umgewandelt. Dieses Grundstück ist als die Eingabedatei für die ab-initio Modellierung Pakete (z.B. DAMMIN/DAMMIF) verwendet, um mit niedriger Auflösung Strukturen der Biomoleküle oder andere Pakete (z.B. SASREF/CORAL) zu erhalten, wenn die hoch aufgelöste Strukturierung der Teile des Die Biomoleküle oder einzelne Biomoleküle des Komplexes ist bekannt. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 2. (A) ein Grundstück von einer Intensität des gestreuten Licht vs. Streuwinkel (Q = 4πsinθ/λ, nm-1) was auf die Qualität der Biomoleküle (niedrige Region) und Form (hohe Region) von Biomolekülen. (B) die Elektronenpaar-Entfernung Verteilung P(R) durch die Streuung der Daten bestimmt empfehlen eine längliche Form von Biomolekülen untersucht (Laminin γ-1, Nidogen-1 und ihrer Anlage). (C) Kratky Grundstück was darauf hindeutet, dass Nidogen-1 und Laminin γ-1 Proteine werden nicht aufgeklappt. (D) Guinier Grundstück für Nidogen-1, Laminin γ-1 und ihre komplexe, unter Angabe des linearen Bereichs zur Bestimmung des Radius der Gyration Datennutzung bei geringem Streuwinkel. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 3. Mit niedriger Auflösung Struktur des Komplexes Nidogen-1 und Laminin γ-1 durch die Analyse der zusammengeführten Datensätze, die mit dem Programm MONSA. Das Farbschema ist dasselbe wie Abbildung 2. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Video 1. Die niedrig aufgelöste Struktur des Nidogen-1 und Laminin γ-1 Komplex. Dieser Film wurde zubereitet PYMOL, verschiedene strukturelle Merkmale des Komplexes zu visualisieren. Die Kristallstruktur des Laminin-Nidogen-Komplexes (PDB ID: 1NPE) als Band-Karikaturen, Hervorhebung der Interaktion für dieses komplexe Websites angezeigt wird. Das Farbschema ist dasselbe wie Abbildung 2. Bitte klicken Sie hier, um dieses Video anzusehen. (Rechtsklick zum download)
Diskussion
Die entscheidenden Schritte der SAXS Datenanalyse beschrieben im Abschnitt "Protokoll" der dieses Papier Include Puffer Subtraktion Guinier Analyse, Kratky, Daten zusammenführen und Analyse P(r) Verteilung. Die ab-initio -Perle-Modellierung ist zu umfangreich, um hier ausführlich behandelt und fällt daher nur kurz.
An Synchrotrons (z.B. DESY in Deutschland, Diamant im Vereinigten Königreich und in Frankreich ESRF), ist es möglich, SAXS Datensammlung für einen winzigen Bruchteil (~ paar µL) jeder Probe als die Brüche sind aus der Spalte "s" eluiert werden d. h. angeschlossenen in-Line (siehe Abbildung 1 ). Die elastisch gestreuten SAXS Daten ist radial gemittelt über die Pakete zur Verfügung gestellt durch die Gerätehersteller oder die Synchrotron bevor Puffer Subtraktion stattfinden kann. Die Ergebnisdaten 1D repräsentiert die Menge an Streulicht (In ich(Q)) auf der Y-Achse und Streuwinkel (Q= 4πsinθ/λ, λ die Wellenlänge des Vorfalls X ist-Strahlung) ist in Abbildung 1dargestellt. Das Programm PRIMUS/qt12 wird verwendet, um direkt jeden beliebigen Hintergrund durch Puffer abziehen und wird in Abschnitt 1.1 beschrieben. Andere Programme wie z. B.; ScÅtter43 (Download verfügbar auf www.bioisis.net) mit einem Tutorial auf https://www.youtube.com/channel/UCvFatdC5HcZOLv6OSjblfeAund BioXtas RAW44 (erhältlich bei https:// bioxtas-raw.readthedocs.io/en/Latest/index.html) als Alternative zu dem ATSAS-Paket genutzt werden.
Die Guinier-Analyse gibt Auskunft auf Probe Aggregation und Homogenität sowie die Makromolekül von Interesse, die anhand der SAXS-Daten aus dem niedrigen s Region14Radius of Gyration (Rg) vorsieht. Ein Grundstück ist konstruiert mit PRIMUS/qt für SAXS Daten aus jeder Konzentration, gefolgt von Kurvenanpassung mit der maximalen Reichweite von bis zu 1,30 für q x Rg. Ein Prozess-Probenvorbereitung soll eine lineare Guinier Handlung in dieser Region (Abb. 2D), während Aggregationsergebnisse in einer nicht-linearen Guinier-15,16 Plot. Wenn die Guinier-Analyse linear ist, kann der Grad der "Unfoldedness" von einem Makromolekül von Interesse mit der Kratky Handlung beobachtet was nützlich ist, wenn die Entscheidung, ob Festkörper-Modellierung oder Ensembles mit niedriger Auflösung Modelle zu konstruieren. Eine kugelförmige Protein erscheint in einem Kratky Plotten eine glockenförmigen Kurve haben Moleküle ausgedehnt oder entfaltet Peptide erscheint plateau oder sogar im Bereich von größeren q erhöhen und fehlt die Glockenform (Abbildung 2).
Erlangung von Rg aus Guinier Analyse berücksichtigt nur Datenpunkte aus der niedrigen Q -Region der 1D Streudiagramm (Abb. 2D), jedoch ist es möglich, fast das gesamte Dataset verwenden, um eine indirekte Fourier-Transformation durchzuführen um die gegenseitige Rauminformationen der ln (I (Q)) vs. (Q) in einer realen Raum Abstand Verteilungsfunktion (P(R)) umwandeln, die über Dmax und Rg informiert (Abb. 2 b) Die Form der Handlung P(R) repräsentiert die grobe Lösung Konformation der Makromolekül Interesse18,19. die Umstellung der Kehrwert Raumdaten auf Real-Daten ist ein wichtiger Schritt, aber eine ausführliche Beschreibung ist nicht in den Anwendungsbereich dieses Papier. Daher beziehen sich auf einen Artikel von Svergun20 , jeden Parameter zu verstehen.
Sobald der Puffer abgezogen bei einzelnen Konzentrationen verarbeitet werden durch Guinier Analyse mit einen konsistenten Wert für R-g, gefolgt von ihrer Faltungsmuster Kratky-Analyse untersucht, diese Daten zusammengeführt werden können. Die zusammengeführten Daten werden Nidogen-1, Laminin γ-1 und ihre Anlage wie oben beschrieben verarbeitet wurden und die sich daraus ergebenden P(r) Grundstücke in Figur 2 bdargestellt. Im Idealfall sollte man auch paar-Entfernung Verteilungsfunktion fallt für jede Konzentration zu bestimmen, ob SAXS Daten für jede Konzentration ähnliche Rg und D-max -Werte bietet berechnen. Wenn die R,g und Dmax über einen weiten Bereich von Konzentrationen ähnlich bleiben, sollte der Benutzer gehen. Es sei darauf hingewiesen, dass je nach Signal, Daten vor dem Zusammenführen der Daten abgeschnitten werden können. Dies ist oft der Fall, wenn die Konzentrationen und/oder Molekulargewicht der Makromoleküle untersucht ist gering.
Mit niedriger Auflösung Form Analyse mit DAMMIN kann in verschiedenen Modi ausgeführt werden (z. B. Fast, Slow, Experten-Modi, etc.). Der schnelle Modus ist ein idealer erster Schritt zu bewerten, ob das P(r) Grundstück gute Qualitätsmodelle bietet. In der Regel mindestens 10 Modelle erhalten Sie für jedes P(r) Grundstück, wenn reproduzierbare Ergebnisse in Bezug auf die niedrige Auflösung Struktur mit einer niedrigen Güte von Fit Parameter mit dem Namen χ (ein Wert von 0,5-1,0 gilt als gut basierend auf unserer umfangreichen Arbeit erhalten werden, zu überprüfen ), ein Wert, der eine Vereinbarung zwischen experimentell ermittelten SAXS Daten und Modell abgeleiteten Daten beschreibt. Zwecks Veröffentlichung wir in der Regel langsam oder Expert Modus verwenden und mindestens 15 Modellen zu berechnen. Neben DAMMIN, eine schnellere Version des es, DAMMIF37, sowie GASBOR38 sind auch Alternativen. Darüber hinaus um Protein-Protein oder Eiweiß-Nuclein-Säure-komplexe zu studieren, ist es möglich, die MONSA Programm35, verwenden die gleichzeitige Montage von SAXS Einzeldaten für Makromoleküle sowie ihre Anlage erleichtert. Weitere Einzelheiten über hochauflösende Modellrechnungen sowie für RNS-Protein-Interaktion-Studien finden Sie in einem kürzlich erschienenen Artikel von Patel Et al.3.
SAXS ist theoretisch einfach, aber zweifellos eine sehr ergänzende Methode zu anderen Strukturbiologie Tools und Ergebnisse in niedriger Auflösung Strukturdaten, die eigenständig verwendet werden können oder in Verbindung mit hochauflösenden Techniken, um Informationen zu erhellen über die makromolekulare Struktur und Dynamik. Solange eine Prozess-Vorbereitung von Makromolekülen und ihre komplexe abgerufen werden kann, können SAXS genutzt werden, um in Lösung Struktur und Interaktionen von jeder Art von biologischen Makromolekül zu studieren. Bei den komplexen hier diskutiert, ist es bemerkenswert, dass weniger als 10 % der insgesamt zugängliche Fläche von Stickstoff-1 und Laminin γ-1 in diesem Komplex begraben, während der Rest der Domänen der beiden Proteine sind frei zugänglich, mit anderen zu interagieren Proteine an die extrazelluläre Matrix zu seiner strukturellen Steifigkeit (Abbildung 3). Erlangung dieser Informationen für einen Komplex mit ~ 240kDa wäre sehr schwierig, mit anderen Strukturbiologie Techniken wie Röntgen-Kristallographie, NMR und Cryo-EM-Mikroskopie.
Aufdeckung Proteinstruktur mittels Röntgenkristallographie oder NMR ist ein von Natur aus zeitaufwändiger Vorgang. Dieser Engpass bei der Strukturaufklärung ist ein Bereich, wo SAXS seine Stärke als strukturelle Technik zeigt; Datenerfassung für einen einzigen SAXS Experiment kann weniger als eine Stunde dauern und mit Hilfe der optimierten Analyse-Software, kann Analysen schnell und effizient erledigt werden. SAXS hat das Potenzial, stark Durchsatz der strukturelle Studien als Stand-alone-Technik erhöhen, denn es Modellstadt mit niedriger Auflösung für die makromolekulare Struktur, bietet bevor Feindaten verfügbar ist. Ein Hindernis für andere strukturelle Techniken ist die Voraussetzung für eine hochreine, konzentrierte Probe für die Datenerfassung, die ein hohes Maß an Protein-Expression und Stabilität über einen langen Zeitraum hinweg erfordert. Während SAXS auch müssen rein und konzentriert sein Proben, die Probenvolumina sind etwa 100 µL SAXS eine relativ kostengünstige Methode zur Analyse im Vergleich zu anderen strukturellen Techniken machen. Darüber hinaus ist SAXS gepaart mit Größe Ausgrenzung Chromatographie immer häufiger die bietet eines zusätzliche Qualitätskontrolle Schritt. Vor kurzem gab es starke Fortschritte in der Kombination von NMR und SAXS Daten mit dem Ensemble Optimierung Methode (EOM)45,46 , flexible Systeme aufzuklären. In einem jüngst veröffentlichten Papier von Mertens und Svergun47beschreiben die Autoren mehrere aktuelle Beispiele von EOM SAXS in Kombination mit NMR, zusammen mit vielen anderen Beispielen von SAXS Daten in Verbindung mit NMR verwendet werden. Werden kontinuierlich Fortschritte auf dem Gebiet der SAXS werden, und neue Techniken entwickelt für SAXS in Verbindung mit verwendet werden, nicht nur komplementär zu anderen strukturellen Techniken. Daher glauben wir, dass die Nachfrage für SAXS wird nur im Laufe der Zeit, besonders in Verbindung mit NMR, dynamische Systeme zu charakterisieren, wo Funktionen durch Flexibilität definiert sind.
Offenlegungen
Autoren haben keine Angaben zu erklären.
Danksagungen
Dieses Projekt wurde von NSERC RGPIN-2018-04994, Campus Alberta Innovationsprogramm (RCP-12-002 C) und Alberta Prion Research Institute unterstützt / Alberta innoviert Bio Solutions (201600018) Stipendien M.O TRP ist ein Canada Research Chair in RNA & Protein-Biophysik (201704) und erkennt NSERC Discovery Grant (RGPIN-2017-04003). TM wird finanziert durch die NSERC Entdeckung Zuschuss an TRP
Materialien
| Name | Company | Catalog Number | Comments |
| HEK 293 EBNA Cell Line | In-Lab availability | - | Cell line used to overexpress protein(s) |
| Ni Sepharose High Performance histidine-tagged protein purification resin | GE Healthcare | 17524801 | Affinity protein purification resin |
| Superdex 200 Increase 10/300 | GE Healthcare | 28990944 | SEC Column |
| ÄKTA Pure FPLC | GE Healthcare | - | FPLC System |
| Nanodrop | Nanodrop | - | Spectrophotometer |
| Basic Reagents (NaCl, Tris-HCl etc.) | |||
| S-MAX3000 | Rigaku | - | SAXS Pinhole Camera System |
| Zetasizer Nano-S | Malvern Instruments Ltd | - | Dynamic Light Scattering instrument |
| 0.1µm Filter | Millipore | JVWP04700 | Used to Concentrate Sample Prior to DLS |
| Thrombin cleavage kit | abcam | ab207000 | Thrombin cleavage to remove His tag |
| Strep-Tactin Sepharose Column | IBA | 2-1201-010 | Strep-Tag Affinity Purification |
| D-desthiobiotin | Sigma-Aldrich | 533-48-2 | Elution of Strep Tag Protein |
| Software | |||
| SAXGUI | Rigaku | - | Data Collection for SAXS and data reduction |
| ATSAS Suit | Franke et al., 2017 | - | SAXS Data Analysis Software program suite |
| PRIMUS | Konarev et al., 2003 | - | Buffer Subtraction |
| GNOM | Svergun, 1992 | - | Rg, Dmax and p(r) Calculation |
| DAMMIF | Franke and Svergun, 2009 | - | Ab initio model calculation |
| DAMAVER | Volkov and Svergun, 2003 | - | Averaged Solution Conformation calculations |
| MONSA | Svergun, 1999 | - | Simultaneous model fitting for the complex |
| GASBOR | Svergun et al., 2001 | - | Alternative Ab initio model calculations |
| DTS Software V6.20 | Malvern Instruments Ltd | - | DLS supplied instrument software |
| PyMOL | Schrodinger, LLC. | - | The PyMOL Molecular Graphics System V2.0 |
| The Protein Data Bank | Berman et al., 2000 | - | PDB ID: 1NPE |
Referenzen
- Svergun, D. I., Koch, M. H. J. Advances in structure analysis using small-angle scattering in solution. Current Opinion in Structural Biology. 12 (5), 654-660 (2002).
- Patel, T. R., Chojnowski, G., Koul, A., McKenna, S. A., Bujnicki, J. M. Structural studies of RNA-protein complexes: A hybrid approach involving hydrodynamics, scattering, and computational methods. Methods. 118, 146-162 (2017).
- Stetefeld, J., McKenna, S. A., Patel, T. R. Dynamic light scattering: a practical guide and applications in biomedical sciences. Biophysical Reviews. 8 (4), 409-427 (2016).
- Uversky, V. N., Gillespie, J. R., Fink, A. L. Why are “natively unfolded” proteins unstructured under physiologic conditions?. Proteins. 41 (3), 415-427 (2000).
- Uversky, V. N., Gillespie, J. R., Millett, I. S., Khodyakova, A. V., Vasiliev, A. M., Chernovskaya, T. V., Abramov, V. M. Natively Unfolded Human Prothymosin α Adopts Partially Folded Collapsed Conformation at Acidic pH. Biochemistry. 38 (45), 15009-15016 (1999).
- Kikhney, A. G., Svergun, D. I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Letters. 589 (19 Pt A), 2570-2577 (2015).
- Schneidman-Duhovny, D., Hammel, M., Tainer, J. A., Sali, A. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Research. 44, 424-429 (2016).
- Svergun, D., Barberato, C., Koch, M. H. J. CRYSOL - a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. Journal of Applied Crystallography. 28 (6), 768-773 (1995).
- Pérez, J., Vachette, P. A Successful Combination: Coupling SE-HPLC with SAXS. Advances in Experimental Medicine and Biology. 1009, 183-199 (2017).
- Patel, T. R., Bernards, C., Meier, M., McEleney, K., Winzor, D. J., Koch, M., Stetefeld, J. Structural elucidation of full-length nidogen and the laminin-nidogen complex in solution. Matrix Biology. 33, 60-67 (2014).
- Meier, M., Moya-Torres, A., Krahn, N. J., McDougall, M. D., Orriss, G. L., McRae, E. K. S. Structure and hydrodynamics of a DNA G-quadruplex with a cytosine bulge. Nucleic Acids Research. , (2018).
- Franke, D., Petoukhov, M. V., Konarev, P. V., Panjkovich, A., Tuukkanen, A., Mertens, H. D. T. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. Journal of Applied Crystallography. 50, 1212-1225 (2017).
- Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., Svergun, D. I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography. 36 (5), 1277-1282 (2003).
- Tuukkanen, A. T., Svergun, D. I. Weak protein-ligand interactions studied by small-angle X-ray scattering. The FEBS Journal. 281 (8), 1974-1987 (2014).
- Rambo, R. P., Tainer, J. A. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 496 (7446), 477-481 (2013).
- Koch, M. H., Vachette, P., Svergun, D. I. Small-angle scattering: a view on the properties, structures and structural changes of biological macromolecules in solution. Quarterly Reviews of Biophysics. 36 (2), 147-227 (2003).
- Putnam, C. D., Hammel, M., Hura, G. L., Tainer, J. A. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Quarterly Reviews of Biophysics. 40 (3), 191-285 (2007).
- Petoukhov, M. V., Svergun, D. I. Applications of small-angle X-ray scattering to biomacromolecular solutions. The International Journal of Biochemistry & Cell Biology. 45 (2), 429-437 (2013).
- Svergun, D. I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. Journal of Applied Crystallography. 25 (4), 495-503 (1992).
- Patel, T. R., Morris, G. A., Zwolanek, D., Keene, D. R., Li, J., Harding, S. E. Nano-structure of the laminin γ-1 short arm reveals an extended and curved multidomain assembly. Matrix Biology: Journal of the International Society for Matrix Biology. 29 (7), 565-572 (2010).
- Patel, T. R., Reuten, R., Xiong, S., Meier, M., Winzor, D. J., Koch, M., Stetefeld, J. Determination of a molecular shape for netrin-4 from hydrodynamic and small angle X-ray scattering measurements. Matrix Biology: Journal of the International Society for Matrix Biology. 31 (2), 135-140 (2012).
- Dzananovic, E., Patel, T. R., Deo, S., McEleney, K., Stetefeld, J., McKenna, S. A. Recognition of viral RNA stem-loops by the tandem double-stranded RNA binding domains of PKR. RNA. 19 (3), 333-344 (2013).
- Meier, M., Patel, T. R., Booy, E. P., Marushchak, O., Okun, N., Deo, S. Binding of G-quadruplexes to the N-terminal recognition domain of the RNA helicase associated with AU-rich element (RHAU). The Journal of Biological Chemistry. 288 (49), 35014-35027 (2013).
- Dzananovic, E., Patel, T. R., Chojnowski, G., Boniecki, M. J., Deo, S., McEleney, K. Solution conformation of adenovirus virus associated RNA-I and its interaction with PKR. Journal of Structural Biology. 185 (1), 48-57 (2014).
- Bacik, J. -. P., Tavassoli, M., Patel, T. R., McKenna, S. A., Vocadlo, D. J., Khajehpour, M., Mark, B. L. Conformational itinerary of Pseudomonas aeruginosa 1,6-anhydro-N-acetylmuramic acid kinase during its catalytic cycle. The Journal of Biological Chemistry. 289 (7), 4504-4514 (2014).
- Deo, S., Patel, T. R., Dzananovic, E., Booy, E. P., Zeid, K., McEleney, K. Activation of 2’ 5’-oligoadenylate synthetase by stem loops at the 5’-end of the West Nile virus genome. PloS One. 9 (3), e92545 (2014).
- Vadlamani, G., Thomas, M. D., Patel, T. R., Donald, L. J., Reeve, T. M., Stetefeld, J., Mark, B. L. The β-lactamase gene regulator AmpR is a tetramer that recognizes and binds the D-Ala-D-Ala motif of its repressor UDP-N-acetylmuramic acid (MurNAc)-pentapeptide. The Journal of Biological Chemistry. 290 (5), 2630-2643 (2015).
- Deo, S., Patel, T. R., Chojnowski, G., Koul, A., Dzananovic, E., McEleney, K., McKenna, S. A. Characterization of the termini of the West Nile virus genome and their interactions with the small isoform of the 2’ 5’-oligoadenylate synthetase family. Journal of Structural Biology. 190 (2), 236-249 (2015).
- Ariyo, E. O., Booy, E. P., Patel, T. R., Dzananovic, E., McRae, E. K., Meier, M., McKenna, S. A. Biophysical Characterization of G-Quadruplex Recognition in the PITX1 mRNA by the Specificity Domain of the Helicase RHAU. PloS One. 10 (12), (2015).
- Hyde, E. I., Callow, P., Rajasekar, K. V., Timmins, P., Patel, T. R., Siligardi, G., Scott, D. J. Intrinsic disorder in the partitioning protein KorB persists after co-operative complex formation with operator DNA and KorA. The Biochemical Journal. 474 (18), 3121-3135 (2017).
- Dzananovic, E., Astha, n. u. l. l., Chojnowski, G., Deo, S., Booy, E. P., Padilla-Meier, P., McKenna, S. A. Impact of the structural integrity of the three-way junction of adenovirus VAI RNA on PKR inhibition. PloS One. 12 (10), (2017).
- Krahn, N., Meier, M., To, V., Booy, E. P., McEleney, K., O’Neil, J. D., Stetefeld, J. Nanoscale Assembly of High-Mobility Group AT-Hook 2 Protein with DNA Replication Fork. Biophysical Journal. 113 (12), 2609-2620 (2017).
- Patel, T. R., Besong, T. M. D., Meier, M., McEleney, K., Harding, S. E., Winzor, D. J., Stetefeld, J. Interaction studies of a protein and carbohydrate system using an integrated approach: a case study of the miniagrin-heparin system. European Biophysics Journal: EBJ. , (2018).
- Svergun, D. I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophysical Journal. 76 (6), 2879-2886 (1999).
- Blanchet, C. E., Svergun, D. I. Small-Angle X-Ray Scattering on Biological Macromolecules and Nanocomposites in Solution. Annual Review of Physical Chemistry. 64 (1), 37-54 (2013).
- Volkov, V. V., Svergun, D. I. Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography. 36, 860-864 (2003).
- Kozin, M. B., Svergun, D. I. Automated matching of high- and low-resolution structural models. Journal of Applied Crystallography. 34 (1), 33-41 (2001).
- Franke, D., Svergun, D. I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. Journal of Applied Crystallography. 42 (Pt 2), 342-346 (2009).
- Svergun, D. I., Petoukhov, M. V., Koch, M. H. J. Determination of Domain Structure of Proteins from X-Ray Solution Scattering. Biophysical Journal. 80 (6), 2946-2953 (2001).
- Baumgartner, R., Czisch, M., Mayer, U., Pöschl, E., Huber, R., Timpl, R., Holak, T. A. Structure of the nidogen binding LE module of the laminin gamma1 chain in solution. Journal of Molecular Biology. 257 (3), 658-668 (1996).
- Stetefeld, J., Mayer, U., Timpl, R., Huber, R. Crystal structure of three consecutive laminin-type epidermal growth factor-like (LE) modules of laminin gamma1 chain harboring the nidogen binding site. Journal of Molecular Biology. 257 (3), 644-657 (1996).
- Takagi, J., Yang, Y., Liu, J. -. H., Wang, J. -. H., Springer, T. A. Complex between nidogen and laminin fragments reveals a paradigmatic beta-propeller interface. Nature. 424 (6951), 969-974 (2003).
- Förster, S., Apostol, L., Bras, W. Scatter: software for the analysis of nano- and mesoscale small-angle scattering. J. Appl. Cryst. 43, 639-646 (2010).
- Hopkins, J. B., Gillilan, R. E., Skou, S. BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Cryst. 50, 1545-1553 (2017).
- Bernadó, P., Mylonas, E., Petoukhov, M. V., Blackledge, M., Svergun, D. I. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc. (17), 5656-5664 (2007).
- Tria, G., Mertens, H. D., Kachala, M., Svergun, D. I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ. 2 (Pt 2), 207-217 (2015).
- Mertens, H. D. T., Svergun, D. I. Combining NMR and small angle X-ray scattering for the study of biomolecular structure and dynamics. Arch Biochem Biophys. 628, 33-41 (2017).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten