
Method Article
Messung der Nukleotidbindung an intakte, funktionelle Membranproteine in Echtzeit
In diesem Artikel
Zusammenfassung
Dieses Protokoll stellt eine Methode zur Messung der Adenin-Nukleotid-Bindung an Rezeptoren in Echtzeit in einer zellulären Umgebung vor. Die Bindung wird als Förster-Resonanzenergietransfer (FRET) zwischen Trinitrophenylnukleotidderivaten und Proteinen, die mit einer nicht-kanonischen, fluoreszierenden Aminosäure markiert sind, gemessen.
Zusammenfassung
Wir haben eine Methode entwickelt, um die Bindung von Adenin-Nukleotiden an intakte, funktionelle Transmembranrezeptoren in einer zellulären oder membranartigen Umgebung zu messen. Diese Methode kombiniert die Expression von Proteinen, die mit der fluoreszierenden nicht-kanonischen Aminosäure ANAP markiert sind, und FRET zwischen ANAP und fluoreszierenden (Trinitrophenyl)-Nukleotidderivaten. Wir präsentieren Beispiele für die Bindung von Nukleotiden an ANAP-markierte KATP-Ionenkanäle , gemessen in unüberdachten Plasmamembranen und exzidierten, von innen nach außen gerichteten Membranpflastern unter Spannungsklemme. Letzteres ermöglicht die gleichzeitige Messung der Ligandenbindung und des Kanalstroms, ein direktes Auslesen der Proteinfunktion. Die Datenverarbeitung und -analyse wird ausführlich diskutiert, zusammen mit potenziellen Fallstricken und Artefakten. Diese Methode liefert reichhaltige mechanistische Einblicke in das ligandenabhängige Gating vonK-ATP-Kanälen und kann leicht für die Untersuchung anderer Nukleotid-regulierter Proteine oder anderer Rezeptoren angepasst werden, für die ein geeigneter fluoreszierender Ligand identifiziert werden kann.
Einleitung
Mehrere wichtige Proteinklassen werden direkt durch die Ligandenbindung reguliert. Diese reichen von löslichen Enzymen bis hin zu in Membranen eingebetteten Proteinen, einschließlich Rezeptor-Tyrosinkinasen, G-Protein-gekoppelten Rezeptoren (GPCRs) und Ionenkanälen. GPCRs und Kanäle machen ~34% bzw. ~15% aller aktuellen Wirkstoffziele aus 1,2. Daher besteht ein erhebliches biochemisches und medizinisches Interesse an der Entwicklung von Methoden, die mechanistische Einblicke in Ligand-Rezeptor-Interaktionen liefern. Herkömmliche Methoden zur Messung der Ligandenbindung, einschließlich Photoaffinitätsmarkierung und Radioligandenbindungsstudien, erfordern große Mengen an teilweise gereinigtem Protein und werden in der Regel unter nicht-physiologischen Bedingungen und Zeitskalen durchgeführt. Eine ideale Methode würde nur geringe Mengen an Protein benötigen, könnte an intakten Proteinen durchgeführt werden, die in einer Zell- oder Membranumgebung exprimiert werden, könnte in Echtzeit überwacht werden und wäre mit dem direkten Auslesen der Proteinfunktion kompatibel.
Der Förster-Resonanzenergietransfer (FRET) ist eine Methode, die die Nähe zwischen zwei fluoreszenzmarkierten Molekülen detektiert3. FRET tritt auf, wenn ein angeregter Donor-Fluorophor Energie auf strahlungsfreie Weise auf ein Akzeptormolekül (typischerweise einen anderen Fluorophor) überträgt. Die Energieübertragung führt zu einer Abschreckung der Donorfluoreszenzemission und einer Sensibilisierung der Akzeptoremission (wenn es sich bei dem Akzeptor um einen Fluorophor handelt). Die Übertragungseffizienz ist abhängig von der 6. Potenz des Abstands zwischen dem Donor und dem Akzeptor. Darüber hinaus müssen sich Donor und Akzeptor in unmittelbarer Nähe befinden (in der Regel weniger als 10 nm), damit FRET auftreten kann. Daher kann FRET genutzt werden, um die direkte Bindung zwischen einem fluoreszenzmarkierten Proteinrezeptor und einem fluoreszierenden Liganden zu messen.
Verschiedene Proteine werden durch die Bindung von intrazellulären oder extrazellulären Adenin-Nukleotiden (ATP, ADP, AMP, cAMP) reguliert oder aktiviert. Viele Transporterproteine benötigen ATP-Hydrolyse für ihren Reaktionszyklus, darunter ATP-bindende Kassettentransporter und P-Typ-ATPasen wie die Na+/K+-Pumpe 4,5. ATP-sensitive K+ (KATP)-Kanäle, der Mukoviszidose-Transmembran-Leitfähigkeitsregulator (CFTR) und zyklisch-nukleotidregulierte Kanäle sind allesamt Ionenkanäle, die durch die Bindung intrazellulärer Adenin-Nukleotide gesteuert werden, wodurch sie äußerst empfindlich auf Veränderungen des zellulären Stoffwechsels und der Signaltransduktion reagieren 6,7,8. Purinerge P2X- und P2Y-Rezeptoren reagieren auf Veränderungen des extrazellulären ATP, das als Neurotransmitter oder als Folge einer Gewebeschädigung freigesetzt werden kann9. Wir haben einen FRET-basierten Assay zur Messung der Adenin-Nukleotid-Bindung an Membranproteine in Echtzeit entwickelt. Wir haben diese Methode bereits angewendet, um die Nukleotidbindung anK-ATP-Kanäle 10,11 zu untersuchen.
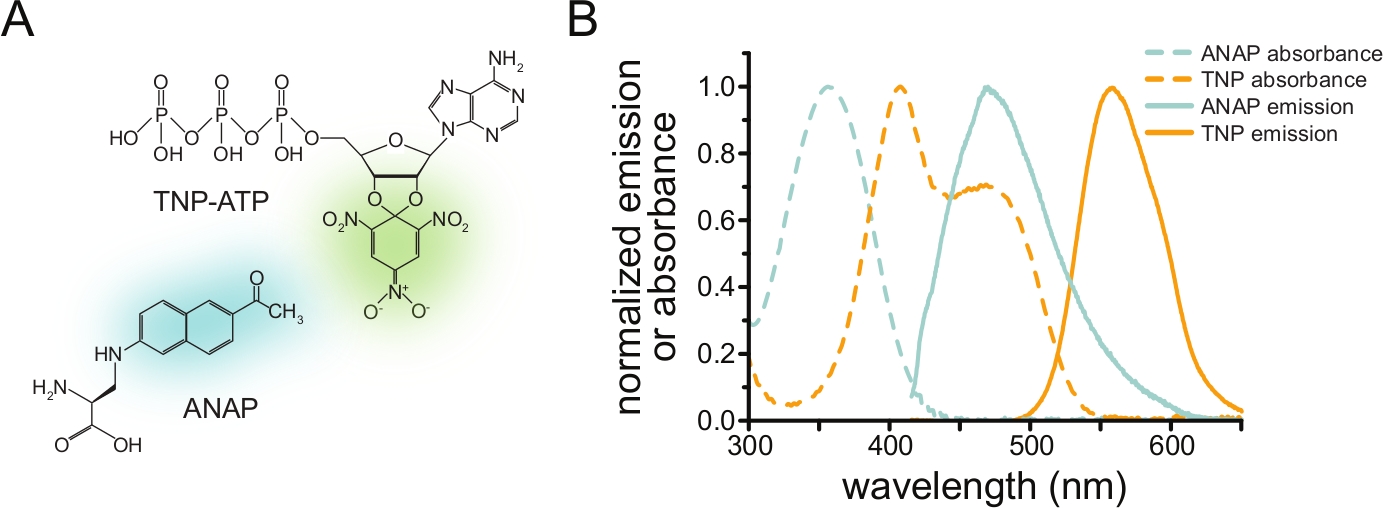
Um die Nukleotidbindung über FRET zu messen, muss ein Protein von Interesse zunächst mit einem Fluorophor markiert werden. Die fluoreszierende Markierung muss ortsspezifisch in das interessierende Protein eingefügt werden, so dass sie nahe genug an der Ligandenbindungsstelle liegt, damit FRET auftreten kann, wobei besonders darauf zu achten ist, dass die Markierung die Gesamtstruktur und -funktion des Proteins nicht beeinträchtigt. Um dies zu erreichen, verwenden wir eine von Chatterjee et al. entwickelte Technik, bei der eine fluoreszierende, nicht-kanonische Aminosäure (l-3-(6-acetylnaphthalen-2-ylamino)-2-aminopropion; ANAP) am gewünschten Standort12. Wir messen die Nukleotidbindung als FRET zwischen ANAP-markiertem Protein und fluoreszierenden Trinitrophenyl (TNP)-Nukleotidderivaten (Abbildung 1A). Das Emissionsspektrum von ANAP überschneidet sich mit dem Absorptionsspektrum von TNP-Nukleotiden, eine Bedingung, die für das Auftreten von FRET erforderlich ist (Abbildung 1B). Hier skizzieren wir zwei verschiedene Arten von Bindungsexperimenten. Im ersten Fall wird die Nukleotidbindung an die intrazelluläre Seite von ANAP-markiertenK-ATP-Kanälen in Zellen gemessen, die durch Beschallung befreit wurden, wobei anhaftende Fragmente der Plasmamembran auf einem Glasdeckglas10,11,13,14 zurückbleiben.
Bei der zweiten Methode wird die Nukleotidbindung an ANAP-markierte KATP-Kanäle in einem Membranfeld unter Spannungsklemme gemessen, was die gleichzeitige Messung von Ionenströmen und Fluoreszenz ermöglicht. Durch die Kombination dieser beiden experimentellen Ansätze können Änderungen in der Bindung direkt mit Änderungen der Kanalfunktion11 korreliert werden. Typische Ergebnisse, mögliche Fallstricke und Datenanalysen werden diskutiert.
Protokoll
1. Vorbereitung von Deckgläsern
HINWEIS: Diese Schritte müssen in einer sterilen Gewebekulturhaube erfolgen. Für die Zubereitung von 10 Gerichten sind Mengenangaben angegeben.
- Legen Sie zehn autoklavierte, 30 mm dicke Borosilikat-Deckgläser einzeln in zehn unbehandelte sterile 35-mm-Schalen und spülen Sie sie einmal mit 2 ml sterilem, destilliertem Wasser aus.
- 1 ml 0,1 Gew.-% Poly-L-Lysin-Lösung wird in steriles, destilliertes Wasser auf ein Gesamtvolumen von 10 ml verdünnt (Endkonzentration 0,01 Gew.-%) Gut mischen, dann 1 ml auf jedes Deckglas pipettieren und 20 Minuten bei Raumtemperatur inkubieren.
- Aspirieren Sie das Poly-L-Lysin und waschen Sie jedes Deckglas zweimal mit mindestens 2 ml sterilem destilliertem Wasser. Mindestens 3 Stunden einwirken lassen, bis sie vollständig getrocknet sind.
2. Aussaat von HEK-293T-Zellen
HINWEIS: Diese Schritte müssen in einer Gewebekulturhaube stattfinden. HEK-293T-Zellen wurden aufgrund ihres geringen Stromhintergrunds und der einfachen Züchtung in Kultur ausgewählt. Dieses Protokoll kann auf andere Zelltypen übertragen werden.
- Spülen Sie einen 80-90%igen konfluenten T75-Kolben mit HEK-293T-Zellen einmal mit 12 ml phosphatgepufferter Kochsalzlösung (PBS), bevor Sie ihn 2-5 Minuten lang mit 2 ml Trypsin inkubieren oder bis die Zellen vollständig abgelöst und fast vollständig dissoziiert sind.
- Resuspendieren Sie die Zellen durch Zugabe von 10 ml Dulbecco's Modified Eagle Medium (DMEM), ergänzt mit 10% fötalem Kälberserum, 100 U/ml Penicillin und 100 μg/ml Streptomycin. Pipettieren Sie vorsichtig gegen den Boden des Kolbens, um verbleibende Zellklumpen aufzubrechen.
- Fügen Sie 2 ml zugesetztes DMEM zu der gewünschten Anzahl von 35-mm-Schalen mit beschichteten Deckgläsern hinzu. Fügen Sie jeder Schale 100 μl resuspendierte Zellen hinzu. Über Nacht bei 37 °C inkubieren.
3. Transfektion
HINWEIS: Diese Schritte müssen in einer Gewebekulturhaube stattfinden. Es werden Mengen für die Transfektion von 10 Gerichten angegeben. Für den ortsspezifischen ANAP-Einbau muss das DNA-Codon an der für die Markierung vorgesehenen Position durch das bernsteinfarbene (TAG) Stoppcodon ersetzt werden. Dieses Konstrukt wird mit zwei Plasmiden co-transfiziert: pANAP und peRF1-E55D12,15. pANAP kodiert für mehrere Kopien eines ANAP-spezifischen tRNA/tRNA-Synthetase-Paares. In Gegenwart von ANAP entsteht durch die Transfektion dieses Plasmids eine mit ANAP geladene tRNA, die das bernsteinfarbene Stoppcodon erkennt. peRF1-E55D kodiert für einen dominanten negativen ribosomalen Freisetzungsfaktor, der die Ausbeute an ANAP-markiertem Protein in voller Länge erhöht.
- Bereiten Sie ein 1,5-ml-Röhrchen mit 10 μg pANAP, 10 μg peRF1-E55D und DNA für das Konstrukt vor, das für die Markierung mit ANAP vorgesehen ist. Auf ein Endvolumen von 500 μl mit unsupplementiertem DMEM bringen.
- In einem separaten Röhrchen werden 3 μl lipidbasiertes Transfektionsreagenz (siehe Materialtabelle) für jeweils 1 μg DNA hergestellt und mit unsupplementiertem DMEM auf ein Endvolumen von 500 μl gebracht.
- Kombinieren Sie die DNA- und Transfektionsreagenzienmischungen in einem einzigen Röhrchen und inkubieren Sie sie 20 Minuten lang bei Raumtemperatur.
- Fügen Sie 400 μl des 1 mM ANAP-Stammes (Trifluoracetatsalz in 30 mM NaOH) zu 20 ml supplementiertem DMEM hinzu, um eine Endkonzentration von 20 μM ANAP zu erreichen. Ersetzen Sie das alte Medium aus den plattierten Zellen durch 2 ml des ANAP-haltigen Mediums pro Schale.
- Pipettieren Sie 10 % der DNA-Transfektionsmischung auf jede Schale. Vor den Experimenten bei 33 °C 2-4 Tage inkubieren. Die Inkubation bei 33 °C verlangsamt die Zellteilung und erhöht die Proteinausbeute pro Zelle16.
4. Experimente mit nicht überdachten Membranen
- Verwenden Sie eine Pinzette, um ein Deckglas mit transfizierten Zellen in kleinere Fragmente zu zerbrechen.
- Befolgen Sie eines der folgenden Verfahren, um das Dach der Zellen zu öffnen.
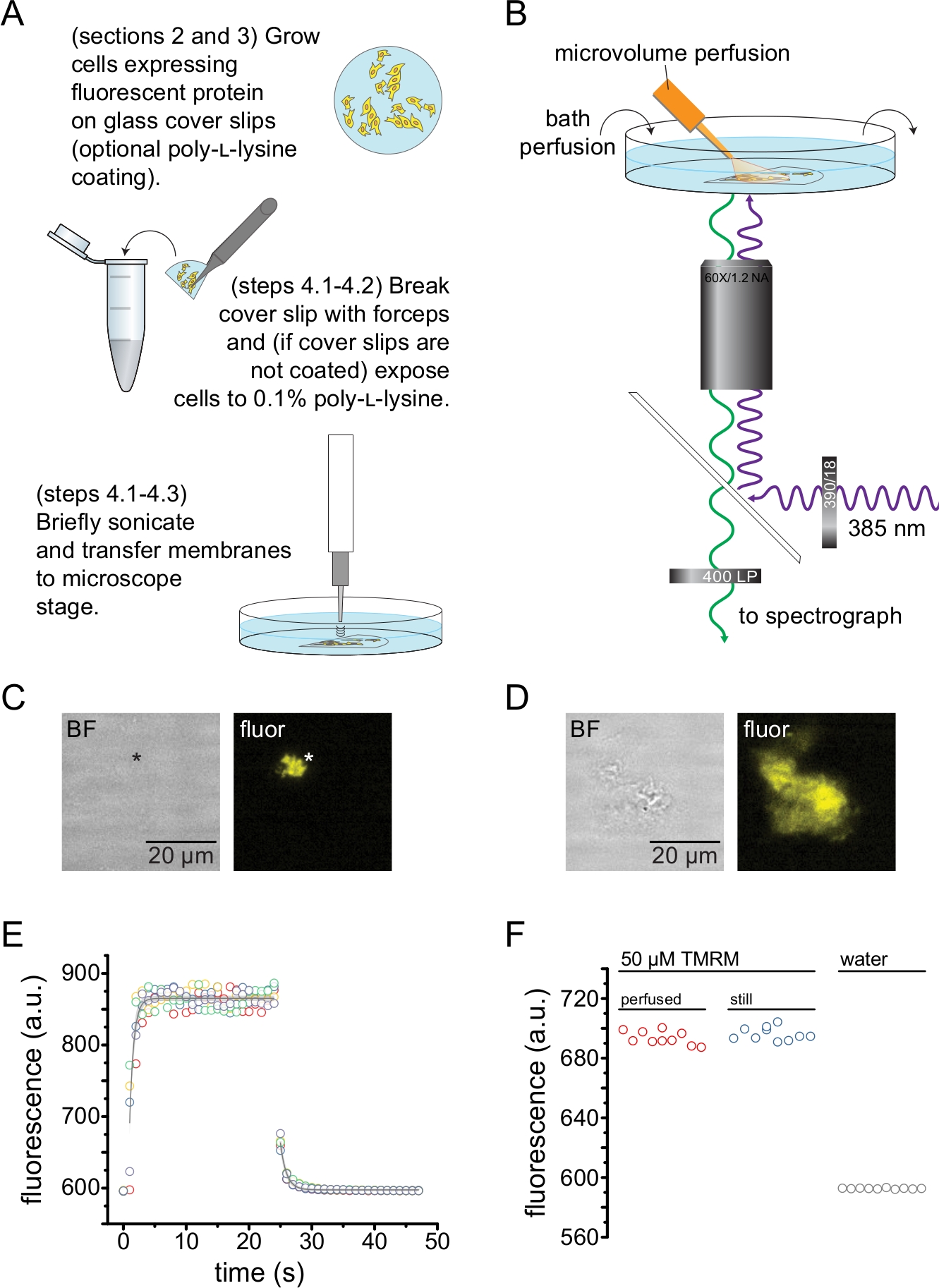
- Wenn Sie vorbeschichtete Deckgläser verwenden, spülen Sie ein Fragment mit PBS aus und legen Sie es dann auf den Boden einer 35-mm-Schale mit 2 ml PBS. Mit einem Sonden-Ultraschallgerät (50 W, 20%-40% Amplitude, 3 mm Sonde), das 3-5 mm über der Probe positioniert ist, wird kurz beschallt, um die Zellen zu öffnen und anhaftende Plasmamembranfragmente zu hinterlassen (Abbildung 2A,C).
Anmerkungen: Die Schallleistung, die Dauer und die Sondenhöhe über der Probe können variiert werden, um eine hohe Ausbeute an Membranen ohne Dach zu erzielen, ohne das Deckglas vollständig zu entblößen. - Wenn Sie keine vorbeschichteten Deckgläser verwenden, spülen Sie ein Deckglasfragment mit PBS aus und tauchen Sie es dann ~30 s lang in ein Röhrchen mit 0,1 % w/v Poly-L-Lysin, bevor Sie es kurz beschallen (wie in Schritt 4.2.1), um die Zellen zu entdachen und ungedeckte/teilweise unüberdachte Plasmamembranfragmente zu hinterlassen (Abbildung 2A, C, D). Es hat sich gezeigt, dass eine kurze Exposition gegenüber Poly-L-Lysin die Haftung auf dem Deckglasverbessert 13.
- Wenn Sie vorbeschichtete Deckgläser verwenden, spülen Sie ein Fragment mit PBS aus und legen Sie es dann auf den Boden einer 35-mm-Schale mit 2 ml PBS. Mit einem Sonden-Ultraschallgerät (50 W, 20%-40% Amplitude, 3 mm Sonde), das 3-5 mm über der Probe positioniert ist, wird kurz beschallt, um die Zellen zu öffnen und anhaftende Plasmamembranfragmente zu hinterlassen (Abbildung 2A,C).
- Das beschallte Fragment wird in eine 35-mm-Abdeckschale mit Glasboden mit 2 ml Badlösung gegeben und auf ein inverses Mikroskop montiert, das mit einem 60-fachen Wasserimmersionsobjektiv mit hoher NA ausgestattet ist. Der Kameraanschluss des Mikroskops ist mit einem Spektrographen in Reihe mit einer hochempfindlichen CCD-Kamera verbunden. Die Badekammer (0,5 – 1 ml/min) mit Puffer mit einer Peristaltikpumpe durchströmen. Die Zusammensetzung des Puffers hängt vom untersuchten Protein ab.
HINWEIS: Wenn der Benutzer keinen Zugang zu einem Objektiv mit großem Arbeitsabstand hat, kann es aufgrund der zusätzlichen Höhe des Deckglases unmöglich sein, sich auf die nicht überdachten Membranfragmente zu konzentrieren. Eine Alternative besteht darin, Zellen direkt auf Schalen mit Poly-L-Lysin-Glasboden zu säen (siehe Materialtabelle für ein Beispiel). Dadurch werden auch mögliche Abbildungsfehler reduziert, die mit der Fokussierung durch zwei Glasscheiben verbunden sind. Diese Aberrationen haben keinen Einfluss auf die Form der aufgenommenen Spektren. - Identifizierung von Membranfragmenten ohne Dach, die den ANAP-markierten Kanal exprimieren, indem nach Kanalfluoreszenz gesucht wird (Abbildung 2C,D).
HINWEIS: Es wird empfohlen, eine zusätzliche Fluoreszenzmarkierung zu verwenden (bei der das Emissionsspektrum vom ANAP-Emissionsspektrum unterscheidbar ist), um Membranen ohne Dach zu identifizieren, die das interessierende Protein enthalten. Die Experimente in Abbildung 2C,D wurden an ANAP-markierten Kanälen mit C-terminalen fluoreszierenden Protein-Tags durchgeführt. - Ziehen Sie die Spektrometermaske teilweise (~10 % anheben) zwischen dem Kameraanschluss des Mikroskops und dem Spektrographen ein. Der Schatten der Maske erscheint auf dem Kamerabild. Richten Sie die Membran ohne Dach an der Spektrometermaske aus, indem Sie den Mikroskoptisch einstellen. Erfassen Sie ein Hellfeld- und Fluoreszenzbild der unüberdachten Membran. Diese werden verwendet, um eine Region von Interesse für die Analyse auszuwählen.
- Bringen Sie die Spitze des Mikrovolumen-Perfusionssystems in die Nähe der nicht überdachten Membran.
HINWEIS: Um die Hintergrundfluoreszenz zu reduzieren, wurde der Abfluss des Perfusionssystems durch eine spezielle Spitze aus Borosilikatglas ersetzt. - Um Fluoreszenzspektren abzubilden, wird die Membran mit einer 385 nm LED durch einen 390/18 nm Bandpass-Anregungsfilter und einen 416 nm Kantendichroit angeregt. Sammeln Sie das emittierte Licht durch einen 400-nm-Langpass-Emissionsfilter (Abbildung 2B).
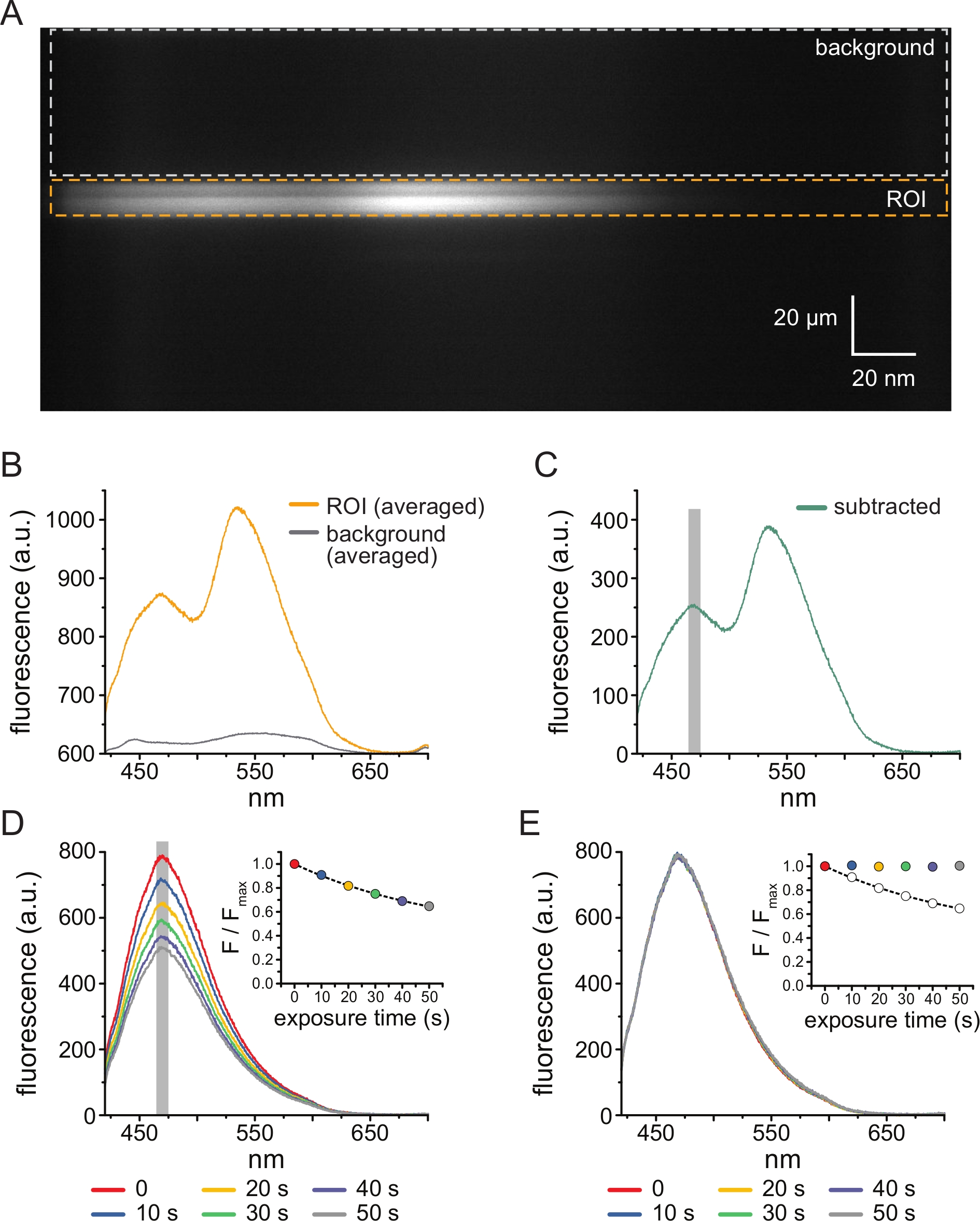
- Setzen Sie die Spektrometermaske ein und stellen Sie sicher, dass das emittierte Licht durchgelassen wird. Spektrometergitter einrasten (300 Rillen/mm). Wenn die Gitter angebracht sind, wird das vom Spektrometer gebeugte Licht auf den Chip der CCD-Kamera projiziert, um Spektralbilder zu erzeugen (Abbildung 3A). Diese Bilder behalten räumliche Informationen in der y-Dimension bei. Die x-Dimension wird durch die Wellenlänge ersetzt.
- Wenn das interessierende Protein mit einem fluoreszierenden Protein markiert ist, können Sie optional ein Spektralbild des fluoreszierenden Proteins mit dem entsprechenden Filtersatz aufnehmen.
- Nehmen Sie zu Beginn des Experiments eine oder mehrere 0,1-10 s-Belichtungen auf, während Sie nukleotidfreie Pufferlösung perfundieren. Diese werden verwendet, um die Daten im weiteren Verlauf des Experiments zu korrigieren und zu normalisieren (siehe Abschnitt 5 unten).
Anmerkungen: Die Wahl der Belichtungszeit hängt von der erreichten Expressionsstufe, der Helligkeit des Fluorophors und der Optik ab. Die Belichtungszeit sollte gewählt werden, um das Signal zu maximieren und die beobachtete Bleichrate zu minimieren. Der in 4.10 angegebene Belichtungszeitbereich eignet sich für Gleichgewichtsbindungsmessungen, kann aber auch für die Messung langsamerer kinetischer Änderungennützlich sein 10. Die Möglichkeit, kurze Belichtungszeiten zu nutzen, um eine schnellere Kinetik zu verfolgen, wird eher durch die Proteinexpression und das Photobleichen als durch die Hardware begrenzt. - Wenden Sie eine Reihe von Konzentrationen von TNP-ATP (normalerweise in Badelösung hergestellt) an, um eine Konzentrations-Wirkungs-Kurve zu erstellen. Durchbluten Sie jede Lösung mindestens 1 Minute lang, um sicherzustellen, dass ein stabiler Zustand erreicht wird, und waschen Sie jede Konzentration mindestens 1 Minute lang mit Badelösung aus.
HINWEIS: Es ist wichtig sicherzustellen, dass das Perfusionssystem schnell ein Gleichgewicht erreichen kann (Abbildung 2E) und die richtige lokale Konzentration von TNP-ATP erreicht (Abbildung 2F). - Belichten Sie (mit der gleichen Dauer wie in Schritt 4.10) bei jeder Konzentration und am Ende jedes Auswaschens.
5. Spektralanalyse
HINWEIS: Diese Anleitung ist für die Verwendung mit dem Analysecode "pcf.m" geschrieben, der auf GitHub zu finden ist. https://github.com/mpuljung/spectra-analysis10. Zusätzlicher und alternativer Code finden Sie unter https://github.com/smusher/KATP_paper_201911. Wir haben die von der Software ausgeführten Vorgänge hier beschrieben, damit der Benutzer seinen eigenen Code erstellen oder die Daten manuell analysieren kann.
- Starten Sie das Analyseprogramm, indem Sie den Namen des Programms ("pcf") in die Befehlszeile eingeben.
- Wenn ein Dialogfeld zum Öffnen einer Datei/eines Ordners mit der Eingabeaufforderung "Dateien für ROI auswählen" geöffnet wird, wählen Sie die Dateinamen aus, die mit den Hellfeld- und Fluoreszenzbildern der unüberdachten Membran verknüpft sind. In der Befehlszeile wird eine Eingabeaufforderung angezeigt, in der Sie den Namen der Ausgabedatei eingeben müssen.
- Geben Sie den Dateinamen ein und drücken Sie die Eingabetaste.
- Wenn die Software die Hellfeld- und Fluoreszenzbilder anzeigt, wählen Sie im Spektralbild eine Region of Interest (ROI) aus, die der Position des Membranfragments ohne Dach oder des herausgeschnittenen Pflasters (siehe Abschnitt 6) entspricht, und folgen Sie den Anweisungen der Software. Wählen Sie einen Hintergrundbereich im selben Spektralbild (der denselben Wellenlängenbereich wie in der ROI darstellt) aus, der einem Abschnitt des Deckglases oder der Schale ohne angeschlossene Membran entspricht (Abbildung 3A). Die Software fordert Sie auf, auf den oberen Rand des ROI zu klicken und die Eingabetaste zu drücken, auf den unteren Rand des ROI zu klicken und die Eingabetaste zu drücken und diesen Vorgang dann für den Hintergrundbereich zu wiederholen.
- Wenn ein Dialogfeld zum Öffnen einer Datei/eines Ordners mit der Eingabeaufforderung "Datei für FP-Spektrum auswählen" geöffnet wird, wählen Sie den Dateinamen aus, der dem Spektrum des fluoreszierenden Proteins (FP) zugeordnet ist (optionaler Schritt 4.9). Wenn kein FP-Spektrum erfasst wurde, wählen Sie eine andere Spektrumsdatei aus. Das FP-Spektrum dient zur Qualitätskontrolle zur Unterscheidung zwischen markiertem Protein und Hintergrundfluoreszenz.
- Wenn sich ein Dialogfeld zum Öffnen einer Datei/eines Ordners mit der Eingabeaufforderung "Dateien für die Analyse auswählen" öffnet, wählen Sie alle Dateien aus, die den ANAP-Spektren entsprechen (von den Schritten 4.10 bis 4.12), einschließlich der Dateien, die für die Bleichkorrektur benötigt werden.
- Wenn sich ein Dialogfeld zum Öffnen einer Datei/eines Ordners mit der Eingabeaufforderung "Dateien für die Bleichsammlung auswählen" öffnet, wählen Sie die Teilmenge der Dateien aus Schritt 5.6 aus, die den Anfangsspektren entspricht, die zu Beginn des Versuchs in nukleotidfreier Lösung aufgenommen wurden, oder den Spektren, die während des Waschens in nukleotidfreier Lösung aufgenommen wurden, um sie zur Korrektur zu verwenden (von den Schritten 4.10 bis 4.12).
- Berechnen Sie den Linienmittelwert für jedes Bild, um Spektren zu erzeugen, d. h. die Intensität für alle Pixel in der y-Dimension eines ROI- oder Hintergrundbereichs bei jeder Wellenlänge zu mitteln. (Abbildung 3B). Subtrahieren Sie das resultierende gemittelte Hintergrundspektrum von dem gemittelten Spektrum, das aus dem ROI gewonnen wurde, um Hintergrundfluoreszenz und Fluoreszenz aus ungebundenem TNP-ATP zu entfernen (Abbildung 3C). Diese Schritte werden von der Software automatisch ausgeführt.
- Bestimmen Sie die ANAP-Intensität für jede Exposition, indem Sie die Intensität eines 5-nm-Fensters mitteln, das um den ANAP-Peak der subtrahierten Spektren zentriert ist (typischerweise ~470 nm, kann aber je nach lokaler Mikroumgebung des ANAP-Rests variieren).
HINWEIS: Abbildung 3D zeigt 6 Spektren, die aus aufeinanderfolgenden 10-Sekunden-Aufnahmen eines Membranfragments ohne Dach gewonnen wurden, das ANAP-markierte Kanäle exprimiert. Der Einschub zeigt die gemittelte Intensität der Spitze jedes Spektrums. Die Software findet automatisch die Spitzenwellenlänge im ersten erfassten Spektrum und verwendet diesen Wert durchgehend. Die Intensität wird von der Software automatisch berechnet. - Normalisieren Sie die ANAP-Intensitäten für jedes Experiment, indem Sie die ANAP-Intensität einer bestimmten Exposition (F) durch die ANAP-Intensität der ersten Exposition in der Zeitreihe dividieren, die in Schritt 4.10 (Fmax) aufgenommen wurde. Auch hier führt die Software diese Berechnungen automatisch durch.
- Führen Sie die folgenden Schritte aus, um Daten abzurufen.
- Um das ANAP-Photobleichen zu korrigieren, passen Sie zunächst einen einzelnen exponentiellen Zerfall (F/Fmax) = A*exp(-t/τ)+(1-A), wobei t die kumulative Belichtungszeit, τ die Zeitkonstante und A die Amplitude) ist, entweder an die Zwischenwaschschritte zwischen TNP-ATP-Anwendungen oder an mehrere Erstbelichtungen an, die vor dem Waschen auf TNP-ATP aufgenommen wurden (Abbildung 3D, Einschub).
HINWEIS: Die Software zeigt diese Passform an und fordert Sie auf, sie zu akzeptieren oder abzulehnen. Wenn die Passform abgelehnt wird, erhalten Sie eine weitere Möglichkeit, Dateien für die Bleichkorrektur auszuwählen. - Teilen Sie die normalisierten (in Schritt 5.10) ANAP-Spektren durch den vorhergesagten Wert der exponentiellen Anpassung aus Schritt 5.11.1 zu jedem Zeitpunkt (Abbildung 3E).
HINWEIS: Für das gezeigte Beispiel beträgt die beobachtete normalisierte Spitzenfluoreszenz bei 50 s 0,65 und die vorhergesagte Fluoreszenz aus der exponentiellen Anpassung 0,64. Um das Ausbleichen zu korrigieren, dividieren Sie den beobachteten Wert (0,65, Abbildung 3E-Einschub, leerer Kreis) durch den vorhergesagten Wert (0,64, Abbildung 3E-Einschub, gestrichelte Linie), um den korrigierten Wert (~1, Abbildung 3E-Einschub, farbiger Kreis) zu erhalten. Wenn die Bleichkorrektur angemessen ist, sollte die Intensität der ANAP bei allen Expositionen, die in Abwesenheit von Nukleotiden aufgenommen wurden, ungefähr gleich sein (Abbildung 3E). Diese Berechnungen werden von der Software automatisch durchgeführt. - Rufen Sie die Ausgabe als Bild ab, in dem die Daten dargestellt werden, und eine Tabelle mit Registerkarten, die die Rohspektren, die subtrahierten Spektren, die für die Photobleichung korrigierten Spektren und die Peakdaten für jede Datei enthält, damit eine weitere Analyse durchgeführt werden kann.
- Um das ANAP-Photobleichen zu korrigieren, passen Sie zunächst einen einzelnen exponentiellen Zerfall (F/Fmax) = A*exp(-t/τ)+(1-A), wobei t die kumulative Belichtungszeit, τ die Zeitkonstante und A die Amplitude) ist, entweder an die Zwischenwaschschritte zwischen TNP-ATP-Anwendungen oder an mehrere Erstbelichtungen an, die vor dem Waschen auf TNP-ATP aufgenommen wurden (Abbildung 3D, Einschub).
6. Patch-Clamp-Fluorometrie-Experimente
- Ziehen Sie Patchpipetten aus dickwandigen Borosilikatglaskapillaren auf einen Widerstand von 1,5 MΩ bis 2,5 MΩ, wenn sie mit Pipettenlösung gefüllt sind. Die Zusammensetzung der Pipettenlösung variiert je nach untersuchtem Protein.
- Übertragen Sie ein Deckglas mit transfizierten Zellen auf eine 35-mm-Abdeckschale mit Glasboden und 2 ml Badlösung und montieren Sie sie auf ein inverses Mikroskop, das mit einem 60-fachen Wasserimmersionsobjektiv mit hoher NA ausgestattet ist. Die Badekammer (0,5 – 1 ml/min) mit einer Peristaltikpumpe mit Badelösung durchströmen. Was die Pipettenlösung betrifft, so variiert die Badelösung je nach untersuchtem Protein.
- Identifizieren Sie eine Zelle, die ANAP-markierte Kanäle exprimiert, indem Sie nach Fluoreszenz an der Zellmembran suchen.
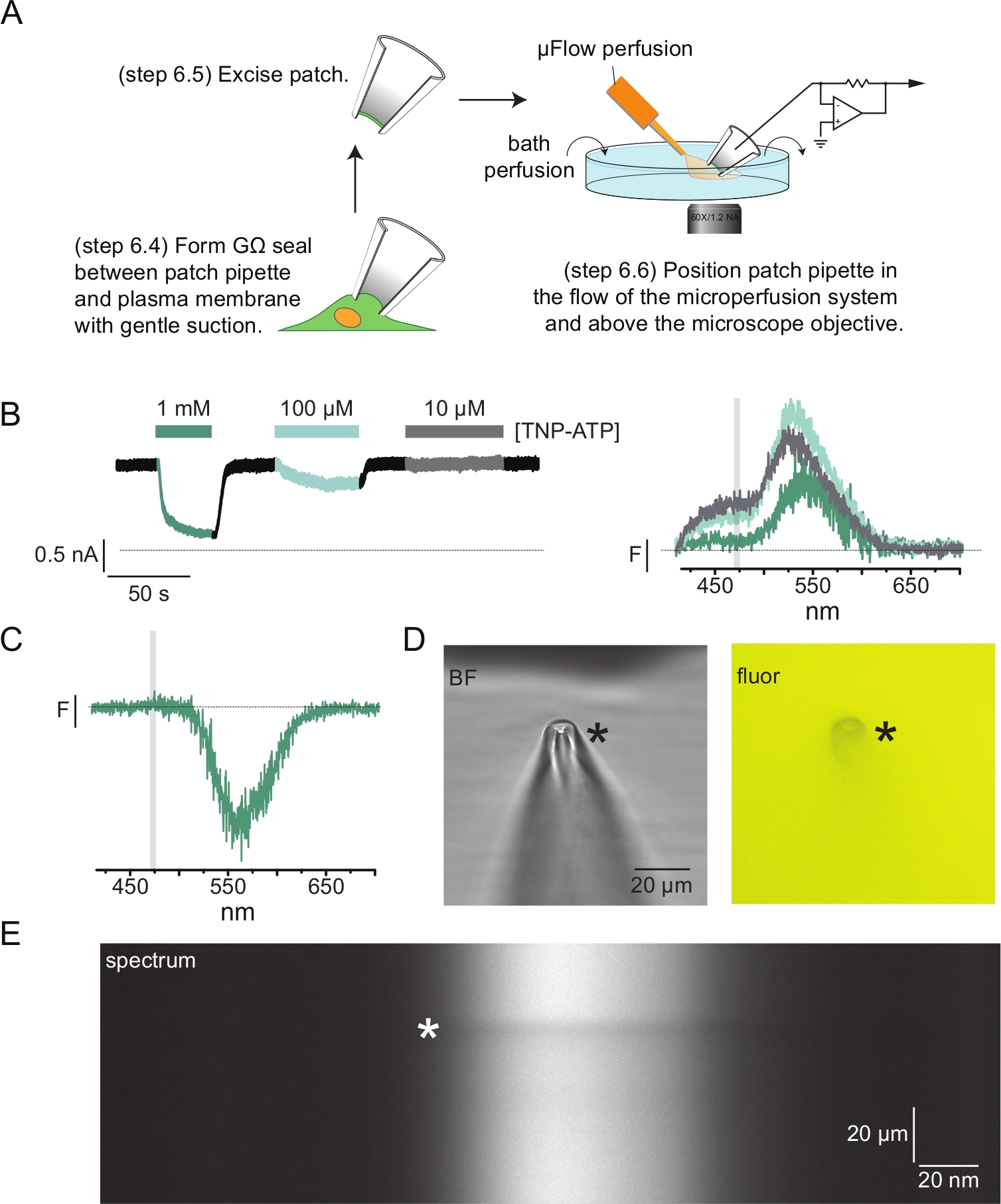
- Füllen Sie eine Patch-Pipette mit Pipettenlösung. Üben Sie sanften positiven Druck auf die Pipette aus und legen Sie sie in die Badkammer. Drücken Sie die Pipette gegen die Membran der Zelle und saugen Sie sanft ab, um eine GΩ-Abdichtung zu erzielen (Abbildung 4A).
- Entfernen Sie das Pflaster, indem Sie den Pipettenhalter schnell von der Zelle wegbewegen (Abbildung 4A).
HINWEIS: Wenn Sie das Pflaster auf diese Weise entfernen, sollte ein von innen nach außen gerichtetes Pflaster entstehen, bei dem die zytosolischen Domänen des Proteins dem Perfusionssystem ausgesetzt sind. Wenn die Position der zu untersuchenden Nukleotidbindungsstelle nicht zytosol ist, ist es notwendig, Outside-Out-Patches oder Ganzzellableitungen zu verwenden, um PCF-Experimente durchzuführen. - Bringen Sie die Spitze der Patch-Pipette in die Nähe der Spitze des Perfusionssystems und überprüfen Sie, ob sich das Pflaster innerhalb des Schlitzes der Spektrometermaske befindet (Abbildung 4A).
- Wenden Sie TNP-ATP und Bildspektren wie in den Schritten 4.10-4.12 an und zeichnen Sie gleichzeitig die Ionenstromantwort auf die Nukleotidapplikation auf.
HINWEIS: Das Pipettenglas kann zu räumlichen Aberrationen und Reflexionen in den aufgenommenen Bildern führen. Diese Aberrationen haben jedoch keinen Einfluss auf die Form der aufgenommenen Spektren, und das reflektierte Anregungslicht kann entweder mit dem Spektrographen oder einem Langpass-Emissionsfilter leicht von der Fluoreszenz getrennt werden. - Analysiere die Spektren. Spektren, die von exzidierten Patches aufgenommen wurden, können aufgrund des Ausschlusses von TNP-ATP aus dem Glas der Patch-Pipette eine Übersubtraktion der ungebundenen TNP-ATP-Fluoreszenz aufweisen (Abbildung 4C-E). Diese Übersubtraktion hat keinen Einfluss auf das ANAP-Emissionsspektrum und kann daher ignoriert werden.
HINWEIS: Da das Fluoreszenzsignal in herausgeschnittenen Pflastern niedriger ist als in nicht überdachten Membranen, ist es wichtig, eine Belichtungszeit zu verwenden, die ein ausreichend hohes Signal-Rausch-Verhältnis ergibt, ohne ANAP zu schnell auszubleichen.
Ergebnisse
Abbildung 2 zeigt den grundlegenden Versuchsaufbau zur Messung der Nukleotidbindung an fluoreszierende Proteine in Membranfragmenten ohne Dach, die durch Beschallung erhalten wurden (Abbildung 2A,B). Es wurden zwei verschiedene Ansätze verwendet, um unüberdachte Membranen zu erhalten: die direkte Kultivierung von Zellen auf Poly-L-Lysin-beschichteten Deckgläsern oder die Kultivierung von Zellen auf unbehandeltem Glas und die kurze Exposition gegenüber Poly-L-Lysin (0,1 % in Wasser) vor dem Abnehmen des Daches. Abbildung 2C zeigt ein typisches Membranfragment ohne Dach einer HEK-293T-Zelle, die K-ATP-Kanäle exprimiert, die mit orange fluoreszierendem Protein (OFP) markiert sind. Unüberdachte Membranen waren in Hellfeldbildern praktisch unsichtbar und wurden durch die Fluoreszenz markierter Membranproteine oder durch Gegenfärbung mit einem Membranfarbstoff wie Octadecylrhodamin B13 identifiziert. Bei der Beschallung von HEK-293T-Zellen wurden neben nicht überdachten Membranen auch teilweise nicht überdachte Zellfragmente erzeugt (Abbildung 2D)10,17. Diese Fragmente waren im hellen Feld sichtbar. Dies kann die Folge von gekräuselten Plasmamembranen sein, die nur schlecht auf dem Deckglas haften. Alternativ können diese Fragmente Vesikel und Membranen aus intrazellulären Organellen enthalten. Daher ist es vorzuziehen, nur Bilder von "echten" Membranen ohne Dach aufzunehmen, da markierte Zielproteine, die mit intrazellulären Membranen assoziiert sind, Zwischenstufen der posttranslationalen Verarbeitung und Assemblierung widerspiegeln können. Die Kultivierung von Zellen auf Poly-L-Lysin-beschichtetem Glas wird empfohlen, da dies bei der Beschallung zu einer höheren Ausbeute an "echten" Membranen ohne Dach führte.
Ein Mikrovolumen-Perfusionssystem wurde auf fluoreszierende Nukleotide angewendet, um die in einem typischen Experiment benötigten Mengen zu minimieren (Abbildung 2B). Die mitgelieferte polyimidbeschichtete Glasspitze wurde in unserem Perfusionsaufbau durch eine handgezogene Borosilikatglasspitze ersetzt, die den Fluoreszenzhintergrund reduzierte. Um die Nukleotidakkumulation um die abgebildeten Membranen ohne Dach zu minimieren, wurde die gesamte Badkammer langsam mit Puffer durchblutet. Daher wollten wir die Geschwindigkeit der Lösungsänderung aus unserem Mikrovolumen-Perfusionssystem messen und überprüfen, ob wir in der Lage waren, die beabsichtigte Ligandenkonzentration in unserem interessierenden Bereich zu erreichen, d.h. dass der Ligand aus unserem Perfusionssystem nicht direkt in das Bademedium verdünnt wurde, bevor er die unüberdachte Membran erreichte. Um diese Möglichkeiten zu kontrollieren, wurde das Ein- und Auswaschen einer 50 μM Lösung von Tetramethylrhodamin-5-maleimid (TMRM) aus unserem Mikrovolumen-Perfusionssystem gemessen, die auf die Oberfläche einer mit Wasser durchbluteten Deckglasbodenschale gerichtet war (Abbildung 2E). Die Kinetik des Lösungsaustauschs war reproduzierbar und gut beschrieben durch einen einzigen exponentiellen Zerfall mit Zeitkonstanten von weniger als 1 s sowohl für das Einwaschen als auch für das Auswaschen. Solche Lösungsaustauschzeiten schränken unsere Fähigkeit ein, die Kinetik der Ligandenbindung und -entbindung in unserem derzeitigen Aufbau zu messen. Um zu überprüfen, ob wir in der Lage waren, die gewünschte Ligandenkonzentration an der Oberfläche des Deckglases zu erreichen, verglichen wir die Fluoreszenzintensität von 50 μM TMRM, die von unserem Mikrovolumenperfusionssystem an das Deckglas abgegeben wurde, mit 50 μM TMRM in einem stillen Bad (Abbildung 2F). Es wurde kein Intensitätsunterschied beobachtet, was bestätigt, dass mit unserem Mikrovolumen-Perfusionssystem geeignete Ligandenkonzentrationen an der Oberfläche des Deckglases erreicht werden können, selbst wenn das Bad durchblutet ist.
Abbildung 3A zeigt ein Spektralbild, das von ANAP-markierten K-ATP-Kanälen in einer unüberdachten Membran einer HEK-239T-Zelle aufgenommen wurde, die bei 5 μMTNP-ATP exponiert wurde. Um solche Bilder zu erhalten, wurde das von der Membran ohne Dach emittierte Licht durch ein Spektrometer in Reihe mit einer CCD-Kamera geleitet. Die emittierte Fluoreszenz wurde von Gittern gebeugt und auf den Kamerachip projiziert, wodurch Spektren erzeugt wurden. Die resultierenden Bilder behalten räumliche Informationen in der y-Dimension bei, aber die x-Dimension wurde durch die Wellenlänge ersetzt. Die Region of Interest (ROI), die der nicht überdachten Membran entspricht, ist orange umrandet. Im Bild sind zwei Bereiche mit hoher Intensität zu erkennen, die der Spitzenemission von ANAP und TNP-ATP entsprechen. Dies zeigte sich am besten in dem in Abbildung 3B dargestellten, wellenlängenweise gemittelten Spektrum (über den gesamten ROI). Der Peak ~470 nm entspricht ANAP, das in KATP eingebaut ist; der Peak ~535 nm entspricht TNP-ATP. Um die Hintergrundfluoreszenz und die direkte Anregung von TNP-ATP in Lösung zu korrigieren, wurde aus jedem Bild eine Hintergrundregion (Abbildung 3A, grau) ausgewählt. Das gemittelte Hintergrundspektrum ist in Abbildung 3B dargestellt. Das endgültige Spektrum wurde erhalten, indem das gemittelte Hintergrundspektrum vom gemittelten ROI-Spektrum subtrahiert wurde (Abbildung 3C).
ANAP ist anfällig für Photobleichartefakte. Abbildung 3D zeigt die Abnahme der maximalen ANAP-Fluoreszenz nach Mehrfachbelichtung. Die Peak-Fluoreszenz von mehreren Aufnahmen in Abwesenheit von TNP-ATP (oder von Waschungen zwischen den TNP-ATP-Konzentrationen) wurde an einen einzigen exponentiellen Zerfall angepasst und dies wurde zur Korrektur von Photobleichartefakten verwendet (Abbildung 3E). Es wird empfohlen, Konzentrations-Antwort-Experimente sowohl von niedrigen zu hohen als auch von hohen bis niedrigen Nukleotidkonzentrationen durchzuführen. Wenn durch die Bleaching-Korrektur keine zusätzlichen Artefakte entstehen, sollten die Ergebnisse vergleichbar sein11.
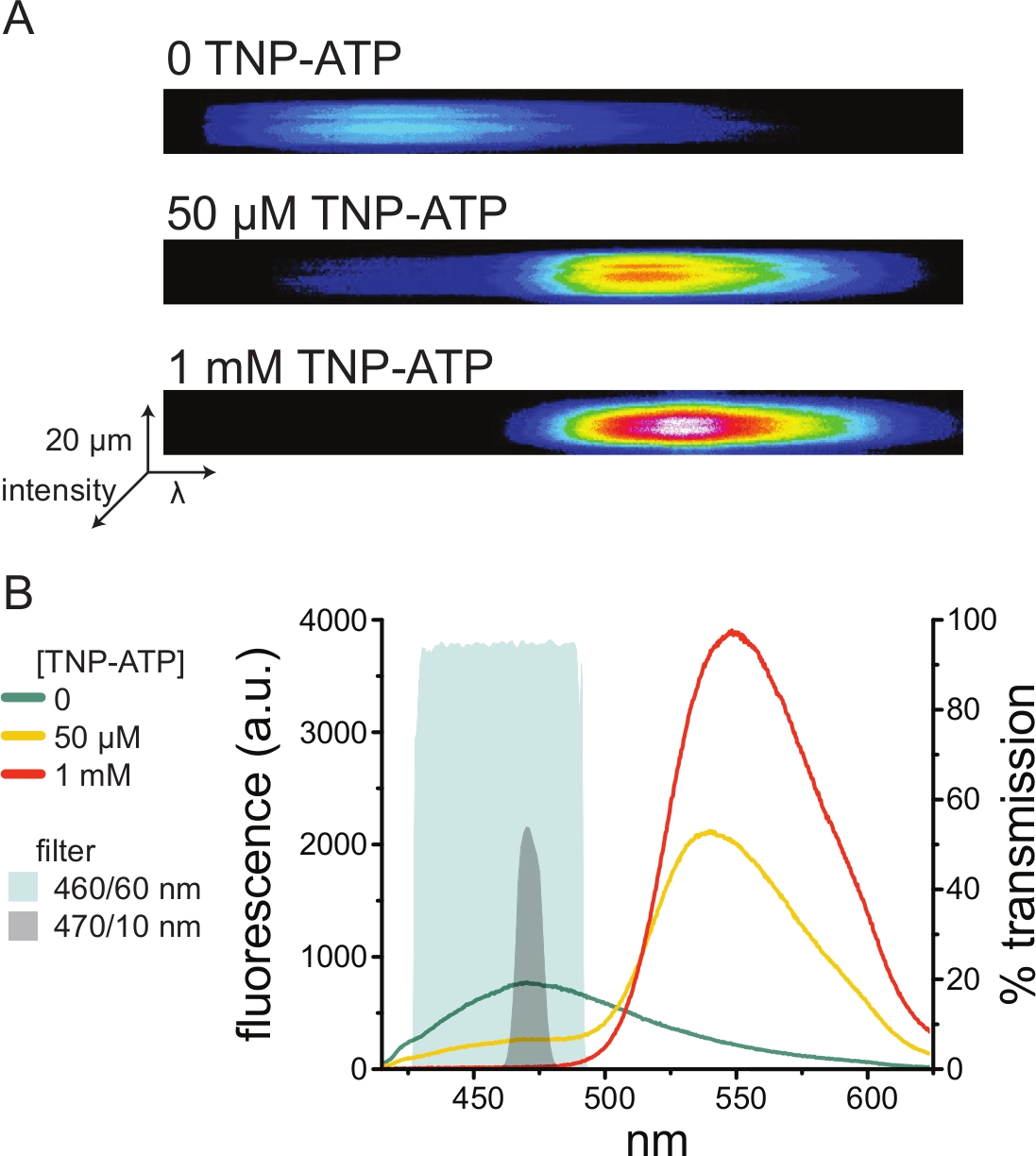
Abbildung 5A zeigt repräsentative Spektralbilder einer Membran ohne Dach, die aus einer Zelle gewonnen wurde, die ANAP-markierte K-ATP-Kanäle in Abwesenheit und Gegenwart vonTNP-ATP exprimiert. Die korrigierten Spektren sind in Abbildung 5B dargestellt. Bei der Betrachtung der Emissionsspektren zeigte sich eine klare Trennung zwischen der Fluoreszenzemission des Donors und des Akzeptors. Da eine unspezifische Bindung von TNP-ATP an naive Plasmamembranen von nicht transfizierten HEK-293T-Zellen beobachtet wurde, wird empfohlen, FRET als Reduktion der Donorfluoreszenz (ANAP) zu quantifizieren10,11. Dieser Peak war spezifisch für den markierten Rezeptor.
Für Liganden, die eine Konformationsänderung ihres Rezeptors induzieren, liefern isolierte Bindungsstudien keine direkten, mechanistisch aussagekräftigen Informationen über den Ligandenbindungsprozess18. Die Konzentrations-Antwort-Beziehung für die Ligandenbindung hängt nicht nur von der intrinsischen Bindungsaffinität ab, sondern auch von der durch die Ligandenbindung induzierten Konformationsänderung und der inhärenten Neigung des Rezeptors, die Konformation in Abwesenheit eines Liganden zu ändern. Um die Prozesse, die den Ligand-Rezeptor-Interaktionen zugrunde liegen, besser zu verstehen, können Bindungsmessungen mit Experimenten kombiniert werden, die eine Ablesung der Proteinfunktion ermöglichen. Hierfür sind Ionenkanäle ein ideales Modellsystem, da ihre Ströme mit Hilfe einer Spannungszange mit einer Zeitauflösung von sub ms bis in den Einzelmolekülbereich gemessen werden können. In der Vergangenheit haben gepaarte Strom- und Fluoreszenzmessungen wichtige Einblicke in das Öffnen und Schließen (Gating) von spannungs- und ligandengesteuerten Ionenkanälengeliefert 19,20,21. Es wurden Experimente durchgeführt, um gleichzeitig ionische Ströme und die Bindung fluoreszierender zyklischer Nukleotide an verschiedene zyklische Nukleotid-regulierte Kanäle zu messen22,23,24. In diesen Studien wurde ein Ligand verwendet, der seine Quantenausbeute bei der Bindung erhöhte. Die Fluoreszenz des ungebundenen Liganden im Lösungsvolumen in der Nähe des Pflasters kann durch die Abbildung der Pflaster mit konfokaler Mikroskopie subtrahiert werden22,23. In unseren Studien wurde die Bindung anhand der Reduktion der ANAP-Fluoreszenz gemessen. Da dieses Signal kanalspezifisch ist und FRET zwischen ANAP und TNP-ATP stark abstandsabhängig ist (halbmaximal bei ~43 Å), konnte eine Kontamination unseres Signals durch unspezifisch gebundene und ungebundene Nukleotide vermieden werden.
Abbildung 4A zeigt ein typisches Patch-Clamp-Fluorometrie-Experiment (PCF). Zwischen einer mit Kochsalzlösung gefüllten Borosilikatglaspipette (verbunden mit einem Spannungszangenverstärker) und einer Zelle, die ANAP-markiertes KATP exprimiert, wurde eine hochohmige Dichtung (GΩ) gebildet. Nach der Versiegelungsbildung wurde die Pipette von der Zelle weggezogen, um den Zugang zu den intrazellulären Nukleotidbindungsstellen zu ermöglichen. Die Pipette wurde dann über dem Mikroskopobjektiv positioniert, auf den Schlitz der Spektrometermaske zentriert und der Ausfluss des Mikrovolumen-Perfusionssystems (modifiziert mit einer Borosilikatglasspitze) in die Nähe der Pipette gebracht (Abbildung 4D). Die Spannung wurde kontrolliert und die Ströme wurden von den Kanälen im Patch gemessen. Repräsentative Ströme und Spektren von ANAP-markierten KATP-Kanälen sind in Abbildung 4B dargestellt, farbkodiert, um die Spektren an die Ströme anzupassen. Die Emissionsspektren wurden für Untergrund und Ausbleichen wie für ungedeckte Membranen korrigiert.

Abbildung 1: ANAP und TNP-ATP ergeben ein geeignetes FRET-Paar. (A) Strukturen von ANAP und TNP-ATP. Die fluoreszierenden Einheiten werden hervorgehoben. (B) Absorptions- und Fluoreszenzemissionsspektren von ANAP und TNP-ATP. Für FRET ist eine Überlappung zwischen ANAP-Emission und TNP-ATP-Absorption erforderlich. Adaptiert von Puljung et al. (veröffentlicht unter der Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)10. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 2: Messung der Nukleotidbindung in Plasmamembranen ohne Dach. (A) Schematische Darstellung für die Herstellung von Plasmamembranen ohne Dach aus adhärenten Zellen, die ein fluoreszierendes Membranprotein exprimieren. Anweisungen werden für Zellen bereitgestellt, die auf Poly-L-Lysin-beschichteten oder unbehandelten Deckgläsern gezüchtet wurden. (B) Versuchsaufbau zur Messung der Nukleotidbindung in ungedeckten Membranen. (C) Hellfeld- und Fluoreszenzbilder einer völlig unbedeckten Plasmamembran, die von einer Zelle stammt, die mit orangefarbenem Fluoreszenzprotein (OFP) markierten KATP-Kanälen exprimiert. Das Sternchen markiert die Position der Membran, die im Hellfeldbild nahezu unsichtbar ist. OFP wurde mit einer breiten 565 nm LED durch einen 531/40 nm Bandpassfilter und 562 nm Kantendichroitik angeregt und das emittierte Licht wurde durch einen 593/40 nm Bandpassfilter gesammelt. (D) Hellfeld- und Fluoreszenzbilder eines teilweise nicht überdachten Membranfragments, das von einer Zelle stammt, die mit orangefarbenem Fluoreszenzprotein (OFP) markierten KATP-Kanälen exprimiert. (E) Lösungsaustauschzeitverlauf, der unter Verwendung des unter B beschriebenen Aufbaus erworben wurde. Fünf technische Wiederholungen sind dargestellt. Das Mikrovolumen-Perfusionssystem wurde mit 50 μM Tetramethylrhodamin-5-maleimid (TMRM) beladen. Das Bad wurde mit einer Geschwindigkeit von ~0,5 ml/min mit Wasser durchblutet. Die Daten der Wash-on- (zunehmende Fluoreszenz) und Wash-out-Zeitverläufe (abnehmende Fluoreszenz) wurden mit einem einzigen exponentiellen Zerfall der Form F = A*exp(-x/τ) + y0 angepasst. Die Zeitkonstante (τ) für das Einwaschen betrug ~0,6 s. Die Zeitkonstante für das Auswaschen betrug ~1,0 s. TMRM wurde mit einer breiten 565 nm LED durch einen 540/25 nm Bandpassfilter und 565 nm Kantendichroitik angeregt und das emittierte Licht durch einen 605/55 nm Bandpassfilter gesammelt. (F) Vergleich der Fluoreszenzintensität einer 50 μM TMRM-Lösung, die unter Verwendung des Mikrovolumen-Perfusionssystems wie in B aufgetragen wurde, und eines Destillierbades mit 50 μM TMRM. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 3: Hintergrundsubtraktion und Bleichkorrektur. (A) Spektralbild (räumliche Information in der y-Dimension, Wellenlänge in der x-Dimension) einer unbedachten Plasmamembran einer Zelle, die ANAP-markierteK-ATP-Kanäle exprimiert. 5 μM TNP-ATP wurde mit dem in Abbildung 2B beschriebenen Aufbau appliziert. Das orangefarbene Kästchen kennzeichnet die Region of Interest (ROI), die der nicht überdachten Membran entspricht. Das graue Kästchen kennzeichnet den Hintergrundbereich, der für die Korrektur des Spektrums verwendet wird. (B) Emissionsspektren, die aus wellenlängenweisen Mittelwerten der ROI und der Hintergrundregionen in A abgeleitet wurden. (C) Spektrum, abgeleitet durch Subtraktion des gemittelten Hintergrundspektrums vom gemittelten ROI-Spektrum in B. Das 5-nm-Fenster um den ANAP-Peak, das zur Bestimmung der durchschnittlichen Intensität verwendet wird, wird als grau schattierter Bereich dargestellt. (D) Spektren, die aus sechs aufeinanderfolgenden 10-Sekunden-Belichtungen einer Plasmamembran ohne Dach einer Zelle aufgenommen wurden, die ANAP-markierte KATP-Kanäle exprimiert. Man beachte die Abnahme der Fluoreszenz, die sich aus der Photobleichung ergibt. Der Einschub zeigt die normierte Peak-Fluoreszenzanpassung mit einem einzigen exponentiellen Zerfall der Form F/Fmax = A*exp(-t/τ) + (1-A). Die Symbole im Einschub sind farbkodiert, um den Spektren zu entsprechen. (E) Die gleichen Spektren wie in D, korrigiert um Photobleichung. Der Einschub zeigt die normalisierte Peak-Fluoreszenz von D als offene Kreise, wobei die korrigierte Peak-Fluoreszenz mit gefüllten Kreisen dargestellt wird. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 4: Simultane Messungen von Nukleotidbindung und Kanalströmen mittels Patch-Clamp-Fluorometrie (PCF). (A) Schematische Darstellung des Versuchsaufbaus zur Messung der Nukleotidbindung und der Ionenströme. (B) Beispielhafte Ströme (links) und Spektren (rechts), die von einem Membranpflaster aufgenommen wurden, das aus einer Zelle herausgeschnitten wurde, die ANAP-markierteK-ATP-Kanäle exprimiert. Die Ströme wurden bei einem Haltepotential von -60 mV aufgezeichnet, bei 20 kHz digitalisiert und bei 5 kHz gefiltert. Der grau schattierte Bereich entspricht dem Wellenlängenbereich, aus dem die ANAP-Intensität quantifiziert wurde. Adaptiert von Usher et al. (veröffentlicht unter der Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)11. (C) Spektrum, das aus einem Membranpflaster gewonnen wurde, das aus einer Zelle herausgeschnitten wurde, die ANAP-markierte K-ATP-Kanäle exprimierte, die 1 mMTNP-ATP ausgesetzt waren. Beachten Sie den negativen Peak, der dem Wellenlängenbereich entspricht, über den TNP-ATP-Fluoreszenz beobachtet wird. Der grau schattierte Bereich bezeichnet den Wellenlängenbereich, der zur Quantifizierung der ANAP-Fluoreszenz verwendet wird, wie in B. Adaptiert von Usher et al. (veröffentlicht unter der Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)11. (D) Hellfeld- und Fluoreszenzbilder einer Patch-Pipette, die bei 1 mM TNP-ATP belichtet wurde. Das Sternchen markiert die Spitze der Pipette. (E) Spektralbild derselben Patch-Pipette in 1 mM TNP-ATP. Das Sternchen markiert die Position der Pipette. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 5: TNP-ATP-Bindung an ANAP-markierteK-ATP-Kanäle . (A) Spektralaufnahmen einer Plasmamembran ohne Dach aus einer Zelle, die ANAP-markierte K-ATP-Kanäle in Abwesenheit von TNP-ATP oder in Gegenwart von 50 μM oder 1 mMTNP-ATP exprimiert. Die Intensitäten werden als Heatmap dargestellt. (B) Wellenlängen-für-Wellenlängen-gemittelte Spektren aus den Bildern in A, die das Löschen der ANAP-Fluoreszenz durch TNP-ATP zeigen. Die schraffierten Bereiche stellen zwei verschiedene Bandpassfilter dar, die zur Messung der ANAP-Löschung verwendet werden können, wenn kein Spektrometer verfügbar ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

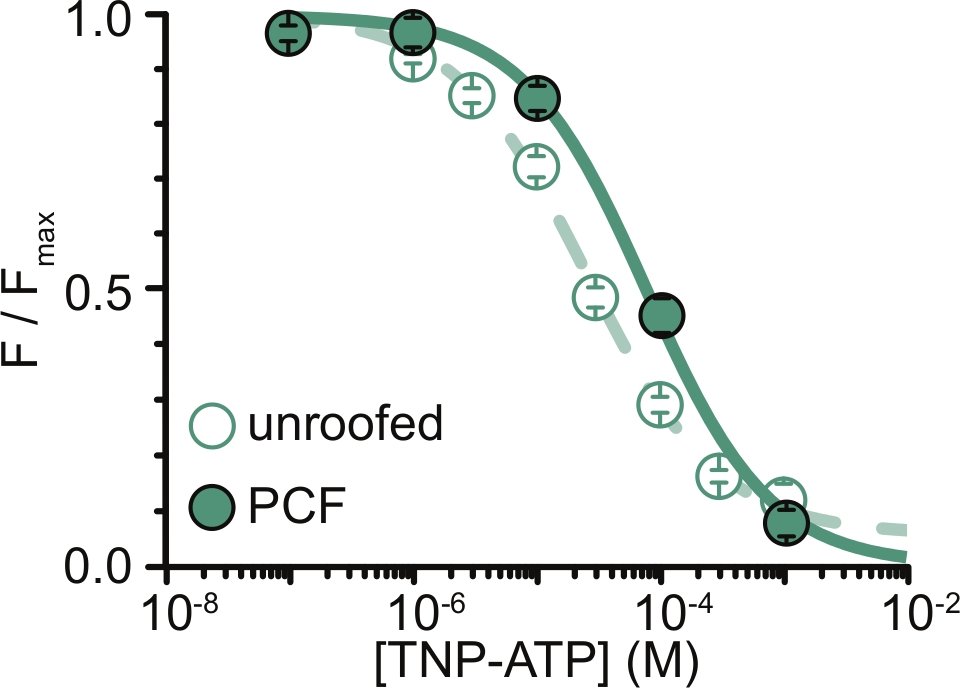
Abbildung 6: Quenching von ANAP-markierten K ATP-Kanälen durchTNP-ATP in ungedeckten Membranen und PCF. Überlagerung von Daten von Usher et al. (veröffentlicht unter der Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)11. Die Daten wurden an die Hill-Gleichung angepasst: F / F max = E max + (1 – E max) / (1+10(EC50 –[TNP-ATP])*h). F ist die gemessene Fluoreszenz, F max ist die maximale Fluoreszenz in Abwesenheit von Nukleotiden, E max ist die maximale Abschreckung bei sättigenden Nukleotidkonzentrationen und h ist die Hill-Steigung. EC50 (die Nukleotidkonzentration, bei der die Abschreckung halb maximal ist) und [TNP-ATP] sind logarithmische Werte. Dachlose Membranen: EC50 = -4,59 (25,7 μM), h = 0,82, Emax = 0,93. PCF: EC50 = -4,11 (77,6 μM), h = 0,87, Emax = 1,00. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Diskussion
Wir haben eine Methode entwickelt, um die Adenin-Nukleotid-Bindung an intakte Membranproteine in Echtzeit zu messen. Unsere Methode baut auf mehreren anderen etablierten Techniken auf, darunter die Markierung von Proteinen mit ANAP unter Verwendung von Amber-Stop-Codon-Suppression 12, Cell Unroofing 14 und Voltage-Clamp-Fluorometrie/PCF 19,20,21,22,23,24,25 . Die Synthese dieser Ansätze ermöglicht die Messung der Nukleotidbindung mit hoher räumlicher und zeitlicher Auflösung. Tatsächlich konnten wir in unserer früheren Arbeit mit diesem Ansatz zwischen verschiedenen Bindungsstellen auf demselben Proteinkomplex unterscheiden10,11. Wichtig ist, dass diese Technik direkt auf kleine Proteinmengen in einer zellulären Umgebung unter Bedingungen angewendet werden kann, die die Proteinfunktion erhalten. Die Verwendung unserer Bindungsmethode in Verbindung mit dem direkten, elektrophysiologischen Auslesen von Ionenkanalströmen ermöglicht es uns, reichhaltige Einblicke in die molekularen Grundlagen des Kanal-Gating11 zu erhalten.
Da es sich bei Spektrometern um ein nicht standardmäßiges Laborgerät handelt, kann die ANAP-Intensität auch relativ isoliert mit Bandpassfiltern überwacht werden. Abbildung 5B zeigt die spektralen Eigenschaften zweier solcher Filter. Der 470/10 nm Bandpassfilter schirmt das Fluoreszenzsignal von TNP-ATP effektiv ab und überlappt sich gut mit der ANAP-Spitzenfluoreszenz. Die maximale Durchlässigkeit dieses Filters beträgt jedoch nur etwa 50 %, was es schwierig machen kann, gute Signale von schwachen Membranen (oder in herausgeschnittenen Membranfeldern unter Spannungsklemme) zu erhalten. Eine weitere Option ist ein 460/60 nm Bandpassfilter. Es gibt etwas mehr Überlappung zwischen dem 460/60-nm-Filter und dem Fuß des TNP-ATP-Emissionspeaks im Vergleich zum 470/10-nm-Filter. Der 460/60-nm-Bandpass hat jedoch eine Transmission von 90-95% über einen weiten Bereich des ANAP-Peaks, was das Fluoreszenzemissionssignal verstärken würde.
ANAP ist ein umweltempfindlicher Fluorophor 12,26,27. Die Spitzenemission und die Quantenausbeute variieren je nach Einbaustelle auf dem interessierenden Protein und können sich ändern, wenn sich die Konformation des Proteins ändert. Solche Veränderungen wären aus den Emissionsspektren sofort ersichtlich, aber nicht so offensichtlich, wenn die ANAP-Intensität mit Filtern gemessen wird. In jedem Fall sind geeignete Kontrollen erforderlich, um nachzuweisen, dass das Fluoreszenzsignal nicht aufgrund von Veränderungen in der lokalen Umgebung von ANAP nach der Nukleotidbindung variiert. Kontrollexperimente mit unmarkierten Nukleotiden können helfen, zu überprüfen, ob Änderungen der ANAP-Intensität das Ergebnis von FRET zwischen ANAP- und TNP-Nukleotiden sind. TNP-Nukleotide können unspezifisch an die Membranen von nicht transfizierten Zellen binden (entweder an die Plasmamembran oder an native Membranproteine)10. Wir quantifizieren die Bindung als Dekrement in der Donorfluoreszenz, da dieses Signal spezifisch für den markierten Kanal ist. Wir empfehlen jedoch, für jedes Agonist/Rezeptor-Paar zusätzliche Kontrollexperimente durchzuführen, z. B. die Nukleotid-Bindungsstelle zu mutieren, falls bekannt, um zu überprüfen, ob die Änderung der Donorfluoreszenz wirklich das Ergebnis einer direkten Bindung an den markierten Rezeptor11 ist. Schließlich empfiehlt es sich, mit Konstrukten zu arbeiten, die neben der ANAP-Markierung auch einen fluoreszierenden Protein-Tag enthalten. Dies hilft bei der Unterscheidung zwischen markierter Rezeptorfluoreszenz und Hintergrund-/Autofluoreszenz. Die Hintergrundfluoreszenz kann durch den Peak und die Form der Emissionsspektren10 von ANAP unterschieden werden, aber solche Bestimmungen können sehr schwierig sein, wenn nur Filtersätze verwendet werden. Darüber hinaus können Zellen und unbedeckte Membranen, die fluoreszierende Rezeptoren exprimieren, mit dem fluoreszierenden Protein-Tag identifiziert werden, ohne ANAP anregen zu müssen und eine übermäßige Photobleichung zu riskieren.
In vielen unserer PCF-Aufzeichnungen beobachteten wir einen starken negativen Peak in unseren Spektren bei hohen TNP-ATP-Konzentrationen (Abbildung 4C). Dieser negative Peak ist ein Artefakt unseres Hintergrundsubtraktionsprotokolls. Abbildung 4D zeigt Hellfeld- und Fluoreszenzbilder einer Patch-Pipette, die mit 1 mM TNP-ATP belichtet wurde. An der Pipettenspitze ist ein Schatten zu erkennen, der sich aus dem Ausschluss von TNP-ATP aus dem Volumen der Pipettenwände ergibt, was innerhalb der Fokusebene am deutlichsten ist. Das Spektralbild in Abbildung 4E zeigt ein dunkles Band, das diesem Schatten entspricht. Wenn ein Bereich oberhalb oder unterhalb dieses dunklen Bandes für die Hintergrundsubtraktion verwendet wird, erzeugt dies eine negative Spitze. Wichtig ist, dass dieser Peak über einen Wellenlängenbereich auftrat, der der TNP-ATP-Emission entsprach und unsere Messungen der ANAP-Abschreckung nicht beeinflusste.
Die größte Einschränkung unserer Experimente bestand darin, eine adäquate Plasmamembranexpression von ANAP-markierten Konstrukten zur Messung der Fluoreszenz zu erhalten. Es war im Allgemeinen einfacher, qualitativ hochwertige Spektren von Membranen ohne Dach zu erhalten als mit PCF, da sie größer sind und wir in der Lage sind, schnell eine ganze Schüssel mit nicht überdachten Membranen zu scannen, im Gegensatz zu PCF, wo Patches nur einzeln erhalten werden können. In unseren Experimenten waren die Daten von Membranen ohne Dach und PCF-Experimenten ähnlich, aber nicht äquivalent (Abbildung 6)11. Es gibt jedoch keinen A-priori-Grund, warum dies eine universelle Beobachtung sein sollte, da Proteine in einer Patch-Pipette in einem anderen funktionellen Zustand sein können als solche in Membranen ohne Dach.
Hier wurde versucht, die Expression unserer ANAP-markierten Konstrukte zu maximieren, insbesondere die Zellkulturtemperatur auf 33 °C zu senken10,11,16. Unserer Erfahrung nach führte der Versuch, Stellen im Protein zu identifizieren, an denen ANAP eine konservative Substitution darstellen würde, nicht durchweg zu Konstrukten, die gut exprimiert wurden. Wir hatten mehr Erfolg damit, ganze Proteinregionen systematisch nach ANAP-Einbaustellen zu durchsuchen und Kandidaten auf Oberflächenexpression zu untersuchen10. Das ANAP-Markierungssystem funktioniert auch in Xenopus laevis-Oozyten, wodurch viel größere Membranflecken herausgeschnitten werden können, wodurch das Signal-Rausch-Verhältnis erhöhtwird 26,27,28.
Während erwartet wird, dass größere Expressionsniveaus zu helleren Signalen führen, hängt die minimale Anzahl von Kanälen, die zur Messung der Fluoreszenz erforderlich sind, von mehreren Faktoren ab, darunter die Helligkeit des Fluorophors, der Grad der Photobleichung, die Intensität des Anregungslichts und die Schärfeebene. Theoretisch könnten Schätzungen durch Korrelation der Fluoreszenzintensität und des Kanalstroms vorgenommen werden, wie bereits gezeigt wurde28,29. Die Zuverlässigkeit solcher Schätzungen erfordert jedoch eine gewisse Kenntnis des Einkanalleitwerts und der Offenheitswahrscheinlichkeit des Kanals. Zusätzlich zu den oben genannten Faktoren wird das Fluoreszenzsignal auch durch Kanäle beeinflusst, die mit Vesikeln oder Abschnitten der Plasmamembran verbunden sind, die mit dem Pipettenglas verbunden sind und nicht unter Spannung stehen.
Diese Methode lässt sich leicht für die Untersuchung anderer nukleotidsensitiver Ionenkanäle verwenden. CFTR ähnelt strukturell der akzessorischen Sulfonylharnstoffrezeptor-Untereinheit von KATP30,31. Wie K,ATP, wird auch das CFTR-Gating durch Nukleotidbindung gesteuert, was es zu einem offensichtlichen zukünftigen Ziel unserer Methodemacht 7. Purinerge P2X-Rezeptoren sind Ionenkanäle, die von extrazellulärem ATP9 gesteuert werden. TNP-ATP wirkt als Antagonist für P2X-Rezeptoren32,33. Daher ist es für die Untersuchung der P2X-Aktivierung nicht nützlich, obwohl es in Wettbewerbsassays mit P2X-Agonisten verwendet werden kann. Alternativ können auch andere fluoreszierende ATP-Derivate mit ausreichender spektraler Überlappung mit der ANAP-Emission verwendet werden, um die Aktivierung zu untersuchen. Alexa-647-ATP ist ein fluoreszierender P2X-Agonist34. Das berechnete R0 zwischen Alexa-647 und ANAP beträgt ~85 Å, was bedeutet, dass eine direkte Bindung an P2X zu einer erheblichen Abschreckung von ANAP führen sollte, die in den Kanal eingebaut ist. Ein derart langes R0 führt jedoch auch zu einer Abschreckung von Alexa-647-ATP, die an benachbarte Untereinheiten gebunden ist, und erhöht die Wahrscheinlichkeit, dass eine unspezifische Nukleotidbindung zu FRET führt. Da die Ligandenbindungsstelle in P2X-Rezeptoren extrazellulär ist, würden Bindungsmessungen an intakten Zellen, in Ganzzell-Spannungsklemmen oder in Outside-Out-Membranpatches durchgeführt. Unsere Methode kann auch erweitert werden, um die Bindung und Aktivierung von elektrogenen und nicht-elektrogenen Transportern und Pumpen zu untersuchen, die für ihren Reaktionszyklus von ATP abhängig sind, sowie von G-Protein-gekoppelten P2Y-Rezeptoren. Obwohl wir diese Methode zur Messung der Adenin-Nukleotid-Bindung (TNP-ATP, TNP-ADP, TNP-AMP) entwickelt haben, kann derselbe Ansatz verwendet werden, um die Bindung an praktisch jeden Rezeptor zu untersuchen, für den ein geeigneter, fluoreszierender Ligand identifiziert wurde.
Offenlegungen
Die Autoren erklären keine Interessenkonflikte.
Danksagungen
Wir bedanken uns bei Raul Terron Exposito für die hervorragende technische Unterstützung. Diese Arbeit wurde vom Forschungsrat für Biotechnologie und Biowissenschaften (BB/R002517/1; MCP und FMA) und dem Wellcome Trust (203731/Z/16/A; SGU)
Materialien
| Name | Company | Catalog Number | Comments |
| T75 tissue-culture treated flask | StarLab | CC7682-4875 | |
| 0.1% w/v poly-L-lysine | Sigma-Aldrich | P8920 | |
| 30 mm borosilicate cover glass slips | VWR | 631-0174 | |
| 35 mm non-treated sterile dishes | CytoOne | CC7672-3340 | |
| 35 mm cover glass bottom dish | WPI | FD35-PDL-100 | |
| Dulbecco’s Modified Eagle Medium (DMEM) | Gibco | 31966021 | |
| Foetal bovine serum (FBS) | Gibco | 10500-064 | |
| Penicillin/Streptomycin | Gibco | 15140-122 | |
| TrypLE select (tryosin) | Gibco | 12563-011 | Trypsin/EDTA reagent |
| Phosphate buffered saline (PBS) | Gibco | 14040-091 | |
| UltraPure distilled water | Invitrogen | 10977-035 | |
| HEK293T cells | ATTC | CRL-3216 | Used between passages 5-30 |
| ANAP-TFA | AsisChem | ASIS-0014 | Reconstituted in 30 mM NaOH to a final concentration of 1 mM |
| pANAP expression plasmid | Addgene | Plasmid #48696 | Encodes tRNA/tRNA synthetase pair for expression of ANAP-tagged protein |
| peRF1-E55D | Chin Lab (MRC Laboratory of Molecular Biology, Cambridge, UK) | Jason Chin: DOI: 10.1021/ja5069728 | Encodes dominant-negative eukaryotic ribosomal release factor |
| TransIT-LT1 | Mirus Bio | MIR 2300 | Lipopolyplex transfection reagent |
| Thick-walled borosilicate glass capillaries | Harvard Apparatus | GC150F-15 | |
| Tetramethylrhodamine-5-maleimide | Sigma-Aldrich | 94506 | |
| TNP-ATP | Jena Bioscience | NU-221L | Delivered at 10 mM in water |
| Nikon Eclipse TE2000-U inverted microscope microscope | Nikon | ||
| 60x water immersion objective (1.4 NA) | Nikon | MRD07602 | |
| 4-Wavelength High-Power LED Head | ThorLabs | LED4D245 | 385/490/565/625 nm LEDs |
| Four-Channel LED Driver | ThorLabs | DC4100 | |
| 390/18 nm band-pass excitation filter | ThorLabs | MF390-18 | For ANAP excitation |
| 400 nm long-pass emission filter | ThorLabs | FEL0400 | For imaging ANAP spectra |
| 416 nm edge dichroic | ThorLabs | MD416 | For imaging ANAP spectra |
| 460/60 nm band-pass emission filter | ThorLabs | MF460-60 | Suggested wide band-pass filter for imaging ANAP fluorescence (Figure 4B) |
| 470/10 nm band-pass emission filter | ThorLabs | FB470-10 | Suggested narrow band-pass filter for imaging ANAP fluorescence (Figure 4B) |
| 531/40 band-pass excitation filter | Brightline | FF01-531/40-25 | For orange fluorescent protein (OFP) excitation |
| 540/25 nm band-pass excitation filter | Chroma | D540/25X | For tetramethylrhodamine-5-maleimide (TMRM) excitation |
| 562 nm edge dichroic | Semrock | FF562-Di03 | For imaging OFP fluorescence |
| 565 nm edge dichroic | Chroma | 565DC | For imaging TMRM fluorescence |
| 593/40 nm band-pass excitation filter | Brightline | FF01-387/11-25 | For imaging OFP fluorescence |
| 605/55 nm band-pass emission filter | Chroma | D605/55M | For imaging TMRM fluorescence |
| IsoPlane-160 Imaging Spectrometer | Princeton Instruments | IsoPlane-160 | |
| PIXIS 400BR_eXcelon Camera | Princeton Instruments | PIXIS: 400BR_eXcelon | |
| Axopatch 200B amplifier | Molecular Devices | Axopatch 200B-2 | |
| Digidata 1440A digitizer | Molecular Devices | Digidata 1440A | |
| Probe sonicator | Sonics & Materials | VC-50 | For unroofing |
| REGLO digital peristaltic pump | Ismatec | ISM 832 | For bath perfusion |
| Microvolume perfusion system | ALA Scientific Instruments | ALA μFlow-8 | For TNP-ATP perfusion |
| pClamp 10.6.2 | Molecular Devices | Recording and analysing currents | |
| Lightfield 5.20.1507 | Princeton Instruments | Acquisition software for images and spectra | |
| Matlab | Mathworks | For data analysis | |
| Python 3.8.1 | Python Software Foundation | For data analysis |
Referenzen
- Garcia, M. L., Kaczorowski, G. J. Ion channels find a pathway for therapeutic success. Proceedings of the National Academy of Sciences of the United States of America. 113 (20), 5472-5474 (2016).
- Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schioth, H. B., Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nature Reviews Drug Discovery. 16 (12), 829-842 (2017).
- Lakowicz, J. R. Principles of fluorescence spectroscopy. 3rd edn. , Springer. (2006).
- Higgins, C. F., Linton, K. J. The ATP switch model for ABC transporters. Nature Structural & Molecular Biology. 11 (10), 918-926 (2004).
- Toyoshima, C., Cornelius, F. New crystal structures of PII-type ATPases: excitement continues. Current Opinion in Structural Biology. 23 (4), 507-514 (2013).
- Craven, K. B., Zagotta, W. N. CNG and HCN channels: two peas, one pod. Annual Review of Physiology. 68, 375-401 (2006).
- Csanady, L., Vergani, P., Gadsby, D. C. Strict coupling between CFTR's catalytic cycle and gating of its Cl- ion pore revealed by distributions of open channel burst durations. Proceedings of the National Academy of Sciences of the United States of America. 107 (3), 1241-1246 (2010).
- Vedovato, N., Ashcroft, F. M., Puljung, M. C. The Nucleotide-Binding Sites of SUR1: A Mechanistic Model. Biophysical Journal. 109 (12), 2452-2460 (2015).
- Burnstock, G. Introduction to the Special Issue on Purinergic Receptors. Advances in Experimental Medicine and Biology. 1051, 1-6 (2017).
- Puljung, M., Vedovato, N., Usher, S., Ashcroft, F. Activation mechanism of ATP-sensitive K(+) channels explored with real-time nucleotide binding. Elife. 8, 41103(2019).
- Usher, S. G., Ashcroft, F. M., Puljung, M. C. Nucleotide inhibition of the pancreatic ATP-sensitive K+ channel explored with patch-clamp fluorometry. Elife. 9, 52775(2020).
- Chatterjee, A., Guo, J., Lee, H. S., Schultz, P. G. A genetically encoded fluorescent probe in mammalian cells. Journal of the American Chemical Society. 135 (34), 12540-12543 (2013).
- Gordon, S. E., Senning, E. N., Aman, T. K., Zagotta, W. N. Transition metal ion FRET to measure short-range distances at the intracellular surface of the plasma membrane. Journal of General Physiology. 147 (2), 189-200 (2016).
- Heuser, J. The production of 'cell cortices' for light and electron microscopy. Traffic. 1 (7), 545-552 (2000).
- Schmied, W. H., Elsasser, S. J., Uttamapinant, C., Chin, J. W. Efficient multisite unnatural amino acid incorporation in mammalian cells via optimized pyrrolysyl tRNA synthetase/tRNA expression and engineered eRF1. Journal of the American Chemical Society. 136 (44), 15577-15583 (2014).
- Lin, C. Y., et al. Enhancing Protein Expression in HEK-293 Cells by Lowering Culture Temperature. PloS One. 10 (4), 0123562(2015).
- Usukura, J., et al. Use of the unroofing technique for atomic force microscopic imaging of the intra-cellular cytoskeleton under aqueous conditions. Journal of Electron Microscopy. 61 (5), 321-326 (2012).
- Colquhoun, D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. British Journal of Pharmacology. 125 (5), 924-947 (1998).
- Mannuzzu, L. M., Moronne, M. M., Isacoff, E. Y. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 271 (5246), 213-216 (1996).
- Zheng, J., Zagotta, W. N. Gating rearrangements in cyclic nucleotide-gated channels revealed by patch-clamp fluorometry. Neuron. 28 (2), 369-374 (2000).
- Zheng, J., Zagotta, W. N. Patch-clamp fluorometry recording of conformational rearrangements of ion channels. Science's STKE. 2003 (176), 7(2003).
- Biskup, C., et al. Relating ligand binding to activation gating in CNGA2 channels. Nature. 446 (7134), 440-443 (2007).
- Kusch, J., et al. Interdependence of receptor activation and ligand binding in HCN2 pacemaker channels. Neuron. 67 (1), 75-85 (2010).
- Wu, S., et al. State-dependent cAMP binding to functioning HCN channels studied by patch-clamp fluorometry. Biophysical Journal. 100 (5), 1226-1232 (2011).
- Cha, A., Bezanilla, F. Characterizing voltage-dependent conformational changes in the Shaker K+ channel with fluorescence. Neuron. 19 (5), 1127-1140 (1997).
- Kalstrup, T., Blunck, R. Dynamics of internal pore opening in K(V) channels probed by a fluorescent unnatural amino acid. Proceedings of the National Academy of Sciences of the United States of America. 110 (20), 8272-8277 (2013).
- Kalstrup, T., Blunck, R. S4-S5 linker movement during activation and inactivation in voltage-gated K(+) channels. Proceedings of the National Academy of Sciences of the United States of America. 115 (29), 6751-6759 (2018).
- Dai, G., Aman, T. K., DiMaio, F., Zagotta, W. N. The HCN channel voltage sensor undergoes a large downward motion during hyperpolarization. Nature Structural & Molecular Biology. 26 (8), 686-694 (2019).
- Liu, C., et al. Patch-clamp fluorometry-based channel counting to determine HCN channel conductance. Journal of General Physiology. 148 (1), 65-76 (2016).
- Hwang, T. C., et al. Structural mechanisms of CFTR function and dysfunction. Journal of General Physiology. 150 (4), 539-570 (2018).
- Puljung, M. C. Cryo-electron microscopy structures and progress toward a dynamic understanding of KATP channels. Journal of General Physiology. 150 (5), 653-669 (2018).
- Kasuya, G., et al. Structural insights into the competitive inhibition of the ATP-gated P2X receptor channel. Nature Communications. 8 (1), 876(2017).
- Virginio, C., Robertson, G., Surprenant, A., North, R. A. Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3 receptors. Molecular Pharmacology. 53 (6), 969-973 (1998).
- Bhargava, Y., Nicke, A., Rettinger, J. Validation of Alexa-647-ATP as a powerful tool to study P2X receptor ligand binding and desensitization. Biochemical and Biophysical Research Communications. 438 (2), 295-300 (2013).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten