
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Ermöglicht Echtzeitkompensation bei schnellen photochemischen Oxidationen von Proteinen zur Bestimmung von Proteintopographie-Änderungen
In diesem Artikel
Zusammenfassung
Die schnelle photochemische Oxidation von Proteinen ist eine aufkommende Technik zur strukturellen Charakterisierung von Proteinen. Verschiedene Lösungsmittelzusätze und Liganden haben unterschiedliche Hydroxyl-Radikal-Aufräumeigenschaften. Um die Proteinstruktur unter verschiedenen Bedingungen zu vergleichen, ist eine Echtzeitkompensation von Hydroxylradikalen, die in der Reaktion erzeugt werden, erforderlich, um die Reaktionsbedingungen zu normalisieren.
Zusammenfassung
Die schnelle photochemische Oxidation von Proteinen (FPOP) ist eine strukturbiologische Massenspektrometrie-Technik, die die lösungsmittelzugängliche Oberfläche von Proteinen untersucht. Diese Technik beruht auf der Reaktion von Aminosäure-Seitenketten mit Hydroxylradikalen, die sich frei in Lösung diffundieren. FPOP erzeugt diese Radikale in situ durch Laserphotolyse von Wasserstoffperoxid, wodurch ein Ausbruch von Hydroxylradikalen entsteht, der in der Größenordnung von einer Mikrosekunde erschöpft ist. Wenn diese Hydroxylradikale mit einer lösungsmittelzugänglichen Aminosäure-Seitenkette reagieren, weisen die Reaktionsprodukte eine Massenverschiebung auf, die durch Massenspektrometrie gemessen und quantifiziert werden kann. Da die Reaktionsgeschwindigkeit einer Aminosäure zum Teil von der durchschnittlichen lösungsmittelzugänglichen Oberfläche dieser Aminosäure abhängt, können gemessene Veränderungen der Oxidationsmenge einer bestimmten Region eines Proteins direkt mit Veränderungen der Lösungsmittelzugänglichkeit dieser Region zwischen verschiedenen Konformationen korreliert werden (z. B. Liganden gebunden versus Ligandenfrei, Monomer vs. Aggregat usw.). FPOP wurde in einer Reihe von Problemen in der Biologie angewendet, einschließlich Protein-Protein-Wechselwirkungen, Protein-Konformationsänderungen, und Protein-Ligand-Bindung. Da die verfügbare Konzentration von Hydroxylradikalen aufgrund vieler experimenteller Bedingungen im FPOP-Experiment variiert, ist es wichtig, die effektive Radikaldosis zu überwachen, der das Proteinanalyt ausgesetzt ist. Diese Überwachung wird effizient durch die Einbindung eines Inline-Dosimeters zur Messung des Signals aus der FPOP-Reaktion erreicht, wobei der Laserflonin in Echtzeit angepasst wird, um die gewünschte Oxidationsmenge zu erreichen. Mit dieser Kompensation können Veränderungen in der Proteintopographie, die Konformationsveränderungen, Ligandenbindungsflächen und/oder Protein-Protein-Interaktionsschnittstellen widerspiegeln, in heterogenen Proben mit relativ geringen Probenmengen bestimmt werden.
Einleitung
Die schnelle photochemische Oxidation von Proteinen (FPOP) ist eine aufkommende Technik zur Bestimmung topografischer Proteinveränderungen durch ultraschnelle kovalente Modifikation der lösungsmittelexponierten Oberfläche von Proteinen, gefolgt von der Detektion durch LC-MS1. FPOP erzeugt eine hohe Konzentration von Hydroxylradikalen in situ durch UV-Laserblitzphotolyse von Wasserstoffperoxid. Diese Hydroxylradikale sind sehr reaktiv und kurzlebig, verbraucht auf etwa einer Mikrosekunde Zeitskala unter FPOP-Bedingungen2. Diese Hydroxylradikale diffundieren durch Wasser und oxidieren verschiedene organische Komponenten in Lösung mit kinetischen Raten im Allgemeinen von schnell (-106 M-1 s-1) bis zu diffusionskontrolliert3. Wenn das Hydroxylradikal auf eine Proteinoberfläche trifft, oxidiert der Radikal die Aminosäure-Seitenketten auf der Proteinoberfläche, was zu einer Massenverschiebung dieser Aminosäure (am häufigsten die Nettozugabe eines Sauerstoffatoms)4. Die Geschwindigkeit der Oxidationsreaktion bei jeder Aminosäure hängt von zwei Faktoren ab: der inhärenten Reaktivität dieser Aminosäure (die von der Seitenkette und dem Sequenzkontext abhängt)4,5 und der Zugänglichkeit dieser Seitenkette zum diffundierenden Hydroxylradikal, die eng mit der durchschnittlichen lösungsmittelzugänglichen Oberfläche6,7korreliert. Alle Standard-Aminosäuren mit Ausnahme von Glycin wurden von diesen hochreaktiven Hydroxylradikalen in FPOP-Experimenten als gekennzeichnet beobachtet, wenn auch mit sehr unterschiedlichen Erträgen; in der Praxis werden Ser, Thr, Asn und Ala in den meisten Proben nur unter hohen radikalen Dosen oxidiert und durch sorgfältige und empfindliche gezielte ETD-Fragmentierungidentifiziert 8,9. Nach der Oxidation werden Proben abgeschreckt, um Wasserstoffperoxid und sekundäre Oxidationsmittel (Superoxid, Singlet-Sauerstoff, Peptidylhydroperoxide usw.) zu entfernen. Die abgeschreckten Proben werden dann proteolytisch verdaut, um Mischungen von oxidierten Peptiden zu erzeugen, wobei die Strukturinformationen als chemischer "Schnappschuss" in den Mustern der Oxidationsprodukte der verschiedenen Peptide eingefroren werden (Abbildung 1). Flüssigkeitschromatographie gekoppelt an Massenspektrometrie (LC-MS) wird verwendet, um die Menge der Oxidation von Aminosäuren in einem bestimmten proteolytischen Peptid basierend auf den relativen Intensitäten der oxidierten und unoxidierten Versionen dieses Peptids zu messen. Durch den Vergleich dieses oxidativen Fußabdrucks desselben Proteins, das unter verschiedenen konformen Bedingungen (z. B. Liganden gebunden im Vergleich zu Liganden-frei) gewonnen wird, können Unterschiede in der Oxidationsmenge einer bestimmten Region des Proteins direkt mit Unterschieden in der lösungsmittelzugänglichen Oberfläche dieser Region6,7korreliert werden. Die Fähigkeit, proteintopografische Informationen zur Verfügung zu stellen, macht FPOP zu einer attraktiven Technologie für die höherrangige Strukturbestimmung von Proteinen, auch bei der proteintherapeutischen Entdeckung und Entwicklung10,11.

Abbildung 1: Übersicht über FPOP. Die Oberfläche des Proteins wird durch hochreaktive Hydroxylradikale kovalent modifiziert. Die Hydroxylradikale reagieren mit Aminosäureseitenketten des Proteins mit einer Geschwindigkeit, die stark durch die Lösemittelzugänglichkeit der Seitenkette beeinflusst wird. Topografische Veränderungen (z. B. durch die Bindung eines Liganden wie oben gezeigt) schützen Aminosäuren im Bereich der Wechselwirkung vor einer Reaktion mit Hydroxylradikalen, was zu einer Abnahme der Intensität des modifizierten Peptids im LC-MS-Signal führt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Verschiedene Bestandteile der FPOP-Lösung (z.B. Liganden, Hilfsstoffe, Puffer) haben unterschiedliche Aufräumaktivitäten gegenüber den Hydroxylradikalen, die bei der Laserphotolyse von Wasserstoffperoxid3erzeugt werden. In ähnlicher Weise kann eine kleine Änderung der Peroxidkonzentration, des Lasereinflusses und der Pufferzusammensetzung die effektive Radikaldosis verändern, was die Reproduktion von FPOP-Daten in den Proben und zwischen verschiedenen Labors zu einer Herausforderung macht. Daher ist es wichtig, die verfügbare Hydroxyl-Radikaldosis mit Protein in jeder Probe mit einem von mehreren verfügbaren Hydroxylradikaldosimetern12,,13,,14,15,16zu vergleichen. Hydroxyl-Radikal-Dosimeter wirken, indem sie mit dem Analyten (und mit allen Scavengern in Lösung) für den Pool von Hydroxylradikalen konkurrieren; die effektive Dosis von Hydroxylradikalen wird durch Messung der Oxidationsmenge des Dosimeters gemessen. Beachten Sie, dass "effektive Hydroxyl-Radikal-Dosis" ist eine Funktion sowohl der anfänglichen Konzentration von Hydroxyl-Radikal erzeugt und die Halbwertszeit des Radikals. Diese beiden Parameter sind teilweise voneinander abhängig, was die theoretische kinetische Modellierung etwas komplex macht (Abbildung 2). Zwei Proben könnten völlig unterschiedliche anfängliche radikale Halbwertszeiten haben, während sie immer noch die gleiche effektive radikale Dosis beibehalten, indem sie die anfängliche Konzentration von Hydroxylradikal gebildet ändern; sie erzeugen immer noch identische Fußabdrücke17. Adenin13 und Tris12 sind praktische Hydroxylradikal-Dosimeter, da ihr Oxidationsgrad durch UV-Spektroskopie in Echtzeit gemessen werden kann, so dass Forscher schnell erkennen können, wenn es ein Problem mit einer wirksamen Hydroxylradikaldosis gibt, und ihr Problem beheben können. Um dieses Problem zu lösen, ist ein Inline-Dosimeter im Strömungssystem direkt nach dem Bestrahlungsort wichtig, das das Signal von Adenin-Absorptionsänderungen in Echtzeit überwachen kann. Dies hilft bei der Durchführung von FPOP-Experimenten in Puffern oder anderen Hilfsstoffen mit sehr unterschiedlichen Konzentrationen von Hydroxyl-Radikal-Aufräumkapazität17. Diese radikale Dosierungskompensation kann in Echtzeit durchgeführt werden, was statistisch nicht unterscheidbare Ergebnisse für den gleichen Conformer ergibt, indem die effektive Radikaldosis angepasst wird.
In diesem Protokoll haben wir detaillierte Verfahren für die Durchführung eines typischen FPOP-Experiments mit radikaler Dosierungskompensation mit Adenin als internes optisches Radikaldosimeter. Diese Methode ermöglicht es den Forschern, Footprints über FPOP-Bedingungen hinweg zu vergleichen, die unterschiedliche Aufräumkapazitäten haben, indem sie eine Kompensation in Echtzeit durchführen.

Abbildung 2: Kinetische Simulation der dosimetriebasierten Kompensation. 1 mM Adenin-Dosimeter-Antwort wird in 5 'M Lysozym-Analyt mit einer anfänglichen Hydroxyl-Radikalkonzentration von 1 mM (▪OH t1/2=53 ns) gemessen und als Zieldosimeter-Antwort (schwarz) eingestellt. Nach Zugabe von 1 mM des Abräumers Excipient Histidin nimmt die Dosimeterreaktion (blau) zusammen mit der Menge der Proteinoxidation proportional ab (Cyan). Die Halbwertszeit des Hydroxylradikals nimmt ebenfalls ab (▪OH t1/2=39 ns). Wenn die Menge an Hydroxylradikal erhöht wird, um eine gleichwertige Ausbeute an oxidiertem Dosimeter in der Probe mit 1 mM Histidinfänger zu geben, wie mit 1 mM Hydroxylradikal in Abwesenheit von Scavenger (rot) erreicht, wird die Menge der Proteinoxidation, die ähnlich auftritt, identisch (Magenta), während die Halbwertszeit des Hydroxylradikals noch weiter abnimmt (▪OH t1/2=29 ns). Angepasst mit Genehmigung von Sharp J.S., Am Pharmaceut Rev 22, 50-55, 2019. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Protokoll
1. Bereiten Sie die optische Bank und die Kapillare für FPOP vor
VORSICHT: KrF-Excimerlaser sind extreme Augengefahren, und direktes oder reflektiertes Licht kann bleibende Augenschäden verursachen. Tragen Sie immer einen geeigneten Augenschutz, vermeiden Sie nach Möglichkeit das Vorhandensein reflektierender Objekte in der Nähe des Strahlwegs, und verwenden Sie technische Steuerelemente, um unbefugten Zugriff auf einen aktiven Laser zu verhindern und streunende Reflexionen zu verhindern.
- Bereiten Sie die optische FPOP-Bank vor.
- Schalten Sie den Laser ein, um sich aufzuwärmen. Stellen Sie den Laser auf externe Trigger, Konstante Energie, kein Gasersatz. Stellen Sie die Laserenergie pro Puls ein (in der Regel zwischen 80-120 mJ/Puls).
- Richten Sie die optische Bank mit der plano-konvexen Linse (30 mm Durchmesser x 120 mm FL unbeschichtet) direkt im Weg des Laserstrahls und einem nicht reflektierenden Backstop ein, um das Licht zu absorbieren, wie in Abbildung 3Adargestellt.

Abbildung 3: Optische Bank für das FPOP-Experiment. (A) Die Probe wird mitH2O2, Adenin-Radikaldosimeter und Glutaminfänger gemischt und in die Spritze geladen. Die Probe wird durch die geschmolzene Kieselsäurekapillare durch den fokussierten Strahlweg eines KrF-Excimer-UV-Lasers geschoben. Das UV-Licht photolyziertH2O2 in Hydroxylradikale, die das Protein und das Adenindosimeter oxidieren. Der Spritzenstrom schiebt die beleuchtete Probe vor dem nächsten Laserpuls aus dem Pfad des Lasers, mit einem unbeleuchteten Ausschlussvolumen zwischen beleuchteten Bereichen. Unmittelbar nach der Oxidation wird die Probe durch ein Inline-UV-Spektrophotometer geleitet, das die UV-Absorption von Adenin bei 265 nm misst. Die Probe wird dann in einen Abschreckpuffer eingelagert, um die verbleibendenH2O2 und Sekundäroxidationsmittel zu eliminieren. (B) Die Spotgröße wird gemessen, nachdem eine farbige Haftnote hinter der Kapillare mit dem Laser bei 248 nm bestrahlt wurde. Die Breite des Spots wird für die Berechnung des Probendurchflusses verwendet, und die Silhouette der Kapillare in der Mitte des Spots wird verwendet, um die optische Bank auszurichten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Schneiden Sie eine geeignete Länge der geschmolzenen Kieselsäurekapillare (360 m Außendurchmesser und 100 m Innendurchmesser) und verbinden Sie mit einer Hülse die gasdichte Spritze mit einem Steckverbinder mit geringem Totvolumen.
- Verbrennen Sie die Polyimidbeschichtung der Kapillare vorsichtig mit einem Butanbrenner an der Stelle, an der das Inline-Dosimeter das Absorptionssignal nach Laserbelichtung der Proben bei 265 nm abliest. Wischen Sie den Schmutz auf der Kapillare vorsichtig mit Methanol auf einem fusselfreien Wisch ab. Die Polyimidbeschichtung an der Stelle des Lasereinfalls kann entweder mit dem Butanbrenner ähnlich oder mit dem Excimerlaser mit geringer Leistung abgebrannt werden.
HINWEIS: Warten Sie, bis die Kapillare abkühlt, da es eine Brandgefahr ist, das Methanol auf der heißen Kapillare zu verwenden. - Platzieren Sie diese Kapillare durch den Strahlweg des Lasers und in das Inline-Dosimeter.
- Drücken Sie den Hebel auf der Oberseite des Inline-Dosimeters, um das Scharnier zu öffnen. Entfernen Sie die Magnethalter. Legen Sie die Kapillare in die bearbeitete Nut des Inline-Dosimeters, indem Sie die Magnethalter verwenden, um die Kapillare an Ort und Stelle zu halten. Schließen Sie das Dosimeterscharnier über die Kapillare, drücken Sie es, bis der Hebel an Ort und Stelle verriegelt ist.
- Klicken Sie mit der Dosimetrie-Software auf die Schaltfläche Flash starten, um mit dem Abfeuern des Excimer-Lasers zu beginnen. Stellen Sie die voreingestellte Laserleistung zwischen 50-100 mJ/Puls auf der Lasersteuerungssoftware selbst ein, und stellen Sie die voreingestellte Wiederholungsrate zwischen 10-20 Hz in der Registerkarte Einstellungen der Dosimetriesoftware ein.
- Fokussieren Sie den Laserstrahl mit einer plano konvexen Linse, die auf einer linearen motorisierten Bühne montiert ist. Messen Sie die Breite und Höhe des Laserflecks an der Position der Kapillare auf einer Haftnote genau mit einem Bremssattel, um den Einfallsflaus (mJ/mm2) zu berechnen, wie in Abbildung 3Bdargestellt.
- Platzieren Sie eine opake Blende in der Nähe der Kapillare, um eine gleichmäßige Beleuchtete Breite der Kapillare zu gewährleisten, unabhängig von Änderungen der Strahlgröße durch Bewegung der Linse oder Änderung der Energie pro Puls desLasers 18.
- Mit dem Laserfeuer, bewegen Sie die motorisierte Bühne durch seinen Bewegungsbereich. Stellen Sie sicher, dass der Strahl auf der Blende zentriert bleibt und die Silhouette der Kapillare durchgehend beobachtet werden kann. Der Durchmesser der Blende muss kleiner sein als die Breite des einschneidenden fokussierten Strahls an jedem Punkt im Bereich der motorisierten Stufe.
- Laufen Sie das Wasser mindestens eine Minute lang mit 20 l/min durch die Kapillare, um die Kapillare zu waschen.
- Klicken Sie auf die Schaltfläche Daten starten + AutoZero auf der Dosimeter-Software, um das Dosimeter auf Wasser zu null enden und mit der Datenerfassung zu beginnen.
HINWEIS: Wenn das Puffersystem für FPOP eine signifikante UV-Absorption von 265 nm hat, sollte das FPOP-System auf dem Puffer auf Null gesetzt werden, nicht wasserfrei.
- Klicken Sie auf die Schaltfläche Daten starten + AutoZero auf der Dosimeter-Software, um das Dosimeter auf Wasser zu null enden und mit der Datenerfassung zu beginnen.
- Legen Sie den berechneten Durchfluss auf der Spritzenpumpe fest.
- Die Durchflussrate der Proteinprobe hängt vom bestrahlten Volumen pro Schuss (VIrr), der Anzahl der Laserschüsse pro Sekunde (R) und der gewünschten unirradiated Exclusion Volume Fraktion (FEx) ab, um laminare Strömungseffekte und Probendiffusion (0,15-0,30 empfohlen)2,19,20zu korrigieren. Berechnen Sie denV-Irr (in L) basierend auf dem Innendurchmesser der Kapillare in mm ( Vd) und der Breite des Laserflecks, der auf die Kapillare eindringt (d. h. die Breite der Blende) in mm (w) mit der folgenden Gleichung:
VIrr = π(d/2)2w - Berechnen Sie den gewünschten Durchfluss (in L/min) basierend auf der folgenden Gleichung:
Durchfluss = 60R[VIrr (1 + FEx)]
- Die Durchflussrate der Proteinprobe hängt vom bestrahlten Volumen pro Schuss (VIrr), der Anzahl der Laserschüsse pro Sekunde (R) und der gewünschten unirradiated Exclusion Volume Fraktion (FEx) ab, um laminare Strömungseffekte und Probendiffusion (0,15-0,30 empfohlen)2,19,20zu korrigieren. Berechnen Sie denV-Irr (in L) basierend auf dem Innendurchmesser der Kapillare in mm ( Vd) und der Breite des Laserflecks, der auf die Kapillare eindringt (d. h. die Breite der Blende) in mm (w) mit der folgenden Gleichung:
2. Herstellung der Proteinlösung für FPOP
- Bereiten Sie das Protein in den zwei oder mehr unterschiedlichen Zubedingungen vor, die verglichen werden sollen (z. B. Ligandengebunden und Ligandenfrei; Aggregat und Monomer; allein und mit einem Protein-Protein-Bindungspartner; etc.), um die Konformationsveränderungen zu erkennen.
- Legen Sie das Gesamtvolumen für FPOP fest, um den Anforderungen des Experiments gerecht zu werden. Die Mindestgrenze hängt in der Regel vom Volumen der Bestrahlungskapillare und dem Material ab, das für eine robuste Detektion und relative Quantifizierung erforderlich ist, und hängt weitgehend vom verwendeten LC-MS/MS-System und dem Verfahren zur Nachetikettierung von Proben ab. Das Gesamtvolumen für FPOP-Lösungen, die in unserer Gruppe üblich sind, beträgt 20 l nach Zugabe von Wasserstoffperoxid. Die Endkonzentration des Proteins beträgt in der Regel 1-10 m, mit 17 mM Glutamin (zur Begrenzung der Lebensdauer des Hydroxylradikals), 1 mM Adenin (als radikales Dosimeter)13,,17 und 10 mM Phosphatpuffer (ein Puffer, der ein schlechter Abräumer von Hydroxylradikalen ist). Die Stichproben werden in der Regel mit mehreren Replikationen erstellt, um eine statistische Modellierung der Ergebnisse zu ermöglichen.
- Für die meisten allgemeinen Zwecke bereiten Sie Proben in Dreifacharbeit in beiden Zuständen vor, sowie mindestens eine Probe, die als No-Laser-Steuerung zur Messung der Hintergrundoxidation verwendet werden kann. Bereiten Sie 18 L dieses FPOP-Lösungsmixes vor.
HINWEIS: Viele Puffer und Additive, die häufig in der Biochemie verwendet werden, sind Hydroxyl-Radikalfänger. Diese Additive und Puffer können verwendet werden; Es kann jedoch zu einer Verringerung der Oxidation durch Hydroxylradikalaufräumung des Puffers kommen. Im Allgemeinen halten Sie alle Zusatzstoffe auf das Minimum, das das biologische System benötigt, um die Proteinoxidationsausbeute zu maximieren. Dimethylsulfoxid sollte aufgrund der Neigung zur Erzeugung von Sekundärradikalen vermieden werden; Dimethylformamid war eine nützliche Alternative in unseren Händen. Bei der Verwendung von Puffern, die starke Hydroxylradikalfänger sind, kann Glutamin oft aus dem FPOP-Lösungsmix ausgeschlossen werden.
- Für die meisten allgemeinen Zwecke bereiten Sie Proben in Dreifacharbeit in beiden Zuständen vor, sowie mindestens eine Probe, die als No-Laser-Steuerung zur Messung der Hintergrundoxidation verwendet werden kann. Bereiten Sie 18 L dieses FPOP-Lösungsmixes vor.
- Bereiten Sie 1 M Wasserstoffperoxid unmittelbar vor dem FPOP-Experiment vor.
HINWEIS: 30 % Wasserstoffperoxid, wie es üblicherweise von Anbietern verkauft wird, enthält einen Stabilisator, der die Haltbarkeit erhöht. Einmal verdünnt, Wasserstoffperoxid sollte schnell verwendet werden, auf jeden Fall innerhalb des gleichen Tages. Wasserstoffperoxid sollte auch regelmäßig auf Zersetzung durch FPOP mit einem Hydroxylradikaldosimeter getestet werden. - Bereiten Sie Mikrozentrifugen-Röhrchen vor, die eine Abschreckungslösung von 25 l mit einer Flasche von 0,5 g/l Methioninamid und 0,5 g/l Kataalase enthalten. Wenn für FPOP ein Probenvolumen von mehr als 20 l verwendet wird, erhöhen Sie das Löschlösungsvolumen proportional.
3. Führen Sie das FPOP-Experiment durch
- Fügen Sie 2 l Wasserstoffperoxid in die 18-L-Lösungsmischung ein. Mischen Sie den Inhalt vorsichtig mit einer Pipette und drehen Sie die Lösung schnell auf den Boden der Mikrozentrifugenrohre. Sofort mit einer gasdichten Spritze sammeln und in die Spritzenpumpe laden.
- Starten Sie den Durchfluss an der Spritzenpumpe mit der in Schritt 1.8.1 ermittelten Durchflussrate (in der Regel zwischen 8-16 l/min), indem Sie auf die Taste Pumpe starten auf der Dosimeter-Software klicken.
- Überwachen Sie den Echtzeit-Adenin-Messwert mit dem Inline-Dosimeter (siehe Materialtabelle) und sammeln Sie die Probe im Abfall. Warten Sie, bis sich das Abs265-Signal stabilisiert hat.
- Klicken Sie auf die Start-Flash-Taste in der Dosimeter-Software, um den Laser mit der voreingestellten Wiederholungsrate und Energie zu feuern.
- Überwachen Sie den Echtzeit-Adenin-Messwert mit einem Inline-Dosimeter (siehe Materialtabelle); der Unterschied in Abs265 mit dem Laser aus und dem Laser auf ist die 'Abs265 Lesung.
HINWEIS: Das Auftreten von hochinstabilen Abs265 Messwerten beim Abfeuern des Lasers in Gegenwart von Wasserstoffperoxid ist auf die Erzeugung von Blasen in Lösung zurückzuführen. Reduzieren Sie den Einfluss des Lasers und/oder die Konzentration von Wasserstoffperoxid, um die Blasen zu beseitigen.
4. Kompensation durchführen
HINWEIS: Verschiedene Liganden, Puffer usw. können unterschiedliche Aufräumkapazitäten gegenüber Hydroxylradikalen haben. Es ist wichtig sicherzustellen, dass vergleichbare effektive Hydroxyl-Radikal-Dosen zur Verfügung stehen, um mit Protein über verschiedene Proben zu reagieren. Dies wird erreicht, indem eine gleiche Hydroxyl-Radikal-Dosimeter-Reaktion zwischen den Proben sichergestellt wird. Mit Adenin-Dosimetrie spiegelt die Veränderung der UV-Absorption bei 265 nm (Abs265) die effektive Hydroxyl-Radikal-Dosis wider; Je größer die "Abs265",desto höher die effektive Hydroxyl-Radikaldosis.
- Vergleichen Sie den mit dem Inline-Dosimeter erhaltenen Messwert "Abs265" mit dem gewünschten Wert von265, der durch vorherige Experimente oder Kontrollen erzielt wurde. Ein wertschwächerer Wert von265 als der gewünschte Wert deutet auf eine unzureichende effektive Dosis von Hydroxylradikalen hin; ein Abs265-Wert deutet auf eine zu hohe effektive Radikaldosis hin. Befindet sich der Wert der Abs265 auf dem gewünschten Niveau, so sammeln Sie die Probe unmittelbar nach der Laserbestrahlung im Löschpuffer17.
- Kompensieren Sie die effektive Radikaldosis, um die Abs265auszugleichen. Diese Kompensation kann auf drei Arten durchgeführt werden: Die Wasserstoffperoxidkonzentration ändern, den Lasereinfluss erhöhen, indem die Laserenergie pro Puls geändert wird, oder den Lasereinfluss durch Änderung der Fokusebene der Fokuslinse erhöhen.
- Um eine große Änderung (>10 mAU) in der Messung von265 zu machen, stellen Sie die Probe mit mehr oder weniger Wasserstoffperoxid um und führen Sie die Probe gemäß Abschnitt 3 erneut aus.
- Passen Sie die Brennweite des Einfallsstrahls an, indem Sie die Position der Fokussierungslinse mit der 50 mm Motorisierten Bühne anpassen, um eine kleine Änderung des Messwerts265 in Echtzeit vorzunehmen. Wenn die Brennebene näher an die Position der Kapillare heranrückt, erhöht sich der Wert von265; Wenn die Brennebene weiter von der Position der Kapillare entfernt wird, wird die Abs265-Messung verringert.
- Überwachen Sie das Adenin -Abs265, um die effektive Menge an Hydroxylradikalen zu messen, die in der Probe nach Derlaserbestrahlung13vorhanden ist. Die Echtzeitüberwachung mit einem Inline-UV-Kapillardetektor ermöglicht eine Echtzeitkompensation gemäß 4.2.2; Stellen Sie die Linsenposition mit der motorisierten Stufe ein, bis der Wert "Abs265" dem gewünschten Messwert entspricht. Postexperimentelle Absorptionsmessungen mit einem UV-Spektralphotometer sind ebenfalls genau, erfordern aber neue Proben für jede effektive Radikaldosis.
5. Verdauen Sie die Proteinproben
HINWEIS: Trypsin wird am häufigsten verwendet, um Proteinproben für FPOP zu verdauen, und ist die Protease in diesem Protokoll verwendet. Es ist eine zuverlässige Protease, die Peptide mit grundlegenden Standorten sowohl an der N- und C-Terminus erzeugt, Förderung multiplizierter Peptidionen bei MS. Darüber hinaus spaltet es nach Lysin und Arginin, zwei Aminosäuren, die nur mäßig reaktiv auf Hydroxylradikale sind; Daher sind Veränderungen im Verdauungsmuster aufgrund der Analytoxidation selten. Andere Proteasen wurden erfolgreich mit FPOP21verwendet, aber es sollte darauf geachtet werden, dass Verdauungsmuster zwischen nicht oxidierten und oxidierten Proben vergleichbar sind.
- Messen Sie das endgültige Volumen der gelöschten FPOP-Probe. 500 mM Tris, pH 8,0 mit 10 mM CaCl2 mit 50 mM Dithiothreitol (DTT) nach Abschreckung auf eine Endkonzentration von 50 mM Tris, 1 mM CaCl2 und 5 mM DTT in die Proteinlösung geben.
- Die Proteinprobe bei 95 °C 15 Minuten erhitzen.
- Die Probe sofort 2 min auf Eis abkühlen.
- Fügen Sie den Proben das Verhältnis von 1:20 Trypsin/Protein gewicht hinzu.
- Das Protein über Nacht bei 37 °C mit Mischen verdauen.
- Beenden Sie die Verdauungsreaktion durch Zugabe von 0,1% Ameisensäure und/oder Erhitzen der Probe auf 95 °C für 10 min.
- Fügen Sie den Proben 2 mM DTT hinzu und erhitzen Sie bei 60 °C 15 min unmittelbar vor LC-MS/MS.
HINWEIS: Während andere Gruppen alkylieren von Thiolen in FPOP-Experimenten berichtet haben, haben wir in unseren Händen Nebenprodukte bei der Alkylierung von oxidierten Proteinen festgestellt (möglicherweise aufgrund einer Reaktion mit nucleophilen Carbonylen, die als kleineres Oxidationsprodukt gebildet wurden). Daher entscheiden wir uns, Alkylierung von Thiolen zu vermeiden, wenn möglich.
6. Flüssigkeitschromatographie-Tandem-Massenspektrometrie (LC-MS/MS)
- Bereiten Sie die mobile Phase A vor, die aus Wasser mit 0,1 % Ameisensäure und mobiler Phase B besteht, bestehend aus Acetonitril mit 0,1 % Ameisensäure.
- Die Probe zuerst auf eine C18-Falle-Säule (300 x 5 mm 100 x Porengröße, 5 m Partikelgröße) legen und mit 2% Lösungsmittel B 3 Minuten lang bei einer Durchflussrate von 5,0 l/min waschen, um Salze und hydrophile kleine Moleküle zu entfernen.
- Trennen Sie dann die Peptide auf C18-Nanosäule (0,75 mm x 150 mm, 2 m Partikelgröße, 100 x Porengröße) bei einer Durchflussrate von 300 nL/min. Der Gradient besteht aus einer linearen Erhöhung von 2 bis 35% Lösungsmittel B über 22 min, ramped auf 95% Lösungsmittel B über 5 min und gehalten für 3 min, um die Säule zu waschen, und dann wieder auf 2% B über 3 min und gehalten für 9 min, um die Säule wieder zu kompensieren.
HINWEIS: Dieser Gradient ist ausreichend für LC-MS/MS der meisten Ein- und Zwei-Protein-FPOP-Mischungen, die eine Peptid-Quantifizierung anstreben. Der Prozentsatz des Lösungsmittels B muss möglicherweise verändert werden, um die Peptidauflösung in seltenen Fällen zu erhöhen, in denen Peptide aufgrund ähnlicher Retentionszeiten und m/z-Werte miteinander stören. Proteome-Skala FPOP22 oder experimentelle Designs, die Peptid-Oxidationsprodukt Isomere1,23,24,25 trennen möchten, können längere LC-Gradienten erfordern und außerhalb des Rahmens dieses Berichts liegen. - Elute die Peptide direkt in die Nanospray-Quelle eines hochauflösenden Massenspektrometers mit einem leitfähigen Nanospray-Emitter.
- Erfassen Sie die Daten im positiven Ionenmodus. Stellen Sie die Sprühspannung auf 2400 V und die Temperatur des Ionenübertragungsrohrs auf 300 °C ein.
- Erfassen Sie die vollständigen MS-Scans von m/z 250 bis 2000 mit einer nominalen Auflösung bei m/z 200 von 60.000, gefolgt von acht nachfolgenden datenabhängigen linearen Ionenfallen MS/MS-Scans auf den acht häufigsten Peptidionen, die kollisionsinduzierte Dissoziation bei 35% normalisierter Energie verwenden, um die Peptide zu identifizieren. Fragmentieren Sie die Peptide bis zu fünf Mal innerhalb von 30 s und übertragen Sie sie dann in eine Ausschlussliste für 60 s.
7. Datenverarbeitung und Berechnung der durchschnittlichen Oxidation von Peptiden
- Bestimmen Sie die Sequenzabdeckung des Proteins, m/z-Werte und Retentionszeiten von nicht oxidierten Peptiden mithilfe der Suchmaschine MS/MS proteomics.
- Stellen Sie die Vorläufermassentoleranz auf 10 ppm ein und lassen Sie bis zu zwei verpasste Dekolleté-Stellen für die Trypsin-verdauten Proben mit Standard-Trypsinspaltungssspezifität zu.
- Stellen Sie die Massetoleranz des Peptidmassenfragments auf 0,4 Daltons ein.
- Berechnen Sie auf Basis des m/z-Verhältnisses der nachgewiesenen unveränderten Peptide und der bekannten Massenverschiebungen der wichtigsten Oxidationsprodukte die m/z der verschiedenen theoretischen Oxidationsprodukte jedes Peptids4,26,27,28,29.
- Identifizieren Sie das extrahierte Ionenchromatogramm dieser m/z-Werte mithilfe von Software, um den massenspektrometrischen Lauf anzuzeigen (Abbildung 4). Identifizieren Sie die Peptidoxidationsprodukte anhand ihres m/z,ihres Ladezustands und der Ähnlichkeit der Elutionszeit mit dem unveränderten Peptid. In unseren Händen, Peptid-Oxidation Seut zwischen 240 Sekunden vor bis 180 Sekunden nach dem unveränderten Peptid mit dem LC-Gradient oben. Da Oxidation oft zu mehreren isomeren Oxidationsprodukten führt, ist es üblich, mehrere teilweise aufgelöste Spitzen in den extrahierten Ionenchromatogrammen von Peptidoxidationsprodukten zu beobachten, wie in Abbildung 4dargestellt. Peptidoxidationsprodukte werden auf der Grundlage der Fläche der Peak(s) in den extrahierten Ionenchromatogrammen quantifiziert.

Abbildung 4: Extrahiertes Ionenchromatogramm eines Peptids und seiner Oxidationsprodukte nach FPOP. Die m/z der Peptidoxidationsprodukte werden auf der Grundlage des m/z des nicht oxidierten Peptids und der bekannten Oxidationsprodukte berechnet; und die Bereiche dieser Peptidprodukte werden bestimmt. Der Bereich der Peptidprodukte wird dann für die Berechnung der durchschnittlichen Oxidationsereignisse pro Peptid verwendet. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
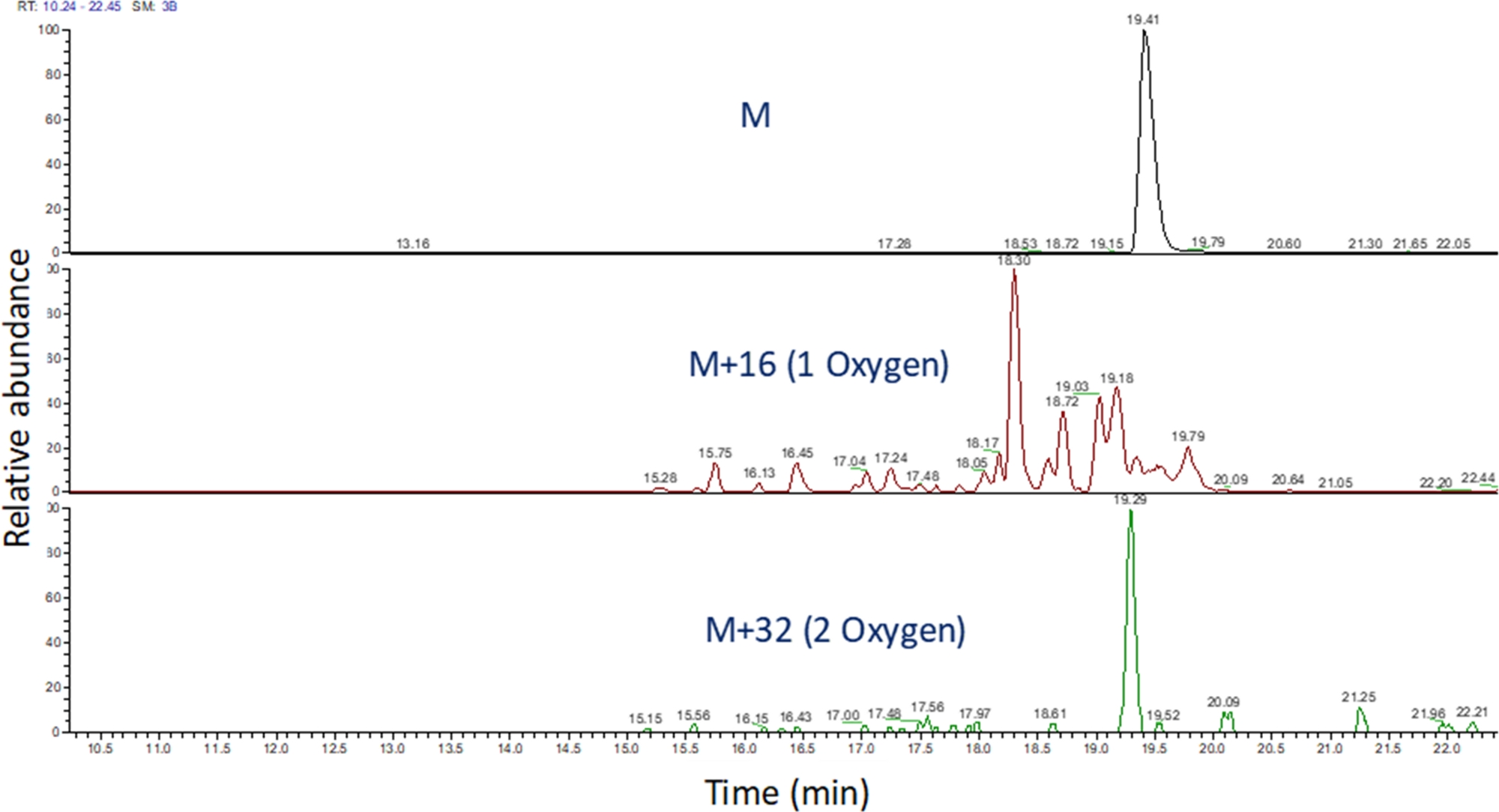{kind=link}
- Berechnen Sie die durchschnittliche Oxidation der Peptide mit der folgenden Gleichung.

wobei P die durchschnittliche Anzahl von Oxidationsereignissen pro Peptidmolekül angibt und ich den Spitzenbereich des nichtoxidierten Peptids (Iunoxidiert)und des Peptids mit n Oxidationsereignissen darstellt. Beachten Sie, dass I(einzeln oxidiert) nicht nur Zusätze eines einzelnen Sauerstoffatoms, sondern auch andere weniger häufige einzelne Oxidationsereignisse umfassen würde, die der Prüfer messen kann (z. B. oxidative Decarboxylierung, Carbonylbildung usw.). 4,26,27,28,29.
Ergebnisse
Der Vergleich des schweren Kettenpeptid-Fußabdrucks des Adalimumab-Biosimilars im Phosphatpuffer und bei einer Erhitzung bei 55 °C für 1 h zeigen interessante Ergebnisse. Der T-Test des Schülers wird zur Identifizierung von Peptiden verwendet, die unter diesen beiden Bedingungen signifikant verändert werden (p ≤ 0,05). Die Peptide 20-38, 99-125, 215-222, 223-252, 260-278, 376-413 und 414-420 zeigen einen erheblichen Schutz vor Lösungsmitteln, wenn das Protein erhitzt wird, um Aggregate zu bilden (
Diskussion
Massenspektrometrie-basierte Strukturtechniken, einschließlich Wasserstoff-Deuterium-Austausch, chemische Vernetzung, kovalente Etikettierung und native Sprühmassenspektrometrie und Ionenmobilität, haben aufgrund ihrer Flexibilität, Empfindlichkeit und Fähigkeit, mit komplexen Mischungen umzugehen, rapide an Popularität gewonnen. FPOP verfügt über mehrere Vorteile, die seine Popularität im Bereich der Massenspektrometrie-basierten Strukturtechniken erhöht hat. Wie die meisten kovalenten Etikettierungsstrategien...
Offenlegungen
Joshua S. Sharp offenbart ein erhebliches finanzielles Interesse an GenNext Technologies, Inc., einem kleinen Unternehmen, das Technologien für die Analyse von Proteinstrukturen mit höherer Ordnung einschließlich Hydroxylradikal-Protein-Fußabdruck kommerzialisieren will.
Danksagungen
Wir würdigen forschungsmittel vom National Institute of General Medical Sciences Grant R43GM125420-01zur Unterstützung der kommerziellen Entwicklung eines FPOP-Tischgeräts und R01GM127267 für die Entwicklung von Standardisierungs- und Dosimetrieprotokollen für Hochenergie-FPOP.
Materialien
| Name | Company | Catalog Number | Comments |
| Adenine | Acros Organics | 147440250 | Soluble in water upto 3.5 mM |
| Aperture | Edmund Optics | 39-905 | 1000 μm Aperture Diameter, Gold-Plated Copper Aperture |
| Aperture holder | Edmund Optics | 53-287 | 25.8mm Outer Diameter, Precision Pinhole Mount |
| Catalse | Sigma Aldrich | C-40 | Catalase from bovine liver, lyophilized powder, ≥10,000 units/mg protein |
| COMPex Pro laser | Coherent | 1113836 | COMPexPRO 102, F-Vversion, KrF laser, No XeCl |
| Dithiotheitol (DTT) | Promega | V3151 | DTT, Molecular Grade (DL-Dithiothreitol) |
| Fraction collector | GenNext Technologies, Inc. | N/A | Automated fraction collector |
| Fused silica capillay | Molex | 1068150023 | Polymicro Flexible Fused Silica Capillary Tubing, Inner Diameter 100 µm, Outer Diameter 375 µm, TSP100375 |
| Glutamine | Acros Organics | 119951000 | L(+)-Glutamine, 99% |
| Holder for lens | Edmund Optics | 03-668 | 53 mm Outer Diameter, Three-Screw Adjustable Ring Mount |
| Hydrogen peroxide | Fisher Scientific | H325-100 | Hydrogen Peroxide, 30% (Certified ACS), Fisher Chemical |
| LC-MS/MS system | Thermo Scientific | IQLAAEGAAPFADBMBCX | Dionex Ultimate 3000 coupled to Orbitap Fusion Tribrid mass spectrometer |
| Mas spec grade Acetonitrile | Fisher Scientific | A955-1 | Acetonitrile, Optima LC/MS Grade, Fisher Chemical |
| Mass spec grade formic acid | Fisher Scientific | A117-50 | Formic Acid, 99.0+%, Optima™ LC/MS Grade, Fisher Chemical |
| Mass spec grade water | Fisher Scientific | W6-4 | Water, Optima LC/MS Grade, Fisher Chemical |
| MES buffer | Sigma Aldrich | M0164 | MES hemisodium salt |
| Methionine amide | Bachem | 4000594.0005 | H-met-NH2.HCl |
| Micro V clamp | Thor Labs | VK250 | Micro V-clamp with stainless steel blades |
| Motorized stage | Edmund Optics | 68-638 | 50mm Travel Motorized Stage System with Manual Control |
| Nano C18 colum | Thermo Scientific | 164534 | Acclaim PepMap 100 C18 HPLC Columns |
| Optical bench | Edmund Optics | 56-935 | 18" x 18" breadboard |
| Pioneer FPOP Module System | GenNext Technologies, Inc. | N/A | Inline FPOP Radical Dosimetry System |
| Post holder | Edmund Optics | 58-979 | 3" Length, ¼-20 Thread, Post Holder |
| Sodium phosphate dibasic | Fisher Scientific | BP331-500 | Sodium Phosphate Dibasic Heptahydrate (Colorless-to-White Crystals), Fisher BioReagents |
| Sodium phosphate monobasic | Fisher Scientific | BP330-500 | Sodium Phosphate Monobasic Monohydrate (Colorless-to-white Crystals), Fisher BioReagents |
| Syringe | Hamilton | 81065 | 100 µL, Model 1710 RN SYR, Small Removable NDL, 22s ga, 2 in, point style 3 |
| Syringe pump | KD Scientific | 788101 | Legato 101 syringe pump |
| Trap C18 column | Thermo Scientific | 160454 | Thermo Scientific Acclaim PepMap 100 C18 HPLC Columns |
| Tris | Sigma Aldrich | 252859 | Tris(hydroxymethyl)aminomethane |
| Trypsin | Promega | V5111 | Sequencing Grade Modified Trypsin |
| UV plano convex lens | Edmund Optics | 84-285 | 30 mm Dia. x 120 mm FL Uncoated, UV Plano-Convex Lens |
Referenzen
- Kaur, P., Kiselar, J., Yang, S., Chance, M. R. Quantitative protein topography analysis and high-resolution structure prediction using hydroxyl radical labeling and tandem-ion mass spectrometry (MS). Molecular & Cellular Proteomics. 14 (4), 1159-1168 (2015).
- Hambly, D. M., Gross, M. L. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. Journal of the American Society for Mass Spectrometry. 16 (12), 2057-2063 (2005).
- Buxton, G. V., Greenstock, C. L., Helman, W. P., Ross, A. B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O- in Aqueous Solution. Journal of Physical and Chemical Reference Data. 17 (2), 513 (1988).
- Xu, G., Chance, M. R. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Analytical Chemistry. 77 (14), 4549-4555 (2005).
- Sharp, J. S., Tomer, K. B. Effects of anion proximity in peptide primary sequence on the rate and mechanism of leucine oxidation. Analytical Chemistry. 78 (14), 4885-4893 (2006).
- Huang, W., Ravikumar, K. M., Chance, M. R., Yang, S. Quantitative mapping of protein structure by hydroxyl radical footprinting-mediated structural mass spectrometry: a protection factor analysis. Biophysical Journal. 108 (1), 107-115 (2015).
- Xie, B., Sood, A., Woods, R. J., Sharp, J. S. Quantitative protein topography measurements by high resolution hydroxyl radical protein footprinting enable accurate molecular model selection. Scientific Reports. 7 (1), 4552 (2017).
- Li, Z., et al. High structural resolution hydroxyl radical protein footprinting reveals an extended Robo1-heparin binding interface. Journal of Biological Chemistry. 290 (17), 10729-10740 (2015).
- Li, X., et al. Structural analysis of the glycosylated intact HIV-1 gp120-b12 antibody complex using hydroxyl radical protein footprinting. Biochemistry. 56 (7), 957-970 (2017).
- Li, K. S., Shi, L., Gross, M. L. Mass spectrometry-based fast photochemical oxidation of proteins (FPOP) for higher order structure characterization. Accounts of Chemical Research. 51 (3), 736-744 (2018).
- Li, J., Chen, G. The use of fast photochemical oxidation of proteins coupled with mass spectrometry in protein therapeutics discovery and development. Drug Discovery Today. 24 (3), 829-834 (2019).
- Roush, A. E., Riaz, M., Misra, S. K., Weinberger, S. R., Sharp, J. S. Intrinsic buffer hydroxyl radical dosimetry using Tris(hydroxymethyl)aminomethane. Journal of the American Society for Mass Spectrometry. 31 (2), 169-172 (2020).
- Xie, B., Sharp, J. S. Hydroxyl radical dosimetry for high flux hydroxyl radical protein footprinting applications using a simple optical detection method. Analytical Chemistry. 87 (21), 10719-10723 (2015).
- Niu, B., Zhang, H., Giblin, D., Rempel, D. L., Gross, M. L. Dosimetry determines the initial OH radical concentration in fast photochemical oxidation of proteins (FPOP). Journal of the American Society for Mass Spectrometry. 26 (5), 843-846 (2015).
- Niu, B., et al. Incorporation of a reporter peptide in FPOP compensates for adventitious scavengers and permits time-dependent measurements. Journal of the American Society for Mass Spectrometry. 28 (2), 389-392 (2017).
- Garcia, N. K., Sreedhara, A., Deperalta, G., Wecksler, A. T. Optimizing hydroxyl radical footprinting analysis of biotherapeutics using internal standard dosimetry. Journal of the American Society for Mass Spectrometry. 31 (7), 1563-1571 (2020).
- Sharp, J. S., Misra, S. K., Persoff, J. J., Egan, R. W., Weinberger, S. R. Real time normalization of fast photochemical oxidation of proteins experiments by inline adenine radical dosimetry. Analytical Chemistry. 90 (21), 12625-12630 (2018).
- Zhang, B., Cheng, M., Rempel, D., Gross, M. L. Implementing fast photochemical oxidation of proteins (FPOP) as a footprinting approach to solve diverse problems in structural biology. Methods. 144, 94-103 (2018).
- Konermann, L., Stocks, B. B., Czarny, T. Laminar flow effects during laser-induced oxidative labeling for protein structural studies by mass spectrometry. Analytical Chemistry. 82 (15), 6667-6674 (2010).
- Gau, B. C., Sharp, J. S., Rempel, D. L., Gross, M. L. Fast photochemical oxidation of protein footprints faster than protein unfolding. Analytical Chemistry. 81 (16), 6563-6571 (2009).
- Li, K. S., et al. Hydrogen-Deuterium exchange and hydroxyl radical footprinting for mapping hydrophobic interactions of human bromodomain with a small molecule Inhibitor. Journal of the American Society for Mass Spectrometry. 30 (12), 2795-2804 (2019).
- Espino, J. A., Jones, L. M. Illuminating biological interactions with in vivo protein footprinting. Analytical Chemistry. 91 (10), 6577-6584 (2019).
- Charvatova, O., et al. Quantifying protein interface footprinting by hydroxyl radical oxidation and molecular dynamics simulation: application to galectin-1. Journal of the American Society for Mass Spectrometry. 19 (11), 1692-1705 (2008).
- Gau, B., Garai, K., Frieden, C., Gross, M. L. Mass spectrometry-based protein footprinting characterizes the structures of oligomeric apolipoprotein E2, E3, and E4. Biochemistry. 50 (38), 8117-8126 (2011).
- Gau, B. C., Chen, J., Gross, M. L. Fast photochemical oxidation of proteins for comparing solvent-accessibility changes accompanying protein folding: Data processing and application to barstar. Biochimica et Biophysica Acta. 1834 (6), 1230-1238 (2013).
- Garrison, W. M. Reaction mechanisms in the radiolysis of peptides, polypeptides, and proteins. Chemical Reviews. 87 (2), 381-398 (1987).
- Xu, G., Chance, M. R. Radiolytic modification of sulfur-containing amino acid residues in model peptides: fundamental studies for protein footprinting. Analytical Chemistry. 77 (8), 2437-2449 (2005).
- Xu, G., Chance, M. R. Radiolytic modification of acidic amino acid residues in peptides: probes for examining protein-protein interactions. Analytical Chemistry. 76 (5), 1213-1221 (2004).
- Xu, G., Takamoto, K., Chance, M. R. Radiolytic modification of basic amino acid residues in peptides: probes for examining protein-protein interactions. Analytical Chemistry. 75 (24), 6995-7007 (2003).
- Misra, S. K., Orlando, R., Weinberger, S. R., Sharp, J. S. Compensated hydroxyl radical protein footprinting measures buffer and excipient effects on conformation and aggregation in an adalimumab biosimilar. AAPS Journal. 21 (5), 87 (2019).
- Simmons, D. A., Konermann, L. Characterization of transient protein folding intermediates during myoglobin reconstitution by time-resolved electrospray mass spectrometry with on-line isotopic pulse labeling. Biochemistry. 41 (6), 1906-1914 (2002).
- Vahidi, S., Konermann, L. Probing the time scale of FPOP (fast photochemical oxidation of proteins): radical reactions extend over tens of milliseconds. Journal of the American Society for Mass Spectrometry. 27 (7), 1156-1164 (2016).
- Chance, M. R. Unfolding of apomyoglobin examined by synchrotron footprinting. Biochemical and Biophysical Research Communications. 287 (3), 614-621 (2001).
- Xu, G., Chance, M. R. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chemical Reviews. 107 (8), 3514-3543 (2007).
- Zhang, Y., Rempel, D. L., Zhang, H., Gross, M. L. An improved fast photochemical oxidation of proteins (FPOP) platform for protein therapeutics. Journal of the American Society for Mass Spectrometry. 26 (3), 526-529 (2015).
- Cornwell, O., Radford, S. E., Ashcroft, A. E., Ault, J. R. Comparing hydrogen deuterium exchange and fast photochemical oxidation of proteins: a structural characterisation of wild-type and ΔN6 β(2)-microglobulin. Journal of the American Society for Mass Spectrometry. 29 (2), 2413-2426 (2018).
- Xie, B., Sharp, J. S. Relative Quantification of sites of peptide and protein modification using size exclusion chromatography coupled with electron transfer dissociation. Journal of the American Society for Mass Spectrometry. 27 (8), 1322-1327 (2016).
- Srikanth, R., Wilson, J., Vachet, R. W. Correct identification of oxidized histidine residues using electron-transfer dissociation. Journal of Mass Spectrometry. 44 (5), 755-762 (2009).
- Li, X., Li, Z., Xie, B., Sharp, J. S. Improved identification and relative quantification of sites of peptide and protein oxidation for hydroxyl radical footprinting. Journal of the American Society for Mass Spectrometry. 24 (11), 1767-1776 (2013).
- Li, X., Li, Z., Xie, B., Sharp, J. S. Supercharging by m-NBA Improves ETD-Based Quantification of Hydroxyl Radical Protein Footprinting. Journal of the American Society for Mass Spectrometry. 26 (8), 1424-1427 (2015).
- Khaje, N. A., Sharp, J. S. Rapid quantification of peptide oxidation isomers from complex mixtures. Analytical Chemistry. 92 (5), 3834-3843 (2020).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten