
Method Article
Anwendungen der viralen RNA/DNA-In-situ-Hybridisierung der nächsten Generation in der Forschung an humanen Immundefizienzviren/Affenimmundefizienzviren
In diesem Artikel
Zusammenfassung
Hier präsentieren wir einen in situ Hybridisierungsassay der nächsten Generation zur Identifizierung spezifischer viraler RNA- oder DNA-Sequenzen in formalinfixierten Paraffin-eingebetteten (FFPE) Geweben. Dieser Ansatz ermöglicht die Visualisierung geringer Kopien von RNA und DNA in weniger als 24 h mit sehr hoher Sensitivität und Spezifität.
Zusammenfassung
Die In-situ-Hybridisierung ist eine leistungsfähige Technik, um spezifische RNA- oder DNA-Sequenzen innerhalb einzelner Zellen in Gewebeschnitten zu identifizieren und wichtige Einblicke in physiologische Prozesse und die Pathogenese von Krankheiten zu liefern. Die In-situ-Hybridisierung (ISH) wird seit vielen Jahren eingesetzt, um die Position von Zellen zu bestimmen, die mit Viren infiziert sind, aber vor kurzem wurde ein ISH-Ansatz der nächsten Generation mit einer einzigartigen Sondendesignstrategie entwickelt, die eine gleichzeitige Signalverstärkung und Hintergrundunterdrückung ermöglicht, um eine Einzelmolekülvisualisierung unter Beibehaltung der Gewebemorphologie zu erreichen. Diese ISH der nächsten Generation basiert auf einem Ansatz wie der verzweigten PCR, wird jedoch in situ durchgeführt und ist einfacher, sensitiver und reproduzierbarer als klassische ISH-Methoden oder In-situ-PCR-Ansätze zum routinemäßigen Nachweis von RNA oder DNA in formalinfixierten, in Paraffin eingebetteten (FFPE) Geweben. In den letzten Jahren hat unser Labor diese ISH-Plattform für den Nachweis von humaner Immundefizienz- (HIV) und Affenimmundefizienz-positiver (SIV) viraler RNA (vRNA) und/oder viraler DNA (vDNA) positiver Zellen in einer Vielzahl von FFPE-Geweben eingesetzt. Mit diesem ausführlichen technischen Manuskript möchten wir unser Wissen und unsere Ratschläge mit allen Personen teilen, die daran interessiert sind, die ISH der nächsten Generation in ihrer Forschung zu nutzen.
Einleitung
ISH ist der experimentelle Ansatz, der verwendet wird, um komplementäre DNA, RNA oder modifizierte Nukleinsäurestränge (d. h. Sonden) auf spezifische DNA- oder RNA-Sequenzen innerhalb einer Zelle oder eines Gewebeabschnitts abzuzielen und zu visualisieren. ISH ermöglicht die spezifische Lokalisierung und Visualisierung spezifischer Nukleinsequenzen in Geweben, was wichtig ist, um das Expressionsniveau, die Organisation, die Verteilung und die Wechselwirkungen zwischen dem Ziel und seiner zellulären Umgebung zu verstehen, was wertvolle Informationen sind, die mit anderen populären Techniken, wie z. B. qPCR, nicht gewonnen werden können. Bis vor kurzem wurde die ISH üblicherweise entweder mit einer markierten komplementären DNA oder einer komplementären RNA (Riboprobe) durchgeführt. Diese Sonden wurden entweder direkt mit radio-, fluoreszierenden oder antigenmarkierten Basen (z. B. 35S, FITC und Digoxigenin) konjugiert und dann entweder mittels Autoradiographie, Fluoreszenzmikroskopie bzw. immunhistochemischer Detektionsansätze im Gewebe lokalisiert und quantifiziert. Obwohl diese In-situ-Technologien nach wie vor wertvolle Ansätze sind, gibt es viel Raum für Verbesserungen, um Ansätze zu entwickeln, die weniger arbeitsintensiv, einfacher, schneller, empfindlicher und spezifisch sind.
Ein alternativer kommerzieller Next-Generation-ISH-Ansatz (z.B. RNAscope Assay), der erstmals 2012 zum Nachweis von Wirts-Boten-RNA (mRNA) beschrieben wurde, basiert auf der verzweigten PCR. Der mRNA-Nachweis wird in FFPE-Zellen und -Geweben durchgeführt, mit einer Sensitivität, die sich der Visualisierung einzelner RNA-Moleküle in einzelnen Zellenannähert 1. Die Spezifität dieses Ansatzes wird unter der einzigartigen Bedingung erreicht, dass zwei Doppel-Z-Zielsonden zusammenhängend an ihre jeweiligen komplementären RNA- (oder DNA-) Sequenzen binden, damit ein Signalvorverstärkersequenziell 1 bindet. Dies ermöglicht die Initiierung einer Signalamplifikationskaskade über nachfolgende Hybridisierungsschritte, ähnlich der verzweigten DNA (bDNA)1,2. Darüber hinaus ist dieser Ansatz bemerkenswert schnell und einfach, mit Ergebnissen, die in nur 1 Tag (<8 h) erzielt werden, ein erheblicher Vorteil im Vergleich zu bis zu 4 Wochen mit alternativen Techniken, einschließlich Radio-ISH 1,2. Diese ISH der nächsten Generation hat der HIV/SIV-Forschung neue Perspektiven und Möglichkeiten eröffnet. Die größten Hindernisse für eine HIV-Heilung sind die Zell- und Gewebereservoire, die in den frühen Stadien der Krankheit angelegt werden 3,4. Das übergeordnete Ziel dieser Technik ist es, die wichtigsten Gewebekompartimente zu identifizieren, zu lokalisieren und letztendlich zu verstehen, die als virales Reservoir fungieren und in einem infizierten Wirt persistent sind. Dies wiederum wird bei der Entwicklung wirksamer Heilungsstrategien gegen HIV helfen.
In diesem Manuskript erläutern wir unser Duplex-RNA/DNA-Multiplex-ISH-Protokoll der nächsten Generation (z. B. RNAscope/DNAscope) im Detail und erklären, wie wir das bestehende RNA-ISH-Protokoll modifiziert haben, um das ISH der nächsten Generation für unsere Proben und spezifischen Ziele zu optimieren. Dieses Protokoll ermöglicht die Visualisierung, Lokalisierung und Quantifizierung von HIV/SIV-viraler RNA und viraler DNA innerhalb von 5-μm-Gewebeschnitten. Die gleichzeitige Visualisierung von vRNA und vDNA erfolgt durch die Kombination von zwei benutzerdefinierten Sondensätzen: einen Sense, der auf den vDNA-kodierenden Strang abzielt (C1 SIVmac239 Gag-Pol-Sense-Sonde [416141-C1]), und einen Anti-Sense, der auf vRNA-Transkripte abzielt (C2 SIVmac239 Vif-Env-Nef-Tar-Anti-Sense-Sonde [416131-C2]), der verschiedene Regionen des viralen Genoms abdeckt (Tabelle 1), wobei zwei verschiedene Visualisierungskanäle, C1 und C2, verwendet werden. In diesem Protokoll ermöglichen uns die Kanäle C1 und C2, Signale in verschiedenen Farben zu visualisieren (d. h. AP in Rot und HRP in Braun) und die Sonden mit unterschiedlichen Ansätzen zu detektieren. Ohne die Verarbeitung und das Schneiden der Gewebefixierung dauert dieser Assay 2 Tage. Hier wird das Duplex-vRNA- und vDNA-in-situ-Hybridisierungsprotokoll vorgestellt, das an Zellpellets oder Gewebeschnitten durchgeführt werden kann.
Protokoll
1. Schnitt- und Folienvorbereitungen
- Schneiden Sie die Paraffinblöcke ab und schneiden Sie mit einem Mikrotom 5 +/-1 μm-Abschnitte. Montieren Sie die Schnitte oder Zellpellets auf geladene Objektträger in einem 40-45 °C RNase-freien Wasserbad. Rutschen über Nacht bei 37 °C oder RT an der Luft trocknen.
HINWEIS: Die Objektträger können bis zu 3 Monate bei Raumtemperatur (RT) und 6 Monate bei 4 °C gelagert werden. - Entparaffinieren Sie FFPE-Objektträger.
- Die Folien 1 h trocken bei 60 °C backen.
- Füllen Sie in einem Abzug zwei Objektträger-Färbeschalen mit ~200 mL frischem Xylol und zwei weitere Färbeschalen mit ~200 mL frischem 100 % Ethanol. Decken Sie die Behälter mit Deckeln ab.
- Legen Sie die Objektträger in ein Gitter und tauchen Sie sie in die erste xylolhaltige Schale. 5-10 min bei RT unter Rühren inkubieren.
- Legen Sie die Objektträger in die zweite xylolhaltige Schale und inkubieren Sie sie 5-10 min lang bei RT unter Rühren.
- Legen Sie die Objektträger sofort in die Schale mit 100% Ethanol. Inkubieren Sie die Objektträger 5-10 Minuten lang bei RT unter Rühren.
- Legen Sie die Objektträger sofort in die zweite Schale mit 100% Ethanol und inkubieren Sie sie 5-10 Minuten lang bei RT unter Rühren.
- Nehmen Sie den Rost vom Ethanol, klopfen Sie vorsichtig auf die Seite des Rosts, um überschüssiges Ethanol zu entfernen, und spülen Sie ihn 5-15 Minuten lang in RNase-freiem Wasser ab.
2. Vorbereitung des Ofens
- Schalten Sie den Hybridisierungsofen ein und stellen Sie die Temperatur auf 40 °C ein.
- Legen Sie ein Tuch oder ein robustes, saugfähiges Papiertuch in ein Tablett und befeuchten Sie es vollständig mit doppelt destilliertem Wasser, um die Feuchtigkeit zu kontrollieren.
- Setzen Sie das abgedeckte Blech in den Ofen ein und schließen Sie die Ofentür. Erwärmen Sie das Tablett vor Gebrauch mindestens 30 min bei 40 °C. Bewahren Sie das Blech bei Nichtgebrauch im Ofen auf.
3. Wärmeinduzierte Epitop-Gewinnung
- Bereiten Sie einen 0,5x Citrat-basierten ISH-Hybridisierungs-Target-Retrieval-Puffer vor (10 nmol/L, pH = 6, siehe Materialtabelle). In einem Becherglas auf der Heizplatte zum Kochen bringen.
- Führen Sie eine hitzeinduzierte Epitop-Entnahme durch, indem Sie die Objektträger für 30 Minuten in einen Siedeziel-Entnahmepuffer legen.
- Nehmen Sie die Objektträger aus dem Zielrückholpuffer und waschen Sie sie sofort in doppelt destilliertem Wasser. Vor dem Trocknen an der Luft 5 Minuten lang in 100 % Ethanol dehydrieren.
- Sobald die Objektträger an der Luft getrocknet sind, tragen Sie einen hydrophoben Barrierestift auf, um den Gewebeabschnitt auf dem Objektträger zu umgeben. Achten Sie darauf, die hydrophobe Barriere vollständig an der Luft trocknen zu lassen.
4. Protease-Vorbehandlung
- Legen Sie die getrockneten Objektträger auf ein verschließbares Objektträgergestell und bereiten Sie dann die Reagenzien für die Proteasevorbehandlung (Proteaseaufschlusslösung, 2,5 μg/ml) vor, indem Sie sie mit sterilem, kaltem PBS im Verhältnis 1:5 verdünnen. Gut mischen.
HINWEIS: Drei verschiedene Protease-Reagenzien mit unterschiedlichen Konzentrationen sind im kommerziell erhältlichen Kit enthalten. Protease III (Standard), Protease IV (stark) und Protease Plus (mild). Testen Sie empirisch die Zeit und Verdünnung der Protease vor der Durchführung in einer Studie, da die optimalen Bedingungen je nach Gewebetyp, Fixierung und Dicke variieren (siehe Diskussion). - Geben Sie die verdünnte Proteaselösung auf Objektträger, um die Gewebeschnitte vollständig zu bedecken. Die Objektträger werden sofort 20 Minuten lang bei 40 °C in einem Ofen (vorbereitet in Schritt 1.4) inkubiert, wobei darauf zu achten ist, dass die Objektträger in der feuchten Hybridisierungsschale verschlossen sind. Lassen Sie die Gewebeschnitte für den Rest des Protokolls nicht austrocknen.
- Spülen Sie sofort 3x aus, indem Sie den arretierbaren Schieberost in eine mit doppelt destilliertem Wasser gefüllte Waschschale tauchen.
- Führen Sie die endogene Peroxidase-Blockade durch, indem Sie die Peroxidaselösung auf jeden Gewebeabschnitt tropfen, um ihn vollständig zu bedecken. Inkubieren Sie die Objektträger 10 Minuten lang bei RT. Wenn Sie fertig sind, spülen Sie die Abschnitte 3x in doppelt destilliertem Wasser ab.
5. Sondenhybridisierung und Signalverstärkung
HINWEIS: Um Verdunstung zu verhindern, stellen Sie sicher, dass das feuchtigkeitskontrollierte Tablett richtig abdichtet, damit das Gewebe während der Inkubationsschritte nicht austrocknet. Stellen Sie die Feuchtigkeitskammer während des Waschvorgangs wieder in den Ofen, um sicherzustellen, dass sie bei 40 °C bleibt.
- Mischen Sie die C2-Sonde und die C1-Sonde im Verhältnis 1:50, indem Sie 1 Volumen der C2-Sonde auf 50 Volumen der C1-Sonde in ein Röhrchen pipettieren, wie vom Hersteller empfohlen. Drehen Sie das Rohr mehrmals um. Die Zielsondenmischung im 40 °C Ofen für ~10 min vorwärmen, um eventuellen Niederschlag vor der Verwendung aufzulösen.
HINWEIS: Die gemischten Zielsonden können bis zu 6 Monate bei 4 °C gelagert werden. - Nehmen Sie die Objektträger aus dem Wasser. Spülen Sie die Objektträger aus und klopfen oder schnippen Sie sie, um überschüssiges Wasser aus den Gewebeabschnitten zu entfernen. Geben Sie die Sonde sofort auf die Objektträger und stellen Sie sicher, dass jeder Gewebeabschnitt vollständig und ohne Luftblasen bedeckt ist. Inkubieren Sie die Sondenmischung über Nacht bei 40 °C in der Feuchtigkeitskammer.
- Nehmen Sie die Objektträger am nächsten Tag aus dem Ofen und legen Sie sie für 5 min bei RT in das Waschtablett mit 0,5x Waschpuffer. Wiederholen Sie den Waschschritt noch einmal.
- Entfernen Sie die Objektträger aus dem Waschpuffer. Spülen Sie die Objektträger aus und klopfen oder schnippen Sie sie, um überschüssigen Waschpuffer aus den Gewebeabschnitten zu entfernen.
- Kommerzielle Abgabe von gebrauchsfertigem AMP 1-Reagenz (2 nmol/L) in Hybridisierungspuffer B (20 % Formamid, 5x SSC, 0,3 % Lithiumdodecylsulfat, 10 % Dextransulfat, Blockierungsreagenzien) auf den Objektträgern, um eine vollständige Abdeckung des Gewebeschnitts ohne Luftblasen zu gewährleisten. 30 min bei 40 °C in der Feuchtigkeitskammer inkubieren. Wiederholen Sie die Schritte 1.6.3-1.6.4 und führen Sie den Waschvorgang jeweils 2 Minuten lang durch.
- Geben Sie das im Handel erhältliche AMP 2 aus. Achten Sie darauf, dass der Gewebeabschnitt vollständig und ohne Luftblasen bedeckt ist. Inkubieren Sie die Objektträger in der Feuchtigkeitskammer für 15 min bei 40 °C. Wiederholen Sie die Schritte 5.3-5.4 und führen Sie den Waschgang jeweils 2 Minuten lang durch.
- Geben Sie handelsübliches AMP 3 aus. Achten Sie darauf, dass der Gewebeabschnitt vollständig und ohne Luftblasen bedeckt ist. Inkubieren Sie die Objektträger in der Feuchtigkeitskammer für 30 min bei 40 °C. Wiederholen Sie die Schritte 5.3-5.4 und führen Sie den Waschgang jeweils 2 Minuten lang durch.
- Geben Sie das im Handel erhältliche AMP 4 aus. Achten Sie darauf, dass der Gewebeabschnitt vollständig und ohne Luftblasen bedeckt ist. Inkubieren Sie die Objektträger in der Feuchtigkeitskammer für 15 min bei 40 °C. Wiederholen Sie die Schritte 5.3-5.4 und führen Sie den Waschgang jeweils 2 Minuten lang durch.
- Geben Sie das im Handel erhältliche AMP 5 aus. Achten Sie darauf, dass der Gewebeabschnitt vollständig und ohne Luftblasen bedeckt ist. Inkubieren Sie die Objektträger in der Feuchtigkeitskammer für 30 min bei 40 °C. Wiederholen Sie die Schritte 5.3-5.4 und führen Sie den Waschgang jeweils 2 Minuten lang durch.
- Geben Sie das im Handel erhältliche AMP 6 aus. Achten Sie darauf, dass der Gewebeabschnitt vollständig und ohne Luftblasen bedeckt ist. Inkubieren Sie die Objektträger in der Feuchtigkeitskammer für 15 min bei 40 °C. Wiederholen Sie die Schritte 5.3-5.4 und führen Sie den Waschgang jeweils 2 Minuten lang durch.
- Spülen Sie die Objektträger vor der Detektion einmal in 1x TBS-Tween 20 (0,05 % v/v). Entfernen Sie die Objektträger aus dem Waschpuffer, spülen Sie sie aus und klopfen oder schnippen Sie auf die Objektträger, um überschüssigen Waschpuffer von den Gewebeabschnitten zu entfernen. Sofort in die mit 1x TBS-Tween-Puffer gefüllte Waschschale legen.
6. Erkennung des Zielsignals von Kanal 1 (C1)
HINWEIS: Dies erfolgt unter Verwendung der roten alkalischen Phosphatase und der schnellen roten Chromogenamplifikation 6 aus 2-Plex-Nachweiskits (siehe Materialtabelle), die alkalische Phosphatase-Markierungen enthalten, und chromogener Nachweis. Es verwendet schnelles Rot als Substrat, um ein rotes Signal zu erzeugen.
- Bereiten Sie die Arbeitslösung Fast Red (FR) mit einer Verdünnung von Fast RED-B auf Fast RED-A im Verhältnis 1:60 vor. Gut mischen. Um den Niederschlag zu reduzieren und ein saubereres Signal zu erhalten, filtrieren Sie die Chromogenlösung mit einer Spritze durch eine 0,45 μm MCE-Membran.
HINWEIS: Verwenden Sie die Fast RED-B-Lösung innerhalb von 5 Minuten. Nicht direkter Sonneneinstrahlung oder UV-Licht aussetzen. - Nehmen Sie die Objektträger aus dem TBS-Tween, spülen Sie sie aus und klopfen oder schnippen Sie auf die Objektträger, um überschüssigen Puffer aus den Gewebeschnitten zu entfernen.
- Geben Sie gemischte, filtrierte FR-Lösung auf jeden Gewebeabschnitt und achten Sie darauf, dass jeder Abschnitt vollständig bedeckt ist. 6-8 Minuten bei RT inkubieren. Unter dem Mikroskop beobachten.
- Spülen Sie die Objektträger in 0,5x Waschpuffer 2x aus. Entfernen Sie die Objektträger aus dem Waschpuffer, spülen Sie sie aus und klopfen oder schnippen Sie auf die Objektträger, um überschüssigen Waschpuffer von den Gewebeabschnitten zu entfernen.
- Geben Sie das im Handel erhältliche AMP 7 aus. Achten Sie darauf, dass der Gewebeabschnitt vollständig und ohne Luftblasen bedeckt ist. Inkubieren Sie die Objektträger 10 Minuten lang bei 40 °C in der Feuchtigkeitskammer. Wiederholen Sie die Schritte 5.3-5.4 und führen Sie den Waschgang jeweils 2 Minuten lang durch.
- Geben Sie handelsübliches AMP 8 aus. Achten Sie darauf, dass der Gewebeabschnitt vollständig und ohne Luftblasen bedeckt ist. Inkubieren Sie die Objektträger in der Feuchtigkeitskammer für 15 min bei 40 °C. Wiederholen Sie die Schritte 5.3-5.4 und führen Sie den Waschgang jeweils 2 Minuten lang durch.
- Geben Sie das im Handel erhältliche AMP 9 aus. Achten Sie darauf, dass der Gewebeabschnitt vollständig und ohne Luftblasen bedeckt ist. Inkubieren Sie die Objektträger in der Feuchtigkeitskammer für 30 min bei 40 °C. Wiederholen Sie die Schritte 5.3-5.4 und führen Sie den Waschgang jeweils 2 Minuten lang durch.
- Geben Sie handelsübliches AMP 10 aus. Achten Sie darauf, dass der Gewebeabschnitt vollständig und ohne Luftblasen bedeckt ist. Inkubieren Sie die Objektträger in der Feuchtigkeitskammer für 30 min bei 40 °C. Wiederholen Sie die Schritte 5.3-5.4 und führen Sie den Waschgang jeweils 2 Minuten lang durch.
7. Erkennung des Zielsignals von Kanal 2 (C2)
HINWEIS: Dies wird mit handelsüblichen Brown HRP- und DAB-Chromogen-Kits durchgeführt (siehe Materialtabelle). Die Amplifikation 10 aus der 2-Plex-Detektion enthält Meerrettichperoxidase-Markierungen, und die chromogene Detektion wird unter Verwendung von DAB durchgeführt, um ein braunes Signal zu erzeugen.
- Für eine optimale Erkennung des DAB-Signals verwenden Sie handelsübliche Kits und befolgen Sie die Anweisungen des Herstellers (siehe Materialtabelle). Unter dem Mikroskop beobachten.
ACHTUNG: DAB ist giftig. Befolgen Sie die entsprechenden Vorsichtsmaßnahmen und Sicherheitsrichtlinien beim Umgang und der Entsorgung dieser Chemikalie.
8. Gegenfärbung und Montage
- Färben Sie die Objektträger mit Hämatoxylin gegen.
- Färben Sie die Objektträger 30 s lang mit 50 % frischem gefiltertem Hämatoxylin, während Sie die Objektträger bewegen. Folien werden violett angezeigt. Spülen Sie sofort unter fließendem Wasser ab, während Sie die Rutschen auf und ab bewegen, bis das Wasser klar ist. Die Gewebeschnitte bleiben violett.
- Um einen besseren Kontrast zu erzielen, legen Sie die gegengefärbten Objektträger 1 Minute lang in destilliertes, mit Lithiumcarbonat gesättigtes Wasser. Spülen Sie mindestens 3x gründlich unter fließendem Wasser ab, während Sie die Objektträger bewegen. Verwenden Sie für die abschließende Spülung doppelt destilliertes Wasser.
- Da FR empfindlich auf organische Lösungsmittel reagiert, müssen Objektträger, die mit FR gefärbt wurden, mit Eindeckmedium auf Wasserbasis bedeckt und über Nacht bei RT getrocknet werden.
- Montieren Sie die Rutschen.
- Stellen Sie sicher, dass die mit Eindeckmedium auf Wasserbasis bedeckten Gewebeschnitte trocken sind.
- Tauchen Sie die Objektträger vor dem Verrutschen der Abdeckung mit Einbettreagenz in Xylol. Achten Sie darauf, Luftblasen zwischen Deckglas und Gewebeteil zu vermeiden oder zu entfernen und lassen Sie es 16 Stunden lang bei RT trocknen.
9. Quantitatives Bildanalyseprotokoll für RNAscope mit CellProfiler5
- Stellen Sie kurz gesagt, dass die Software Hämatoxylin- und FR-Flecken in separate Bilder trennt. Identifizieren und messen Sie die interessierenden Objekte: Zellkerne, Virionen, positive Zellen und aggregierte FR-positive Färbung. Speichern Sie die Messungen in einer CSV-Datei und speichern Sie das analysierte Bild.
- Wählen Sie die Option "Farben entmischen", wodurch die Flecken getrennt und der ursprüngliche Region of Interest (ROI) in separate Hämatoxylin- und FR-Bilder aufgeteilt wird.
- Wenn Hämosiderin, Tätowierung oder ähnliche Merkmale die Analyse beeinträchtigen, fügen Sie einen zweiten "Unmix"-Schritt mit Hämatoxylin, FR und DAB hinzu. Verwenden Sie das zweite FR-Bild, um die intensivsten FR-Pixel zu finden. Dieses zweite Bild wird als Maske für echte FR-Färbung verwendet.
- Optional können Sie die gefärbten Bilder glätten, bevor Sie sie mit einem Schwellenwert versehen. Dies ist eine empirische Entscheidung und nicht immer notwendig. Begrenzen Sie die einzelnen gefärbten Bilder mit "IdentifyPrimaryObjects", um positive Pixel auszuwählen.
- Identifizieren Sie die drei verschiedenen Arten von Objekten (Virion, Virionaggregate, produktive Zelle).
- Stellen Sie sicher, dass es sich bei den Kernen um Objekte mit einem Durchmesser von 4 bis 100 Pixeln (px) handelt, die mit Hämatoxylin gefärbt sind. Klumpen und füllen Sie Löcher nach dem Schwellenwert. "IntenseFastRed"-Objekte haben einen Durchmesser von 4 bis 100 px und umfassen Virionen, positive Zellen und aggregierte positive Färbungen, wie sie bei follikulären dendritischen Zellen (FDCs) in B-Zell-Follikeln (BCF) zu sehen sind. Dieses Bild wird verwendet, um falsch positive Ergebnisse (z. B. Hämosiderin) herauszufiltern.
- Stellen Sie sicher, dass es sich bei den kleinen FR-positiven Werten um Objekte mit einem Durchmesser von 2 bis 12 px handelt. Diese Messung umfasst Virionen und vDNA+-Zellen. Verwerfen Sie alle Objekte außerhalb dieses Bereichs. Klumpen und füllen Sie Löcher nach dem Schwellenwert.
- Stellen Sie sicher, dass FR große positive Objekte: 9-100 px Durchmesser haben. Diese Messung umfasst vRNA+-positive Zellen und aggregierte positive Färbung. Nach dem Schwellenwert sinkt es. Die Größe von kleinen und großen FR-positiven Objekten kann sich überlappen. Sie werden in einem späteren Schritt getrennt.
HINWEIS: Die Software (z. B. Cellprofiler) verwendet Pixel für die Objektgrößen, und die hier verwendeten werden von Dias abgeleitet, die mit 0,2510 μm/Pixel (40x) gescannt wurden.
- Definieren und Extrahieren von Ergebnissen.
- Nachdem alle Objekte identifiziert wurden, definieren und extrahieren Sie die Ergebnisse für die Anzahl der Virionen, produktiven infizierten Zellen und Zellkerne.
- Identifizieren Sie Virionen, indem Sie FastRedSmallPositives maskieren, wobei das IntenseFastRed-Objekt so eingestellt ist, dass falsch positive Ergebnisse (z. B. Hämosiderin) entfernt werden.
- Identifizieren Sie als Nächstes die positiven Zellen und aggregieren Sie die FR-positive Färbung. Entfernen Sie falsch positive Ergebnisse und Virionen aus FastRedLargePositives, indem Sie sie mit IntenseFastRed maskieren und das Virionobjekt festlegen.
- Extrahieren Sie positive Zellen aus den verfeinerten FastRedLargePositives , indem Sie sie erneut mit Nuclei maskieren. Teilen Sie Objekte, die sich nicht mehr berühren, und filtern Sie die Ergebnisse nach Objektbereich, wobei kleine Objekte (≤6 px) entfernt werden. Dadurch werden die Flecken entfernt, die durch das Maskieren der Kernüberlappung entstehen. Das Ergebnis sind die positiven Zellen.
- Definieren Sie abschließend die aggregierten FR-Positivwerte. In diesem Schritt wird IdentifyTertiaryObjects verwendet, um Objekte zu suchen, die in einem größeren, übergeordneten Objekt enthalten sind. In diesem Fall ist der verfeinerte FastRedLargePositives-Objektsatz das übergeordnete Element, und positive Zellen werden subtrahiert.
- Zählen Sie die Anzahl der Virionen und positiven Zellen.
- Messen Sie die aggregierte positive Fläche und rechnen Sie sie von Pixel in mm2 um. Optional können Sie die von Virionen und positiven Zellen eingenommene Fläche in mm2 aufzeichnen, wenn für die Analyse eine Standardzell- und Virionengröße anstelle von direkten Zählungen verwendet werden muss.
- Überlagern Sie die positiven Objekte mit dem Originalbild und speichern Sie das Ergebnis.
HINWEIS: Die ISH-Daten werden zum besseren Verständnis und zur Erleichterung des Vergleichs mit qPCR-Daten durch die Anzahl der Virionen pro 106 Kerne (Zellen) und die Anzahl der produktiv infizierten vRNA+-Zellen pro 106 Kerne (Zellen) angegeben, aber die Ergebnisse können auch nach Gewebefläche in mm2 angegeben werden.
Ergebnisse
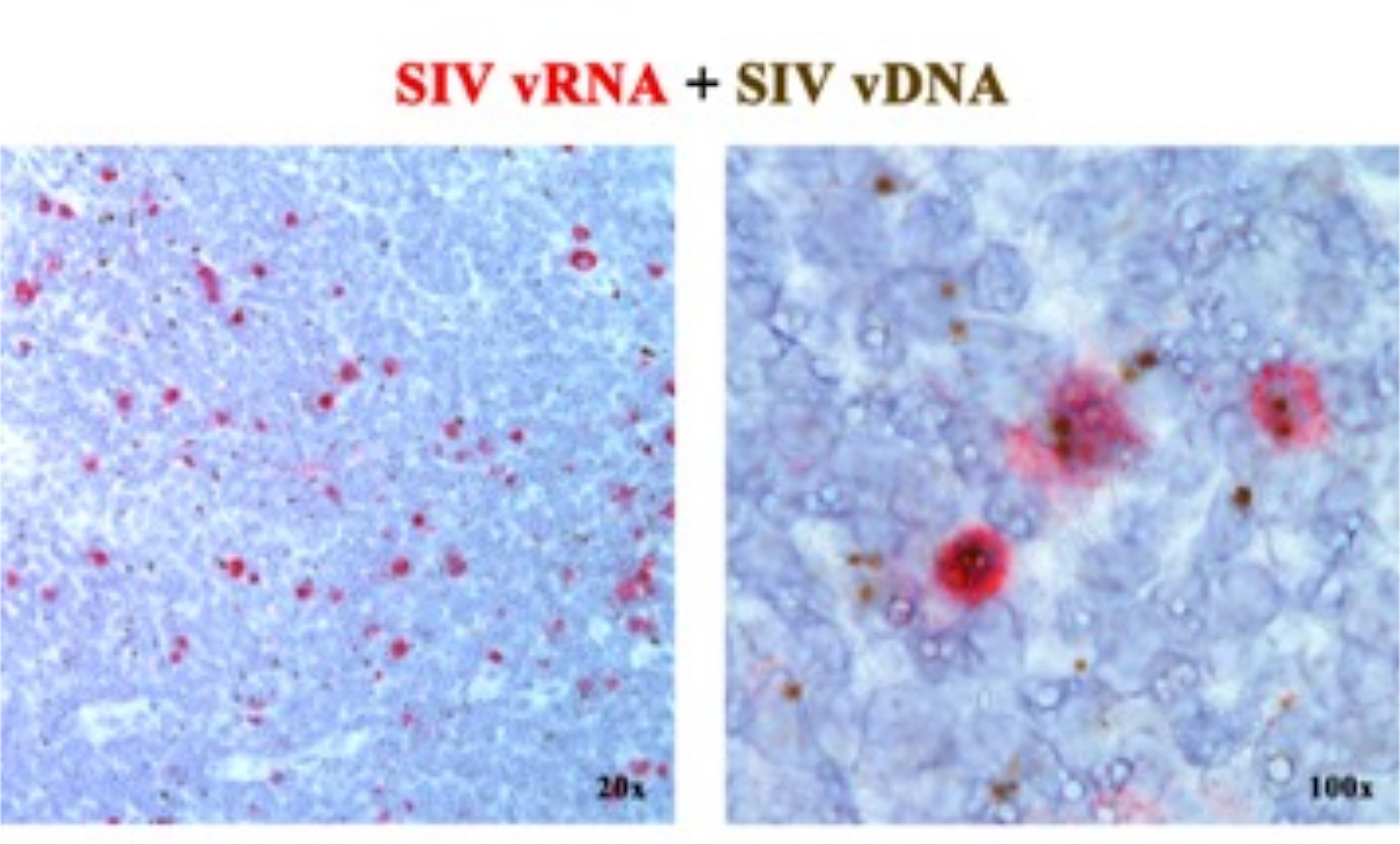
In einem früheren Manuskript 2,6,7,8,9,10,11 haben wir berichtet, dass die ISH-Plattformen der nächsten Generation, die entweder vRNA oder vDNA nachweisen, kombiniert werden können, indem Sense-Sonden (vDNA) verwendet werden, die auf den 5'-gag-pol-Teil des SIV/HIV-Genoms abzielen, und Antisense-Sonden (vRNA), die auf Gene in der 3'-Hälfte des Genoms abzielen (vif, vpx, vpr, tat, env und nef) sowie das TAR-Element im 5'-Genom (Tabelle 1). Dieser Ansatz unterscheidet transkriptionell aktive Zellen (vRNA+, vDNA+) von transkriptionell inaktiven (mutmaßlich latent infizierten Zellen) oder Zellen, die transkriptionell inkompetente Proviren (vRNA-, vDNA+) im selben Gewebe beherbergen, Abschnitt2 (Abbildung 1).

Abbildung 1: Nachweis von viraler RNA und vDNA im selben Gewebeschnitt. Kombination aus RNA-Hybridisierung (rot) und DNA-Hybridisierung (braun) in einem akut SIV-infizierten RM-Lymphknoten, die die Fähigkeit zum Nachweis von vRNA und vDNA im selben Gewebeschnitt demonstriert und einen leistungsstarken Ansatz zur Identifizierung von transkriptionell stummen vDNA+ vRNA-Zellen in situ bietet. Diese Abbildung wurde von Deleage C. et al.2 modifiziert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
| Single Plex RNA/DNAscope-Sondensets | |||
| Name | ACD Katalog # | Anzahl der ZZ | Beschreibung |
| SIVmac239 (Anti-Sense) | 312811 | 83 | Anti-Sense-Sonde, die innerhalb von 1251 bis 9420 bp von D01065.1 zielt (gag, pol, vif, vpx, vpr, tat, env und nef) |
| SIVmac239 (Sense) | 314071 | 83 | Messsonde für den Reverse-Strang innerhalb von 1251-9420bp von D01065.1 (gag, pol, vif, vpx, vpr, tat, env und nef) |
| V-HIV1-Klade A (Anti-Sense) | 416101 | 80 | Anti-Sense-Sonde, die innerhalb von 879-7629bp des HIV-1-Konsenses der Klasse A zielt (gag, pol, vif, vpr, tat, rev, vpu, env und nef) |
| V-HIV1-Klade A (Sense) | 426341 | 80 | Sense-Sonde, die auf den reversen Strang innerhalb von 879-7629bp des HIV-1-Konsenses der Klasse A abzielt (gag, pol, vif, vpr, tat, rev, vpu, env und nef) |
| V-HIV1-Klade B (Anti-Sense) | 416111 | 78 | Anti-Sense-Sonde, die innerhalb von 854-8291bp von AF324493.2, HIV-1 Clade B NL4-3 (gag, pol, vif, vpr, tat, rev, vpu, env und nef) zielt |
| V-HIV1-Klade B (Sense) | 425531 | 78 | Sense-Sonde, die auf den reversen Strang innerhalb von 854-8291bp von AF324493.2, HIV-1 Clade B NL4-3 abzielt (gag, pol, vif, vpr, tat, rev, vpu, env und nef) |
| V-HIV1-Klade D (Anti-Sense) | 416121 | 76 | Anti-Sense-Sonde, die innerhalb von 894-7697 bp des HIV-1-Konsenses der Klasse D zielt (gag, pol, vif, vpr, tat, rev, vpu, env, nef) |
| V-HIV1-Klade D (Sense) | 426351 | 76 | Sense-Sonde, die auf den reversen Strang innerhalb von 894-7697bp des HIV-1 Clade D Konsensus abzielt (gag, pol, vif, vpr, tat, rev, vpu, env, nef) |
| Multi-Plex RNA/DNAscope Sonden-Sets | |||
| Name | ACD Katalog # | Anzahl der ZZ | Beschreibung |
| V-SIVmac239-Knebel-Pol-Sense-C1 | Artikel-Nr.: 416141-C1 | 40 | Sense-Sonde, die auf den Reverse-Strang innerhalb von 1251-4093bp von D01065.1 abzielt (gag und pol) |
| V-SIVmac239-vif-env-nef-tar-C2 (Anti-Sense) | Artikel-Nr.: 416131-C2 | 47 | Anti-Sense-Sonde, die innerhalb von 5381 bis 10257 bp von D01065.1 zielt (vif, vpx, vpr, tat, env, nef und das TAR-Element) |
| V-HIV1-Clade_B-Knebel-Pol-Sinn-C1 | Artikel-Nr.: 444051-C1 | 40 | Sense-Sonde, die auf den Reverse-Strang innerhalb von 854-3940bp von AF324493,2, HIV-1 Clade B NL4-3 abzielt (gag und pol) |
| V-HIV1-Clade_B-vif-vpr-tat-rev-vpu-env-nef-tar-C2 (Anti-Sense) | Artikel-Nr.: 444061-C2 | 40 | Anti-Sense-Sonde, die innerhalb von 5042-9673 bp von AF324493,2, HIV-1 Clade B NL4-3 (vif, vpr, tat, env, nef und das TAR-Element) zielt |
| V-HIV1-Clade_C-Knebel-Pol-Sinn-C1 | Artikel-Nr.: 444021-C1 | 48 | Sense-Sonde, die auf den reversen Strang innerhalb von 888-5032bp der HIV-1-Klade-C-Konzensussequenz abzielt (gag und pol) |
| V-HIV1-Clade_C-vif-vpr- rev-vpu-env-nef-tar-C2 (Anti-Sense) | Artikel-Nr.: 444041-C2 | 49 | Anti-Sense-Sonde, die innerhalb von 5078-9698 bp der HIV-1-Klade-C-Konsenssequenz zielt (vif, vpr, tat, env, nef und das TAR-Element) |
| V-HIV1-Clade_AE-Knebel-Pol-Sinn-C1 | Artikel-Nr.: 444011-C1 | 55 | Sense-Sonde, die auf den reversen Strang innerhalb von 890-4812bp von AF259954.1, HIV-1 Clade AE abzielt (gag und pol) |
| V-HIV1-Clade_AE-vif-vpr-tat-rev-vpu-env-nef-tar-C2 (Anti-Sense) | Artikel-Nr.: 444031-C2 | 57 | Anti-Sense-Sonde, die innerhalb von 5052-9694 bp von AF259954.1, HIV-1 Clade AE (vif, vpr, tat, env, nef und das TAR-Element) zielt |
Tabelle 1: Liste der Sonden für vRNA und vDNA von HIV-1 und SIV.
Diskussion
Die In-situ-Hybridisierung ist ein sorgfältiger Assay, der Strenge und grundlegende Kenntnisse in Nukleinsäurechemie, Zellbiologie und Histologie erfordert, um jeden kritischen Schritt anpassen zu können, um ein Ziel in einer gut konservierten Umgebung zu lokalisieren. In dieser Diskussion möchten wir die kritischen Schritte hervorheben, bei denen die Fehlerbehebung entscheidend ist, um genaue und interpretierbare Ergebnisse zu erhalten.
Die Fixierung und Verarbeitung des Gewebes ist von entscheidender Bedeutung und sollte im Voraus angegangen werden, um sicherzustellen, dass der Assay die besten Ergebnisse liefert. Neutral gepuffertes PFA-Fixiermittel (4 % frisch zubereitet) ist optimal für den Duplex-Assay. Der Assay kann jedoch auch an gefrorenem Gewebe (OCT) mit den entsprechenden Fixationsbedingungen nach der Kryosektion durchgeführt werden.
Die Vorbehandlung der Gewebeschnitte ist ein entscheidender Schritt. In diesem Assay gibt es zwei Vorbehandlungsschritte: Der erste ist eine hitzeinduzierte Epitop-Entnahme (HIER). Dieser Schritt ist wichtig für die Umkehrung von Methylenbrückenvernetzungen und die Wiederherstellung von Proteinstrukturen, die in fixierten Geweben benötigt wird. Die Wirksamkeit dieser Behandlung hängt von der Zeit, der Temperatur, der Art des Rückholpuffers und dem pH-Wert ab. Die zweite Vorbehandlung ist eine Protease-induzierte Epitop-Retrieval (PIER). In diesem Schritt werden Peptide gespalten, das Antigen oder die Nukleotide freigelegt, und es werden Enzyme wie Proteinase K, Trypsin und Pepsin verwendet. Dies ist ein äußerst empfindlicher Schritt, der sowohl die Gewebemorphologie als auch das Zielmolekül schädigen könnte. Die Konzentration des Enzyms sowie die Zeit und Temperatur der Inkubation sind bei diesem Prozess entscheidend. Eine Überverdauung führt zu einer schlechten Kernabgrenzung und Schwierigkeiten bei den Quantifizierungsschritten. Es ist von entscheidender Bedeutung, ein Gleichgewicht zwischen einem optimalen Zugang zum RNA/DNA-Ziel und Vorbehandlungsbedingungen zu finden, die das Gewebe oder das Zielmolekül von Interesse nicht schädigen. Jeder Gewebetyp hat eine unterschiedliche Empfindlichkeit gegenüber jeder dieser Vorbehandlungen und jeder Parameter (Enzymkonzentration, Zeit, Temperatur) sollte empirisch getestet werden.
Die Stringenz des Waschpuffers basiert auf drei Hauptparametern: Temperatur, Konzentration von Salzen und Reinigungsmittel sowie Zeit. Der Waschpuffer ist ein salzhaltiger Natriumcitratpuffer (SSC), und die Salzkonzentration innerhalb des Puffers steuert die Stringenz während der Waschschritte. In ihrem Protokoll empfiehlt ACD, den Waschpuffer in einer Endkonzentration von 0,1x SSC, 0,03 % Lithiumdodecylsulfat zu verwenden. Bei der Arbeit an der DNAscope- und Multiplex-Optimierung stellten wir fest, dass die Verwendung des Waschpuffers in einer Endkonzentration von 0,05x SSC zu besseren Ergebnissen bei der Visualisierung des DNA-Signals führte und erheblich dazu beitrug, die unspezifische Off-Target-Hybridisierung, die sich aus der Inkubation der Sense-Sonde über Nacht ergab, zu reduzieren.
Die Wahl des Nachweisansatzes, Chromogen (rot oder braun) oder Fluoreszenz, muss je nach Gewebetyp und Ziel durchdacht werden, bevor mit dem Assay begonnen wird. Der chromogene Ansatz von Rot ergibt einen schönen Kontrast, da Rot nicht natürlich in Geweben vorkommt. Braunes Chromogen liefert ähnliche Ergebnisse wie rotes Chromogen. Es ist jedoch wichtig zu bedenken, dass einige im Gewebe vorhandene Blutabbauprodukte eine ähnliche Farbe haben und Tätowierfarbe bei der Quantifizierung nur schwer vom braunen Signal zu trennen ist. Ein Fluoreszenzdetektionsansatz wird eine klare Unterscheidung verschiedener zellulärer Marker ermöglichen und das Multiplexing wird einen perfekten Assay bieten, um die Zellen, die vRNA und/oder vDNA beherbergen, zu phänotypisieren.
Mehrere Kontrollen sind notwendig, um die Spezifität der Sonden und die Qualität des Assays sicherzustellen. Jede neu entwickelte Sonde muss an bekannten Positiv- und Negativkontrollgeweben oder Zellpellets getestet werden. Wir erzeugen oft Plasmide, die unsere Zielsequenz enthalten, und führen Transfektion in Zelllinien durch, um positive Kontrollen zu erzeugen. Für jeden Lauf fügen wir ein bekanntes negatives Gewebe (HIV- oder SIV-negativ), eine Kontrolle ohne Sonde, die nur das Sondenverdünnungsmittel enthält, und eine RNase-behandelte Kontrolle hinzu, um die Qualität und Spezifität des Assays sicherzustellen.
Die Quantifizierung ist ein äußerst wichtiger Schritt und sollte mit den entsprechenden Werkzeugen und Algorithmen auf der Grundlage der gestellten Frage durchgeführt werden. In diesem Manuskript haben wir eine Bildanalysesoftware (z.B. Cellprofiler) vorgestellt, die wir nach Abwägung mehrerer Optionen ausgewählt haben. Wir waren der Meinung, dass diese Software die beste Software für unsere Bedürfnisse ist, aber es gibt zahlreiche Bildanalyse-Softwareprogramme, die verwendet werden könnten.
Offenlegungen
Die Autoren haben nichts offenzulegen.
Danksagungen
Dieses Projekt wurde vollständig mit Bundesmitteln des National Cancer Institute, National Institutes of Health, unter der Vertragsnummer finanziert. HHSN261200800001E und durch den Oregon National Primate Research Center NIH Grant Award P51OD011092 (J.D.E). Der Inhalt dieser Veröffentlichung spiegelt nicht unbedingt die Ansichten oder Richtlinien des Department of Health and Human Services wider, noch impliziert die Erwähnung von Handelsnamen, kommerziellen Produkten oder Organisationen eine Billigung durch die US-Regierung. Der Duplex wurde mit Hilfe von Advanced Cell Diagnostics entwickelt.
Materialien
| Name | Company | Catalog Number | Comments |
| ACD HybEZII Hybridization system (110V) with ACD EZ-Batch Slides system | ACD | 321710 | Hybridization oven |
| CAT Hematoxylin | Biocare medical | CATHE-GAL | colorstain |
| Clear-Mount | ELECTRON MICROSCOPY SCIENCES | 17985-15 | mounting reagent for red chromogen |
| Immpact DAB Peroxidase Kit | Vector | SK-4105 | Used to reveal HRP - DAB (Brown) to replace the DAB coming in the ACD kit |
| lithium carbonate | Fisher chemical | L119-500 | bluing solution |
| paraformaldehyde | ELECTRON MICROSCOPY SCIENCES | 15714-S | for tissue fixation (4%) |
| PBS | life technology | 14190-136 | |
| Permount Mounting Medium | ThermoFisher Scientific | SP15-100 | mounting regaent for brown chromogen |
| Prolong Gold | ThermoFisher Scientific | P36930 | mounting regaent for fluorescence |
| ribonucleases A | ThermoFisher Scientific | 12091039 | for RNAse treatment in DNAscope protocol |
| ribonucleases T1 | Roche | R1003 | for RNAse treatment in DNAscope protocol |
| RNAscope 2.5, 2-plex detection reagent | ACD | 322430 | Brown and red kit chromogen detection |
| RNAscope Target Retrieval Reagents | ACD | 322000 | retrieval buffer |
| SuperFrost Plus Glass Slides | ThermoFisher Scientific | 12-550-17 | |
| TBS | BOSTON BIOPRODUCTS | BM-301-4L | for washes |
| TSA Plus Fluorescence palette kit (Cy3, Cy5, TMR, Fluorescein) | Perkin elmer | NEL760001KT | HRP Fluorescence detection |
| Tween 20 | SIGMA | P1379-1L | for washes |
| XYLENE 20LT | ThermoFisher Scientific | AC422680200 |
Referenzen
- Wang, F., et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. Journal of Molecular Diagnosis. 14 (1), 22-29 (2012).
- Deleage, C., et al. Defining HIV and SIV Reservoirs in Lymphoid Tissues. Pathogens and Immunity. 1 (1), 68-106 (2016).
- Sengupta, S., Siliciano, R. F. Targeting the Latent Reservoir for HIV-1. Immunity. 48 (5), 872-895 (2018).
- Churchill, M. J., Deeks, S. G., Margolis, D. M., Siliciano, R. F., Swanstrom, R. HIV reservoirs: what, where and how to target them. Nature Reviews Microbiology. 14 (1), 55-60 (2016).
- McQuin, C., et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biology. 16 (7), e2005970 (2018).
- Deleage, C., Chan, C. N., Busman-Sahay, K., Estes, J. D. Next-generation in situ hybridization approaches to define and quantify HIV and SIV reservoirs in tissue microenvironments. Retrovirology. 15 (1), 4 (2018).
- Deleage, C., Turkbey, B., Estes, J. D. Imaging lymphoid tissues in nonhuman primates to understand SIV pathogenesis and persistence. Current Opinion in Virology. 19, 77-84 (2016).
- Estes, J. D., et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nature Medicine. 23 (11), 1271-1276 (2017).
- Mavigner, M., et al. Simian Immunodeficiency Virus Persistence in Cellular and Anatomic Reservoirs in Antiretroviral Therapy-Suppressed Infant Rhesus Macaques. Journal of Virology. 92 (18), (2018).
- Peterson, C. W., et al. Differential impact of transplantation on peripheral and tissue-associated viral reservoirs: Implications for HIV gene therapy. PLoS Pathogen. 14 (4), e1006956 (2018).
- Deleage, C., et al. Impact of early cART in the gut during acute HIV infection. Journal of Clinical Investigation Insight. 1 (10), e87065 (2016).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten