
Method Article
Genotypisierung und Quantifizierung der In-Situ-Hybridisierungsfärbung in Zebrafischen
In diesem Artikel
Zusammenfassung
Gen-Editing-Technologien haben es Forschern ermöglicht, Zebrafischmutanten zu generieren, um die Genfunktion relativ einfach zu untersuchen. Hier bieten wir einen Leitfaden zur parallelen Embryo-Genotypisierung und Quantifizierung von In-situ-Hybridisierungssignalen in Zebrafischen. Dieser unvoreingenommene Ansatz bietet eine höhere Genauigkeit bei phäotypischen Analysen auf der Grundlage der In-situ-Hybridisierung.
Zusammenfassung
Die In-situ-Hybridisierung (ISH) ist eine wichtige Technik, die es Forschern ermöglicht, die mRNA-Verteilung vor Ort zu untersuchen und ist seit Jahrzehnten eine kritische Technik in der Entwicklungsbiologie. Traditionell stützten sich die meisten Genexpressionsstudien auf die visuelle Auswertung des ISH-Signals, eine Methode, die anfällig für Verzerrungen ist, insbesondere in Fällen, in denen Probenidentitäten von vornherein bekannt sind. Wir haben bereits über eine Methode berichtet, um diese Verzerrung zu umgehen und eine genauere Quantifizierung der ISH-Signale bereitzustellen. Hier stellen wir eine einfache Anleitung zur Anwendung dieser Methode vor, um die Expressionsniveaus von Genen von Interesse an ISH-gefärbten Embryonen zu quantifizieren und diese mit ihren entsprechenden Genotypen zu korrelieren. Die Methode ist besonders nützlich, um räumlich eingeschränkte Genexpressionssignale in Proben gemischter Genotypen zu quantifizieren und bietet eine unvoreingenommene und genaue Alternative zu den traditionellen visuellen Scoring-Methoden.
Einleitung
Die Einführung von Genom-Editing-Technologien (ZFN, TALENs und in jüngerer Zeit CRISPR/Cas9) hat zu einem massiven Anstieg der Anzahl von Laboratorien auf der ganzen Welt geführt, die diese Systeme nutzen, um die Funktion bestimmter Gene in vivo zu untersuchen. Insbesondere Zebrafische sind genetisch manipuliert und viele Mutanten wurden in den letzten1,2erzeugt. Für Entwicklungsbiologen ist die In-situ-Hybridisierung (ISH) eine der häufigsten Methoden zur Bewertung der phäotypischen Folgen von Genmutationen in der embryonalen Entwicklung. In Ermangelung offensichtlicher morphologischer Defekte, die homozygote Mutanten von ihrem wilden Typ oder heterozygoten Geschwistern trennen, ist es wichtig, verschiedene Genotypen korrekt identifizieren zu können.
Die klassische ISH stützt sich auf qualitative Analysen von Signalintensitäten, um Rückschlüsse auf regulatorische Wechselwirkungen zwischen dem Gen von Interesse und ausgewählten Markergenen zu ziehen. Obwohl diese Analysen nützlich sind, leiden sie unter technischen Schwankungen und können durch die Erwartungen der Forscher verzerrt sein. So wurde eine Methode entwickelt, um die Genexpression nach der Abbildung von ISH-gefärbten Embryonen zu quantifizieren, ohne vorher den entsprechenden Genotyp zu kennen. Es folgte eine effiziente DNA-Extraktion und Genotypisierung, die es uns ermöglichte, den Genotyp quantitativ mit der Genexpression3zu korrelieren. Während die Genotypisierung von Embryonen nach ISH vor4,5verwendet wurde, wurde die bildbasierte Quantifizierung von ISH-Mustern außer einigen Studien nicht weit verbreitet6,7. Die beliebtesten Alternativen basieren auf visueller Bewertung oder Zählung von ISH-befleckten Zellen8,9,10, beide anfällig für schlechte Reproduzierbarkeit und Forscher-Bias. Diese Methode ist besonders nützlich, um Veränderungen in Genen mit expressionischen Mustern zu untersuchen, die räumlich eingeschränkt sind, wie runx1 oder gata2b, beide ausgedrückt in einer begrenzten Teilmenge von Aortenbodenzellen, die als hämogenes Endothel11,12bezeichnet werden.
Hier wollen wir einen praktischen Leitfaden für die Umsetzung der Quantifizierung durch Bildanalyse mit Fidschi13, sowie das DNA-Extraktions- und Genotypisierungsprotokoll zur Verfügung stellen. Dies soll unsere zuvor veröffentlichte Methode3visuell veranschaulichen. Unsere Methode ermöglicht eine genaue Darstellung der von der ISH nachgewiesenen Variation der Genexpression und eine unvoreingenommene Zuordnung der Genexpressionsniveaus zu bestimmten Genotypen.
Protokoll
Die Verfahren, an denen Tierquäler beteiligt sind, sind durch das Animals (Scientific Procedures) Act 1986 geregelt und vom Innenministerium und der örtlichen Tierschutz- und Ethikprüfstelle genehmigt worden.
1. Bild ISH-gefärbte Embryonen
- Bereiten Sie eine Glycerinlösung vor (50% –80% in 1x PBS Puffer) und mischen Sie die Lösung zu homogenisieren (z. B. in einer Walze für mindestens 5 min lassen). Diese Lösung kann monatelang bei Raumtemperatur aufbewahrt werden.
- Nach In-situ-Hybridisierung14,15,16,17, übertragen Sie Embryonen auf die Glycerinlösung mit einer 3 ml Pasteur Pipette und lassen Sie sich für mindestens 5 min begleichen. Wenn bildgebende Embryonen älter als 24 hpf (Stunden nach der Befruchtung), können sie wie beschrieben18gebleicht werden.
- Bereiten und beschriften Sie genügend PCR-Röhren, um ISH-gefärbte Embryonen nach der Bildgebung zu übertragen.
- Fügen Sie 100% Glycerin an den Boden des Brunnens in einem Glas-Depressionsschlitten mit einer 3 ml Pasteur Pipette.
- Mit einer 3 ml Pasteur Pipette, übertragen Sie einen einzelnen ISH-gebeizt embryo auf die Glasrutsche und orientieren Sie sich nach Bedarf unter einem Stereomikroskop mit digitaler Kamera und Boden- und Oberbeleuchtung.
HINWEIS: Verwenden Sie Gel-Ladepipettenspitzen, um die Embryonen für die Bildgebung zu positionieren, aber andere Werkzeuge (z. B. Zangen, Sezieren der Nadel) sind gleichermaßen ausreichend. - Passen Sie mit dem ersten Embryo die Beleuchtungs- und Belichtungszeit bei der gewünschten Vergrößerung an. Verwenden Sie diese Bedingungen für ALLE Embryonen im selben Experiment (d. h., wenn 40 Embryonen aus einem heterozygoten Mutanten inkreuz, stellen Sie sicher, dass die Beleuchtung, Belichtungszeit und Vergrößerung für alle gleich sind).
- Bild so viele Embryonen wie erforderlich. Beschriften Sie jedes Bild mit einer eindeutigen Nummer. Übertragen Sie den Embryo nach der Bildgebung in ein PCR-Rohr/eine Platte, die mit der gleichen Zahl gekennzeichnet ist.
HINWEIS: Bilder sollten als TIF-Dateien gespeichert werden, aber auch andere Formate sind ausreichend.- Entfernen Sie bei Bedarf überschüssiges Glycerin in den PCR-Röhren/Platten.
HINWEIS: An dieser Stelle können die Embryonen mehrere Wochen bei Raumtemperatur in den PCR-Röhren gelagert werden.
- Entfernen Sie bei Bedarf überschüssiges Glycerin in den PCR-Röhren/Platten.
2. Extrahieren Sie DNA und Genotyp der ISH-gefleckten Embryonen
HINWEIS: Verwenden Sie hier eine zuverlässige und kostengünstige Methode, um genomische DNA auf Basis der HoTSHOT-Methode19 mit einer DNA-Extraktionseffizienz von 95%-100%3zu isolieren.
- Nachdem die Bildgebung abgeschlossen ist, fügen Sie jedem Rohr 40-75 L alkalischen Lysepuffer (z. B. HoTSHOT) hinzu.
- Bei 95 °C ca. 30 min inkubieren und die Rohre auf 4 °C abkühlen, bevor ein gleiches Volumen des Neutralisationspuffers hinzugefügt wird. Eine nächtliche Inkubation bei 4 °C kann die PCR-Effizienz verbessern.
HINWEIS: An dieser Stelle kann die genomische DNA zur Genotypisierung verwendet oder bei -20 °C gespeichert werden, bis sie benötigt wird. - Genotypproben mit einer geeigneten Methode (z.B. HRMA, RFLP)3,20,21, wie für die Mutation von Interesse erforderlich.
- Beachten Sie den Genotyp, der jeder Probe entspricht (z. B. mithilfe einer Tabellenkalkulationssoftware).
3. Quantifizieren Sie die Pixelintensität von ISH-gefärbten Embryonen (Bildanalyse mit Fidschi-Software)
- Um die In-situ-Hybridisierungs-Signalintensität (ISH) zu quantifizieren, konvertieren Sie alle Bilder in 8-Bit-Graustufen wie beschrieben3. Wenn die Bilder als gespeichert wurden. TIF-Dateien, verwenden Sie ein Fidschi-Makro für Batch-Konvertierung3. Alternativ können Sie Bilder in andere Formate konvertieren (z.B. . JPG) zu . TIF mit entsprechender Software und dann konvertieren in 8-Bit-Graustufen mit Fidschi. Der Einfachheit halber ist hier das Schritt-für-Schritt-Verfahren, das zuvor3veröffentlicht wurde, mit einigen Änderungen.
- Öffnen Sie Bilder in Fidschi, und kehren Sie das Bild mit Bearbeiten > Invertierenum . Ändern Sie dann den Bildtyp in 8-Bit(Bild > Typ > 8-Bit).
- Zeichnen Sie mit dem Polygonauswahlwerkzeug den Bereich von Interesse (ROI) manuell auf dem Bild um den Bereich, der das Signal enthält.
- Drücken Sie t, um den ROI-Manager zu öffnen. Verwenden Sie den Befehl "Messen" des ROI-Managers, um die Intensität des ROI zu messen. Kopieren Sie den Mittelwert aus dem Ergebnisfenster in eine Tabellenkalkulationssoftware.
- Bewegen Sie den gleichen ROI, um die gleiche Größe und Form wie die ursprüngliche Region zu gewährleisten, in eine Region des Zebrafisches, die keine Färbung enthält. Wiederholen Sie Schritt 3.4, um den Hintergrund zu messen.
- Um die mittlere Pixelintensität des ISH-Signals zu erhalten, subtrahieren Sie den mittleren Intensitätswert des Hintergrundbereichs von dem des gebeizten Bereichs für jeden Embryo.
- Weisen Sie jeden Intensitätswert einem Genotyp zu (ab Schritt 2.3).
4. Analysieren Sie die Ergebnisse mit geeigneten statistischen Tests
- Zeichnen Sie alle Werte in einem Q-Q-Diagramm, um Abweichungen von der Normalverteilung zu identifizieren.
HINWEIS: Die Normalverteilung kann auch mit dem Kolmogorov-Smirnov-Test oder dem Shapiro-Wilk-Test überprüft werden. Bei großen Stichprobengrößen besteht jedoch ein hohes Risiko von Fehlalarmen in diesen Tests. - Wenn starke Abweichungen von der Normalverteilung bestehen, transformieren Sie alle Werte (mit ln- oder sqrt-Funktionen), um sicherzustellen, dass sie normal verteilt sind, bevor Sie fortfahren.
- Analysieren Sie die Unterschiede zwischen den Werten (gegebenenfalls transformiert), die jedem Genotyp zugewiesen sind (wt vs. heterozygote vs. mutant) mit 2-tailed ANOVA mit 95% Konfidenzniveau, wobei die Gleichheit von Abweichungen mit einem Levene-Test und einer Welch-Korrektur berücksichtigt wird. Für paarweise Vergleiche zwischen jedem Paar genotypen verwenden Sie Tukeys (gleiche Varianzen) oder Games-Howell (ungleiche Varianzen) Post-hoc-Test.
- Wenn die Werte trotz der Transformation nicht normal verteilt sind, verwenden Sie einen nicht-parametrischen Test (Kruskall-Wallis), um die Unterschiede zwischen den rangierten Werten und den Mehrfachvergleichstest eines Dunnnach-hoc-Mahnfachs mit der Bonferroni-Korrektur für paarweise zu analysieren. Vergleiche.
- Zeichnen Sie die nicht transformierten Werte (ab Schritt 3.6) als Punktdiagramme für die beste Darstellung der Ergebnisse.
Ergebnisse
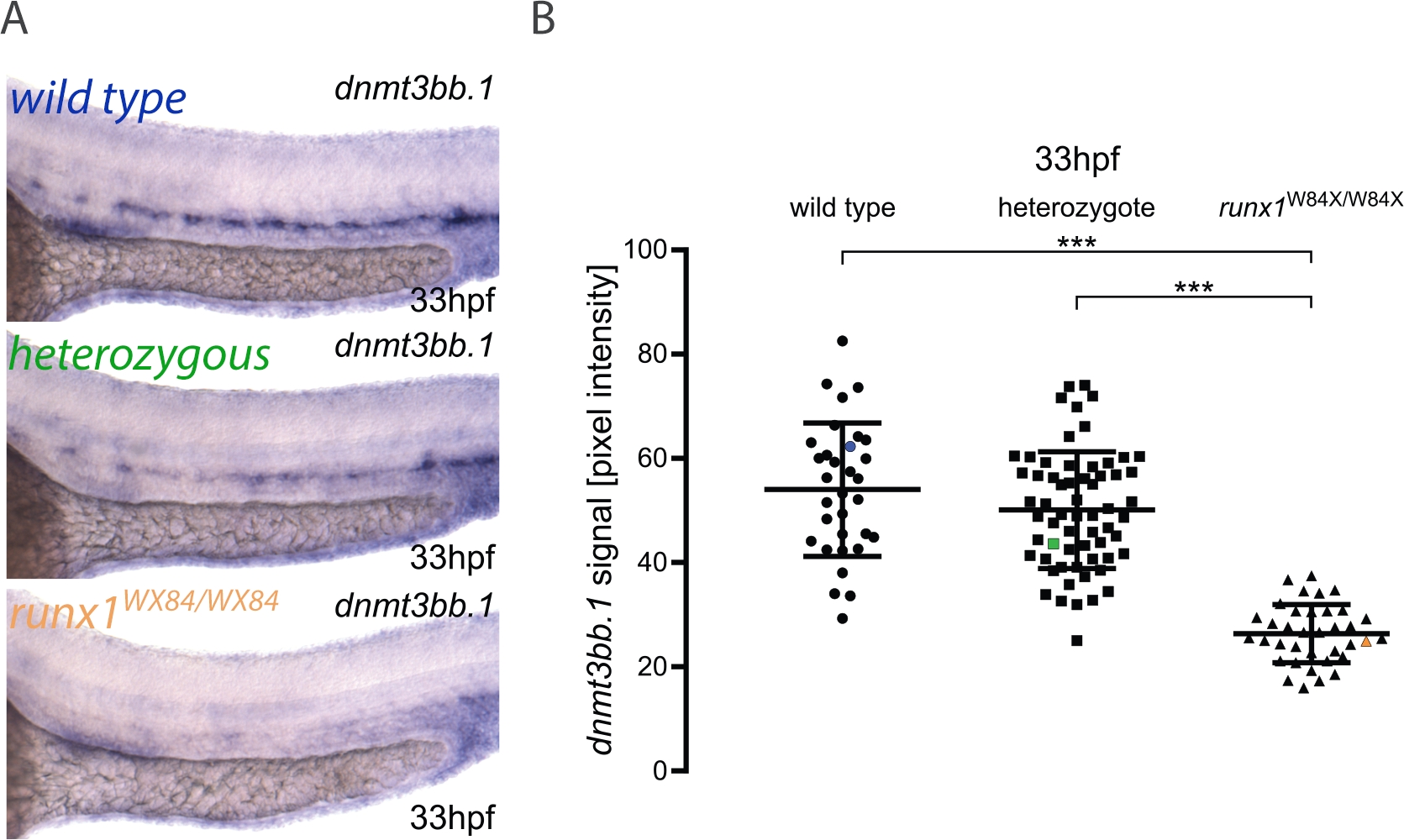
Hier beschreiben wir die praktische Anwendung der Pipeline für Bildquantifizierung und Embryo-Genotypisierung, wie an anderer Stelle veröffentlicht3. Der Workflow für die Methode ist in Abbildung 1dargestellt. Um zu veranschaulichen, wie diese Methode verwendet wird, wurde ISH für dnmt3bb.1 in 33 hpf Embryonen aus einem runx1W84X/+22 incross durchgeführt ( Abbildung2). 130 Embryonen wurden unter den gleichen Beleuchtungsbedingungen wie im Protokoll beschrieben und mit einer eindeutigen Nummer gekennzeichnet. Nach der Bildgebung wurde jeder Embryo zur Genotypisierung in ein PCR-Röhrchen übertragen. Zu diesem Zeitpunkt wurde die Bildanalyse durchgeführt, um jedem Bild einen Pixelintensitätswert zuzuweisen. Der Genotyp wurde dann dem entsprechenden Bild zugeordnet und die Pixelintensitätswerte nach ihrem Genotyp für statistische Analysen gruppiert. Eine Abnahme des dnmt3bb.1-Ausdrucks wurde in runx1W84X/W84X Mutanten (Abbildung 2A,B)3, in Übereinstimmung mit früheren Beobachtungen5festgestellt. Interessanterweise zeigten runx1W84X/+ heterozygote Embryonen keine signifikanten Unterschiede in der dnmt3bb.1-Expression (Abbildung 2A,B) im Vergleich zu ihren Wildtyp-Geschwistern, was darauf hindeutet, dass eine Kopie von Runx1 ausreicht, um den dnmt3bb.1-Ausdruck auf entsprechenden Ebenen aufrechtzuerhalten.
Viele Zebrafischmutanten zeigen keinen embryonalen Phänotyp, der sonst mit anderen Funktionsverlusttechnologien wie Morpholino-Oligonukleotiden (MOs) nachgewiesen werden kann. Diese Diskrepanz kann auf eine Reihe von Ursachen zurückgeführt werden, einschließlich Off-Target-Effekte, mütterliche Proteinkompensation, ein hypomorphes Allel23 oder das kürzlich entdeckte Phänomen der genetischen Kompensation24,25,26,27. In diesem Beispiel haben wir gefragt, ob der Runx1-Ausdruck in lmo4auob100 Mutanten reduziert oder verloren gegangen ist, da zuvor veröffentlichte Daten mit einem lmo4a MO darauf hindeuteten, dass runx1 in lmo4a Morphanten28verringert wird. Hier ergab die Analyse keine signifikanten Unterschiede in der Runx1-Expression zwischen Wildtyp und lmo4auob100 homozygotmutants3 (Abbildung 3A,B). Weitere Analysen durch einzelne Embryonen qPCR zeigten, dass eine kleine, aber signifikante Abnahme der Runx1-Expression bei lmo4auob100 Mutanten zu verzeichnen war (Abbildung 3C). Daher ist es möglich, dass die Bildquantifizierung möglicherweise nicht in der Lage ist, kleine Unterschiede in den Ausdrucksebenen zu erkennen. Alternativ ist der Mangel an Unterschieden zwischen den genotypen, die wir entdeckt haben, real und die qPCR-Experimente erkennen Veränderungen der Runx1-Expression in anderen Geweben wie dem Telenchephalon, wo sowohl Lmo4a als auch Runx1 exprimiert werden. Die Forscher sollten ihre Ergebnisse immer mit einer unabhängigen Methode wie qPCR verifizieren, aber idealerweise durch Durchflusszytometrie für das Gewebe von Interesse anreichern.
In seltenen Fällen, in denen die ISH einen hohen Hintergrund hat(Abbildung 3D), ist der Pixelintensitätswert dieses Bereichs so hoch, dass die Subtraktion vom Signalwert eine negative Zahl erzeugt und in solchen Fällen diese Embryonen von der Analyse ausgeschlossen würden. Unserer Erfahrung nach trat dies bei etwa 0,4% der runx1-sonsiertenEmbryonen3 auf, kann aber zwischen Experimenten, Sonden oder Chargen von Reagenzien variieren. Obwohl dies eine Einschränkung der Methode sein könnte, ist es sehr unwahrscheinlich, dass die niedrige Frequenz des hohen Hintergrunds die Gesamtergebnisse beeinflusst.
Um die Wirkung der Auswahl verschiedener Bereiche für Hintergrundkorrekturen zu testen, haben wir zunächst die Pixelintensität des runx1 ISH-Signals in 28-hpf-Embryonen unter Verwendung verschiedener Bereiche für Hintergrundkorrekturen gemessen (Abbildung 4). Es wurden vier verschiedene Regionen ausgewählt: zwei im Stammbereich (R1 und R2), eine im Dotterbereich (ungefärbt, aber wahrscheinlich Hintergrundfärbung) und eine kleinere Fläche, die vor dem ROI liegt (R4, Abbildung 4B). Die Messung der Pixelintensität in diesen Regionen zeigte einen relativ stabilen Unterschied in der Intensität zwischen ROI und beiden Hintergrundbereichen (Abbildung 4C). Allerdings zeigte R3 immer sehr hohe Werte (über denen im ROI). Nach Inversion und Konvertierung in 8-Bit erscheint der Dotterbereich sehr hell und eignet sich daher nicht für die Verwendung als Hintergrundkorrektur. R2 war näher am ROI, enthielt aber ein ISH-Signal, und die Verwendung von randaliertem Signal verringerte die mittlere Pixelintensität im Vergleich zu R1 (weiter dorsal, weg vom ISH-Signal) oder R4. Daher sind entweder R1 oder R4 geeignete Bereiche, die für die Hintergrundkorrektur verwendet werden können (obwohl die Fläche von R4 kleiner als die von R1 ist). Als Nächstes wollten wir vergleichen, wie sich die Verwendung von R1 oder R4 auf die Ergebnisse beim Vergleich des runx1-Ausdrucks auswirkt. Dazu haben wir dll4+/- Heterozygoten29 gekreuzt und den Runx1-Ausdruck in zufällig ausgewähltem Wildtyp und dll4-/-Embryonen analysiert (Abbildung 4E). Obwohl die Verwendung von R1 oder R4 für die Hintergrundkorrektur einzelne Werte beeinflusste, unterschieden sich die mittleren Pixelintensitäten innerhalb desselben Genotyps nicht signifikant (Abbildung 4E). Darüber hinaus ergibt der Vergleich der Runx1-Expression immer noch ähnliche mittlere Intensitätswerte zwischen Genotypen, die entweder R1- oder R4-Bereiche als Hintergrundkorrektur verwenden(-R1=16,3 bzw.R4=18,2). Zusammengenommen kamen wir zu dem Schluss, dass die Wahl des Hintergrundbereichs zwar wichtig ist, die Hauptkriterien jedoch darin besteht, dass sie keine Dotterregionen (anfällig für die Anhäufung von Hintergrundfärbungen) umfasst und dass sie keine (spezifische) Färbung enthalten sollte, die die Pixelintensitätswerte des Hintergrunds verzerren könnte.

Abbildung 1: Workflow des Parallelbildquantitierungs- und Genotypisierungsprotokolls. Embryonen, die aus einem Fisch-Heterozygoten für ein mutiertes Allel gesammelt wurden, werden mit einem Standard-ISH-Protokoll auf das gemessene Gen untersucht. Nach der Bildgebung wird die genomische DNA mithilfe des HotSHOT-Protokolls extrahiert, indem der Lysepuffer direkt in eine 0,2 ml PCR-Röhre in den Embryo aufgenommen wird, gefolgt von einer 30 min Inkubation bei 95 °C. Diese DNA wird zur Genotypisierung der Embryonen durch PCR, PCR und Restriktionsfragmentlänge Polymorphismus (RFLP), KASP-Assays oder jede andere geeignete Methode verwendet. Parallel dazu werden die Bilder für jeden Embryo invertiert und in 8-Bit-Graustufen umgewandelt. ROIs gleicher Form und Größe, die das ISH-Signal (gelb) und den Hintergrund (blau) enthalten, werden manuell ausgewählt und gemessen. Die Messungen, die den entsprechenden Genotypen zugeordnet sind, werden statistisch analysiert. Abbildung angepasst von Dobrzycki et al.3Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Die Bildquantifizierung in runx1-Mutanten zeigt einen reduzierten Dnmt3bb.1-Ausdruck durch ISH. (A) Beispielbilder von ISH in 33 hpf wild type (blau), runx1+/W84X (grün) und runx1W84X/W84X (orange) Embryonen, die dnmt3bb.1-Expression in der dorsalen Aorta zeigen. (B) Die Pixelintensitätswerte von dnmt3bb.1 mRNA in runx1W84X/W84XEmbryonen (n=36) sind im Vergleich zu Wildtypen (n=32) und Heterozygoten (n=62) (ANOVA, p < 0.001) signifikant verringert. Die Variationskoeffizienten betragen 24 %, 22 % bzw. 21 % für Wildtyp-, Heterozygot- bzw. Mutantengruppen. Blauer, grüner und orangefarbener Datenpunkt entspricht den Beispielbildern aus Panel A. Die Balken stellen den Mittelwert s.d. ***p<0.001 (Games-Howell post-hoc-Test) dar. Abbildung angepasst von Dobrzycki et al.3Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Messung der Runx1-Expressionswerte durch ISH in lmo4auob100Mutanten. (A) Repräsentative Bilder der ISH für runx1 in 28 hpf wild type (blau), heterozygot (grün) und lmo4auob100/uob100 (orange) Embryonen, die den Ausdruck in der dorsalen Aorta zeigen. (B) Quantifizierung des runx1 mRNA-Signals, von DER ISH, von 28 hpf wild type (n=15), heterozygot lmo4a+/- (het) (n=34) und lmo4auob100/uob100 mutant (n=18) embryonen aus einer Kupplung zeigt keinen signifikanten Unterschied in der Runx1-Pixelintensität zwischen den verschiedenen Genotypen (ANOVA,> p 0.6). Blauer, grüner und orangefarbener Datenpunkt entspricht den Beispielbildern aus Panel A. Die Balken stellen den Mittelwert s.d. (C) Boxplots dar, die normalisierte Runx1 mRNA-Werte (2-Ct)in einem einzelnen Wildtyp (blau; n=12) und lmo4auob100/uob100 (mut, orange; n=12) Embryonen anzeigen, gemessen mit qRT-PCR, die im Vergleich zum Wildtyp verringerte Runx1-Werte in den Mutanten aufweisen. *p < 0,05 (t-Test). (D) Beispiel für ein ISH-Experiment an einem 28-hpf-Embryo (gefärbt für runx1, gelbe Pfeilspitzen) mit hohem Hintergrund. Abbildung angepasst von Dobrzycki et al.3Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Auswirkungen der Hintergrundintensitätskorrektur auf die Messergebnisse. (A) Repräsentatives Bild der Runx1 ISH Färbung in einem wilden Embryo bei 28 hpf. (B) Gleiches Bild nach Inversion und Konvertierung in 8-Bit. Der Bereich von Interesse (ROI) wird grün hervorgehoben und vier verschiedene Bereiche, die für die Hintergrundkorrektur (R1-R4) verwendet werden, sind gelb hervorgehoben. (C) Rohpixelintensitätsmessungen in allen in Panel B dargestellten Bereichen. Beachten Sie, dass die Intensität in R3 (Dotter) konstant höher ist als das tatsächliche ISH-Signal im ROI (n=11). (D) Runx1-Ausdrucksebenen im ROI mit R1-, R2- und R4-Hintergrundbereichen. Bereiche für ROI, R1, R2 und R3 bis 28500 Pixel; R4 bis 8500 Pixel. Beachten Sie, dass R3-Hintergrund für diesen Vergleich nicht verwendet wurde, da die Hintergrundkorrektur (ROI-R3) konsistent negative Werte lieferte. (E) Runx1-Expressionsebenen im Wildtyp und dll4-/- Mutanten mit R1 oder R4 zur Hintergrundkorrektur (n=10 für jede Probe). Die statistische Analyse in den Panels D und E wurde mit einem nicht-parametrischen Kruskal-Wallis-Test durchgeführt, wobei davon ausgegangen wird, dass die Pixelintensitätswerte nicht normal verteilt sind. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Bei der Verwendung dieser Methode zur Quantifizierung der Genexpression sollten einige Faktoren berücksichtigt werden. Die bildgebenden Bedingungen müssen während des gesamten Experiments beibehalten werden (z. B. Beleuchtung, Belichtungszeiten und Embryopositionierung), um die Variabilität zwischen den Messungen zu reduzieren. Ein kritischer Punkt ist es, eine Überfärbung der Proben zu vermeiden, da Unterschiede in der Färbung zwischen den Proben maskiert werden können. So konnte beispielsweise die Abnahme der VegfA-Expression in Abwesenheit von Eto2 in Xenopus laevis Embryonen30 nur durch sorgfältige Überwachung der Färbung über einen Zeitraum von 24 Stunden nachgewiesen werden. Daher ist es eine gute Praxis, empirisch angemessene Färbeniveaus für jedes Gen zu bestimmen, das seine Expression am besten darstellt, ohne die Sättigung zu erreichen. Überfärbung erhöht auch künstlich die Hintergrundpixelintensität in den konvertierten 8-Bit-Graustufenbildern und verzerrt die Quantifizierungsergebnisse. In extremen Fällen kann der Hintergrundpegel in embryonalen Geweben höher sein als das ISH-Signal im ausgewählten ROI, und diese Proben sollten von der Analyse ausgeschlossen werden. Ein ähnliches Phänomen wurde beobachtet, als wir die Eignung des ungefärbten Dotters auf Hintergrundkorrektur testeten (Abbildung 4). Nach Inversion und Konvertierung in 8-Bit werden die dunkleren Pixel im Dotterbereich heller als das ISH-Signal im Embryo und machen die Hintergrundkorrigiertwerte negativ. Vermeiden Sie daher die Verwendung des Dotters zur Hintergrundkorrektur. Die Messung des Hintergrundsignals in pigmentierten Bereichen im Embryo (z.B. die Augen oder der dorsale Teil des Stammes ab 26/28 hpf) verzerrt gleichermaßen die Quantitationsergebnisse und sollte ebenfalls vermieden werden. Es gibt Protokolle zum Bleichen von Zebrafischembryonen, entweder vor oder nach der ISH18 und Bleichembryonen, die älter als 24 hpf sind, bevor eine Bildgebung empfohlen wird.
Da diese Methode auf der Messung der Pixelintensität in einem definierten Bereich vor einer Hintergrundpixelintensität in einem äquivalenten nicht-gefärbten Bereich beruht, ist es nicht geeignet, allgegenwärtige oder nahezu allgegenwärtig exprimierte Gene wie sie sind zu quantifizieren. Stattdessen eignet es sich gut zur Messung der Expression von Genen mit einer räumlich begrenzten Verteilung, in der ein Bereich zur Messung der Hintergrundpixelintensität leicht identifiziert werden kann. Unsere zusätzliche Analyse legt nun nahe, dass die Verwendung einer kleineren Fläche (3-4x kleiner) für die Hintergrundkorrektur ähnliche Ergebnisse liefert wie die Verwendung eines gleichwertigen Bereichs wie der ROI. Dies erweitert die Anwendbarkeit der Methode auf Gene, die in größeren räumlichen Bereichen exprimiert werden (und daher größere ROIs für Intensitätsmessungen erfordern), solange man deutlich ungefärbte Bereiche des Embryos zur Hintergrundkorrektur verwenden kann.
Schließlich schlagen wir vor, dass die Genotypisierung parallel oder nach der Bildquantifizierung durchgeführt wird, um die Voreingenommenheit der Experimentatoren zu minimieren. Wenn ein zweiter Experimentator aufgefordert wird, die Quantifizierung auf anonymisierten Proben zu wiederholen und mit dem ersten Satz von Messungen zu vergleichen, wird auch dazu beitragen, die Voreingenommenheit der Experimentatoren zu verringern. Wenn die zu quantifizierenden Bilder aus einem Vergleich zwischen Behandlungen stammen, die keine Genotypisierung erfordern (z. B. Wildtyp vs. chemischer Inhibitor oder Wildtyp vs. MO-Knockdown), sollte der Experimentator, der die Messungen durchführt, für die Identität des Beispiel.
Offenlegungen
Die Autoren erklären, dass sie keine konkurrierenden finanziellen Interessen haben.
Danksagungen
Wir danken den Mitarbeitern von Biomedical Services in Oxford und Birmingham für die ausgezeichnete Zebrafischzucht. T.D. wurde von einem Wellcome Trust Chromosom and Developmental Biology PhD Scholarship (#WT102345/Z/13/Z) gefördert. R.M. und M.K. wurden von der British Heart Foundation (BHF IBSR Fellowship FS/13/50/30436) gefördert und sind dankbar für ihre großzügige Unterstützung. R.M. unterstützt das BHF Centre of Research Excellence (RE/13/1/30181), Oxford.
Materialien
| Name | Company | Catalog Number | Comments |
| 0.2 mL PCR tubes (8-strips with lids) | StarLab | A1402-3700 | 96-well plates are equally appropriate for sample handling but beware of cross contamination between samples |
| 3 mL Pasteur pipettes | Alpha Laboratories | LW4114 | |
| Cavity slides | Brand | BR475535-50EA | |
| Digital Camera (Qimaging Micropublisher 5.0) | Qimaging | ||
| Eppendorf Microloader tips | Eppendorf | 10289651 | the tips are used to orient the embryos for imaging in glycerol |
| Excel | Microsoft | ||
| F3000 Fiber Optic Cold Light Source | Photonic | ||
| Fiji | |||
| Glycerol | Sigma | G5516-1L | |
| Graphpad Prism 8.01 | GraphPad Software, Inc. | we prefer to use Graphpad, but other statistics software packages are also suitable (e.g. SigmaPlot or SPSS) | |
| HotSHOT alkaline lysis buffer | 25 mM NaOH, 0.2 mM disodium EDTA, pH 12 | ||
| HotSHOT neutralization buffer | Tris HCl 40 mM, pH 5 | ||
| PBS (10X) pH 7.4 | Thermofisher | 70011044 | |
| Stereomicroscope with illumination stand (Nikon SMZ800N) | Nikon | ||
| Thermocycler | Thermofisher |
Referenzen
- Varshney, G. K., et al. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Research. 25 (7), 1030-1042 (2015).
- Varshney, G. K., et al. CRISPRz: a database of zebrafish validated sgRNAs. Nucleic Acids Research. 44 (D1), D822-D826 (2016).
- Dobrzycki, T., Krecsmarik, M., Bonkhofer, F., Patient, R., Monteiro, R. An optimised pipeline for parallel image-based quantification of gene expression and genotyping after in situ hybridisation. Biology Open. 7 (4), bio031096 (2018).
- Bresciani, E., et al. CBFbeta and RUNX1 are required at 2 different steps during the development of hematopoietic stem cells in zebrafish. Blood. 124 (1), 70-78 (2014).
- Gore, A. V., et al. Epigenetic regulation of hematopoiesis by DNA methylation. Elife. 5, e11813 (2016).
- Fan, Y., et al. Tissue-Specific Gain of RTK Signalling Uncovers Selective Cell Vulnerability during Embryogenesis. PLoS Genetics. 11 (9), e1005533 (2015).
- Wen, B., et al. GATA5 SUMOylation is indispensable for zebrafish cardiac development. Biochimica et Biophysica Acta. 1861 (7), 1691-1701 (2017).
- Espin-Palazon, R., et al. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell. 159 (5), 1070-1085 (2014).
- Peterkin, T., Gibson, A., Patient, R. Redundancy and evolution of GATA factor requirements in development of the myocardium. Developmental Biology. 311 (2), 623-635 (2007).
- Genthe, J. R., Clements, W. K. R-spondin 1 is required for specification of hematopoietic stem cells through Wnt16 and Vegfa signaling pathways. Development. 144 (4), 590-600 (2017).
- Kalev-Zylinska, M. L., et al. Runx1 is required for zebrafish blood and vessel development and expression of a human RUNX1-CBF2T1 transgene advances a model for studies of leukemogenesis. Development. 129 (8), 2015-2030 (2002).
- Butko, E., et al. Gata2b is a restricted early regulator of hemogenic endothelium in the zebrafish embryo. Development. 142 (6), 1050-1061 (2015).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Lleras Forero, L., et al. Segmentation of the zebrafish axial skeleton relies on notochord sheath cells and not on the segmentation clock. Elife. 7, (2018).
- Jowett, T., Yan, Y. L. Double fluorescent in situ hybridization to zebrafish embryos. Trends in Genetics. 12 (10), 387-389 (1996).
- Thisse, C., Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nature Protocols. 3 (1), 59-69 (2008).
- Narayanan, R., Oates, A. C. Detection of mRNA by Whole Mount in situ Hybridization and DNA Extraction for Genotyping of Zebrafish Embryos. Bio-protocol. , e3193 (2019).
- Monteiro, R., Pouget, C., Patient, R. The gata1/pu.1 lineage fate paradigm varies between blood populations and is modulated by tif1gamma. EMBO JOURNAL. 30 (6), 1093-1103 (2011).
- Truett, G. E., et al. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques. 29 (1), 52-54 (2000).
- Wilkinson, R. N., Elworthy, S., Ingham, P. W., van Eeden, F. J. A method for high-throughput PCR-based genotyping of larval zebrafish tail biopsies. Biotechniques. 55 (6), 314-316 (2013).
- Parant, J. M., George, S. A., Pryor, R., Wittwer, C. T., Yost, H. J. A rapid and efficient method of genotyping zebrafish mutants. Developmental Dynamics. 238 (12), 3168-3174 (2009).
- Jin, H., et al. Definitive hematopoietic stem/progenitor cells manifest distinct differentiation output in the zebrafish VDA and PBI. Developement. 136 (4), 647-654 (2009).
- Stainier, D. Y. R., et al. Guidelines for morpholino use in zebrafish. PLoS Genetics. 13 (10), e1007000 (2017).
- El-Brolosy, M. A., Stainier, D. Y. R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genetics. 13 (7), e1006780 (2017).
- Rossi, A., et al. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 524 (7564), 230-233 (2015).
- Ma, Z., et al. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature. 568 (7751), 259-263 (2019).
- El-Brolosy, M. A., et al. Genetic compensation triggered by mutant mRNA degradation. Nature. 568 (7751), 193-197 (2019).
- Meier, N., et al. Novel binding partners of Ldb1 are required for haematopoietic development. Development. 133 (24), 4913-4923 (2006).
- Kettleborough, R. N., et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature. 496 (7446), 494-497 (2013).
- Leung, A., et al. Uncoupling VEGFA functions in arteriogenesis and hematopoietic stem cell specification. Developmental Cell. 24 (2), 144-158 (2013).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten