
Method Article
Kartierung der Struktur-Funktions-Beziehungen ungeordneter onkogener Transkriptionsfaktoren mittels Transkriptomanalyse
In diesem Artikel
Zusammenfassung
Intrinsisch ungeordnete Domänen sind wichtig für die Funktion des onkogenen Fusionstranskriptionsfaktors. Um diese Proteine therapeutisch anzugreifen, ist ein detaillierteres Verständnis der Regulationsmechanismen dieser Domänen erforderlich. Hier verwenden wir Transkriptomik, um wichtige strukturelle Merkmale der intrinsisch gestörten EWS-Domäne im Ewing-Sarkom abzubilden.
Zusammenfassung
Viele Krebsarten sind durch chromosomale Translokationen gekennzeichnet, die zur Expression onkogener Fusionstranskriptionsfaktoren führen. Typischerweise enthalten diese Proteine eine intrinsisch ungeordnete Domäne (IDD), die mit der DNA-Bindungsdomäne (DBD) eines anderen Proteins verschmolzen ist, und orchestrieren weit verbreitete transkriptionelle Veränderungen, um Malignität zu fördern. Diese Fusionen sind oft die einzige wiederkehrende genomische Aberration bei den von ihnen verursachten Krebsarten, was sie zu attraktiven therapeutischen Zielen macht. Das Targeting onkogener Transkriptionsfaktoren erfordert jedoch ein besseres Verständnis der mechanistischen Rolle, die IDDs mit geringer Komplexität in ihrer Funktion spielen. Die N-terminale Domäne von EWSR1 ist eine IDD, die an einer Vielzahl von onkogenen Fusionstranskriptionsfaktoren beteiligt ist, einschließlich EWS/FLI, EWS/ATF und EWS/WT1. Hier untersuchen wir mit hilfe der RNA-Sequenzierung die strukturellen Merkmale der EWS-Domäne, die für die Transkriptionsfunktion von EWS/FLI beim Ewing-Sarkom wichtig sind. Die erste shRNA-vermittelte Depletion der endogenen Fusion aus Ewing-Sarkomzellen gepaart mit ektopischer Expression einer Vielzahl von EWS-mutanten Konstrukten wird durchgeführt. Dann wird RNA-Sequenzierung verwendet, um die Transkriptome von Zellen zu analysieren, die diese Konstrukte exprimieren, um die funktionellen Defizite zu charakterisieren, die mit Mutationen in der EWS-Domäne verbunden sind. Durch die Integration der transkriptomischen Analysen mit zuvor veröffentlichten Informationen über EWS/FLI-DNA-Bindungsmotive und genomische Lokalisierung sowie funktionelle Assays zur Transformationsfähigkeit konnten wir strukturelle Merkmale von EWS/FLI identifizieren, die für die Onkogenese wichtig sind, und einen neuartigen Satz von EWS/FLI-Zielgenen definieren, die für das Ewing-Sarkom entscheidend sind. Diese Arbeit demonstriert die Verwendung der RNA-Sequenzierung als Methode, um die Struktur-Funktions-Beziehung der intrinsisch gestörten Domäne onkogener Transkriptionsfaktoren abzubilden.
Einleitung
Eine Untergruppe von Krebsarten, einschließlich vieler Malignome der Kindheit und Jugend, sind durch chromosomale Translokationen gekennzeichnet, die neuartige Fusionsonkogene1,2,3,4,5,6erzeugen. Die resultierenden Fusionsproteine fungieren häufig als onkogene Transkriptionsfaktoren und orchestrieren weit verbreitete Veränderungen in der Transkriptionsregulation, um die Tumorgenese zu fördern7,8. Krebsarten mit diesen Translokationen besitzen gewöhnlich eine ansonsten ruhige Mutationslandschaft mit wenigen wiederkehrenden genomischen Aberrationen, abgesehen von der pathognomonischen Fusion4,9. Daher ist das direkte Targeting des Fusionsproteins eine attraktive therapeutische Strategie bei diesen Erkrankungen. Diese onkogenen Transkriptionsfaktoren bestehen jedoch üblicherweise aus einer wenig komplexen, intrinsisch ungeordneten, transkriptionsaktivierenden Domäne, die mit einer DNA-bindenden Domäne (DBD)10,11,12,13,14verschmolzen ist. Sowohl die intrinsisch ungeordneten Domänen (IDDs) als auch die DBDs dieser Proteine haben sich mit herkömmlichen pharmakologischen Ansätzen als schwierig erwiesen. Die Entwicklung neuartiger therapeutischer Ansätze erfordert daher ein detaillierteres molekulares Verständnis der Mechanismen, die von diesen Fusionen verwendet werden, um die Genexpression abnormal zu regulieren.
Der N-terminale IDD-Anteil von EWSR1 wird üblicherweise mit einer DBD bei Krebs verschmolzen, einschließlich EWS / FLI bei Ewing-Sarkom, EWS / WT1 bei diffusem kleinzelligem Rundzelltumor und EWS / ATF1 bei klarzelligen Sarkomen von Weichteilen10. Die mechanistische Rolle der EWS IDD in jeder dieser Fusionen ist unvollständig verstanden. Die EWS/ETS-Fusionsfamilie, insbesondere EWS/FLI, ist die bisher funktionell am besten charakterisierte. EWS/FLI koordiniert genomweite epigenetische und transkriptionelle Veränderungen, die zur Aktivierung und Unterdrückung von Tausenden von Genen führen7,11,15,16. Studien haben gezeigt, dass die IDD sowohl für die Rekrutierung von transkriptionellen Co-Aktivatoren (wie p300, WDR5 und dem BAF-Komplex) als auch von Co-Repressoren (wie dem NuRD-Komplex)11,15,17wichtig ist. Die Fusion der EWS-IDD mit dem C-terminalen Teil von FLI1 verleiht der ETS-DBD von FLI1 eine neuartige DNA-Bindungsspezifität, so dass das Fusions-Onkoprotein (EWS/FLI) zusätzlich zum Konsensus-ETS-Motiv18,19,20an sich wiederholende GGAA-Mikrosatellitenregionen des Genoms bindet. In Kombination mit der Co-Activator-Rekrutierungsfunktion fördert diese emergente DNA-Bindungsaktivität von EWS/FLI die De-novo-Enhancer-Bildung an GGAA-Mikrosatelliten, die distal zu Transkriptionsstartstellen (TSS) ("Enhancer-ähnliche" Mikrosatelliten) sind, und rekrutiert RNA-Polymerase II, um die Transkription an GGAA-Mikrosatelliten in der Nähe von TSS ("Promotor-ähnliche" Mikrosatelliten) zu fördern11,15,16,21.
Zusammengenommen führten uns diese Daten zu der Hypothese, dass diskrete Elemente innerhalb des EWS-Bereichs zur Rekrutierung verschiedener Co-Regulatoren für verschiedene Arten von EWS/FLI-Bindungsstellen beitragen. Die Unterscheidung dieser Elemente innerhalb des EWS-Teils von EWS/FLI und ihre Funktionsweise wurde jedoch durch die stark repetitive und ungeordnete Natur der Domäne behindert. Hier verwenden wir ein zuvor veröffentlichtes Knockdown-Rescue-System in Ewing-Sarkomzellen, um diese Elemente in der EWS-IDD funktionell abzubilden. In diesem System wird EWS/FLI mit hilfe einer shRNA depletiert, die auf die 3'UTR des FLI1-Gens abzielt, und die Expression wird mit unterschiedlichen EWS/FLI-mutierten cDNA-Konstrukten gerettet, denen die 3'UTR7,17,22fehlen . Diese Experimente konzentrierten sich auf Konstrukte mit verschiedenen Deletionen, um die Struktur-Funktions-Beziehung zwischen der EWS-IDD und wichtigen onkogenen Phänotypen abzubilden, einschließlich der Aktivierung eines GGAA-Mikrosatelliten-Reporterkonstrukts, Koloniebildungsassays und der gezielten Validierung von EWS/FLI-aktivierten und -unterdrückten Genen7,17,22 . Diese Studien konnten jedoch keine diskreten Subdomänen innerhalb der EWS-IDD in EWS/FLI finden, die für die Aktivierung oder Repression einzigartig wichtig sind. Alle getesteten Konstrukte waren entweder in der Lage, spezifische Zielgene sowohl zu aktivieren als auch zu unterdrücken, was zu einer effizienten Koloniebildung führte, oder in der Lage, eines der EWS/FLI-Zielgene zu regulieren, was zum Verlust der Koloniebildung führte7,17,22.
Transkriptomische Analysen, die durch die weit verbreitete Einführung der Next-Generation-Sequenzierung ermöglicht werden, werden häufig verwendet, um Genexpressionssignaturen unter zwei Bedingungen zu vergleichen, häufig im Rahmen von Screening- oder deskriptiven Studien. Stattdessen wollten wir die Möglichkeit nutzen, genomweite Expressionsdaten mittels RNA-Sequenzierung (RNA-seq) zu erfassen, um die Beiträge von IDDs zur Transkriptionsfaktorfunktion zu charakterisieren. In diesem Fall wird RNA-seq mit dem Knockdown-Rescue-System gepaart, um die Struktur-Funktions-Beziehung der EWS-Domäne zu erforschen. Dieser Ansatz ist auf andere Fusionstranskriptionsfaktoren anwendbar, einschließlich anderer EWS-Fusionen oder Wildtyp-Transkriptionsfaktoren mit schlecht verstandener Funktion, und hat mehrere Vorteile gegenüber den anderen Assays, die für funktionelle Kartierungsstudien verwendet werden, wie Reporter-Assays oder gezielte qRT-PCR. Dazu gehören das Testen struktureller Funktionsdeterminanten im relevanten Chromatinkontext, die Fähigkeit, mehrere Arten von Antwortelementen in einem Assay zu testen (d.h. aktiviert und unterdrückt, GGAA-Mikrosatellit und Nicht-Mikrosatellit usw.) und die daraus resultierende Fähigkeit, Teilfunktionen besser zu erkennen.
Die erfolgreiche Implementierung dieses Ansatzes hängt von einem zellbasierten System ab, das die interessierenden Phänotypen (in diesem Fall A673-Zellen mit shRNA-vermittelter EWS/FLI-Depletion) und einem Panel von Mutantenkonstrukten in einem für das zellbasierte System geeigneten Expressionsvektor erfasst (in diesem Fall pMSCV-hygro mit verschiedenen 3x-FLAG-markierten EWS/FLI-Mutanten, die durch retrovirale Transduktion abgegeben werden sollen). Die virale Transduktion von CRISPR-basierten Depletionskonstrukten, shRNA-basierten Depletion-Konstrukten und cDNA-Expressionskonstrukten mit geeigneter Selektion zur Erzeugung stabiler Zelllinien wird gegenüber der transienten Transfektion empfohlen. Die nachgelagerte Interpretation der Ergebnisse wird verstärkt, wenn die transkriptomischen Daten mit anderen Daten im Zusammenhang mit der Lokalisierung des Transkriptionsfaktors und anderen phänotypischen Auslesungen, sofern verfügbar, gepaart werden können.
In diesem Artikel wenden wir diesen Ansatz an, um die Aktivität der DAF-Mutante von EWS/FLI14zu charakterisieren. Die DAF-Mutante hat 17 Tyrosin-zu-Alanin-Mutationen in den sich wiederholenden Regionen der EWS IDD von EWS/FLI14. Diese spezielle EWS-Mutante wurde zuvor berichtet und ist nicht in der Lage, die Reportergenexpression zu aktivieren, wenn sie mit dem ATF1 DBD14verschmolzen wird. Vorläufige qRT-PCR-Daten deuteten jedoch darauf hin, dass diese Mutante in der Lage war, die Transkription des EWS/FLI-Ziels NR0B123zu aktivieren. Der hier beschriebene transkriptomische Ansatz ermöglichte den erfolgreichen Nachweis der Teilfunktion der DAF-Mutante. Durch die Kombination dieser transkriptomischen Daten mit Informationen über EWS/FLI-Bindungs- und Erkennungsmotive zeigen wir weiter, dass die DAF-Mutante ihre Funktion bei GGAA-Mikrosatellitenwiederholungen beibehält. Diese Ergebnisse identifizieren DAF als die erste partiell funktionelle EWS/FLI-Mutante und heben die Funktion an Nicht-Mikrosatellitengenen als wichtig für die Onkogenese hervor (wieberichtet 23). Dies zeigt die Leistungsfähigkeit dieses transkriptomischen Struktur-Funktions-Mapping-Ansatzes, um Einblicke in die Funktion onkogener Transkriptionsfaktoren zu geben.
Protokoll
1. In-vitro-Konstrukt-Panel einrichten
HINWEIS: Dieser Schritt variiert je nach dem zu analysierenden Protein.
- Bereiten Sie Aliquots des Virus für die Depletion und Expressionskonstrukte nach Bedarf vor.
- Säen Sie eine 10 cm große Gewebekulturschale mit 3-5 x 106 HEK293-EBNA- oder HEK293T-Zellen für jedes Konstrukt, das für die virale Transduktion benötigt wird. Lassen Sie die Zellen über Nacht in Dulbeccos Modified Eagle Media (DMEM) haften, ergänzt mit 10% fetalem Rinderserum (FBS), Penicillin / Streptomycin / Glutamin (P / S / Q) und 0,3 mg / ml G418.
HINWEIS: HEK293-EBNA- und HEK293T-Zellen werden für die Virusproduktion empfohlen, da sie einfach zu züchten sind, eine hohe Transfektionseffizienz aufweisen und rekombinante Proteine aus episomalen Plasmiden effizient exprimieren. Die Zellen sollten am Tag der Transfektion zwischen 50-70% konfluent sein. - Bereiten Sie für jedes virale Transduktionskonstrukt eine Transfektionsmischung vor. Kombinieren Sie 2 ml reduzierte Serummedien mit 90 μL Transfektionsreagenz.
HINWEIS: Es wird empfohlen, reduzierte Serummedien vorzuwärmen. - Fügen Sie jeweils 10 μg eines viralen Verpackungsplasmids (z. B. Gag-Pol), eines viralen Hüllplasmids (z. B. VSV-G) und eines der CRISPR-basierten Depletion, shRNA-basierten Depletion oder cDNA-Expressionskonstrukte (z. B. pMKO oder pMSCV) zur Transfektionsmischung hinzu. Durch sanftes Pipettieren gut mischen.
- Lassen Sie die Transfektionsmischung 20 Minuten bei Raumtemperatur ruhen. Entfernen Sie HEK293-EBNA-Wachstumsmedien aus Gewebekulturschalen und fügen Sie 3 ml DMEM hinzu, das mit 10% FBS, P / S / Q und 10 mM Natriumpyruvat ergänzt wird. Zu jedem Gericht 2 ml Transfektionsmischung tropfenweise hinzufügen. Lassen Sie die Zellen in Transfektionsmedien über Nacht in einem Inkubator bei 37 °C und 5%CO2 sitzen.
- Am nächsten Morgen werden 20 ml DMEM-Medien mit 10% FBS, P / S / Q-Supplementierung und 10 mM Natriumpyruvat hinzugefügt. Inkubieren Sie die zellen darin bei 37 °C und 5% CO2 über Nacht.
- Ersetzen Sie am nächsten Morgen die Medien durch 5 ml virale Sammelmedien (VCM) (DMEM ergänzt mit 10% wärmeinaktiviertem FBS, P / S / Q und 20 mM HEPES).
- Nach 4 h VCM von Platten sammeln und in einem 50 ml konischen Rohr auf Eis bei 4 °C lagern. Ersetzen Sie durch 5 ml frisches VCM.
- Nach 4 h VCM von Platten in demselben konischen 50-ml-Rohr sammeln und bei 4 °C auf Eis lagern. Ersetzen Sie durch 8 ml frisches VCM für die Sammlung über Nacht.
- Am Morgen VCM von Platten sammeln und im 50 ml konischen Rohr auf Eis bei 4 °C lagern. Ersetzen Sie durch 5 ml frisches VCM.
- Nach 4 h VCM von platten sammeln und im 50 ml konischen Rohr bei 4°C auf Eis lagern. Ersetzen Sie durch 5 ml frisches VCM. Nach 4 h VCM von den Platten sammeln und in das konische 50-ml-Rohr geben.
- Aliquot sammelt sich nach der Filtration durch einen 0,45 μm Filter aus 50 mL Rohren in Kryotuben (2 mL pro Aliquot). Virale Aliquots bis zur Anwendung bei -80 °C lagern.
HINWEIS: Das Protokoll kann hier pausiert werden, und die viralen Aliquots können bis zur Gebrauchsfertigkeit gespeichert werden.
- Säen Sie eine 10 cm große Gewebekulturschale mit 3-5 x 106 HEK293-EBNA- oder HEK293T-Zellen für jedes Konstrukt, das für die virale Transduktion benötigt wird. Lassen Sie die Zellen über Nacht in Dulbeccos Modified Eagle Media (DMEM) haften, ergänzt mit 10% fetalem Rinderserum (FBS), Penicillin / Streptomycin / Glutamin (P / S / Q) und 0,3 mg / ml G418.
- Samenzellen in der entsprechenden Dichte in einer 10 cm gewebekulturellen Schale. Ziel 50% Konfluenz. Lassen Sie die Zellen über Nacht haften, indem Sie sie bei 37 °C mit 5% CO2 in den Inkubatorgeben.
HINWEIS: Für A673-Zellen sind dies 5 x 106 Zellen in 10 ml DMEM-Medien mit 10% FBS, P / S / Q-Supplementierung und 10 mM Natriumpyruvat. Diese Bedingungen können je nach Wachstumsrate der verwendeten Zellen variieren. - Dezimieren Sie den endogenen Faktor von Interesse. Wenn zellen das endogene Protein von Interesse nicht erschöpft haben müssen, fahren Sie mit Schritt 1.4 fort.
- Tauen Sie virales Aliquot zur Transduktion von shRNA oder CRISPR-Konstrukt auf, das auf das protein von Interesse abzielt. Gefrorene Aliquots in einem 37 °C warmen Wasserbad schnell auftauen.
- Zu jedem viralen Aliquot 2,5 μL 8 mg/ml Polybrenn geben und durch sanftes Pipettieren mischen. Entfernen Sie die Medien von den Zellplatten und geben Sie vorsichtig virales Aliquot auf die 10 cm große Platte, indem Sie entlang der Seite der Platte pipettieren. Rocken Sie die Platte, um die 2 ml viralen Aliquot zu verteilen.
- Bei 37 °C im Gewebekultur-Inkubator für 2 h inkubieren. Schaukeln Sie die Platte alle 30 Minuten, um zu verhindern, dass Bereiche der Platte austrocknen.
- Fügen Sie 5 ml DMEM-Medien mit 10% FBS, P / S / Q-Supplementierung und 10 mM Natriumpyruvat mit 5 μL 8 mg / ml Polybrenn hinzu. Lassen Sie die Zellen über Nacht inkubieren.
- Am Morgen Medien aus Zellen entfernen und Zellen in Medien übergehen, die mit einem Selektionsreagenz ergänzt werden. Wenn Sie Zellen passieren, säen Sie sie so aus, dass sie 48-72 h lang wachsen und 50% Konfluenz erreichen können.
HINWEIS: Für A673-Zellen mit pSRP-iEF-2 werden die Zellen in einem 1:5-Split ausgesät und für 72 h mit 2 μg/ml Puromycin ausgewählt.
- Transduce cDNA-Ausdruckskonstrukte.
- Überprüfen Sie die Zellen, um 50-70% Konfluenz zu bestätigen.
- Auftauen des viralen Aliquots(s) zur Transduktion von cDNA-Konstrukt(en) von Interesse. Gefrorene Aliquots in einem 37 °C warmen Wasserbad schnell auftauen. 2,5 μL 8 mg/ml Polybrenn zu jedem viralen Aliquot geben und durch vorsichtiges Pipettieren mischen.
- Entfernen Sie die Medien aus den plattierten Zellen und geben Sie vorsichtig virales Aliquot auf die 10 cm große Platte, indem Sie entlang der Seite der Platte pipettieren. Rocken Sie die Platte, um die 2 ml viralen Aliquot zu verteilen.
- Bei 37 °C im Gewebekultur-Inkubator für 2 h inkubieren. Schaukeln Sie die Platte alle 30 Minuten, um zu verhindern, dass Bereiche der Platte austrocknen.
- Fügen Sie 5 ml DMEM-Medien mit 10% FBS, P / S / Q-Supplementierung und 10 mM Natriumpyruvat mit 5 μL 8 mg / ml Polybrenn hinzu. Lassen Sie die Zellen über Nacht inkubieren.
- Am Morgen Medien aus Zellen entfernen und Zellen in Doppelselektionsmedien durchgehen. Wachsen und passieren Sie Zellen nach Bedarf für 7-10 Tage, um eine doppelte Selektion und Expression des cDNA-Konstrukts zu ermöglichen.
HINWEIS: Diese Aufteilung dieser Passage kann eine Optimierung für verschiedene Zelllinien erfordern. Für A673-Zellen mit pSRP-iEF-2 und einem pMSCV-Hygro-Konstrukt werden Zellen ohne Spaltung in 2 μg/ml Puromycin und 100 μg/ml Hygromycin übergeben.
2. Sammeln Sie Zellen, validieren Sie die Expression von Konstrukten und richten Sie korrelative phänotypische Assays ein
- Nach 7-10 Tagen doppelter Selektion sammeln Sie zellen in einem 15 ml konischen Rohr. Zählen Sie die gesammelten Zellen mit einem Hämozytometer. Aliquot sammelte Zellen für die RNA-Sequenzierung und zur Validierung der Expression von cDNA-Konstrukten.
HINWEIS: Richten Sie alle korrelativen phänotypischen Assays ein, die für die untersuchte Forschungsfrage erforderlich sind. Koloniebildende Assays sind ein Beispiel für einen korrelativen phänotypischen Assay, der hier verwendet wird.- Sammeln Sie zwischen 5 x 105 und 1 x 106 Zellen für die RNA-Sequenzierung und 2 x 106 Zellen für die Proteinextraktion. Pelletzellen durch Zentrifugation bei 1.000 x g bei 4 °C für 5 min und den Überstand entfernen.
- Waschen Sie das Pellet mit 1 ml kaltem PBS. Pellet durch Zentrifugation bei 1.000 x g bei 4 °C für 5 min und Überstand entfernen. Schockfrostpellets in flüssigem Stickstoff und Lagerung bei -80 °C.
- Richten Sie alle korrelativen Assays mit den verbleibenden Zellen ein.
HINWEIS: Das Protokoll kann hier mit gesammelten Proben pausiert werden, die im -80 °C Gefrierschrank aufbewahrt werden.
- Validieren Sie den Knockdown des interessierenden Proteins (falls verwendet) und die Expression des Konstruktpanels.
- Auftauen von Zellpellets zur Proteinextraktion auf Eis. Resuspendieren Sie Zellen in eiskaltem 500 μL Kernextraktionspuffer (20 mM HEPES pH 7,9, 140 mM NaCl, 10% Glycerin, 1,5 mM MgCl2,1 mM EDTA, 1 mM DTT, 1% IGEPAL) mit Proteaseinhibitor. Lassen Sie es für 5 min auf Eis sitzen.
- Pelletkerne durch Zentrifugation bei 1.000 x g bei 4 °C für 5 min und Überstand entfernen. Kerne in 500 μL eiskaltem Kernextraktionspuffer (20 mM HEPES pH 7,9, 140 mM NaCl, 10% Glycerin, 1,5 mMMgCl2,1 mM EDTA, 1 mM DTT, 1% IGEPAL) mit Proteaseinhibitor waschen.
- Pelletkerne durch Zentrifugation bei 1.000 x g bei 4 °C für 5 min und den Überstand entfernen. Resuspend-Kerne in 200 μL kaltem RIPA-Puffer mit Protease-Inhibitor (Anpassen des Volumens des RIPA-Puffers entsprechend der Pelletgröße). Lassen Sie es 45-60 Minuten auf Eis sitzen, mit kräftigem Wirbeln alle 15 Minuten.
- Pelletzellabfälle durch Zentrifugation bei 16.000 x g bei 4 °C für 45-60 min. Den Überstand aufbewahren und in ein frisches kaltes Röhrchen geben
- Bereiten Sie Proben für die SDS-PAGE-Elektrophorese vor, indem Sie 5-10 μg Protein mit 1x Ladepuffer für 5 min sieden. Führen Sie ein SDS-PAGE-Gel nach Bedarf für das gewünschte Protein aus.
- Transfer zu einer Nitrocellulose- oder PVDF-Membran nach Bedarf für das interessierende Protein. Blockieren und mit den entsprechenden primären und sekundären Antikörpern abtupfen, um den Knockdown des endogenen Proteins (falls verwendet) und die ektopische Expression des cDNA-Konstrukts zu bestätigen.
HINWEIS: Das Protokoll kann hier angehalten werden.
- RNA extrahieren. Bewerten Sie die Qualität und Quantität der RNA.
- Zellenpellets auf Eis auftauen. Extrahieren Sie die Gesamt-RNA mit einem Extraktionskit auf Basis von Siliziumdioxid-Spinsäulen gemäß den Anweisungen des Herstellers.
- Kurz gesagt, lysieren Sie die Zellen mit dem Lysepuffer aus dem Kit. Tragen Sie das Lysat entweder auf eine Silica-Spin-Säule mit einem kurzen Spin bei >13000 U / min für 30-60 Sekunden auf oder entfernen Sie gDNA, indem Sie das Lysat auf eine gDNA-Entfernungssäule mit einem kurzen Spin bei >13000 U / min für 30-60 Sekunden auftragen.
- Führen Sie einen DNA-Aufschluss an der Säule durch, wenn Lysat direkt auf eine Siliziumdioxid-Spinsäule aufgetragen wurde. Wenn Sie eine gDNA-Entfernungssäule verwenden, tragen Sie das Eluat auf eine Silica-Spin-Säule mit einem kurzen Spin bei >13000 U / min für 30-60 s auf.
- Waschen Sie RNA auf der Säule gemäß den Anweisungen des Herstellers. Elute RNA in 30 μL Elutionspuffer.
- Beurteilen Sie die RNA-Qualität und -Quantität mit einem Fluorometer oder einem anderen vergleichbaren Instrument. Stellen Sie sicher, dass das Verhältnis 260/280 nahe bei 2 liegt und dass mindestens 2,5 μg RNA zur Sequenzierung eingereicht werden müssen.
HINWEIS: Wenn Replikate gesammelt werden, muss jedes Replikat mit demselben RNA-Extraktionsprotokoll verarbeitet werden. - Verwenden Sie ein kleines Aliquot RNA, um den stabilen Knockdown des interessierenden Proteins zu bestätigen, falls erforderlich, durch qRT-PCR. Die restliche RNA-Probe wird bei -80 °C gelagert.
- Sammeln Sie biologische Replikate, indem Sie die Schritte 1-2 wiederholen, bis 3-4 vollständige RNA-Sätze gesammelt wurden. Stellen Sie sicher, dass jedes Replikat eine ausreichende Expression von cDNA-Konstrukten und einen stabilen Knockdown des endogenen Proteins (falls verwendet) aufweist.
3. Sequenzierung der nächsten Generation
- Senden Sie extrahierte RNA zur Sequenzierung unter Verwendung einer Next-Generation-Sequenzierungsplattform mit einem Ziel von 50 Millionen 150 Basenpaaren (bp) gepaarten Endlesevorgängen. Befolgen Sie die Anweisungen der Einrichtung, die die Proben verarbeitet. Wählen Sie für poly-adenylierte RNAs und strangspezifische Sequenzierung.
4. Alignment und Transcript Counting Pipeline
HINWEIS: Bei diesem Protokoll wird davon ausgegangen, dass nach der Übermittlung und Verarbeitung des Beispiels für jedes Beispiel ein Satz gepaarter FASTQ-Dateien zurückgegeben wird. Diese Dateien werden häufig mit dem Suffix "fastq.gz" komprimiert. Die weitere Analyse dieser FASTQ-Dateien erfordert den Zugriff auf eine HPC-Einrichtung (High Performance Computing), auf der ein Linux-Betriebssystem ausgeführt wird.
- Dateien übertragen
- Öffnen Sie ein Terminal für die HPC-Umgebung mit PuTTY. Erstellen Sie ein Verzeichnis für die Analyse namens "Projekt".
- Navigieren Sie zum Verzeichnis "path_to/project" und erstellen Sie ein neues Verzeichnis für die komprimierten rohen fastq.gz Dateien namens "fastq". Erstellen Sie auch ein Verzeichnis namens "getrimmt". Dies ist in Abbildung S1A-C dargestellt.
- Übertragen Sie die komprimierten rohen fastq.gz Dateien mit WinSCP oder einem ähnlichen Programm aus dem lokalen Speicher in das Verzeichnis "path_to/project/fastq/". Überprüfen Sie, ob für jedes Sample eine "R1"- und eine "R2"-Datei vorhanden ist, wie in Abbildung S1B dargestellt.
- Optional: Installieren Sie bei Bedarf TrimGalore. Legen Sie das Verzeichnis, das die ausführbare trim_galore datei enthält, in der Umgebungsvariablen PATH unter Linux fest.
HINWEIS: Lesevorgänge und Adapter von geringer Qualität sind mit TrimGalore beschnitten. TrimGalore ist bei https://github.com/FelixKrueger/TrimGalore erhältlich. - Optional: Navigieren Sie zum Verzeichnis für heruntergeladene Softwarepakete (z.B. "path_to/Software"). Laden Sie das neueste TrimGalore-Paket mit dem Befehl "curl -fsSL https://github.com/FelixKrueger/TrimGalore/archive/[version].tar.gz -o trim_galore-[version].tar.gz" herunter.
- Optional: Entpacken Sie die tar.gz Datei. Verwenden Sie den Befehl "tar -xvzf trim_galore-[version_number].tar.gz".
- Optional: Machen Sie TrimGalore ausführbar. Verwenden Sie den Befehl "chmod a+x path_to/software/TrimGalore-[version]/trim_galore". Stellen Sie sicher, dass sich dieses neue Verzeichnis im PATH befindet. Verwenden Sie den Befehl "export PATH=path_to/software/TrimGalore-[version]:$PATH".
- Navigieren Sie zu path_to/project/fastq/. Verwenden Sie TrimGalore, um die Lesevorgänge in niedriger Qualität aus den fastq.gz Dateien mit dem befehl in Abbildung S1C zu kürzen.
HINWEIS: Zusätzliche Flags für diesen Befehl können relevant sein und finden Sie hier: https://github.com/FelixKrueger/TrimGalore/blob/master/Docs/
Trim_Galore_User_Guide.md - Suchen Sie im Verzeichnis path_to/project/trimmed nach den zugeschnittenen fastq.gz Dateien. Stellen Sie sicher, dass sie sample1_R1_val_1.fq.gz und sample1_R2_val_2.fq heißen.gz
- Richten Sie beschnittene FASTQ-Dateien mit STAR aus und generieren Sie Transkriptzahlen.
HINWEIS: STAR ist verfügbar unter https://github.com/alexdobin/STAR)- Optional: Installieren Sie STAR Version 2.6 oder höher. Legen Sie die ausführbare STAR-Datei im Pfad fest.
- Optional: Navigieren Sie zum Verzeichnis für heruntergeladene Softwarepakete (z.B. "path_to/Software").
- Optional: Laden Sie das STAR-Paket mit dem Befehl "curl -SLO https://github.com/alexdobin/STAR/archive/[version].tar.gz" herunter. Entpacken Sie die tar.gz Datei.
- Optional: Verwenden Sie den Befehl "tar -xzf [version].tar.gz". Machen Sie STAR ausführbar. Verwenden Sie den Befehl "chmod a+x path_to/software/STAR-[version]/bin".
- Optional: Stellen Sie sicher, dass sich dieses neue Verzeichnis im Pfad befindet. Verwenden Sie den Befehl "export PATH=path_to/software/STAR-[version_number]/bin/linux_x86_64_static:$PATH".
HINWEIS: Das STAR-Handbuch finden Sie unter: (https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf). - Stellen Sie sicher, dass es einen Genomindex gibt, der mit STAR verwendet werden kann. Legen Sie dies in einem Verzeichnis ab, das vom Verzeichnis path_to/project/ getrennt ist. Wenn zuvor ein Index für frühere Experimente generiert wurde, verwenden Sie diesen. Alternativ können Sie einen entsprechenden vorgenerierten Index verwenden, falls hier verfügbar: http://refgenomes.databio.org/. Andernfalls erstellen Sie einen neuen Index mit dem Befehl "STAR--runMode genomeGenerate" unter Verwendung der Anweisungen im STAR-Handbuch.
HINWEIS: Für den Rest dieses Protokolls wird der Pfad zum STAR-Index als "path_to/STAR_index" bezeichnet. - Navigieren Sie zum Verzeichnis path_to/project/. Erstellen Sie ein neues Verzeichnis mit dem Namen "STAR_output", wie in Abbildung S1Dgezeigt.
- Navigieren Sie zum Verzeichnis path_to/project/trimmed/. Verwenden Sie den Befehl in Abbildung S1D, um STAR auszuführen, um die zugeschnittenen fastq.gz Dateien auszurichten.
HINWEIS: Dieser Schritt ist der rechenintensivste und es wird empfohlen, dies auf einem HPC-Cluster mit mehreren Threads (z. B. >16) durchzuführen, die für die Ausrichtung vorgesehen sind. Abhängig von der Anzahl der Stichproben und den verfügbaren Rechenressourcen kann dieser Schritt viele Stunden bis Tage dauern. - Die erforderliche Ausgabe für die nächsten Schritte, die die Anzahl pro Transkript enthalten, finden Sie an der folgenden Stelle: path_to/project/STAR_output/sampleN_ReadsPerGene.out.tab.
Hinweis: In der Datei ReadsPerGene.out.tab enthält Spalte 1 Informationen über das gezählte Feature. Spalte 2 enthält die Anzahl der unstrandeten Lesevorgänge, Spalte 3 die Anzahl der vorwärts gestrandeten Lesevorgänge und Spalte 4 die Anzahl der umgekehrten gestrandeten Lesevorgänge. Die ersten vier Zeilen dieser Datei enthalten Informationen zu den ausgerichteten Lesevorgängen, die nicht auf ein einzelnes Gen ausgerichtet waren. Dieses Protokoll erfordert die unstrandete Leseanzahl. - Verwenden Sie RStudio (vorzugsweise) oder R in der HPC-Umgebung, um die Daten aus Zeile 5 und darunter für die Spalten 1 und 2 für jedes Beispiel zu kompilieren. Setzen Sie das Arbeitsverzeichnis in R auf "project".
- Lesen Sie jede ReadsPerGene.out.tab-Datei mit dem Befehl in Abbildung S2A. Nehmen Sie für die erste Spalte nur die Zeichen vor dem "." in der Spalte "Ensembl-Gen-ID", um die nachgelagerte Verarbeitung zu vereinfachen.
- Kompilieren Sie die Anzahl aus allen Beispielen in einen Datenrahmen namens "totcts" mithilfe der Befehle in Abbildung S2B. Speichern Sie diese neue Tabelle mit rohen Zähldaten als tabulatorgetrennte .txt Datei, d.h. sample_counts.txt, falls gewünscht, mit dem Befehl "write.table".
HINWEIS: Die Reihenfolge der Ensembl-Gen-ID ist für jede ReadsPerGene.out.tab-Datei in allen Stichproben gleich.
5. Differentielle Expression und Downstream-Analyse
- Normalisieren Sie für Batch-Effekte zwischen Samples mit ComBat.
HINWEIS: Es gibt zwei mögliche Variablen, die Veränderungen in der Genexpression erklären, die erste ist das verwendete Konstrukt (d.h. die Probe) und die zweite sind externe Faktoren, die mit dem Durchgang von Zellen durch die Zeit (d.h. der Charge) verbunden sind. Ein Schritt zum Normalisieren von Proben für Batch-zu-Batch-Variationen mit dem R-Paket ComBat wird empfohlen.- Installieren Sie bei Bedarf und laden Sie die Bibliotheken für sva, DESeq2, AnnotationDBI, org. Hs.b.db, pheatmap, RColorBrewer, genefilter, Cairo, ggplot2, ggbiplot, rgl und reshape2 wie in Abbildung S2Cgezeigt. Verwenden Sie für die Installation den Befehl "install.packages" oder Bioconductor gemäß der Dokumentation für jedes Paket.
- Filtern Sie die Daten zunächst nur auf die Gene, die mindestens eine Zählung pro Lesevorgang haben. Speichern Sie diese neue Tabelle, um die Filterung zu kennzeichnen, wie in Abbildung S2D.
HINWEIS: Häufig haben viele Gene sehr niedrige oder keine Lesezahlen. - Bereiten Sie eine zweite Tabelle für die Batchnormalisierung mit dem Namen "vars" vor, wie in Abbildung S2E gezeigt. Legen Sie die Zeilennamen auf die eindeutigen Namen der einzelnen Beispiele fest. Legen Sie die Spaltennamen auf "sample", "batch" und "construct" fest.
- Weisen Sie allen Proben eine eindeutige Nummer in der Spalte "Probe" von 1 bis n zu, wobei n die Anzahl der Proben ist. Weisen Sie allen Proben in der Spalte "Charge" Chargennummern zu, sodass sowohl condition-a_1 als auch condition-b_1 1 und condition-a_2 und condition-b_2 2 zugewiesen werden. Weisen Sie allen Stichproben in der Spalte "Konstrukt" alle Bedingungsbezeichnungen zu, sodass Bedingung a alle "A" und Bedingungsproben b alle "B" sind.
- Definieren Sie auch die Batchvariable und eine bestimmte Nullmodellmatrix für ComBat, wie in Abbildung S2Fgezeigt. Führen Sie ComBat mit dem in Abbildung S2F definiertenBefehl aus.
- Kuratieren Sie die Daten weiter, indem Sie auf die nächste ganze Zahl runden. Entfernen Sie auch Gene mit einem negativen Wert. Verwenden Sie die in Abbildung S3Agezeigten Befehle.
HINWEIS: Die Ausgabe der Batch-Normalisierung hat nicht-ganzzahlige Lesezahlen und einige Gene mit negativen Werten. Dieser Schritt ist erforderlich, da die downstreamielle Differentialausdrucksanalyse keine negativen Lesevorgänge unterstützt. - Definieren Sie das differenzielle Ausdrucksprofil für jedes Konstrukt mit DESeq2.
- Geben Sie den Versuchsentwurf für DESeq2 ein, wie in Abbildung S3B gezeigt. Erstellen Sie ein DESeqDataSet (dds) mit der DESeqDataSetFromMatrix-Funktion, schätzen Sie die Größenfaktoren, und führen Sie DESEq2 aus, wie in Abbildung S3Bgezeigt.
HINWEIS: Es ist zwingend erforderlich, dass die für "Bedingung" eingegebenen Spaltendaten in der gleichen Reihenfolge wie die Spalte in der Zählmatrix vorliegen. - Um die Qualität der Analyse zu bewerten, extrahieren Sie die von DESeq2 verwendeten rlog-normalisierten Zählungen, wie in Abbildung S3B dargestellt.
HINWEIS: Während der Analyse transformiert DESeq2 die Zählungen mit einem "regularisierten Log", rlog, Transformation, um die Proben-zu-Probe-Unterschiede für Gene mit niedriger Anzahl (geringe Information) zu schrumpfen, um Unterschiede in Genen mit höheren Anzahlen über Proben hinweg (hohe Information) zu erhalten. - Wenn Sie die Ergebnisse für jedes Transkriptionsprofil aus den Ergebnissen von DESeq2 extrahieren, führen Sie paarweise Vergleiche in Bezug auf die Knockdown-Bedingung oder den leeren Basislinienvektor durch, wie in Abbildung S3Cgezeigt. Ergänzen Sie diese Ergebnisse mit den HGNC-Gensymbolen, wie in Abbildung S3D gezeigt.
- Extrahieren Sie in Abbildung S3EDaten aus den DESeq2-Ergebnissen. Exportieren Sie als einzelne Datei mit der Ensembl-Gen-ID, dem HGNC-Symbol, der Basismittelwertexpression und differenziellen Expressionsdaten für alle Konstrukte mit log2FoldChange und rohen und angepassten p-Werten.
HINWEIS: Die Verwendung eines angepassten p-Wertes < 0,05 ist der empfohlene Grenzwert für den Differentialausdruck. - Bewerten Sie die erfolgreiche Batch-Normalisierung und die Ähnlichkeit innerhalb der Stichprobe. Überprüfen Sie das Sample Clustering mit PCA und Sample-to-Sample-Abstandsdiagrammen unter Verwendung der rlog-normalisierten Zählungen unter Verwendung des in den Abbildungen S4A-B gezeigtenCodes.
- Geben Sie den Versuchsentwurf für DESeq2 ein, wie in Abbildung S3B gezeigt. Erstellen Sie ein DESeqDataSet (dds) mit der DESeqDataSetFromMatrix-Funktion, schätzen Sie die Größenfaktoren, und führen Sie DESEq2 aus, wie in Abbildung S3Bgezeigt.
- Verwenden Sie die differenziellen Ausdrucksprofile, um Vulkandiagramme mithilfe des Codes in Abbildung S4C zu generieren. Bewerten Sie Veränderungen in der Genexpression über Konstrukte hinweg.
- Verwenden Sie die normalisierten Rlog-Zählungen und die hierarchische Clusterbildung, um Gensignaturen zu identifizieren, die für die verschiedenen Konstrukte eindeutig sind. Verwenden Sie den in Abbildung S4Dgezeigten Code.
- Extrahieren Sie die 1000 variabelsten Gene über alle Konstrukte hinweg in einer Matrix. Verwenden Sie pheatmap, um eine unbeaufsichtigte hierarchische Clusterung Ihrer Proben basierend auf diesen Genen durchzuführen.
- Extrahieren Sie die interessierenden Cluster aus dem Dendrogramm, indem Sie entscheiden, auf welcher Ebene der dendrogrammcluster von Interesse erscheinen. Setzen Sie "k" gleich der Anzahl der Cluster auf dieser Ebene. Stellen Sie die Heatmap nach Cluster neu, um zu bestimmen, welche Cluster von Interesse sind, wie in Abbildung S5 gezeigt.
- Exportieren Sie die Liste der Gene, die jedem Cluster zugeordnet sind, wie in Tabelle S1 gezeigt. Verwenden Sie diese Informationen, um die Gene in Clustern von Interesse zu bestimmen.
- Identifizieren Sie die biologischen Rollen für verschiedene identifizierte Cluster von Genen und vergleichen Sie zwischen den Klassen. Dies kann mit einer Vielzahl von bioinformatischen Werkzeugen durchgeführt werden. ToppGene24 kommt hier zum Einsatz und ist online frei verfügbar.
HINWEIS: Es gibt viele kostenlose Tools, die nur eine Liste von Genen benötigen, um sie zu kopieren und in ein Feld auf einer Website einzufügen. Wählen Sie die Analysewerkzeuge, die für die untersuchten Forschungsfragen am besten geeignet sind. - Wenn Daten über die genomische Bindung verfügbar sind, die die Transkriptionsausgabe für den Transkriptionsfaktor von Interesse antreibt, vergleichen Sie optional die Transkriptionsantwort an Genen, die mit verschiedenen Bindungselementen assoziiert sind, um die Mutantenfunktion weiter zu bewerten.
6. Vergleich mit relevanten Phänotypen
- Vergleichen Sie die korrelativen Phänotypen mit den generierten transkriptomischen Profildaten und interpretieren Sie sie entsprechend.
Ergebnisse
Vorläufige qRT-PCR-Daten deuteten darauf hin, dass eine EWS/FLI-Mutante namens DAF mit spezifischen Tyrosin-zu-Alanin-Mutationen in der sich wiederholenden und ungeordneten Region des EWS die Fähigkeit zur Aktivierung von EWS/FLI-Zielgenen aufrechterhielt, aber kritische Zielgene nicht unterdrückenkonnte 23. Um die Beziehung zwischen diesen Resten im EWS-Bereich und der EWS/FLI-Funktion besser zu verstehen, wurde das oben beschriebene und in Abbildung 1 dargestellte Protokoll verwendet. A673 Ewing-Sarkomzellen wurden viral mit einer shRNA transduziert, die auf die 3'UTR von FLI1abzielt, was zur Erschöpfung des endogenen EWS/FLI führte. Nach vier Tagen der Selektion wurde die EWS/FLI-Funktion mit viraler Transduktion verschiedener 3XFLAG-markierter EWS/FLI-Mutantenkonstrukte gerettet, mit leerem Vektor als Kontrolle für keine Rettung. Eine nicht-funktionelle Mutante, der die EWS-Domäne fehlt, genannt Δ22, wurde als Negativkontrolle verwendet und Wildtyp-EWS/FLI, genannt wtEF, wurde als Positivkontrolle verwendet (Abbildung 2A). DAF wurde als Testkonstrukt verwendet, obwohl bei Bedarf mehr als ein Testkonstrukt verwendet werden kann. Die Zellen wurden für weitere 10 Tage ausgewählt, um die Konstruktexpression zu stabilisieren, und dann für RNA (mit einem gDNA-Entfernungsschritt), Protein- und Koloniebildungsassays gesammelt. Vier Replikate wurden gesammelt und repräsentative qRT-PCR und Western Blots, die einen effektiven Knockdown und eine effektive Rettung zeigen,sind in Abbildung 2B-Ddargestellt. Es sollte beachtet werden, dass DAF-gerettete Zellen keine Kolonien bilden konnten, wie in Abbildung 2Egezeigt, was auf eine beeinträchtigte onkogene Transformation hindeutet.
Nach Abschluss der Replikationsvalidierung und der phänotypischen Assays wurde die RNA beim Institut für Genomische Medizin am Nationwide Children's Hospital zur Bibliotheksvorbereitung und Next-Generation-Sequenzierung mit ~ 50 Millionen 150-bp-Paired-End-Lesevorgängen eingereicht. Die Daten wurden als fastq.gz-Dateien zurückgegeben. Low-Quality-Reads wurden mit TrimGalore aus diesen Dateien abgeschnitten und STAR wurde verwendet, um Reads auf das menschliche Genom hg19 auszurichten und die Reads pro Gen zu zählen. hg19 wurde aus Gründen der Kompatibilität mit den anderen kuratierten Datensätzen für EWS/FLI verwendet, die in der downstreamn Analyse verwendet wurden. Diese Lesezählungen wurden zu einer einzigen Zählmatrix für alle Stichproben zusammengefasst, von denen die ersten 6 Zeilen in Abbildung 3 dargestelltsind.
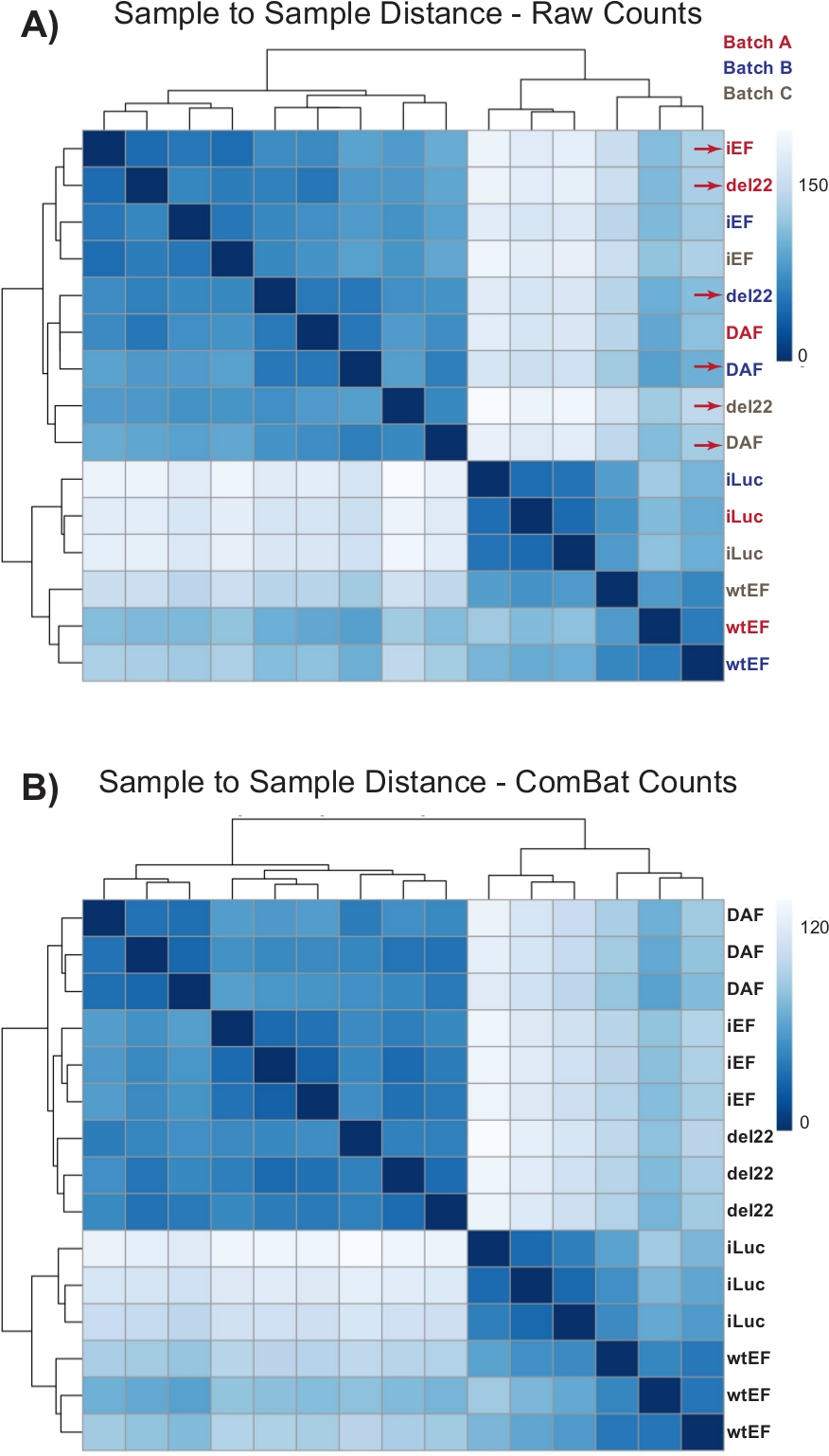
Die Zählungen wurden zunächst ohne Batch-Normalisierung durch DESeq2 durchgeführt, jedoch zeigte die visuelle Inspektion des Proben-zu-Probe-Abstands potenzielle verwirrende Batch-Effekte, wie in Abbildung 4Amit roten Pfeilen hervorgehoben. Dies ist wahrscheinlich auf die biologische Variabilität zurückzuführen, die durch die Passage von Zellen in Kultur und Unterschiede in der Verarbeitung jeder Charge eingeführt wurde. Die Normalisierung für Batch-Effekte wurde mit ComBat durchgeführt und wird im Allgemeinen empfohlen. Die Sample-to-Sample-Abstände der batch-normalisierten Daten sind in Abbildung 4B dargestellt. Nach der Batch-Normalisierung wurde DESeq2 verwendet, um Transkriptionsprofile für die drei Konstrukte (wtEF, Δ22 und DAF) relativ zur Baseline zu generieren. Beachten Sie, dass, während "elterliche" A673-Zellen (Mock-Knockdown und Mock-Rettung, hier "iLuc" genannt) in die Differentialanalyse einbezogen wurden, die Referenz für dieses Experiment die Zellen mit EWS / FLI-depleted sind, die als iEF-Zellen bezeichnet werden. Das Transkriptionsprofil kann hier für das endogene Protein generiert werden, indem die iLuc-Probe mit iEF verglichen wird, und dies kann für das Verständnis der Funktionsweise des Rettungssystems von Interesse sein, aber das ist nicht das Ziel dieser speziellen Analyse. Die für die Mutanten generierten Transkriptionsprofile umfassen positive (wtEF) und negative (Δ22) Kontrollen in Bezug auf iEF, so dass diese als Benchmarks für andere Mutanten fungieren sollten. Dies ist wichtig, da die Positivkontrolle in diesem Beispiel die Funktion des endogenen EWS/FLI nicht vollständig rekapituliert hat, wie an anderer Stellediskutiert 7,23.
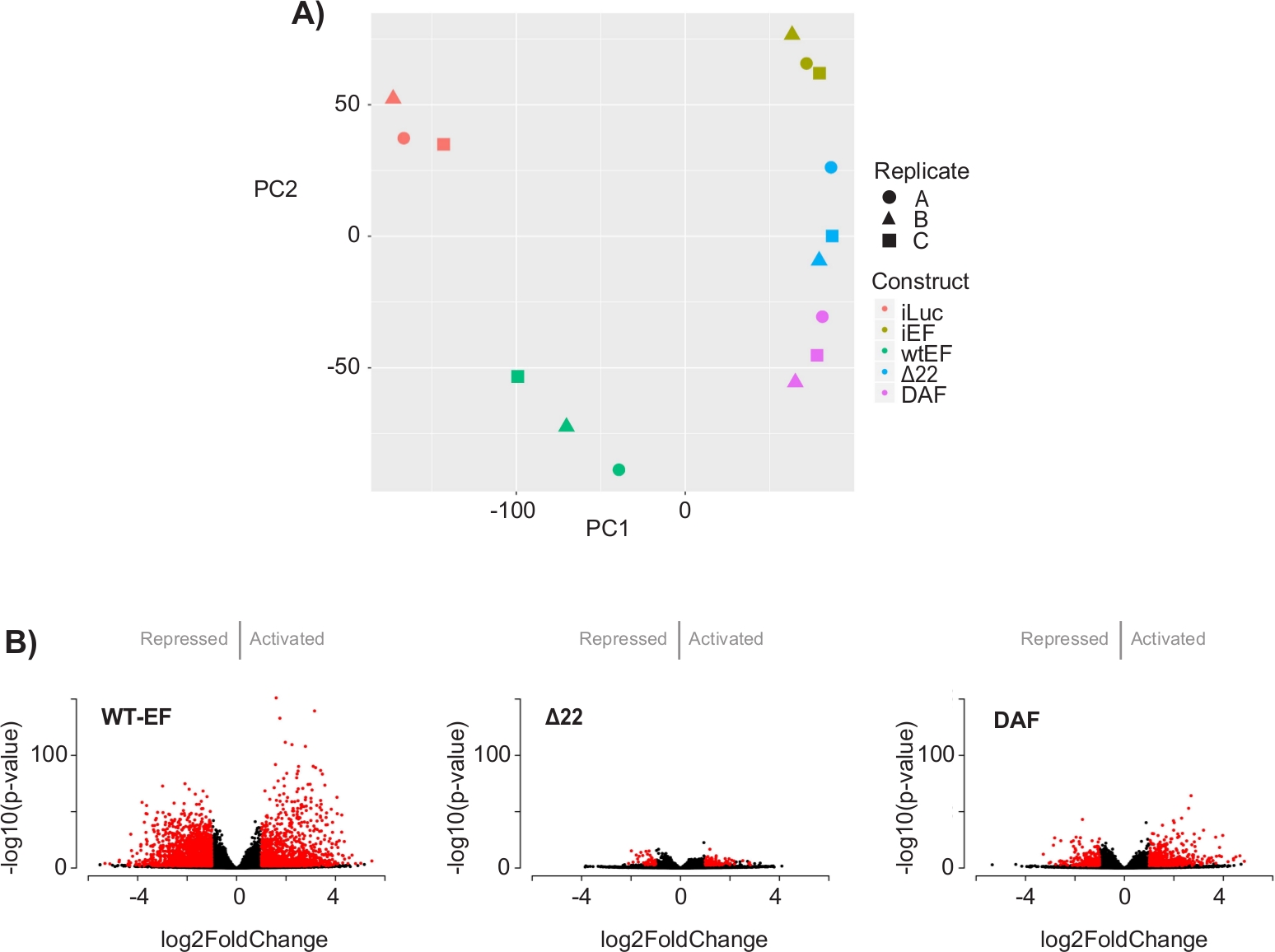
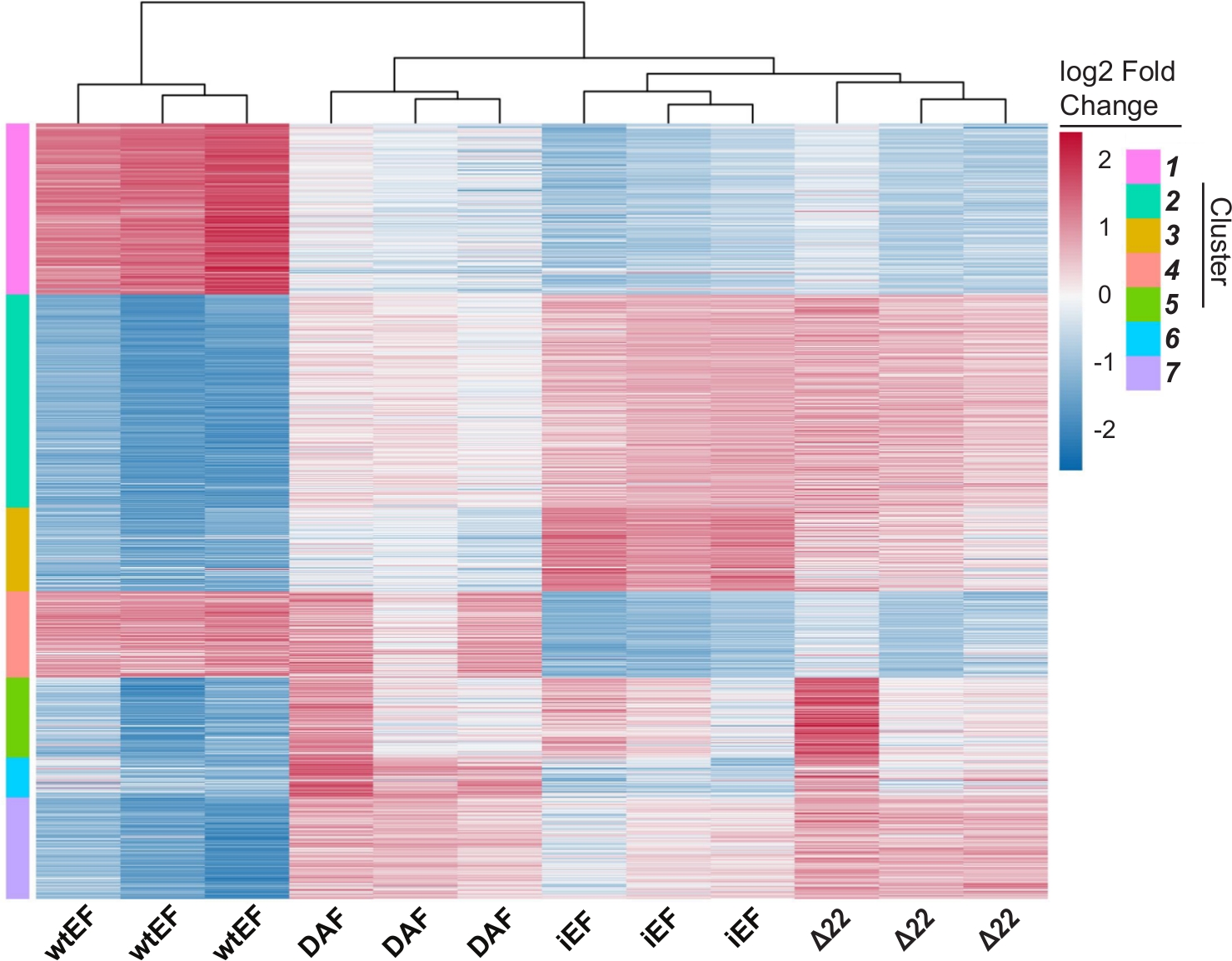
Die Hauptkomponentenanalyse (PCA) in Abbildung 5 legt nahe, dass das Transkriptionsprofil von DAF zwischen wtEF und Δ22 liegt, was die Teilfunktion bestätigt. Darüber hinaus zeigte die hierarchische Clusterung der 1000 unterschiedlichsten Gene über Proben hinweg, dass DAF die EWS/FLI-Zielgene nicht unterdrückte und die Genaktivierungsaktivität nur teilweise beibehielt, wie in Abbildung 6A und Abbildung S5gezeigt. Die ToppGene-Analyse deutete darauf hin, dass sich die Klassen von Genen, die DAF aktiviert, funktionell von jenen EWS/FLI-aktivierten Zielen unterscheiden, bei denen DAF nicht funktionsfähig ist (Abbildung 6B). Interessanterweise scheint die Funktion aktivierter Gene, die durch wtEF, aber nicht durch DAF gerettet wurden, mit der Transkriptionskontrolle und der Chromatinregulation zusammenzuhängen. Basierend auf den Ergebnissen der Koloniebildungsassays sollten die Gene aus dieser Kerngensignatur weiter auf ihre Rolle in der EWS/FLI-vermittelten Onkogenese analysiert werden. Die Bedeutung der EWS/FLI-vermittelten Genrepression wurde bereits beschrieben17.
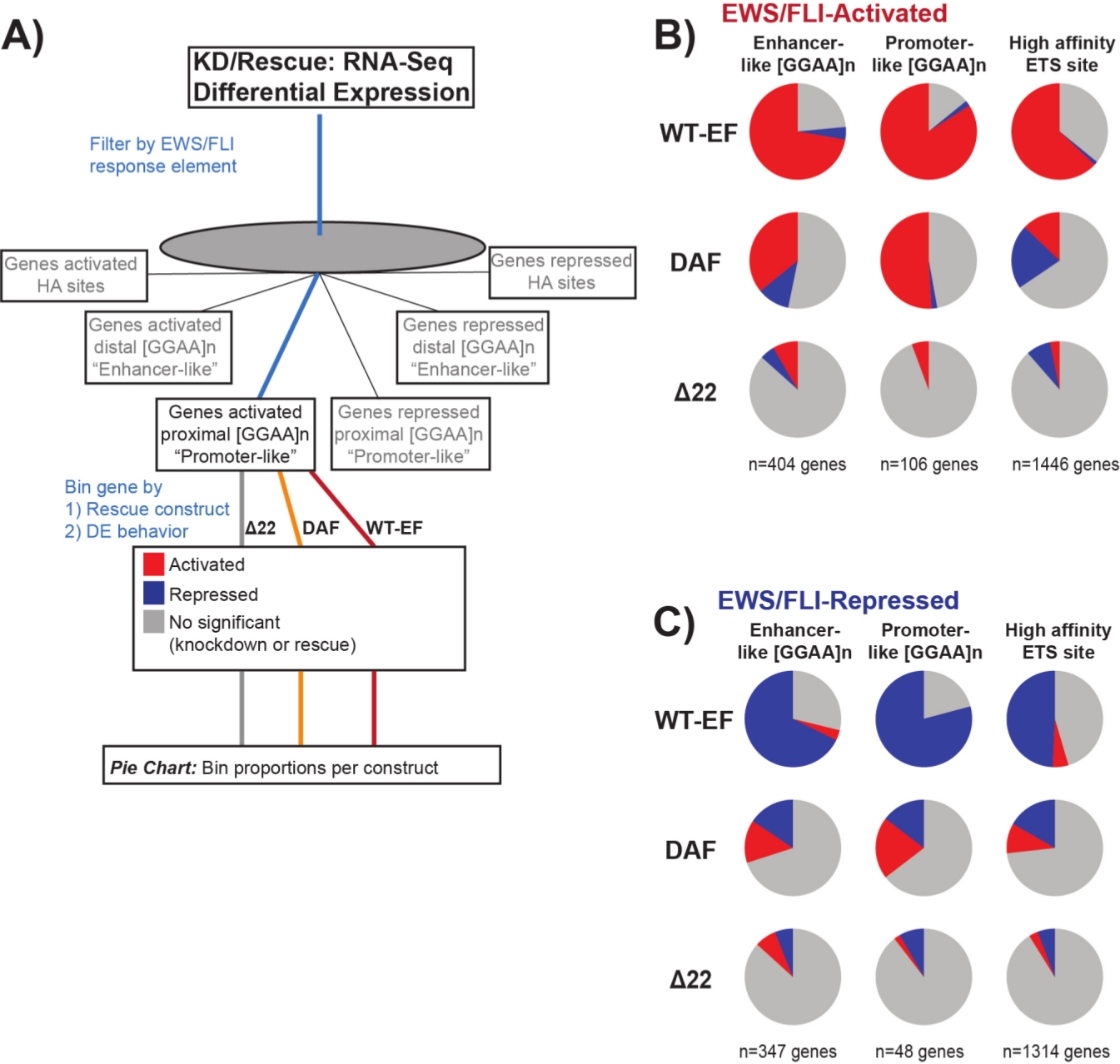
Es ist bekannt, dass EWS/FLI eine einzigartige Bindungsaffinität für GGAA-Mikrosatelliten-Wiederholungselemente19,22besitzt und dass die Bindung an diesen Elementen die nachgeschaltete Genregulation11,15,18,20,22steuert. Diese Mikrosatelliten wurden entweder als mit Aktivierung oder Repression assoziiert und entweder proximal zu (< 5 kb) TSS oder distal zu (> 5 kb) TSS25charakterisiert. Darüber hinaus gibt es EWS/FLI-regulierte Gene mit hochaffinen (HA) ETS-Motiven in der Nähe von TSS23. Um die Eigenschaften der DAF-Funktion weiter zu analysieren und welche Arten von EWS/FLI-aktivierten Genen DAF retten konnte, wurde die differentielle Expression von Genen analysiert, die mit diesen verschiedenen Klassen assoziiert sind. Interessanterweise war DAF am ehesten in der Lage, GGAA-Mikrosatelliten-aktivierte Gene zu retten, aber nicht in der Lage, aktivierte Gene in der Nähe einer HA-Stelle zu retten, wie in Abbildung 7 zu sehen ist. Wie beim hierarchischen Clustering zu sehen ist, gelingt es DAF nicht, die EWS/FLI-vermittelte Repression über Motivklassen hinweg zu retten. Diese Daten deuten darauf hin, dass DAF genügend strukturelle Merkmale von EWS beibehält, um an GGAA-Mikrosatelliten zu binden und von ihnen zu aktivieren, sowohl proximal als auch distal zu TSS. Dies ergibt sich wahrscheinlich aus der intakten SYGQ-Domäne, die als wichtig für die EWS /FLI-Aktivität bei GGAA-Wiederholungen angesehen wird11. Diese Daten deuten auch darauf hin, dass die spezifischen Tyrosine, die in DAF mutiert sind, eine wichtige, aber wenig verstandene Rolle bei der EWS/FLI-vermittelten Genregulation von HA-Standorten sowie bei der Genrepression spielen, was einen wichtigen Bereich weiterer Untersuchungen hervorhebt.

Abbildung 1: Workflow. Darstellung des schrittweisen Vorgehens zur Durchführung eines Struktur-Funktions-Mappings durch Transkriptomics. Zellen wurden zuerst vorbereitet, um die Reihe von Konstrukten auszudrücken, die für die Struktur-Funktions-Abbildung erforderlich sind. Nach der Expression wurden Zellen für RNA und Protein geerntet und auf korrelative Phänotypen untersucht. Die Expression der Konstrukte wurde validiert, und dieser Prozess wurde 3-4 Mal wiederholt, um unabhängige biologische Replikate zu sammeln. RNA wurde dann für die Next-Generation-Sequenzierung (NGS) eingereicht. Wenn Daten empfangen wurden, wurden die Daten auf Qualität getrimmt, ausgerichtet und die Anzahl pro Transkript berechnet. Batch-Effekte wurden kontrolliert und transkriptomische Signaturen und differentielle Expression wurden mit DESeq2 bestimmt. Hierarchisches Clustering und Downstream-Analyse, die andere -omics-Datensätze und verschiedene Pathway- oder Funktionsanalysen integrieren, können integriert werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Validierung von Konstruktexpression und korrelativen Assays. (A) Schematische Darstellung der in diesem Beispiel getesteten Konstrukte. (B) Validierung des Knockdowns von endogenem EWS/FLI und Expression von 3X-FLAG-markierten Konstrukten durch Immunoblot. (C,D) Validierung der Konstruktaktivität an einem EWS/FLI (C) aktivierten Zielgen, NR0B1, und (D) unterdrücktem Zielgen, TGFBR2, durch qRT-PCR. Die Daten werden als mittlere +/- Standardabweichung dargestellt. P-Werte wurden mit einem tukey's honest significance test berechnet. * p < 0,05, ** p < 0,01, *** p < 0,005 (E) Koloniezählungen aus Soft-Agar-Assays, die zur Beurteilung der Transformationsaktivität von Konstrukten durchgeführt wurden. P-Werte wurden mit einem tukey's honest significance test berechnet. * S. < 0,05, ** S. < 0,01, *** S. < 0,005. Diese Abbildung stammt aus Theisen, et al.23Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Endgültige zusammengestellte Zähldaten für die Analyse. Screenshot der ersten 6 Zeilen der Zähldatei mit Genzählungen für alle Proben, die batch normalisiert und analysiert werden sollen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Heatmaps der Sample-to-Sample-Entfernung. (A) Stichproben-zu-Stichproben-Entfernungsdiagramm, das die Stichproben-Clusterung der Rohzählungsdaten zeigt. Proben, die sowohl nach Batch als auch nach Stichprobe gruppiert sind, sind mit roten Pfeilen gekennzeichnet. (B) Stichproben-zu-Stichproben-Abstandsdiagramm nach Batch-Normalisierung mit ComBat. Hier gruppieren sich Samples aus allen Replikaten, unabhängig vom Batch. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5: Ergebnisse der Differentialexpressionsanalyse. (A) Das Diagramm der Prinzipkomponentenanalyse (PCA) der transkriptomischen Signaturen, die für alle Proben generiert wurden, zeigt eine starke Intra-Stichproben-Clusterbildung und zeigt, dass DAF zwischen den positiven (wtEF) und negativen (Δ22) Kontrollen intermediär ist. (B) Vulkandiagramme, die den -log(p-Wert) zeigen, der gegen den log2FoldChange für Gene in jedem Konstrukt aufgetragen wird. Gene mit einem angepassten p-Wert < 0,05 und einem |log2(FoldChange)| > 1 gelten als signifikant und sind rot dargestellt. Panel 5B ist adaptiert von Theisen, et al.23Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 6: Hierarchisches Clustering zur Identifizierung von Genklassen. (A) Hierarchische Clusterbildung der 1000 am meisten variablen Gene über alle Konstrukte hinweg und die Baseline, iEF, zeigt, dass DAF die EWS/FLI-vermittelte Genaktivierung teilweise rettet. (B) Genontologie (molekulare Funktion) resultiert aus ToppGene, die die funktionelle Anreicherung von EWS/FLI-aktivierten Genen zeigen, die entweder durch DAF gerettet oder nicht gerettet werden. Panel 6B ist adaptiert von Theisen, et al.23Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 7: Detaillierte Analyse verschiedener Transkriptionsfaktor-Antwortelemente auf verschiedene Konstrukte: (A) Schematische Darstellung der Datenverarbeitung, die zur Generierung von Panels (B) und (C) verwendet wird, indem andere verfügbare Datensätze mit den transkriptomischen Profilen hier integriert werden. (B,C) Zusammenstellung, die die Rettung verschiedener Klassen von direkten EWS/FLI- (B) aktivierten und (C) unterdrückten Zielen zeigt. Eingeschlossen waren nur gene mit nachweisbarer differentieller Expression durch endogenes EWS/FLI. In jedem Kreisdiagramm zeigt Grau den Teil der Gene an, die nicht durch das Konstrukt gerettet werden. Rot stellt den Anteil der Gene dar, die differentiell aktiviert sind, und Blau stellt den Anteil der Gene dar, die differentiell unterdrückt werden. Diese Abbildung stammt aus Theisen, et al.23Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Abbildung S1: Laden der fastq.gz-Dateien in die HPC-Umgebung, Trimmen und Ausrichten. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
{kind=link}
Abbildung S2: Sortieren von Lesezahlen über Stichproben hinweg und Ausführen der Batchnormalisierung mit ComBat. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
{kind=link}
Abbildung S3: Ausführen von DESeq2 und Extrahieren der Ergebnisse der Differentialexpressionsanalyse. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
{kind=link}
Abbildung S4: Analyse der Ausgabe. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
{kind=link}
Abbildung S5: Hierarchisches Clustering zur Identifizierung von Genklassen: Hierarchisches Clustering der Top 1000 der meisten variablen Gene über alle Konstrukte hinweg und die Baseline, iEF, sortiert in k Cluster. In diesem Fall ist k=7, aber dieser Parameter wird vom Benutzer festgelegt, wie in Abbildung S4Dgezeigt. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
{kind=link}
Tabelle S1: Liste der Gene (Ensembl-Gen-ID) mit Clusterannotation. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Diskussion
Die Untersuchung der biochemischen Mechanismen onkogener Transkriptionsfaktoren ist von entscheidender Bedeutung, um die von ihnen verursachten Krankheiten zu verstehen und neue therapeutische Strategien zu entwickeln. Dies gilt insbesondere für Malignome, die durch chromosomale Translokationen gekennzeichnet sind, die zu Fusionstranskriptionsfaktoren führen. Den domänen, die in diesen chimären Proteinen enthalten sind, könnten sinnvolle Wechselwirkungen mit regulatorischen Domänen in den Wildtypproteinen fehlen, was die Fähigkeit erschwert, Strukturfunktionsinformationen im Kontext der Fusion zu interpretieren26,27,28. Darüber hinaus sind viele dieser onkogenen Fusionen durch intrinsisch ungeordnete Domänen mit geringer Komplexität gekennzeichnet10,13,29,30.
Die EWS-Domäne ist ein Beispiel für eine solche intrinsisch ungeordnete Domäne, die an einer Vielzahl von onkogenen Fusionen beteiligt ist10. Die intrinsisch ungeordnete und sich wiederholende Natur hat die Bemühungen behindert, die molekularen Mechanismen der EWS-Domäne zu verstehen. Frühere Bemühungen, die Strukturfunktion zu untersuchen, haben weitgehend auf die Verwendung verschiedener Mutanten im Kontext von Reportergen-Assays oder in Zellhintergründen zurückgegriffen, die den relevanten zellulären Kontext nicht rekapitulieren oder keine strukturellen Variationen aufweisen, die eine sinnvolle Teilfunktion erzeugen11,17,25. Die hier vorgestellte Methode befasst sich mit diesen Problemen. Das Struktur-Funktions-Mapping wird in einem krankheitsrelevanten Zellkontext durchgeführt, und die Sequenzierung der nächsten Generation ermöglicht die transkriptomische Profilierung, um die Transkriptionsfaktorfunktion im Setting des nativen Chromatins zu bewerten. Im speziellen Fall der DAF-Mutante von EWS/FLI wurde berichtet, dass DAF in Reporterassays mit isolierten Antwortelementen wenig Aktivität zeigte, aber Aktivität im Kontext des vollständigen Genpromotors zeigte, entweder in einem Reporter-Assay oder in nativem Chromatin, was auf einen interessanten Phänotyp23hindeutet. Die Verwendung der hier beschriebenen Methode löst direkter die Frage, welche Art von regulatorischen Elementen im gesamten Genom im Krankheitsumfeld am besten anspricht. Durch das gleichzeitige Testen aller Kandidaten-Zielgene in ihrem nativen Chromatinkontext ist es wahrscheinlicher, dass ein transkriptomischer Ansatz Konstrukte mit Partieller Funktion identifiziert.
Die inhärente Stärke der Verwendung eines krankheitsrelevanten Zellhintergrunds ist vielleicht die größte Einschränkung dieser Technik. Einer der wichtigsten Faktoren ist die Auswahl des geeigneten Zellsystems für diese Experimente. Viele Zelllinien, die von Malignomen mit pathognomonischen Transkriptionsfaktoren abgeleitet sind, tolerieren den Knockdown dieses Transkriptionsfaktors nicht ohne weiteres, und in vielen Fällen, insbesondere bei pädiatrischen Krebsarten, bleibt die wahre Ursprungszelle umstritten und die Expression des Onkogens in anderen Zellhintergründen ist unerschwinglich toxisch31,32 . In diesen Fällen kann es hilfreich sein, Experimente in einem anderen Zellhintergrund durchzuführen, solange der Forscher bei der Interpretation der Ergebnisse Vorsicht walten lässt und relevante Befunde in einem krankheitsrelevanteren Zelltyp entsprechend validiert.
Es ist von entscheidender Bedeutung, die Stabilität und die phänotypischen Folgen der Expression des Onkogens sorgfältig zu validieren und nur Proben zur Sequenzierung einzureichen, die strenge Kriterien erfüllen. Dazu gehörten Western Blot zur Bestätigung von Knockdown und Rettung sowie qRT-PCR einer kleinen Anzahl bekannter Zielgene zur Validierung der Positivkontrolle (Abbildung 2). Ebenso ist es wichtig, die Chargenvariabilität so gering wie möglich zu halten, indem die Zell- und RNA-Präparationen in jeder Charge so ähnlich wie möglich durchgeführt werden.
Die hier beschriebene Methode wird besonders leistungsfähig, wenn sie mit anderen Arten von genomischen Daten gepaart wird, die auf die genomweite Funktion des untersuchten Transkriptionsfaktors hinweisen. Zukünftige Richtungen für diese Art der Struktur-Funktions-Analyse würden sich um ChIP-seq und ATAC-seq erweitern, um die Bindung des Transkriptionsfaktors und alle induzierten Veränderungen der Chromatin-Zugänglichkeit zu bestimmen. Als Suite kann diese Art von Daten darauf hinweisen, wo verschiedene strukturelle Komponenten eines onkogenen Transkriptionsfaktors zu verschiedenen Aspekten der Funktion beitragen (z. B. DNA-Bindung vs. Chromatinmodifikation vs. Co-Regulator-Rekrutierung). Insgesamt kann die Verwendung von NGS-basierten Ansätzen zur Abbildung der Struktur-Funktions-Beziehungen von Fusionstranskriptionsfaktoren neue Erkenntnisse über die biochemischen Determinanten der onkogenen Funktion dieser Proteine liefern. Dies ist wichtig, um das Verständnis der von ihnen verursachten Krankheiten zu fördern und die Entwicklung neuer therapeutischer Strategien zu ermöglichen.
Offenlegungen
SLL erklärt einen Interessenkonflikt als Mitglied des Beirats und Anteilseigner von Salarius Pharmaceuticals. SLL ist auch ein gelisteter Erfinder auf United States Patents No. US 7,393,253 B2, "Methods and compositions for the diagnosis and treatment of Ewing's sarcoma", und US 8,557,532, "Diagnosis and treatment of drug-resistant Ewing's sarcoma". Dies ändert nichts an unserer Einhaltung der JoVE-Richtlinien zur Weitergabe von Daten und Materialien.
Danksagungen
Diese Forschung wurde von der High Performance Computing Facility am Abigail Wexner Research Institute am Nationwide Children's Hospital unterstützt. Diese Arbeit wurde von den National Institutes of Health National Cancer Institute [U54 CA231641 zu SLL, R01 CA183776 zu SLL] unterstützt; Alex's Lemonade Stand Foundation [Young Investigator Award an ERT]; Pelotonia [Stipendium für ERT]; und das National Health and Medical Research Council CJ Martin Overseas Biomedical Fellowship [APP1111032 an KIP].
Materialien
| Name | Company | Catalog Number | Comments |
| Wet Lab Reagents | |||
| anti-FLI rabbit pAb | Abcam | ab15289 | 1:500 |
| anti-lamin B1 rabbit pAb | Abcam | ab16048 | 1:2000 |
| Cell-based system for introduction of mutant constructs | Determined by cell system used | ||
| Cryotubes | For viral aliquots | ||
| DMEM | Corning Cellgro | 10-013-CV | For viral production |
| Fetal bovine serum | Gibco | 16000-044 | For viral production |
| G418 | ThermoFisher | 10131027 | For viral production |
| HEK293-EBNAs | ATCC | CRL-10852 | For viral production |
| HEPES | Gibco | 15630106 | |
| Hygromycin B | ThermoFisher | 10687010 | |
| M2 anti-FLAG mouse mAb | Sigma | F3165 | 1:2000 |
| Near IR-secondary antibodies | Li-Cor | ||
| Optimem | Gibco | 31985062 | For viral production |
| Penicillin/Streptomycin/Glutamine | Gibco | 10378-016 | For viral production |
| Polybrene | Sigma | TR-1003-G | For viral transduction |
| Puromycin | Sigma | P8833 | Stored at 2 mg/mL stock |
| RNeasy Plus kit | Qiagen | 74136 | Has gDNA removal columns |
| Selection reagents | As dictated by cell system used | ||
| Sodium Pyruvate | Gibco | 11360-070 | For viral production |
| Tissue culture media | Determined by cell system used | ||
| TransIT-LT1 | Mirus | MIR 2304 | For viral production |
| Software | |||
| Access to HPC environment | |||
| AnnotationDbi | 1.38.2 | ||
| Cairo | 1.5-10 | ||
| DESeq2 | 1.16.1 | ||
| genefilter | 1.58.1 | ||
| ggbiplot | 0.55 | ||
| ggplot2 | 3.1.1 | ||
| org.Hs.eg.db | 3.4.1 | ||
| pheatmap | 1.0.12 | ||
| PuTTY | |||
| R | 3.4.0 | ||
| RColorBrewer | 1.1-2 | ||
| reshape2 | 1.4.3 | ||
| rgl | 0.100.19 | ||
| R-studio | |||
| STAR | Version 2.6 or later | ||
| sva | 3.24.4 | ||
| TrimGalore! | |||
| WinSCP |
Referenzen
- Miettinen, M., et al. New fusion sarcomas: histopathology and clinical significance of selected entities. Human Pathology. 86, 57-65 (2019).
- Knott, M. M. L., et al. Targeting the undruggable: exploiting neomorphic features of fusion oncoproteins in childhood sarcomas for innovative therapies. Cancer and Metastasis Reviews. 38, 625-642 (2019).
- Yoshihara, K., et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 34, 4845-4854 (2015).
- Duesberg, P. H. Cancer genes generated by rare chromosomal rearrangements rather than activation of oncogenes. Medical Oncology and Tumor Pharmacotherapy. 4, 163-175 (1987).
- Dupain, C., Harttrampf, A. C., Urbinati, G., Geoerger, B., Massaad-Massade, L. Relevance of Fusion Genes in Pediatric Cancers: Toward Precision Medicine. Molecular Therapy - Nucleic Acids. 6, 315-326 (2017).
- Mitelman, F., Johansson, B., Mertens, F. The impact of translocations and gene fusions on cancer causation. Nature Reviews Cancer. 7, 233-245 (2007).
- Smith, R., et al. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing's sarcoma. Cancer Cell. 9, 405-416 (2006).
- Davicioni, E., et al. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Research. 66, 6936-6946 (2006).
- Gröbner, S. N., et al. The landscape of genomic alterations across childhood cancers. Nature. 555, 321-327 (2018).
- Kim, J., Pelletier, J. Molecular genetics of chromosome translocations involving EWS and related family members. Physiological Genomics. 1, 127-138 (1999).
- Boulay, G., et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell. 171, 163-178 (2017).
- Lessnick, S. L., Braun, B. S., Denny, C. T., May, W. A. Multiple domains mediate transformation by the Ewing’s sarcoma EWS/FLI-1 fusion gene. Oncogene. 10, 423-431 (1995).
- Leach, B. I., et al. Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure. 21, 176-183 (2013).
- Ng, K. P., et al. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proceedings of the National Academy of Sciences U.S.A. 104, 479-484 (2007).
- Riggi, N., et al. EWS-FLI1 Divergent Chromatin Remodeling Mechanisms to Directly Activate or Repress Enhancer Elements in Ewing Sarcoma. Cancer Cell. 26, 668-681 (2014).
- Tomazou, E. M., et al. Epigenome Mapping Reveals Distinct Modes of Gene Regulation and Widespread Enhancer Reprogramming by the Oncogenic Fusion Protein EWS-FLI1. Cell Reports. 10, 1082-1095 (2015).
- Sankar, S., et al. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene. 32, 5089-5100 (2013).
- Gangwal, K., et al. Microsatellites as EWS/FLI response elements in Ewing's sarcoma. Proceedings of the National Academy of Sciences U.S.A. 105, 10149-10154 (2008).
- Gangwal, K., Close, D., Enriquez, C. A., Hill, C. P., Lessnick, S. L. Emergent Properties of EWS/FLI Regulation via GGAA Microsatellites in Ewing's Sarcoma. Genes & Cancer. 1, 177-187 (2010).
- Guillon, N., et al. The Oncogenic EWS-FLI1 Protein Binds In Vivo GGAA Microsatellite Sequences with Potential Transcriptional Activation Function. PLoS One. 4, 4932(2009).
- Chong, S., et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 361, (2018).
- Johnson, K. M., et al. Role for the EWS domain of EWS/FLI in binding GGAA-microsatellites required for Ewing sarcoma anchorage independent growth. Proceedings of the National Academy of Sciences U.S.A. 114, 9870-9875 (2017).
- Theisen, E. R., et al. Transcriptomic analysis functionally maps the intrinsically disordered domain of EWS/FLI and reveals novel transcriptional dependencies for oncogenesis. Genes & Cancer. 10, 21-38 (2019).
- Chen, J., Bardes, E. E., Aronow, B. J., Jegga, A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Research. 37, 305-311 (2009).
- Johnson, K. M., Taslim, C., Saund, R. S., Lessnick, S. L. Identification of two types of GGAA-microsatellites and their roles in EWS/FLI binding and gene regulation in Ewing sarcoma. PLOS One. 12, 0186275(2017).
- Kim, P., Ballester, L. Y., Zhao, Z. Domain retention in transcription factor fusion genes and its biological and clinical implications: a pan-cancer study. Oncotarget. 8, 110103-110117 (2017).
- de Mendíbil, I. O., Vizmanos, J. L., Novo, F. J. Signatures of Selection in Fusion Transcripts Resulting from Chromosomal Translocations in Human Cancer. PLOS One. 4, 4805(2009).
- Frenkel-Morgenstern, M., Valencia, A. Novel domain combinations in proteins encoded by chimeric transcripts. Bioinformatics. 28, 67-74 (2012).
- Hegyi, H., Buday, L., Tompa, P. Intrinsic Structural Disorder Confers Cellular Viability on Oncogenic Fusion Proteins. PLoS Computational Biology. 5, 1000552(2009).
- Latysheva, N. S., Babu, M. M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Research. 44, 4487-4503 (2016).
- Deneen, B., Denny, C. T. Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene. 20, 6731-6741 (2001).
- Kendall, G. C., et al. PAX3-FOXO1 transgenic zebrafish models identify HES3 as a mediator of rhabdomyosarcoma tumorigenesis. eLife. 7, 33800(2018).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten