
Method Article
Verwendung der Open-Source MALDI TOF-MS IDBac Pipeline zur Analyse von mikrobiellen Protein- und Spezialisierten Metabolitendaten
In diesem Artikel
Zusammenfassung
IDBac ist eine Open-Source-Pipeline für Massenspektrometrie, die Daten sowohl aus intakten Proteinen als auch aus spezialisierten Metabolitenspektren integriert, die auf Zellmaterial gesammelt werden, das aus Bakterienkolonien abgekratzt wurde. Die Pipeline ermöglicht es Forschern, schnell Hunderte bis Tausende von Bakterienkolonien in vermeintliche taxonomische Gruppen zu organisieren und sie auf der Grundlage der spezialisierten Metabolitenproduktion weiter zu differenzieren.
Zusammenfassung
Um die Beziehung zwischen bakterieller Phylogenie und spezialisierter Metabolitenproduktion von Bakterienkolonien, die auf Nährstoffagar wachsen, zu visualisieren, haben wir IDBac entwickelt – eine kostengünstige und hochdurchsatzbasierte Matrix-unterstützte Laserdesorption/Ionisierung. Zeit-des-Flug-Massenspektrometrie (MALDI-TOF MS) Bioinformatik-Pipeline. IDBac Software ist für Nicht-Experten entwickelt, ist frei verfügbar, und in der Lage, ein paar bis Tausende von Bakterienkolonien zu analysieren. Hier stellen wir Verfahren zur Vorbereitung von Bakterienkolonien für die MALDI-TOF MS-Analyse, MS-Instrumentenbedienung sowie Datenverarbeitung und Visualisierung in IDBac vor. Insbesondere weisen wir Anwender an, Bakterien auf Basis von Protein-MS-Fingerabdrücken in Dendrogramme zu gruppieren und interaktiv Metabolite Association Networks (MANs) aus spezialisierten Metabolitendaten zu erstellen.
Einleitung
Ein großes Hindernis für Forscher, die die bakterielle Funktion untersuchen, ist die Fähigkeit, schnell und gleichzeitig die taxonomische Identität eines Mikroorganismus und seine Fähigkeit zur Herstellung spezialisierter Metaboliten zu bewerten. Dies hat signifikante Fortschritte beim Verständnis der Beziehung zwischen bakterieller Phylogenie und spezialisierter Metabolitenproduktion in der Mehrheit der aus der Umwelt isolierten Bakterien verhindert. Obwohl MS-basierte Methoden, die Protein-Fingerabdrücke verwenden, um Bakterien zu gruppieren und zu identifizieren, gut beschrieben sind1,2,3,4, wurden diese Studien im Allgemeinen an kleinen Gruppen von Isolaten durchgeführt, artspezifisch. Wichtig ist, dass Informationen über die spezialisierte Metabolitenproduktion, ein wichtiger Treiber der mikrobiellen Funktion in der Umwelt, in diesen Studien nicht berücksichtigt wurden. Silva et al.5 lieferten vor kurzem eine umfassende Geschichte, die die Unternutzung von MALDI-TOF MS zur Analyse spezialisierter Metaboliten und den Mangel an Software zur Linderung aktueller Bioinformatik-Engpässe detailliert darlegte. Um diese Mängel zu beheben, haben wir IDBac geschaffen, eine Bioinformatik-Pipeline, die sowohl lineare als auch reflekronische Modi von MALDI-TOF MS6integriert. Dies ermöglicht es Benutzern, bakterielle Isolate schnell zu visualisieren und zu unterscheiden, die sowohl auf Protein- als auch auf spezialisierten Metaboliten-MS-Fingerabdrücken basieren.
IDBac ist kostengünstig, mit hohem Durchsatz und für den Laienbenutzer konzipiert. Es ist frei verfügbar (chasemc.github.io/IDBac) und erfordert nur Zugriff auf ein MALDI-TOF-Massenspektrometer (für die spezielle Metabolitenanalyse ist ein Reflekronmodus erforderlich). Die Probenvorbereitung basiert auf der einfachen Methode der "erweiterten direkten Übertragung"7,8 und die Daten werden mit aufeinanderfolgenden linearen und reflekronischen Erfassungen an einem einzigen MALDI-Zielpunkt gesammelt. Mit IDBac ist es möglich, die vermeintliche Phylogenie und spezialisierte Metabolitenproduktion von Hunderten von Kolonien in weniger als vier Stunden zu analysieren, einschließlich Probenvorbereitung, Datenerfassung und Datenvisualisierung. Dies stellt einen erheblichen Zeit- und Kostenvorteil gegenüber herkömmlichen Methoden zur Identifizierung von Bakterien (z. B. Gensequenzierung) und zur Analyse des metabolischen Outputs (Flüssigchromatographie-Massenspektrometrie [LCMS] und ähnliche chromatographische Methoden) dar.
Mithilfe von Daten, die in der Linearmodusanalyse gewonnen wurden, verwendet IDBac hierarchisches Clustering, um die Verwandtheit von Proteinspektren darzustellen. Da die Spektren meist ionisierte ribosomale Proteine darstellen, bieten sie eine Darstellung der phylogenetischen Vielfalt, die in einer Probe vorhanden ist. Darüber hinaus enthält IDBac Daten im Reflectron-Modus, um spezielle Metaboliten-Fingerabdrücke als Metabolit-Assoziationsnetzwerke (MANs) anzuzeigen. MANs sind zweiteilige Netzwerke, die eine einfache Visualisierung der gemeinsamen und einzigartigen Metabolitenproduktion zwischen bakteriellen Isolaten ermöglichen. Die IDBac-Plattform ermöglicht es Forschern, sowohl Protein- als auch spezialisierte Metabolitendaten im Tandem, aber auch einzeln zu analysieren, wenn nur ein Datentyp erfasst wird. Wichtig ist, dass IDBac Rohdaten von Bruker- und Xiamen-Instrumenten sowie txt, tab, csv, mzXML und mzML verarbeitet. Dadurch entfällt die manuelle Konvertierung und Formatierung von Datensätzen und das Risiko von Benutzerfehlern oder falscher Verarbeitung von MS-Daten wird erheblich reduziert.
Protokoll
1. Herstellung der MALDI-Matrix
- Bereiten Sie 10 mg/ml MALDI-grade und/oder rekristallisierte-Cyano-4-Hydroxycinnamsäure (CHCA) in MS-lösemitteln vor: 50% Acetonitril (ACN), 47,5% Wasser (H2O), 2,5% Trifluoressigsäure (TFA). Beispiel: 100 l Lösung = 50 l ACN + 47,5 l H2O + 2,5 l TFA + 1 mg CHCA
- Bereiten Sie mindestens 1 L Matrixlösung pro MALDI-Plattenfleck und Wirbel vor oder beschallen Sie sie bis zur Lösung (ca. 5 min Beschallung oder keine sichtbaren Feststoffe).
VORSICHT: TFA ist eine starke Säure, die in einer chemischen Rauchhaube behandelt werden sollte, während sie die richtige persönliche Schutzausrüstung trägt, da sie Haut, Augen und Atemwege mit Kontakt oder Inhalation beschädigen kann.
HINWEIS: CHCA ist hygroskopisch und lichtempfindlich und sollte in Bernsteinfläschchen in einem Trockenhaus gelagert werden. Es stehen viele MALDI-Matrixoptionen zur Verfügung. CHCA ist am häufigsten für Protein-Profilierung von Bakterien, sondern arbeitet auch für spezialisierte Metaboliten-Analyse. Die Matrixauswahl hängt von den individuellen Benutzer-/Experimentanforderungen ab.
- Bereiten Sie mindestens 1 L Matrixlösung pro MALDI-Plattenfleck und Wirbel vor oder beschallen Sie sie bis zur Lösung (ca. 5 min Beschallung oder keine sichtbaren Feststoffe).
2. Herstellung von MALDI-Zielplatten
HINWEIS: Siehe Sauer et al.7, für weitere Details.
- MalDI-Platte mit Methanol (HPLC-Grade oder höher) abspülen und mit weichen Papiertüchern trocken wischen. Verwenden Sie bei der Reinigung von Zielplatten keine Schleifbürsten, da dies die Oberfläche der Zielplatte dauerhaft beschädigen kann.
- Weisen Sie Protein- und spezialisierte Metaboliten-Kalibrantenzusflecken zu. Organisieren Sie Kalibrierungspunkte gleichmäßig über die Probenpopulation hinweg, um MALDI-Plattenunregelmäßigkeiten und Instrumentendrift im Laufe der Zeit zu berücksichtigen. Weisen Sie der Studie eine angemessene Anzahl von Medien/Matrix-leeren Spots zu; Diese Spots enthalten nur Medien und Matrix oder nur Matrix.
- Mit einem sterilen Zahnstocher einen kleinen Teil einer Bakterienkolonie an die entsprechende Stelle auf der MALDI-Platte übertragen. Verteilen Sie die Bakterienkolonie gleichmäßig über die Stelle. Der Spot sollte so flach wie möglich erscheinen.
HINWEIS: Es wird einfacher sein, Bakterienkolonien abzuflachen, die mehr Schleimhaut/Amorphsind sind. Vermeiden Sie für starrere/feste Kolonien, sichtbare Zellmassencluster am MALDI-Spot zu lassen (Abbildung 1).

Abbildung 1: MALDI-Zielplatte mit zwei verschiedenen Isolaten vor Zugabe von Ameisensäure und MALDI-Matrix (obere 3 Flecken - Bacillus sp.; unten 3 Flecken - Streptomyces sp.). Für beide stellt Spalte 3 eine überschüssige Stichprobe dar. Spalte 2 stellt die entsprechende Menge an Stichproben dar; Spalte 1 stellt eine unzureichende Stichprobe für die MALDI-Analyse dar. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Bereiten Sie eine Matrix/Mediensteuerung vor, indem Sie einen sterilen Zahnstocher verwenden, um eine minimale Menge an Agar/Medien auf die entsprechende Stelle(n) auf der MALDI-Platte zu übertragen.
- Überlagern Sie 1 l von 70% Massenspektrometrie-Grade-Ameisensäure auf jeden Probenfleck, einschließlich der Matrix-Kontrollpunkte. Säure vollständig in einer chemischen Rauchhaube (ca. 5 min) trocknen lassen.
VORSICHT: Ameisensäure ist eine ätzende Chemikalie und sollte in chemischen Rauchhauben behandelt werden. Es kann Atemwege beschädigen, wenn es eingeatmet wird. - Fügen Sie jedem Probenfleck sowie den Matrix-/Medienkontrollpunkten 1 L der vorbereiteten MALDI-Matrixlösung hinzu. Matrixlösung vollständig trocknen lassen (ca. 5 min).
HINWEIS: Es ist möglich, die Platte in einem Trockenschrank, im Dunkeln zu lagern, bis sie auf einem MALDI-TOF-Massenspektrometer analysiert werden kann. Zulässige Lagerzeiten können je nach Probenstabilität variieren. - Fügen Sie den zugewiesenen Kalibrierpunkten 0,5 – 1,0 L-Kalibranten hinzu, gefolgt von einer MALDI-Matrixlösung von 1 L. Pipette die resultierende Lösung nach oben und unten zu mischen. Lassen Sie alle Flecken vor der Einführung in das MALDI-TOF-Massenspektrometer vollständig trocknen.
HINWEIS: Das Protein und die spezialisierten Metabolitenkalibrame sollten innerhalb von 30 min der MALDI-Analyse zugesetzt werden, da beide anfällig für Abbau sind.
3. Datenerfassung
HINWEIS: Die allgemeinen Parameter für die Datenerfassung sind in Tabelle 1aufgeführt.
| Parameter | protein | Spezialisierter Metabolit |
| Massenstart (Da) | 1920 | 60 |
| Massenende (Da) | 21000 | 2700 |
| Massenverformung (Da) | 1900 | 50 |
| Aufnahmen | 500 | 1000 |
| Frequenz (Hz) | 2000 | 2000 |
| Lasergröße | groß | mittel |
| MaxStdDev (ppm) | 300 | 30 |
Tabelle 1.
- Nach den für das eingesetzten Instrument spezifischen Protokollen können sowohl Protein- als auch spezielle Metabolitenkalibrierungen eingerichtet werden.
- Testen Sie einige separate Zielpunkte, um die optimale Laserleistung und Detektorverstärkung zu bestimmen, die beim Erfassen von Spektren verwendet werden kann (dies variiert von Tag zu Tag und nach Instrument).
ANMERKUNG: Abbildung 2A und Abbildung 3A zeigen optimale Spektren, während Abbildung 2D und Abbildung 3D Beispiele für Spektren von schlechter Qualität sind.

Abbildung 2: Beispiel-Proteinspektren, die den Effekt der Änderung der Laserleistung und der Detektorverstärkung anzeigen. Spectra-Qualität ist am besten in Panel Aund verringert sich bis unzureichende Spektrenqualität in den Panels C und D. Während das Spektrum in Panel B zu verwendbaren Spitzen führen kann, zeigt Panel A optimale Daten an. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Beispiel spezialisierte Metabolitenspektren, die den Effekt der Änderung der Laserleistung und der Detektorverstärkung anzeigen. Die Spektrenqualität ist in Panel A am besten und verringert sich bis zur unzureichenden Spektrenqualität in den Panels C und D. Während das Spektrum in Panel B zu verwendbaren Spitzen führen kann, zeigt Panel A optimale Daten an. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Erfassen Sie Spektren, speichern Sie Proteinspektren in einem Ordner und spezialisierte Metabolitenspektren in einem zweiten, separaten Ordner.
4. Reinigung der MALDI Zielplatte (angepasst von Sauer et al.7)
- Entfernen Sie die MALDI-Zielplatte aus ihrem Halter und spülen Sie sie mit Aceton ab.
- Waschen Sie mit einer nicht abrasiven Flüssigseife, um Spurenproteine und Lipide zu entfernen, und mit weichen Papiertüchern/Weichborsten zahnbürsten.
- Spülen Sie mit entionisiertem Wasser für ca. 2 min, um Seife vollständig zu entfernen.
- Beschallen Sie die Zielplatte in Wasser (HPLC-Grad oder höher) für 5 min.
- Spülen Sie die Zielplatte mit Wasser (HPLC-Grad oder höher).
- Spülen Sie die Zielplatte mit Methanol (HPLC-Grad oder höher).
5. Installieren der IDBac-Software
- Laden Sie die IDBac-Software herunter.
HINWEIS: Permanente, versionierte Sicherungen stehen ebenfalls zum Download zur Verfügung (siehe Tabelle der Materialien). - Doppelklicken Sie auf die heruntergeladene "Install_IDBac.exe", um das Installationsprogramm zu initiieren und den Anweisungen auf dem Bildschirm zu folgen.
6. Beginnend mit Rohdaten
HINWEIS: Detaillierte Erläuterungen und Anweisungen zu jedem Datenverarbeitungsschritt sind in IDBac eingebettet, die Hauptanalysen und interaktiven Eingaben werden jedoch im Folgenden beschrieben.
- Doppelklicken Sie auf die IDBac-Desktopverknüpfung, um IDBac zu starten. IDBac wird standardmäßig auf der Registerkarte Einführung geöffnet.
- Verwenden Sie die Schaltfläche Nach Updates suchen, um sicherzustellen, dass die aktuellste Version von IDBac verwendet wird (erfordert Internetzugang). Wenn eine neuere Version verfügbar ist, lädt IDBac das Update automatisch herunter und installiert es, woraufhin IDBac einen Neustart anfordert.
- Klicken Sie auf die Registerkarte Start mit Rohdaten und wählen Sie im Menü den Datentyp aus, der mit IDBac verwendet werden soll. folgen Sie den Anweisungen in der App.
- Geben Sie beim Einrichten der Konvertierung und Verarbeitung von Datendateien einen beschreibenden Namen für das Experiment ein, wenn Sie dazu aufgefordert werden (siehe Abbildung 4). Experimente werden später alphabetisch angezeigt, so dass eine hilfreiche Strategie darin besteht, Experimentnamen mit einem Gruppenattribut zu beginnen (z. B. "bacillus-trials_experiment-1"; "bacillus-trials_experiment-2").

Abbildung 4: IDBac-Datenkonvertierungs- und Vorverarbeitungsschritt. IDBac konvertiert Raw-Spektren in das offene mzML-Format und speichert mzML, Spitzenlisten und Beispielinformationen in einer Datenbank für jedes Experiment. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
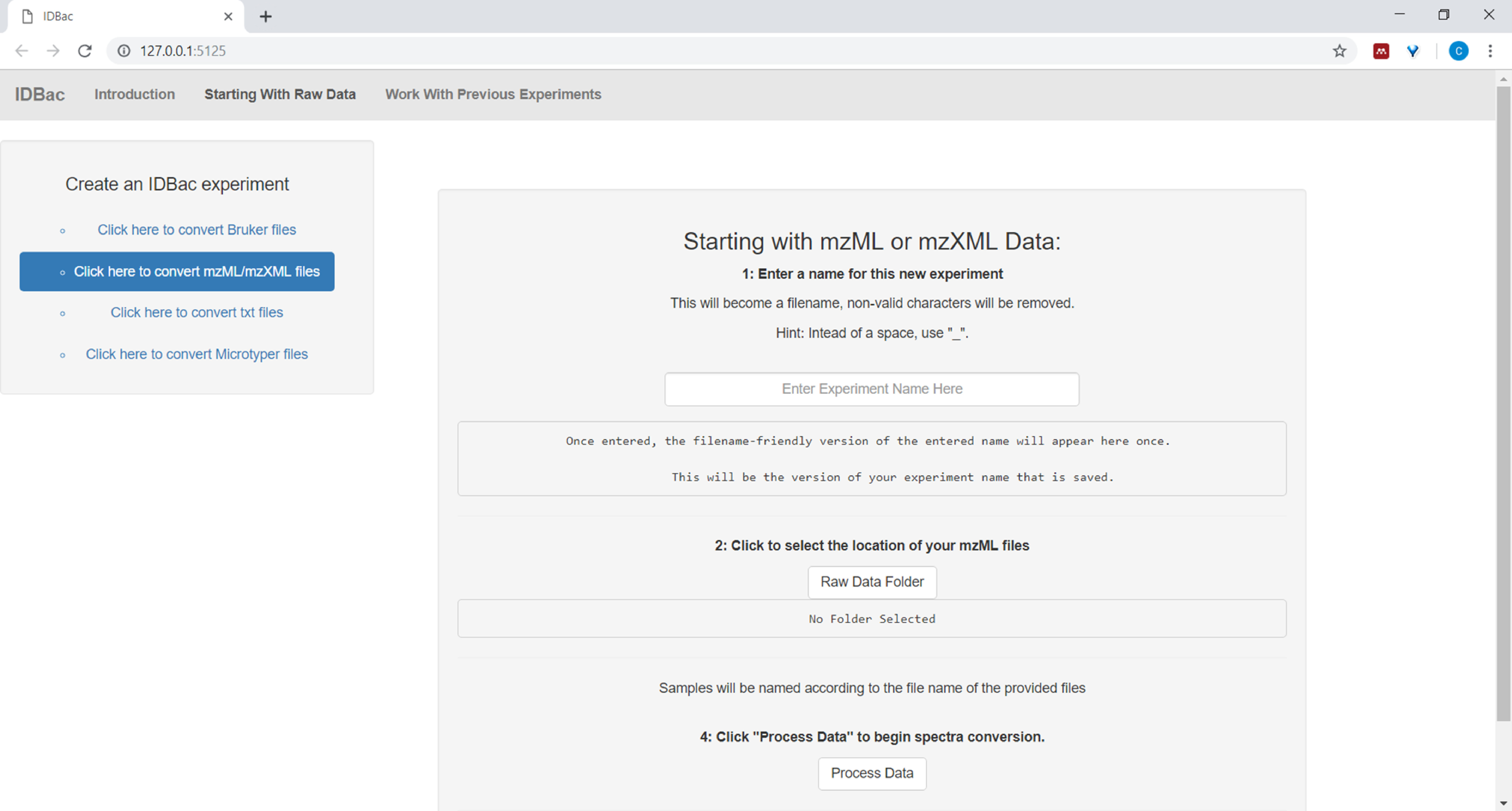{kind=link}
7. Arbeiten mit früheren Experimenten
- Nachdem Sie Dateien konvertiert und mit IDBac verarbeitet haben oder wenn sie ein Experiment erneut analysieren möchten, navigieren Sie zur Seite Arbeiten mit vorherigen Experimenten und wählen Sie ein Experiment aus, mit dem Sie arbeiten möchten (Abbildung 5).

Abbildung 5: Seite "Arbeiten mit früheren Experimenten". Verwenden Sie die Seite "Arbeiten mit früheren Experimenten" von IDBac, um ein Zuanalyse- oder Änderungsexperiment auszuwählen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
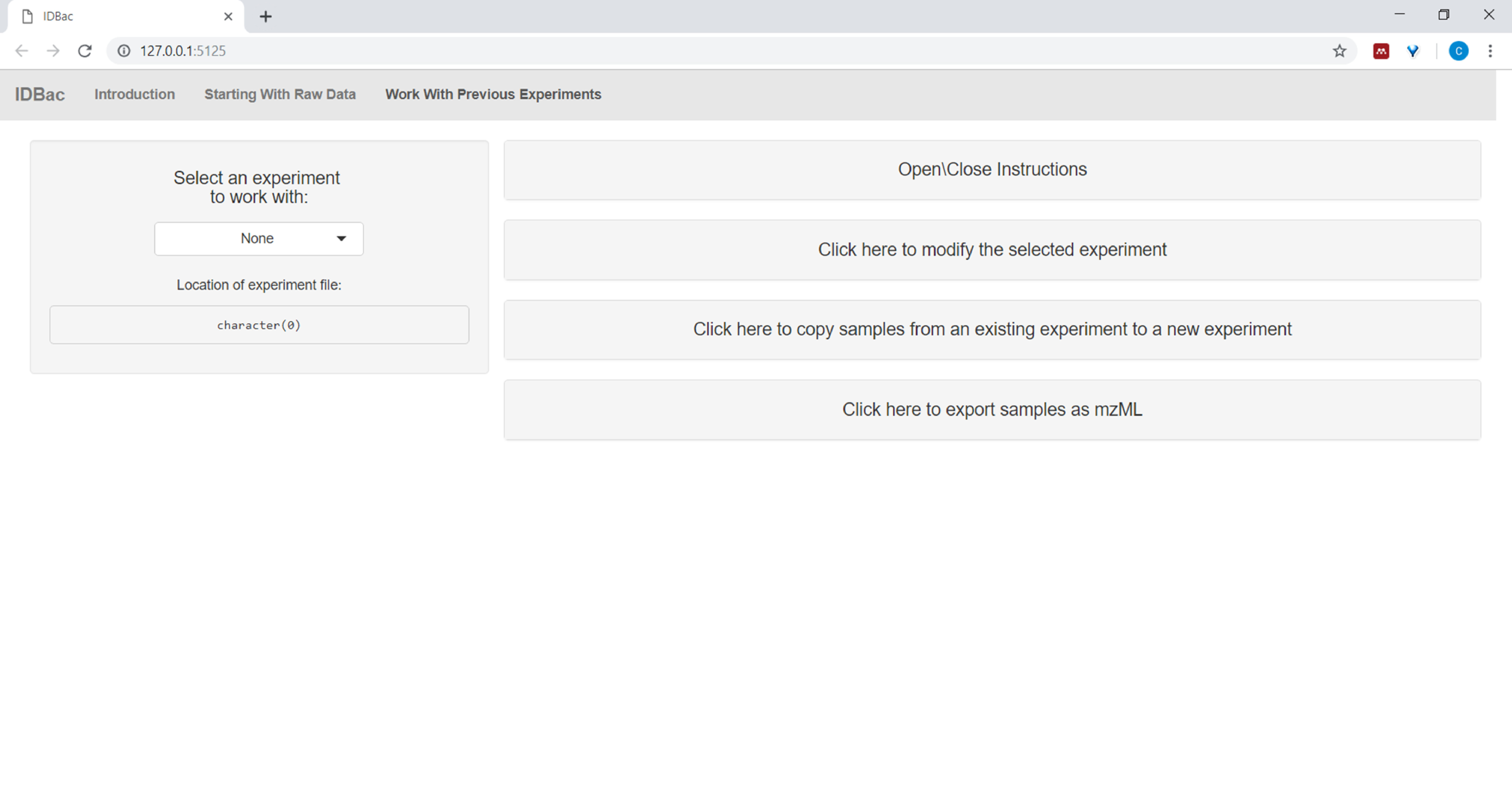{kind=link}
- (Optional) Fügen Sie Informationen zu Beispielen hinzu, indem Sie das Menü Klicken Sie hier, um das ausgewählte Experiment zuändern. Geben Sie Informationen in die automatisch ausgefüllte Kalkulationstabelle ein, und drücken Sie Speichern (Abbildung 6). Mit dieser Option kann der Benutzer Daten während der Analysen farblich kodieren.

Abbildung 6: Eingabe von Beispielinformationen. Auf der Seite "Arbeiten mit früheren Experimenten" können Benutzer Informationen zu Beispielen wie taxonomische Identität, Sammelort, Isolationsbedingungen usw. eingeben. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- (Optional) Übertragen Sie alle oder eine Teilmenge von Proben in ein neues oder ein anderes Experiment, indem Sie auf Samples aus früheren Experimenten auf neue/andere Experimente übertragen und den bereitgestellten Anweisungen folgen ( Abbildung7).

Abbildung 7: Übertragen von Daten. Die Seite "Arbeiten mit früheren Experimenten" enthält die Möglichkeit, Daten zwischen bestehenden Experimenten und zu neuen Experimenten zu übertragen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
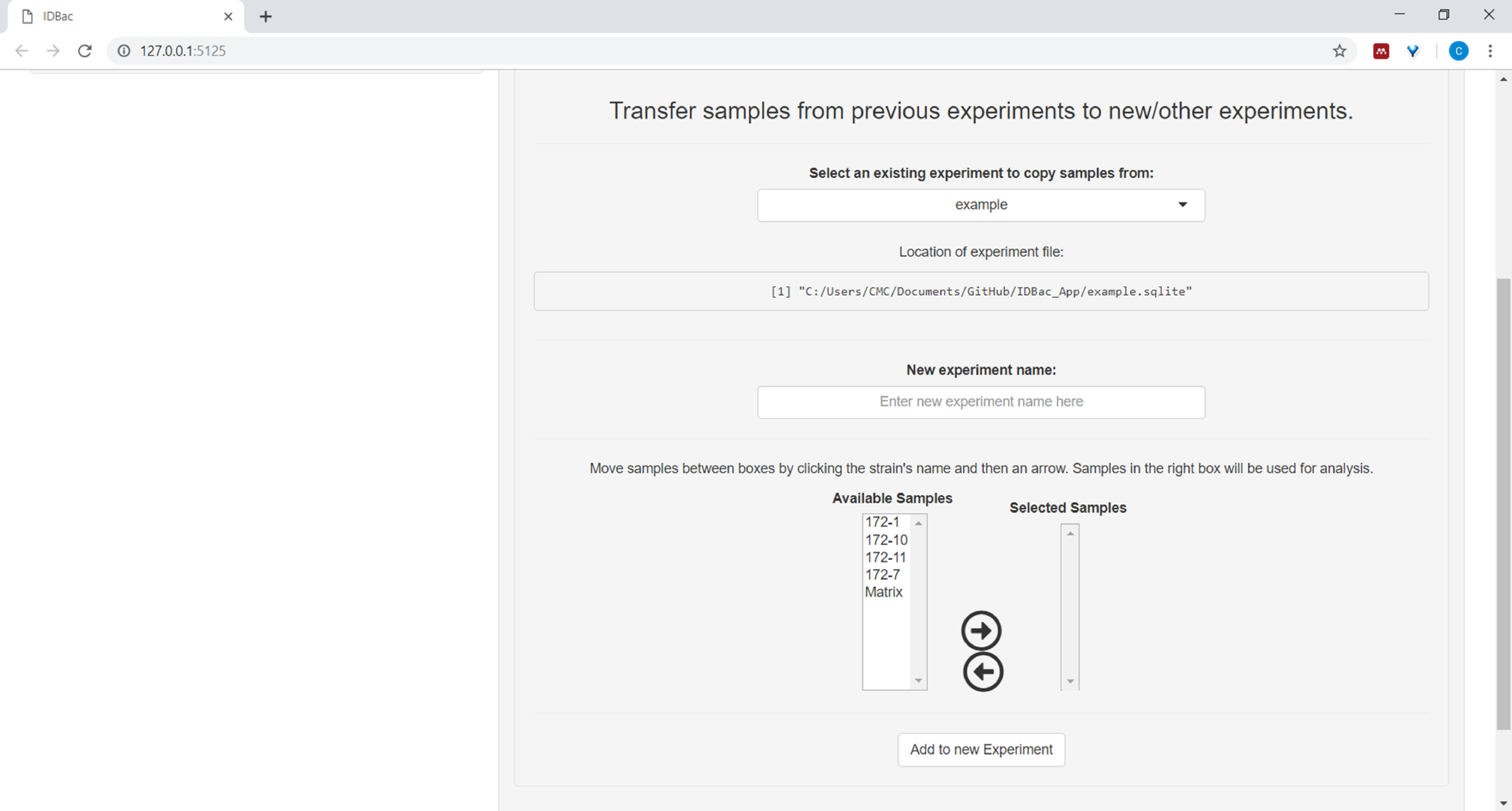{kind=link}
- Wenn Sie mit der Analyse beginnen können, stellen Sie sicher, dass das Experiment ausgewählt ist, mit dem Sie arbeiten möchten. Wählen Sie entweder Protein daten Analysis oder Small Molecule Data Analysisaus.
8. Einrichten von Proteindatenanalysen und Erstellen von Spiegeldiagrammen
- Wenn Sie Proteindaten analysieren, navigieren Sie zuerst zur Seite Proteindatenanalyse. Wählen Sie die Einstellungen für die Spitzenauswahl aus und bewerten Sie die Proteinspektren von Proben über die angezeigten Spiegeldiagramme (Abbildung 8).
HINWEIS: In den Spiegeldiagrammen bedeutet ein roter Peak das Vorhandensein dieses Peaks nur im oberen Spektrum, während blaue Spitzen diejenigen darstellen, die in beiden Spektren auftreten.

Abbildung 8: Wählen Sie aus, wie Spitzen für die Analyse beibehalten werden. Nach der Auswahl eines zu analysierenden Experiments können Benutzer auf der Seite "Proteindatenanalyse" und anschließend das Menü "Wählen, wie Peaks für die Analyse beibehalten werden" Einstellungen wie das Signal-Rausch-Verhältnis für die Beibehaltung von Spitzen auswählen. Das angezeigte Spiegeldiagramm (oder Dendrogramm) wird automatisch aktualisiert, um die ausgewählten Einstellungen widerzuspiegeln. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
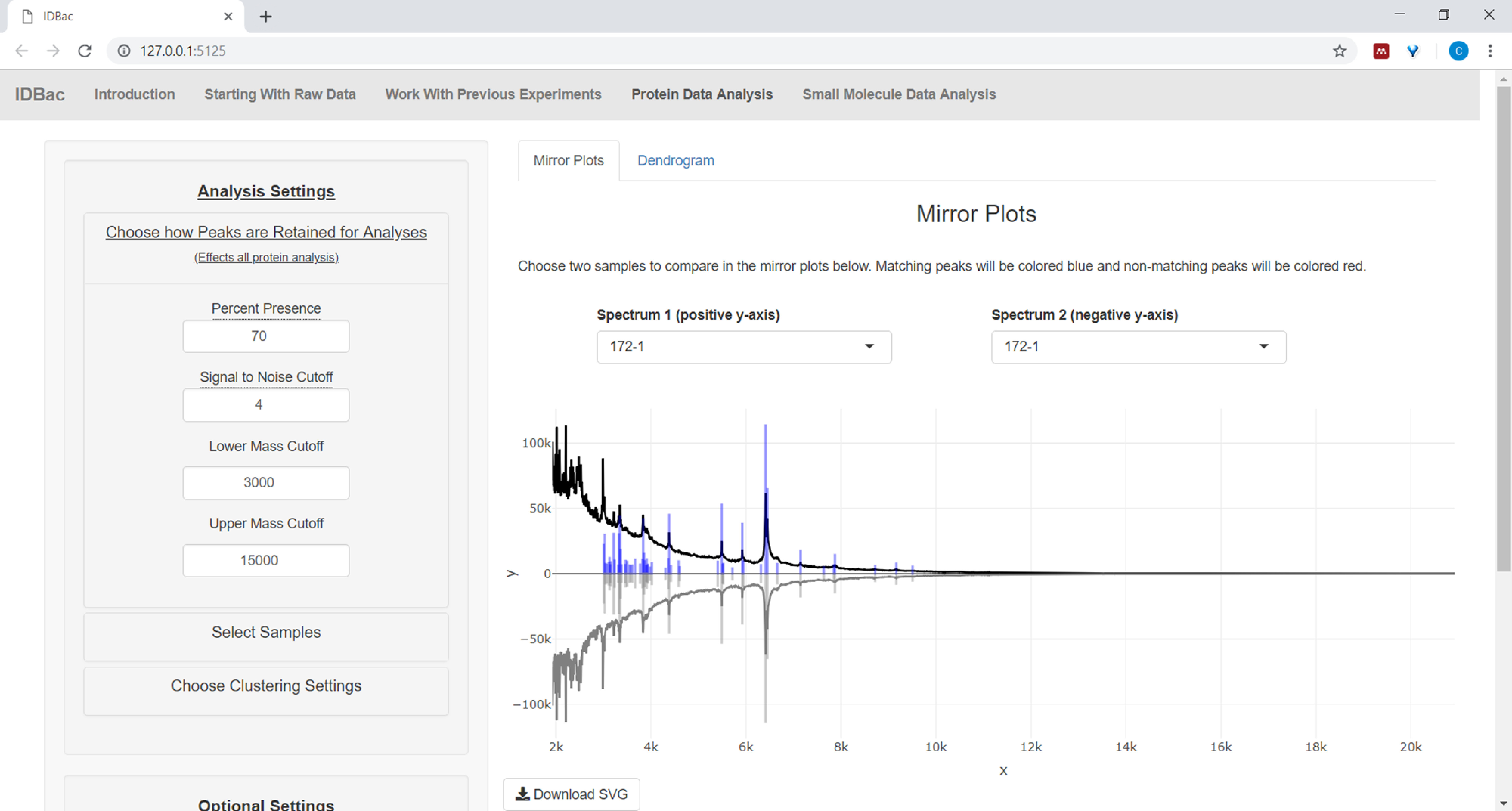{kind=link}
- Passen Sie den Prozentsatz der Replikationen an, in denen ein Peak vorhanden sein muss, damit er für Analysen einbezogen werden kann (z. B. wenn der Schwellenwert auf 70 % festgelegt ist und ein Peak in mindestens 7 von 10 Replikationen auftritt, wird er einbezogen).
- Wenn Sie die Spiegeldiagramme als visuelle Führung verwenden, passen Sie das Signal an die Geräuschabschaltung an, die die "echtsten" Spitzen und das geringste Rauschen beibehält, und beachten Sie, dass mehr Replikationen und ein höherer Wert für die "prozentuale Spitzenpräsenz" die Auswahl eines niedrigeren Signals zum Rauschenabschalten ermöglichen.
- Geben Sie die unteren und oberen m/z-Cutoffs an und diktieren Sie den Bereich der Massenwerte innerhalb jedes Spektrums, der für weitere Analysen von IDBac verwendet werden soll.
9. Clustering-Proben mit Proteindaten
- Wählen Sie auf der Seite Proteindatenanalyse die Registerkarte Dendrogram aus. Dies ermöglicht das Gruppieren von Stichproben in ein Dendrogramm nach vom Benutzer ausgewählten Entfernungsmaßen und Clusteringalgorithmen.
- Klicken Sie im Menü auf Samples auswählen, und folgen Sie den Anweisungen zum Auswählen von Beispielen, die in die Analysen einbezogen werden sollen. Nur Proben, die Proteinspektren enthalten, werden im Feld Verfügbare Proben angezeigt (Abbildung 9).

Abbildung 9: Wählen Sie Beispiele aus dem ausgewählten Experiment aus, die in das angezeigte Dendrogramm aufgenommen werden sollen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Verwenden Sie die Standardwerte, oder wählen Sie unter Clustereinstellungenauswählen die gewünschten Entfernungs- und Clusteralgorithmen aus, die auf die Generierung des Dendrogramms angewendet werden sollen.
- Wählen Sie Präsenz/Abwesenheit als Eingabe aus. Wenn Sie sich über die Spitzenhöhen der Proben (z. B. nach Durchführung einer Studie zur Beurteilung der Variabilität der Spitzenintensität) sicher sind, wählen Sie intensitätsintensiv als Eingabe aus.
HINWEIS: Zum Zeitpunkt der Veröffentlichung bietet IDBac Flexibilität bei den Einstellungen für Clustering, indem es sich darauf verlässt, dass Benutzer die entsprechenden Kombinationen auswählen. Wenn diese Optionen nicht vertraut sind, wird vorgeschlagen, entweder A: "cosine" Entfernung und "Durchschnittlich (UPGMA)" Clustering zu koppeln; oder B: "Euklidische" Entfernung und "Ward.D2"-Clustering. - Um Bootstrap-Werte auf dem Dendrogramm anzuzeigen, geben Sie unter Bootstrapseine Zahl zwischen 2 und 1000 ein.
- Kopieren Sie beim Melden von Ergebnissen den Text im Absatz Vorschläge für die Meldung von Proteinanalysen. Dadurch werden die benutzerdefinierten Einstellungen angegeben, die das spezifische Dendrogramm generiert haben.
10. Anpassen des Proteindendrogramms
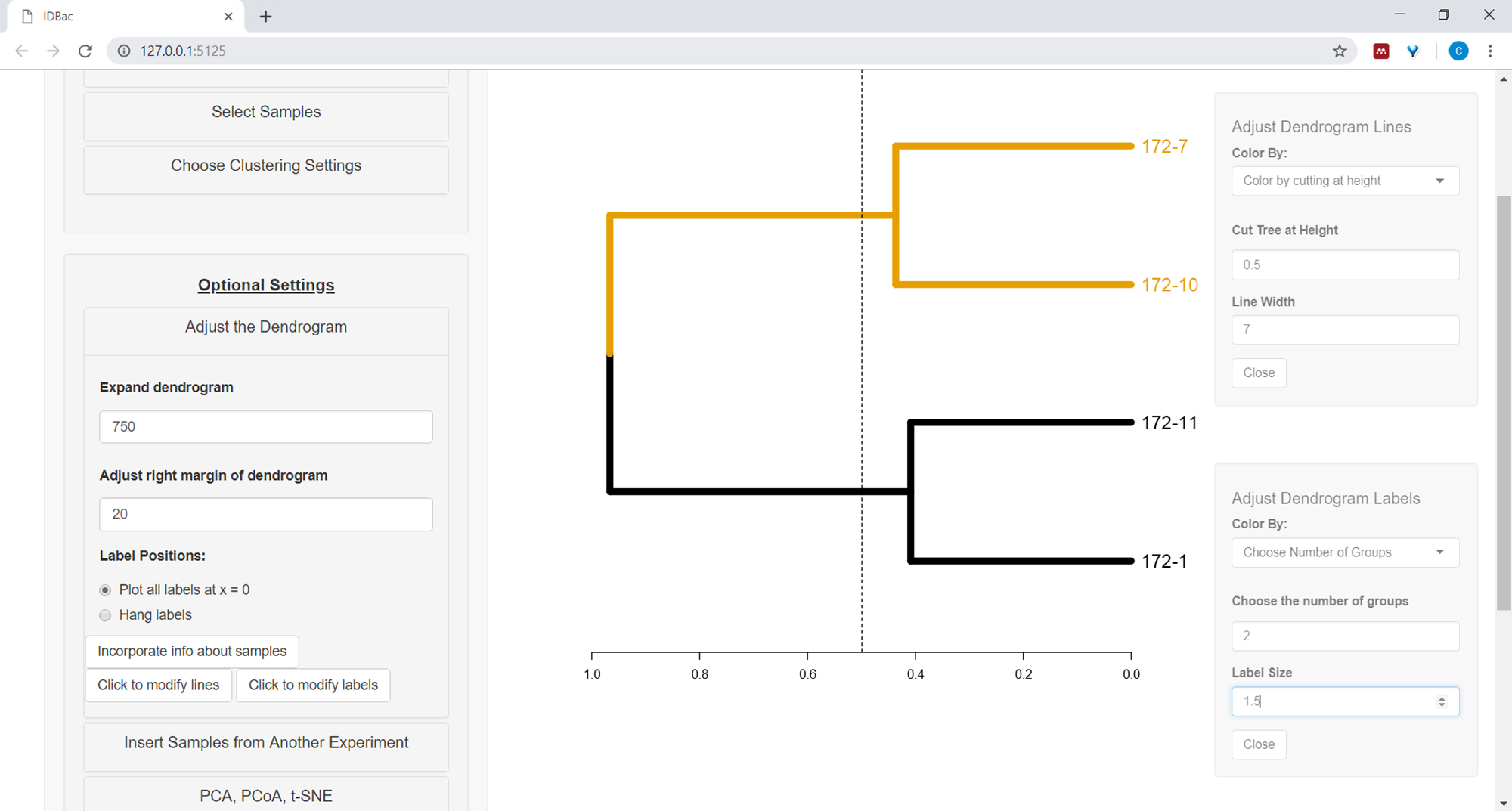
- Um mit der Anpassung des Dendrogramms zu beginnen, öffnen Sie das Menü Das dendrogramm anpassen (Abbildung 10).

Abbildung 10: Passen Sie das Dendrogramm an. IDBac bietet einige Optionen zum Ändern, wie das Dendrogramm aussieht, diese können im Menü "Anpassen des Dendrogramms" gefunden werden. Dazu gehören das Färben von Ästen und Beschriftungen mit k-Mitteln oder durch "Schneiden" des Dendrogramms in einer vom Benutzer bereitgestellten Höhe. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Um die Linien und/oder Beschriftungen des Dendrogramms zu färben, wählen Sie die entsprechende Schaltfläche aus: Klicken Sie hier, um Linien zu ändern, oder klicken Sie, um Beschriftungen zu ändern, und wählen Sie die gewünschten Optionen aus.
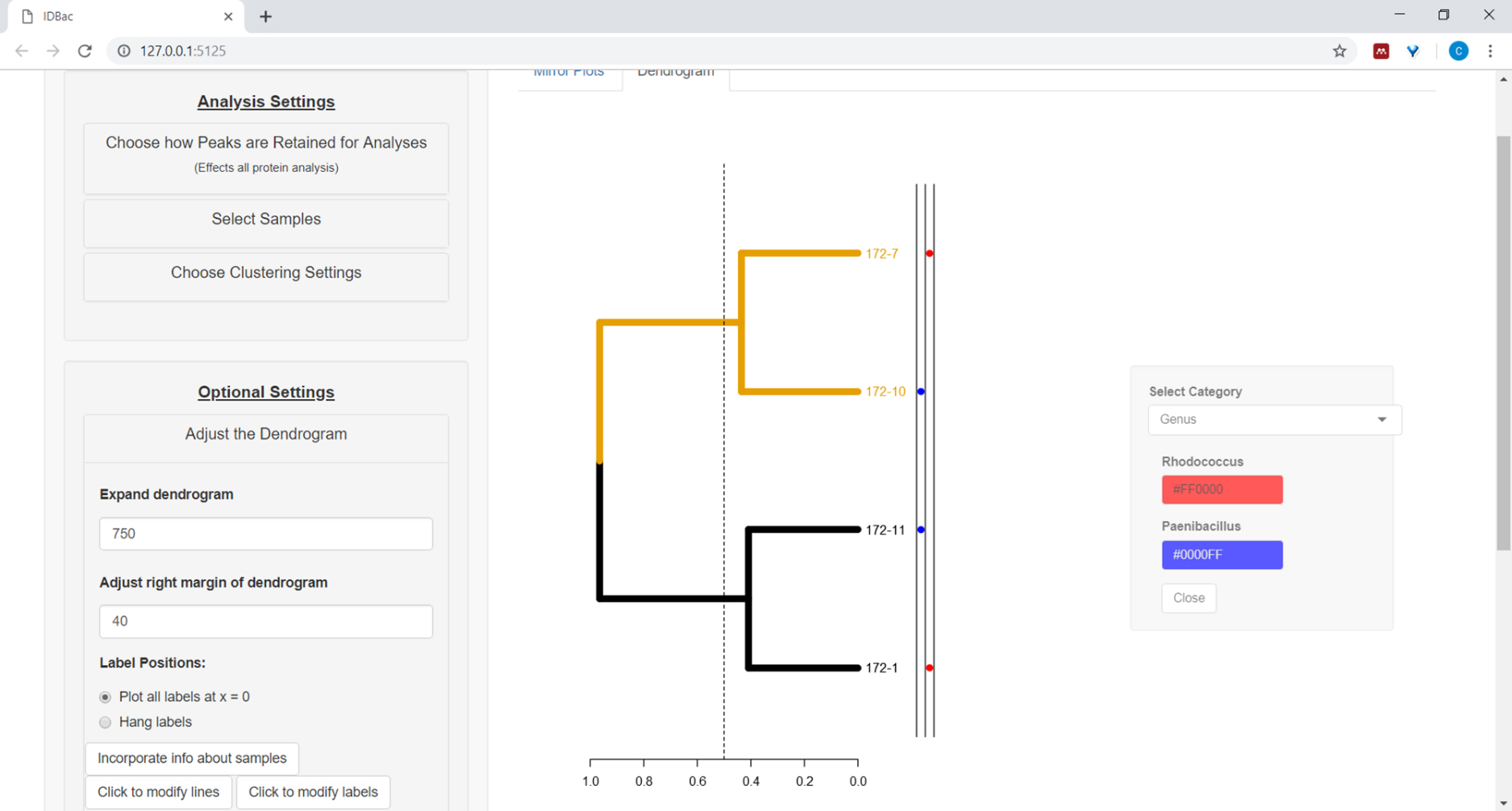
- Um Informationen aus der Kalkulationstabelle neben dem Dendrogramm zu zeichnen (siehe Schritt 7.2), wählen Sie die Schaltfläche Informationen zu Beispielenintegrieren . Dadurch wird ein Bereich geöffnet, in dem sich eine Kategorie (Spalte in der Kalkulationstabelle) basierend auf den eingegebenen Werten selbst auffüllt (Abbildung 11).

Abbildung 11: Integrieren von Informationen zu Beispielen. Im Menü "Das Dendrogramm anpassen" befindet sich die Option "Informationen über Samples integrieren". Wenn Sie diese Option auswählen, können Sie Informationen zu Samples neben dem Dendrogramm zeichnen. Beispielinformationen werden auf der Seite "Arbeiten mit früheren Experimenten" eingegeben. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
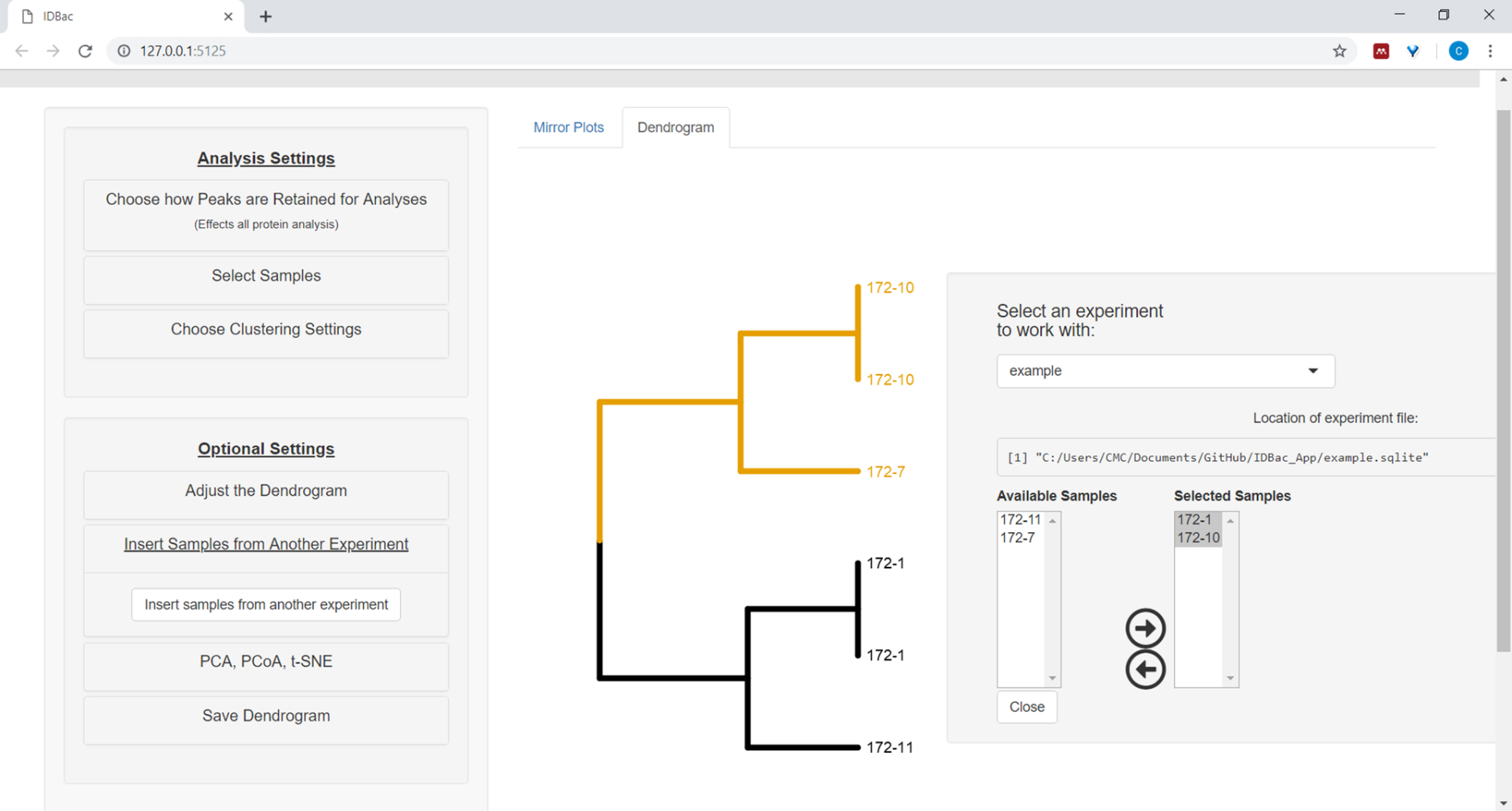
11. Fügen Sie Proben aus einem separaten Experiment in das Dendrogramm ein
- Um Beispiele aus einem anderen Experiment einzufügen, wählen Sie die Menüschaltfläche Samples aus einem anderen Experimenteinfügen aus. Folgen Sie den Anweisungen im neu geöffneten Panel (Abbildung 12).

Abbildung 12: Einfügen von Beispielen aus einem anderen Experimentmenü. Manchmal ist es hilfreich, Proben aus einem anderen Experiment zu vergleichen. Verwenden Sie das Menü "Samples aus einem anderen Experiment einfügen", um Beispiele auszuwählen, die in das aktuell angezeigte Dendrogramm aufgenommen werden sollen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
12. Analyse spezialisierter Metabolitendaten und Metaboliten-Assoziationsnetzwerke (MANs)
- Fahren Sie mit der Seite Metabolite Association Network (Small-Molecule) fort. Diese Seite ermöglicht die Datenvisualisierung durch Prinzipkomponentenanalyse (PCA) und MANs, die zweiseitige Netzwerke verwenden, um die Korrelation von kleinen Molekül-m/z-Werten mit Proben anzuzeigen.
- Wenn ein Proteindendrogramm erstellt wurde (Abschnitt 9), wird es auch auf dieser Seite angezeigt. Klicken Sie auf das Dendrogramm, um ausgewählte, zu analysierende Beispiele hervorzuheben. Wenn keine Proben hervorgehoben oder kein Proteindendrogramm erstellt wurde, erscheint ein MAN einer zufälligen Teilmenge oder alle Proben respektvoll (Abbildung 13).

Abbildung 13: Seite "Kleinmoleküldatenanalyse". Wenn ein Dendrogramm aus Proteinspektren erstellt wurde, wird es auf der Seite "Kleine Moleküldatenanalyse" angezeigt. Auf dieser Seite werden auch Metabolite Associate Networks (MANs) und Principle Components Analysis (PCA) für kleine Moleküldaten angezeigt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Um eine Matrix/Medienleerung im MAN zu subtrahieren, öffnen Sie das Menü Wählen Sie ein Sample aus, das subtrahiert werden soll, und wählen Sie das entsprechende Beispiel aus, das als Leerzeichen verwendet werden soll.
- Öffnen Sie das Menü MAN-Einstellungen anzeigen/ausblenden, um die gewünschten Werte für den Prozentsatz der Spitzenpräsenz in Replikationen, Signal-zu-Rauschen und oberen und unteren Massen-Cutoffs auszuwählen, wie dies bei Proteinspektren in Abschnitt 9 der Effekt geschehen ist. Verwenden Sie die kleinen Molekülspiegeldiagramme, um die Auswahl dieser Einstellungen zu steuern.
- Wählen Sie "Aktuelle Netzwerkdaten herunterladen", um die aktuell angezeigten Daten des MAN zu speichern. Diese Daten können in anderen Netzwerkanalysesoftware als IDBac verwendet werden.
- Kopieren Sie den Text im Absatz Vorschläge für die Meldung der MAN-Analyse, um Ergebnisse zu melden. Dies stellt die benutzerdefinierten Einstellungen bereit, die zum Generieren des erstellten MAN verwendet werden.
13. Gemeinsame Nutzung von Daten
- Jedes IDBac -Experiment wird als einzelne SQLite-Datenbank gespeichert. Es enthält die konvertierten mzML-Rohspektren, erkannte Peaks und alle Benutzereingabeinformationen zu Samples. Um ein IDBac-Experiment freizugeben, geben Sie einfach die SQlite-Datei mit demselben Namen wie das Experiment weiter (der Speicherort der Datei wird auf der Seite Arbeiten mit vorherigen Experimenten angezeigt).
Ergebnisse
Wir analysierten sechs Stämme von Micromonospora Chokoriensis und zwei Stämme von Bacillus subtilis, die zuvor charakterisiert wurden6, mit Daten zur Verfügung doI: 10.5281/zenodo.2574096. Folgende Anweisungen auf der Registerkarte Start mit Rohdaten haben wir die Option Klicken Sie hier, um Bruker-Dateien zu konvertieren, ausgewählt und den von IDBac bereitgestellten Anweisungen für jedes Dataset gefolgt (Abbildung 14).
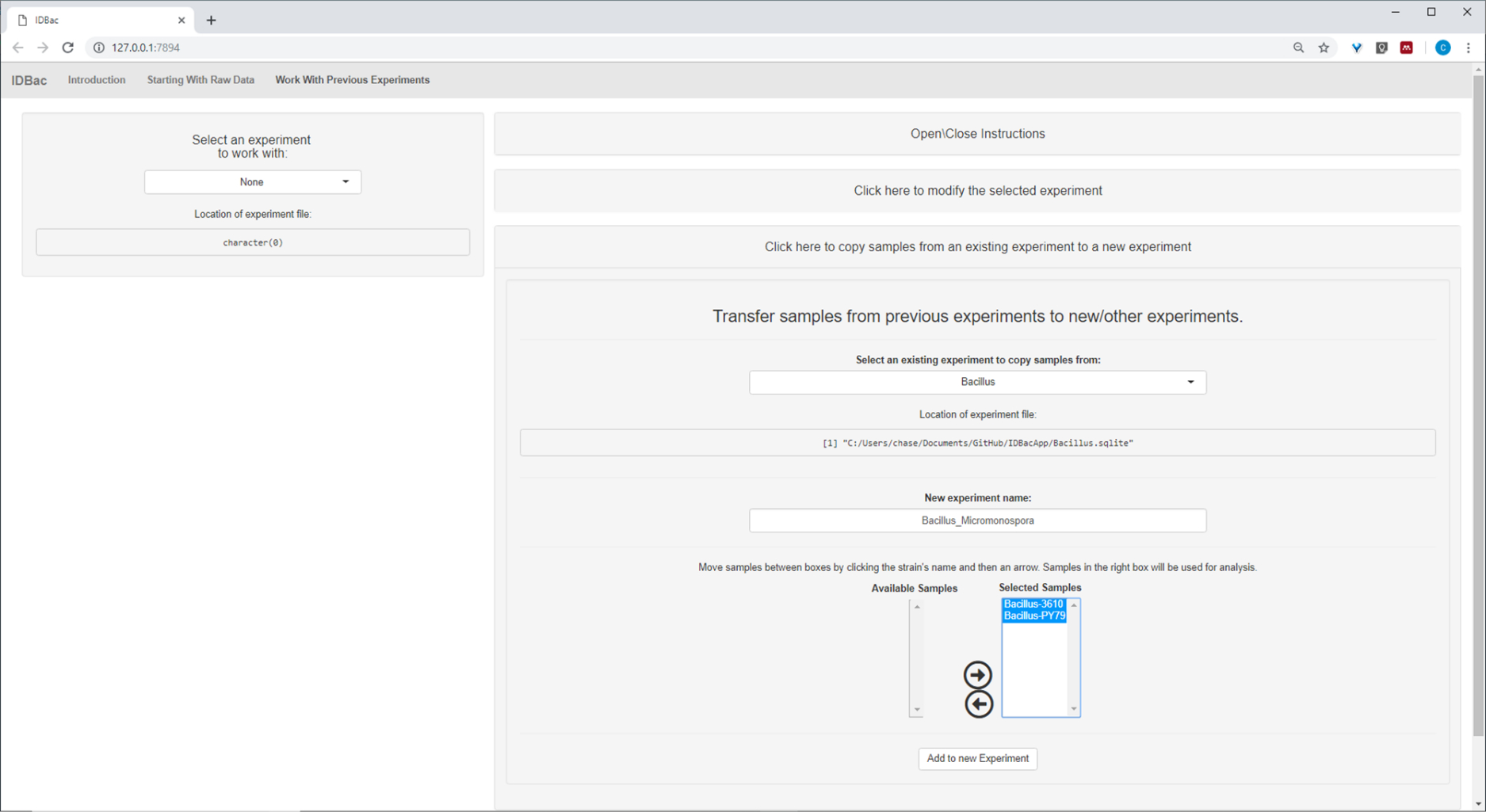
Nachdem die automatisierteKonvertierung und die Vorverarbeitung/Spitzenschritte abgeschlossen waren, machten wir einen neuen kombinierten IDBac-Experiment, indem wir Proben aus den beiden Experimenten in ein einziges Experiment übertrugen, das sowohl Bacillus als auch Mikromonosopora-Proben (Abbildung 15). Die resultierende Analyse umfasste den Vergleich von Proteinspektren mithilfe von Spiegeldiagrammen, wie in Abbildung 16dargestellt, was für die Bewertung der Spektrenqualität und das Anpassen von Spitzenauswahleinstellungen nützlich war. Abbildung 17 zeigt einen Screenshot der Proteinclustering-Ergebnisse mit ausgewählten Standardeinstellungen. Das Dendrogramm wurde durch Anpassen des Schwellenwerts auf dem Diagramm (erscheint als gepunktete Linie) eingefärbt. Bemerkenswert ist die klare Trennung zwischen Gattungen, mit M. chokoriensis und B. subtilis isoliert Clustering getrennt.
Abbildung 18, Abbildung 19und Abbildung 20 unterstreichen die Möglichkeit, MANs von vom Benutzer ausgewählten Regionen zu generieren, indem Sie auf das Proteindendrogramm klicken und es ziehen. Damit konnten wir schnell MANs erstellen, um nur die B. subtilis Stämme zu vergleichen (Abbildung 18), nur die M. chokoriensis Stämme (Abbildung 19), und alle Stämme gleichzeitig (Abbildung 20). Die primäre Funktion dieser Netzwerke besteht darin, den Forschern einen umfassenden Überblick über den Grad der spezialisierten Metabolitenüberlappung zwischen Bakterien zu geben. Mit diesen Daten in der Hand haben die Forscher nun die Fähigkeit, fundierte Entscheidungen aus nur einer kleinen Menge an Material zu treffen, das aus einer Bakterienkolonie abgekratzt wurde.

Abbildung 14: Spectra-Verarbeitung. Heruntergeladene Bruker autoFlex Spektren wurden mit IDBac konvertiert und verarbeitet. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 15: Kombiniertes IDBac-Experiment. Da die Spektren Micromonospora und Bacillus auf verschiedenen MALDI-Zielplatten gesammelt wurden, wurden die beiden Experimente anschließend zu einem einzigen Experiment -"Bacillus_Micromonsopora" zusammengefasst. Dies geschah im Bereich "Arbeiten mit früheren Experimenten" nach Anweisungen im Menü "Proben aus früheren Experimenten auf neue/andere Experimente übertragen". Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 16: Vergleich. Mikromonspora- und Bacillus-Spektren wurden anhand der Spiegeldiagramme auf der Seite "Proteindatenanalyse" verglichen. Letztendlich wurden Standard-Spitzeneinstellungen ausgewählt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 17: Hierarchisches Clustering. Hierarchisches Clustering, mit Standardeinstellungen, korrekt gruppiert Bacillus und Micromonospora Isolate. Das Dendrogramm wurde durch "Schneiden" des Dendrogramms in beliebiger Höhe (angezeigt als gestrichelte Linie) und 100 Bootstraps, die verwendet wurden, um Vertrauen in verzweigte Zuzweigungen zu zeigen, eingefärbt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
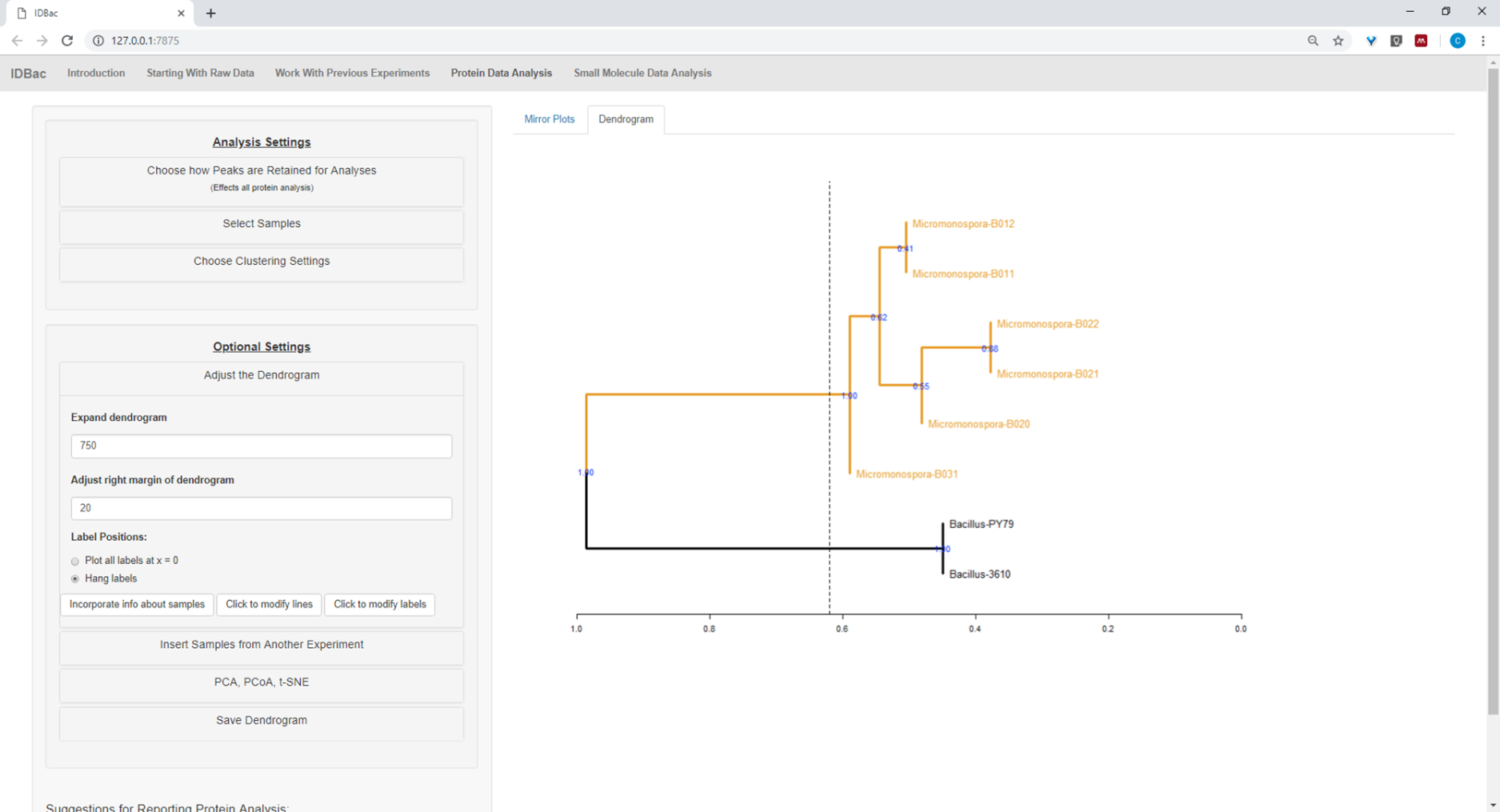{kind=link}

Abbildung 18: MAN erstellt durch die Auswahl der Bacillus sp. Stämme aus dem Protein Dendrogramm zeigte Differentialproduktion von spezialisierten Metaboliten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 19: MAN entstand durch die Auswahl der sechs Micromonospora sp. Stämme aus dem Protein Dendrogramm zeigte differentiale Produktion von spezialisierten Metaboliten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
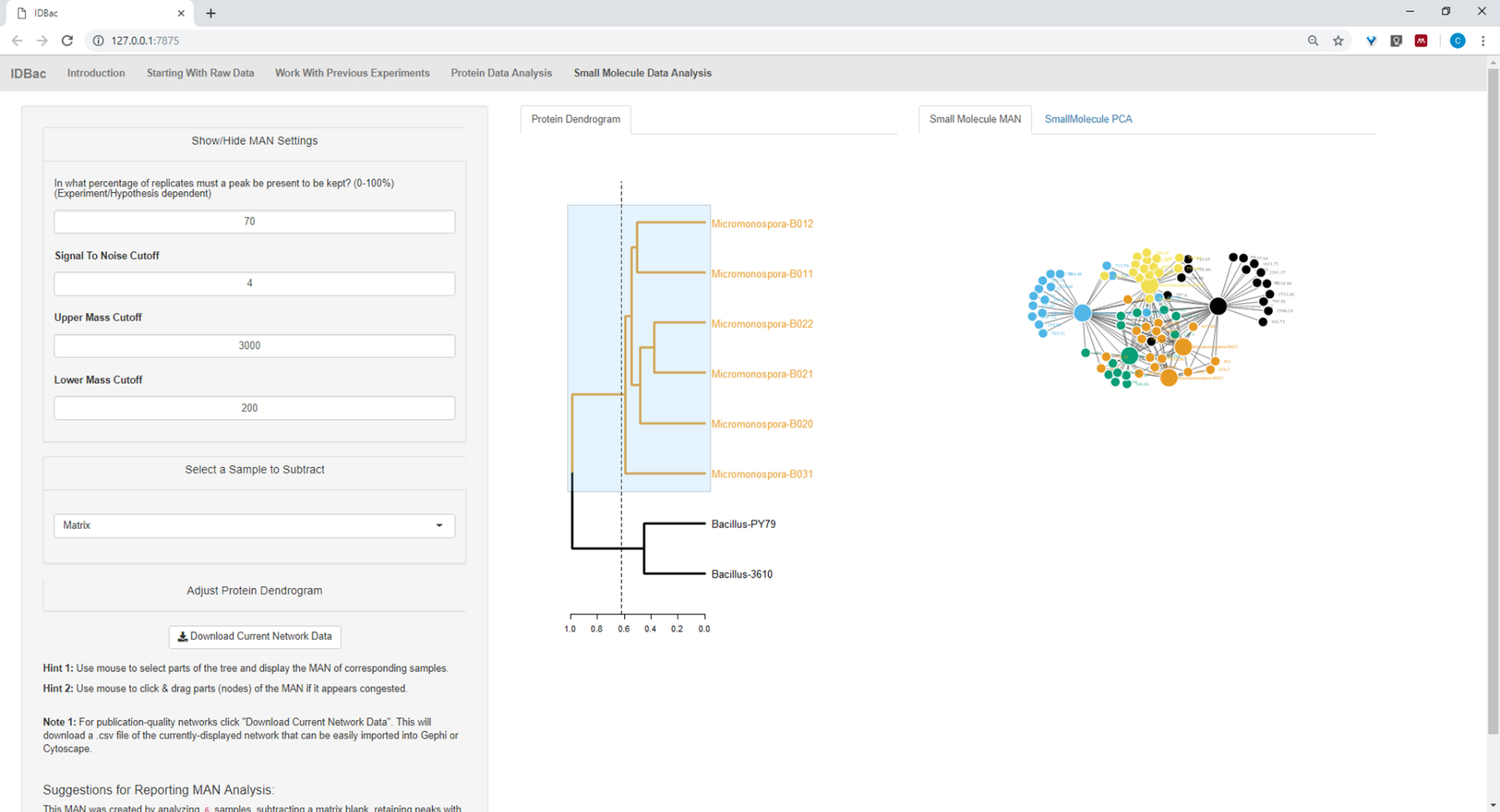{kind=link}

Abbildung 20: MAN von Bacillus sp. und Micromonospora sp. Stämmen, die eine differenzielle Produktion von spezialisierten Metaboliten zeigen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
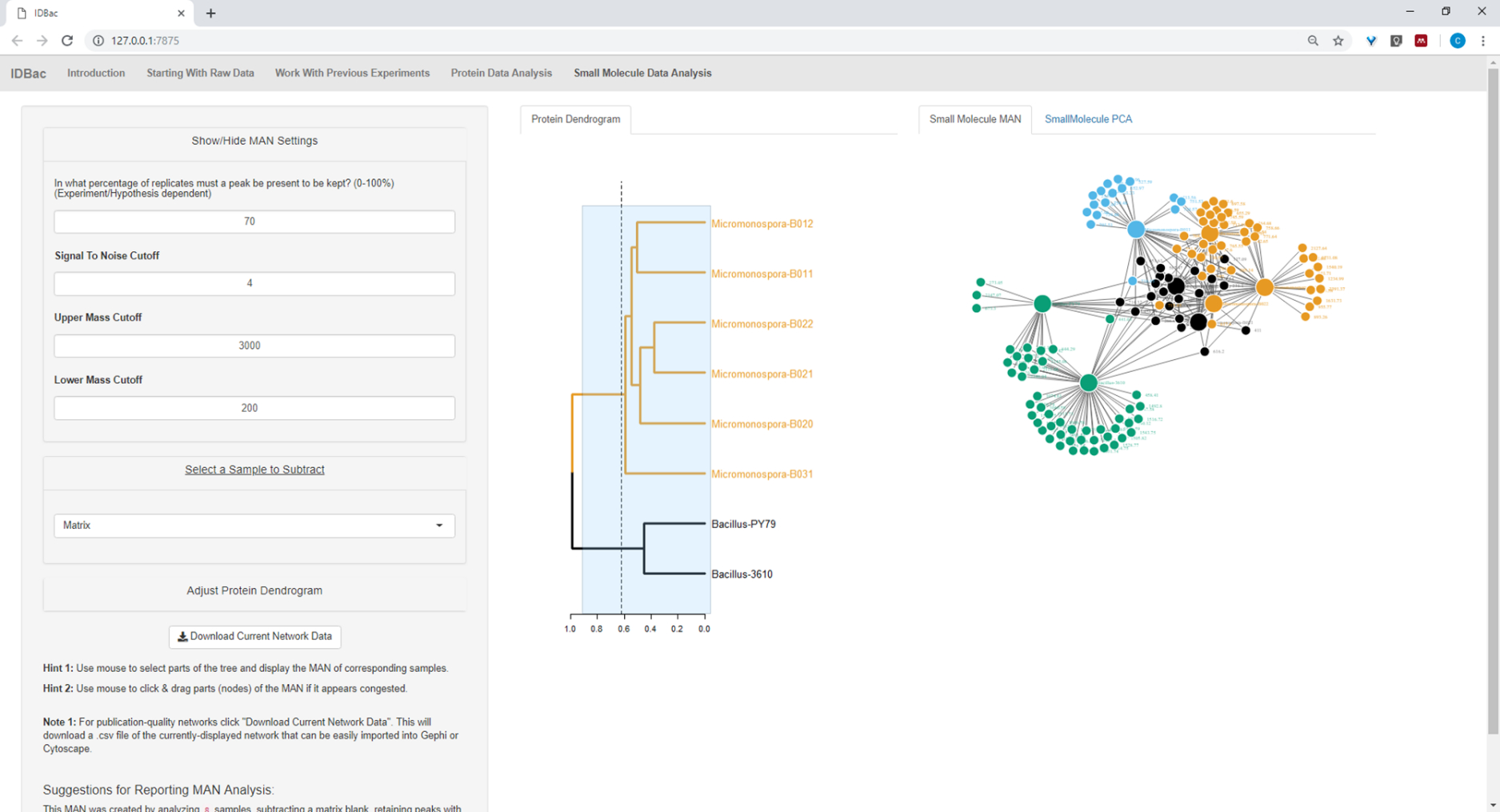{kind=link}
Diskussion
Das IDBac-Protokoll beschreibt die Erfassung und Analyse von bis zu 384 bakteriellen Isolaten in 4 h durch einen einzigen Forscher. Mit IDBac ist es nicht notwendig, DNA aus bakteriellen Isolaten zu extrahieren oder spezielle Metabolitenextrakte aus flüssigen Fermentationsbrühen zu erzeugen und sie mit chromatographischen Methoden zu analysieren. Stattdessen werden Protein- und spezialisierte Metabolitendaten gesammelt, indem Material aus Bakterienkolonien einfach direkt auf eine MALDI-Zielplatte verteilt wird. Dies reduziert den Zeit- und Kostenaufwand für alternative Techniken wie 16S rRNA-Gensequenzierung und LCMS9erheblich.
Es ist wichtig, der MALDI-Platte einen Matrixrohling und Kalibrierflecken hinzuzufügen, und wir empfehlen die Verwendung einer geeigneten Anzahl von Replikationen, um Reproduzierbarkeit und statistisches Vertrauen zu gewährleisten. Die Anzahl der Replikationen ist experimentabhängig. Wenn ein Benutzer beispielsweise beabsichtigt, Tausende von Kolonien von einer Sammlung von Umweltdiversitätsplatten zu unterscheiden, können weniger Replikationen erforderlich sein (unser Labor sammelt drei technische Replikationen pro Kolonie). Wenn ein Benutzer eine benutzerdefinierte Datenbank mit Stämmen aus bestimmten bakteriellen Taxa erstellen möchte, um schnell Unterartenklassifikationen unbekannter Isolate zu bestimmen, dann sind weitere Replikationen angebracht (unser Labor sammelt acht biologische Replikationen pro Dehnung).
IDBac ist ein Werkzeug zur schnellen Differenzierung hochverwandter bakterienisolate auf der Grundlage vermeintlicher taxonomischer Informationen und spezialisierter Metabolitenproduktion. Es kann orthogonale Methoden wie eingehende genetische Analysen, Studien mit Metabolitenproduktion und -funktion oder die Charakterisierung der spezialisierten Metabolitenstruktur durch Kernspinresonanzspektroskopie und/oder LC-MS/MS.
Die spezialisierte Metabolitenproduktion (IDBac MANs) ist sehr anfällig für bakterielle Wachstumsbedingungen, insbesondere mit verschiedenen Medien, was eine potenzielle Einschränkung der Methode darstellt. Diese Eigenschaften können jedoch vom Benutzer ausgenutzt werden, da IDBac leicht MANs erzeugen kann, die die Unterschiede in der spezialisierten Metabolitenproduktion unter einer Vielzahl von Wachstumsbedingungen zeigen. Es ist wichtig zu beachten, dass zwar spezialisierte Metaboliten-Fingerabdrücke je nach Wachstumsbedingung variieren können, wir aber zuvor gezeigt haben, dass Protein-Fingerabdrücke über diese Variablen hinweg relativ stabil bleiben (siehe Clark et al.6). Im Umgang mit Umweltdiversitätsplatten empfehlen wir, bakterielle Isolate vor der Analyse zu reinigen, um mögliche Beiträge aus benachbarten bakteriellen Quergesprächen zu reduzieren.
Schließlich ist das Fehlen einer durchsuchbaren öffentlichen Datenbank mit Protein-MS-Fingerabdrücken ein großes Manko bei der Verwendung dieser Methode zur Klassifizierung unbekannter Umweltbakterien. Wir haben IDBac in diesem Sinne erstellt und die automatisierte Konvertierung von Daten in ein von der Community akzeptiertes Open-Source-Format (mzML)10,11,12 integriert und die Software so konzipiert, dass das Suchen, Teilen und Erstellen von benutzerdefinierte Datenbanken. Wir sind dabei, eine große öffentliche Datenbank (>10.000 voll charakterisierte Stämme) zu erstellen, die die Klassifizierung einiger Isolate auf Artenebene ermöglicht, einschließlich Links zu GenBank-Beitrittsnummern, wenn verfügbar.
IDBac ist Open Source und der Code ist für jedermann verfügbar, um seine Datenanalyse- und Visualisierungsanforderungen anzupassen. Wir empfehlen den Benutzern, eine umfangreiche Literatur (Sauer et al.7, Silva et al.5) zu konsultieren, um ihre experimentellen Ziele zu unterstützen und zu entwerfen. Wir veranstalten ein Diskussionsforum unter: https://groups.google.com/forum/#!forum/idbac und eine Möglichkeit, Probleme mit der Software zu melden unter: https://github.com/chasemc/IDBacApp/issues.
Offenlegungen
Die Autoren haben nichts zu verraten.
Danksagungen
Diese Arbeit wurde vom National Institute of General Medical Sciences Grant R01 GM125943, National Geographic Grant CP-044R-17 unterstützt; Zuschuss des isländischen Forschungsfonds 152336-051; und University of Illinois at Chicago Startup Funds. Außerdem danken wir den folgenden Mitwirkenden: Dr. Amanda Bulman für die Unterstützung bei den Parametern für den Erwerb von MALDI-TOF MS-Proteinen; Dr. Terry Moore und Dr. Atul Jain zur Rekristallisation von Alpha-Cyano-4-Hydroxycinnamic acid matrix (CHCA).
Materialien
| Name | Company | Catalog Number | Comments |
| Acetonitrile | Fisher | 60-002-65 | LC-MS Ultra CHROMASOLV |
| Autoflex Speed LEF MALDI-TOF instrument | Bruker Daltonics | ||
| Bruker Daltonics Bacterial test standard | Fisher | NC0884024 | Bruker Daltonics 8604530 |
| Bruker Peptide Calibration standard | Fisher | NC9846988 | Bruker Daltonics 8206195 |
| Formic Acid | Fisher Chemical | A117-50 | 99.5+%, Optima LC/MS Grade |
| MALDI-TOF target Plate | Bruker Daltonics | ||
| Methanol | Fisher Chemical | A456-500 | Optima LC/MS Grade |
| Toothpicks | any is ok | ||
| Trifluoroacetic acid | Fisher | AC293810010 | 99.5%, for biochemistry, ACROS Organics |
| Water | VWR | 7732-18-5 | LC-MS |
| α-Cyano-4-hydroxycinnamic acid | Sigma | 28166-41-8 | (C2020-25G) ≥98% (TLC), powder |
Referenzen
- Sandrin, T. R., Goldstein, J. E., Schumaker, S. MALDI TOF MS profiling of bacteria at the strain level: A review. Mass Spectrometry Reviews. 32 (3), 188-217 (2013).
- Cain, T. C., Lubman, D. M., Weber, W. J., Vertes, A. Differentiation of bacteria using protein profiles from matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Communications in Mass Spectrometry. 8 (12), 1026-1030 (1994).
- Holland, R. D., Wilkes, J. G., et al. Rapid identification of intact whole bacteria based on spectral patterns using matrix-assisted laser desorption/ionization with time-of-flight mass spectrometry. Rapid Communications in Mass Spectrometry. 10 (10), 1227-1232 (1996).
- Rahi, P., Prakash, O., Shouche, Y. S. Matrix-assisted laser desorption/ionization time-of-flight mass-spectrometry (MALDI-TOF MS) based microbial identifications: challenges and scopes for microbial ecologists. Frontiers in Microbiology. 7, 1359 (2016).
- Silva, R., Lopes, N. P., Silva, D. B. Application of MALDI mass spectrometry in natural products analysis. Planta Medica. 82, 671-689 (2016).
- Clark, C. M., Costa, M. S., Sanchez, L. M., Murphy, B. T. Coupling MALDI-TOF mass spectrometry protein and specialized metabolite analyses to rapidly discriminate bacterial function. Proceedings of the National Academy of Sciences of the United States of America. 115 (19), 4981-4986 (2018).
- Freiwald, A., Sauer, S. Phylogenetic classification and identification of bacteria by mass spectrometry. Nature Protocols. 4 (5), 732-742 (2009).
- Schulthess, B., Bloemberg, G. V., Zbinden, R., Böttger, E. C., Hombach, M. Evaluation of the Bruker MALDI Biotyper for identification of Gram-positive rods: development of a diagnostic algorithm for the clinical laboratory. Journal of Clinical Microbiology. 52 (4), 1089-1097 (2014).
- Schumann, P., Maier, T. MALDI-TOF mass spectrometry applied to classification and identification of bacteria. Methods in Microbiology. 41, 275-306 (2014).
- Chambers, M. C., Maclean, B., et al. A cross-platform toolkit for mass spectrometry and proteomics. Nature Biotechnology. 30 (10), 918-920 (2012).
- Kessner, D., Chambers, M., Burke, R., Agus, D., Mallick, P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 24 (21), 2534 (2008).
- Martens, L., Chambers, M., et al. mzML-a community standard for mass spectrometry data. Molecular & Cellular Proteomics. 10 (1), (2011).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten