
Method Article
Hochdurchsatz-Zellprofilierung gezielter Proteinabbauverbindungen mit HiBiT-CRISPR-Zelllinien
In diesem Artikel
Zusammenfassung
Dieses Protokoll beschreibt den quantitativen lumineszierenden Nachweis der Proteinabbaukinetik in lebenden Zellen, die unter Verwendung von CRISPR/Cas9 entwickelt wurden, um Antikörper freies endogenes Protein-Detektions-Tag zu exprimieren, das mit einem Zielprotein fusioniert ist. Detaillierte Anweisungen zum Berechnen und Abrufen der quantitativen Abbauparameter, Rate, Dmax, DC 50 und Dmax50 sind enthalten.
Zusammenfassung
Gezielte Proteinabbauverbindungen, einschließlich molekularer Klebstoffe oder Proteolyse, die auf Chimären abzielen, sind eine aufregende neue therapeutische Modalität in der Entdeckung niedermolekularer Wirkstoffe. Diese Klasse von Verbindungen induziert den Proteinabbau, indem sie das Zielprotein und die E3-Ligase-Maschinenproteine, die für die Ubiquitinierung und schließlich den Abbau des Zielproteins über den Ubiquitin-Proteasomalen Weg (UPP) erforderlich sind, in die Nähe bringt. Die Profilierung des Zielproteinabbaus im Hochdurchsatz bleibt jedoch angesichts der Komplexität der zellulären Signalwege, die für den Abbau erforderlich sind, eine große Herausforderung. Hier stellen wir ein Protokoll und eine Screening-Strategie vor, die auf der Verwendung von CRISPR / Cas9-endogener Markierung von Zielproteinen mit dem 11-Aminosäuren-HiBiT-Tag basiert, der sich mit hoher Affinität zum LgBiT-Protein ergänzt, um ein lumineszierendes Protein zu erzeugen. Diese CRISPR-Zielzelllinien mit endogenen Tags können verwendet werden, um den verbindungsinduzierten Abbau entweder in echtzeitfähigen, kinetischen lebenden Zell- oder Endpunktlytmodi zu messen, indem das Lumineszenzsignal mit einem lumineszierenden plattenbasierten Lesegerät überwacht wird. Hier skizzieren wir die empfohlenen Screening-Protokolle für die verschiedenen Formate und beschreiben auch die Berechnung der wichtigsten Degradationsparameter von rate, Dmax, DC 50, Dmax50 sowie Multiplexing mit Zelllebensfähigkeitsassays. Diese Ansätze ermöglichen eine schnelle Entdeckung und Triaging von Verbindungen im Frühstadium unter Beibehaltung der endogenen Expression und Regulation von Zielproteinen in relevanten zellulären Hintergründen, was eine effiziente Optimierung der führenden therapeutischen Verbindungen ermöglicht.
Einleitung
Der gezielte Proteinabbau hat sich zu einem der am schnellsten wachsenden Bereiche in der Entdeckung niedermolekularer Wirkstoffe entwickelt, was durch den therapeutischen Erfolg immunmodulatorischer molekularer Klebstoffverbindungen (z. B. IMiD) zur Krebsbehandlung und vielversprechende frühe klinische Studiendaten von Proteolysis Targeting Chimera-Verbindungen 1,2,3,4,5,6,7,8, 9,10,11,12. Gezielte Proteinabbauverbindungen wirken, indem sie ein Zielprotein mit E3-Ligase-Maschinenproteinen in die Nähe bringen 1,2,3,4,5,6,7,8,9,10,11,12 . Diese verbindungsinduzierte Rekrutierung des Zielproteins an die E3-Ligase führt zur Ubiquitinierung und zum Abbau des Zielproteins über den Ubiquitin-Proteasomenweg (UPP)1,2,3,4,5,6,7,8,9,10,11,12 . In der Vergangenheit stützten sich niedermolekulare Wirkstoffforschungsprogramme auf erste biochemische Assays, um die Aktivität zu bewerten und Verbindungen in die Rangordnung zu ordnen. Dies stellt jedoch eine erhebliche Herausforderung für gezielte Proteinabbauer dar, deren endgültige Aktivität, der Abbau über das Proteasom, von einer Kaskade zellulärer Ereignisse abhängt 1,2,4,5,6,11,12,13,14,15,16,17, 18. Die vielfältigen Signalwege und die Komplexität von Proteinkomplexen, die für den erfolgreichen Zielabbau erforderlich sind, erfordern zelluläre Assay-Ansätze für ein frühzeitiges Screening und Triaging erster Verbindungen. Derzeit mangelt es an Technologien zur Überwachung des Zielproteinabbaus im Hochdurchsatz im Kontext der zellulären Umgebung14. Hier werden wir Protokolle für die Echtzeitbewertung der kinetischen Lebendzell- oder Endpunkt-lytischen Abbauaktivität unter Verwendung von CRISPR/Cas9 endogen markierten HiBiT-Zielzelllinien 18,19,20 vorstellen, um den Verlust des Zielproteins durch Lumineszenzmessung nach Behandlung mit den Degradierverbindungen10,11,18,19 zu überwachen.
Um einen erfolgreichen Abbau therapeutischer Ziele zu erreichen und das medikamentöse Proteom zu erweitern, haben sich zahlreiche Ansätze und Arten von Degradern herausgebildet, die auf eine breite Palette von Proteinen zur Zerstörung abzielen können, einschließlich solcher, die an oder in der Plasmamembran, Lysosomen, mitochondrialen Membranen, Zytoplasma und dem Zellkernlokalisiert sind 21–57. Die beiden Hauptklassen von Verbindungen, die am ausführlichsten untersucht wurden, sind molekulare Klebstoffe und Proteine, die auf Cimeras 2,4,5,6,7,12,26 abzielen. Molekulare Klebstoffe sind monovalent, daher typischerweise kleiner und ermöglichen eine neuartige Protein-Protein-Interaktionsschnittstelle mit einem Zielprotein bei Bindung an eine E3-Ligasekomponente 2,12,26. Sie sind am häufigsten Degrader, die an die Cereblon (CRBN) E3-Ligasekomponente 2,12,26,55,56,57 binden. In jüngster Zeit zeigen aufregende neue Beispiele, die andere E3-Ligasemaschinen wie DCAF15 58,59,60 und CDK/Cyclin-Rekrutierung auf DDB145 verwenden, das Potenzial für die Erweiterung dieser Klasse von Verbindungen. Im Gegensatz dazu sind PROTACs größere, bivalente Moleküle, bestehend aus einem Zielbindungsliganden, meistens einem Inhibitor, der über einen chemischen Linker zu einem E3-Ligasengriff 1,3,4,5,7,13 überbrückt wird. Als solche sind diese Verbindungen in der Lage, direkt sowohl an die E3-Ligase als auch an das Zielprotein 1,3,4,5,7,13 zu binden. Es wurde gezeigt, dass zahlreiche Proteine über diese bivalenten Moleküle abgebaut werden, und die am häufigsten verwendeten E3-Ligasengriffe rekrutieren entweder CRBN oder Von Hippel Lindau (VHL)1,3,4,5,7,13. Die Anzahl der verfügbaren Griffe für die E3-Ligase-Rekrutierung in Chimären, die auf das Proteolysedesign abzielen, wächst jedoch schnell und erweitert die Fähigkeiten dieser Klasse von Verbindungen mit dem Potenzial, verschiedene Zielklassen abzubauen und die Zell- oder Gewebetypspezifität zu verbessern 24,48,61,62 . In Kombination mit der minimalen Anforderung, ein Zielprotein zu aktivieren, selbst mit marginaler Affinität, versprechen Abbauverbindungen die Erweiterung des medikamentösen Proteoms.
Die Charakterisierung der zellulären Dynamik des Proteinverlusts sowie der potenziellen Proteinrückgewinnung nach der Behandlung ist entscheidend für das Verständnis der Funktion und Wirksamkeit von Abbauverbindungen. Während es möglich ist, endogene Proteinspiegeländerungen in relevanten zellulären Systemen mit Western-Blot-Antikörper-Assays oder Massenspektrometrie zu untersuchen, sind diese Ansätze schwer an Hochdurchsatz-Screening-Formate anzupassen, haben eine begrenzte Quantifizierungsfähigkeit oder die Fähigkeit, kinetische Veränderungen zu vielen Zeitpunkten zu messen14. Um diesen Herausforderungen zu begegnen, haben wir ein plattenbasiertes zelluläres Lumineszenzsystem zur Überwachung von Veränderungen der endogenen Proteinspiegel entwickelt, das genomische Insertion über CRISPR / Cas9 des 11-Aminosäuren-Tags HiBiT in die Loci eines wichtigen Abbauziels18,19,20 nutzt. Dieses Peptid ergänzt sich mit hoher Affinität zu seinem Bindungspartner LgBiT, um in Gegenwart seines Substrats 18,19,20,63 helle Lumineszenz zu erzeugen, wodurch diese markierten endogenen Proteine in Zellen oder Lysaten leuchten18,19,20,63 . Die mit einem Luminometer gemessenen relativen Lichteinheiten (RLUs) sind direkt proportional zu den markierten Zielproteinspiegeln 18,19,20,63. Mit der Entwicklung stabilisierter Luciferase-Substrate sind kinetische Proteinspiegelmessungen in Echtzeit über 24-48 h Zeiträume möglich 18,53,64. Dies ermöglicht die Bestimmung eines vollständigen Abbauprofils für jedes gegebene Ziel bei jeder gegebenen Verbindungskonzentration, einschließlich der quantitativen Analyse der anfänglichen Abbaurate, des Abbaumaximums (Dmax) und der Wiederfindung nach der Behandlung von Verbindungen18,53. Beim Screening großer Bibliotheken von Abbausubstanzen kann die Endpunktanalyse jedoch auch problemlos im 384-Well-Format bei verschiedenen Wirkstoffkonzentrationen und bestimmten Zeitpunkten durchgeführt werden.
Die in diesem Manuskript vorgestellten Protokolle stellen zelluläre Screening-Strategien für gezielte Proteinabbauverbindungen dar, die für alle Arten von Degradern anwendbar sind. Die Verwendung von HiBiT-CRISPR-Zelllinien zusammen mit diesen Protokollen ist jedoch nicht auf den Proteinabbau beschränkt, sondern sie sind allgemeine Werkzeuge zur Überwachung eines endogenen Zielproteinspiegels, der nach der Behandlung moduliert werden könnte, um den Einfluss von Verbindungen oder sogar Resistenzmechanismen zu untersuchen 20,65,66. Eine Voraussetzung für diese lumineszenzbasierten Detektionsmethoden ist eine CRISPR-endogen markierte HiBiT-Zielzelllinie, die entscheidend ist, da sie eine empfindliche Lumineszenzdetektion ermöglicht und gleichzeitig die endogene Zielexpression und die native Promotorregulation18,19,20 beibehält. Signifikante Fortschritte wurden bei der Verwendung von CRISRP/Cas9 für das Einfügen genomischer Tags erzielt, insbesondere in Bezug auf die Skalierbarkeit 20 und mit der hohen Empfindlichkeit der Detektion in verschiedenen Formaten, einschließlich CRISPR-Pools oder Klonen mit heterozygoten oder homozygoten allelischen Insertionen18,19,20. Die Verwendung der exogenen Expression von HiBiT oder anderer Reporterfusionen in Zellen anstelle einer endogenen Markierung ist möglich, aber bei Systemen mit Proteinüberexpression14,18 ist erhebliche Vorsicht geboten. Diese können zu Artefakten im Verständnis der wahren Wirkstoffpotenz und Proteinwiederherstellungsdynamikführen 14,18, einschließlich potenzieller transkriptioneller Rückkopplungsschleifen, die nach dem Abbau des Ziels aktiviert werden. Darüber hinaus könnten Verbindungen im Frühstadium mit geringer Potenz übersehen werden und sich im Screening als falsch negativ darstellen. Da Proteinverlust durch verbindungsinduzierte Toxizität und Zelltod entstehen könnte, enthalten die hier beschriebenen Protokolle dringend empfohlene, aber optionale Zelllebensfähigkeits-Lumineszenz- oder Fluoreszenzassays gepaart mit dem Abbauprotokoll. Das Protokoll besteht aus zwei Hauptabschnitten, dem lytischen Endpunkt und dem kinetischen Screening lebender Zellen. In jedem dieser Abschnitte sind Optionen für Multiplex-Zelllebensfähigkeitsmessungen in Endpunkt- oder kinetischen Formaten enthalten. Die Überwachung der Veränderungen des markierten endogenen Proteins erfordert eine Ergänzung mit LgBiT in Zellen. Daher verweist die kinetische Screening-Sektion auf wichtige Protokolle für die Einführung dieser, die durch transiente oder stabile Expression erreicht werden können und für die Durchführung der Lebendzelllumineszenzmessungen unerlässlich sind. Alle hier vorgestellten Ansätze ermöglichen eine schnelle Rangordnung und Aktivitätsbewertung von Verbindungen, was ein frühes Screening von Verbindungen und eine schnellere Identifizierung von Bleiabbauern ermöglicht.
Dieses Protokoll ist für die Untersuchung von Abbauverbindungen in Verbindung mit einer HiBiT-CRISPR-Zelllinie konzipiert. Protokolle für die Erzeugung von HiBiT-CRISPR-Insertionen für zahlreiche Ziele wurden in mehreren neueren Publikationen 18,19,20 beschrieben.
Protokoll
1. Endpunktabbaustudien mit HiBiT CRISPR-Zielproteinen im lytischen Format mit optionaler Zelllebensfähigkeitsfluoreszenzanalyse
- Herstellung und Beschichtung von Säugetier-Adhärenten- oder Suspensionszelllinien
- Stellen Sie die Zelldichte auf 2,22 x 105/ml durch Verdünnung in geeigneten Zellmedien ein, die für Passaging und Zellwachstum verwendet werden.
- Dosieren Sie die Zellen in Platten mit mindestens 3 Vertiefungen pro Versuchs- und Kontrollbedingung. Geben Sie 90 μL (20.000 Zellen) pro Vertiefung der Zellsuspension in 96-Well-weiße Platten. Für das 384-Well-Format 36 μL (8.000 Zellen) pro Vertiefung der Zellsuspension in weiße 384-Well-Platten dosieren.
- Herstellung und Zusatz von Verbindungen
- Herstellung von seriell verdünnten PROTAC- oder Degrader-Testverbundplatten bei 1.000x Endkonzentration in 100% DMSO. Dann verdünnen Sie es auf die 10-fache Endkonzentration im Zellkulturmedium. Fügen Sie dem Medium ein gleiches Volumen DMSO hinzu, um es als DMSO-Steuerelement ohne Verbindung zu verwenden.
- Für das 96-Well-Format fügen Sie 10 μL 10x Compound- und Kontrolllösungen zu 90 μL Zellen hinzu. Für das 384-Well-Format fügen Sie 4 μL 10x Compound- und Kontrolllösungen zu 36 μL Zellen hinzu.
- Inkubieren Sie die Platten in einem Inkubator bei 37 °C und 5%CO2 für die gewünschte Zeit oder unter Bedingungen, die für ihr Wachstum optimal sind.
HINWEIS: Da es sich um einen Endpunkttest handelt, erfordert das Testen mehrerer Zeitpunkte die Vorbereitung separater Abbauplatten für jeden Zeitpunkt, wie in Schritt 1.1.2 beschrieben. Die Inkubationszeiten zum Nachweis des durch Verbindungen vermittelten Abbaus sind sehr variabel und hängen wahrscheinlich auch von der Konzentration der Verbindung ab. Vorgeschlagene Anfangszeitpunkte wären 6 h und 24 h. - Wenn Sie die Endpunkt-Lumineszenzerkennung ohne die optionale Messung der Zelllebensfähigkeit messen, fahren Sie direkt mit Schritt 1.3 fort. Wenn Sie Multiplexing mit Zelllebensfähigkeitsmessung durchführen, fahren Sie mit dem nächsten Abschnitt 1.4 fort.
- Lytische Messung von Zellen
- Unmittelbar vor den lytischen HiBiT-Messungen 2x lytisches Detektionsreagenz durch Zugabe von 20 μL lytischem Substrat und 10 μL LgBiT-Protein pro 1 ml des lytischen Puffers vorbereiten. Bereiten Sie genügend 2x-Detektionsreagenz für die Anzahl der zu untersuchenden Vertiefungen vor, einschließlich eines zusätzlichen Volumens zur Berücksichtigung von Pipettierfehlern (d. h. Anzahl der Vertiefungen + 10%).
- Fügen Sie vorbereitetes lytisches Detektionsreagenz zu den Zellen hinzu. Für das 96-Well-Format fügen Sie 100 μL 2x lytisches Detektionsreagenz zu jeder Vertiefung hinzu, die 100 μL Zellen enthält. Für das 384-Well-Format fügen Sie 40 μL 2x lytisches Detektionsreagenz zu jeder Vertiefung hinzu, die 40 μL Zellen enthält. Mischen Sie die Platte auf einem Mikroplatten-Wirbelmischer für 10-20 min bei 350 U/min.
- Messen Sie die Lumineszenz auf einem Luminometer, das die Lumineszenz in einer 96- oder 384-Well-Platte lesen kann.
- Optionales Zelllebensfähigkeitsmultiplexing
HINWEIS: Dieser Schritt wird mit einem handelsüblichen CellTiter-Fluor (CTF)-Kit durchgeführt (siehe Materialtabelle).- 30-40 min vor der gewünschten Endpunktmessung eine 6x Reagenzlösung zum Nachweis der Zelllebensfähigkeit vorbereiten, indem 10 μL des Substrats zu 2 ml des Assay-Puffers gegeben werden. Bereiten Sie genügend 6x Reagenz für jede zu untersuchende Vertiefung vor, einschließlich eines zusätzlichen Volumens für Pipettierfehler (d. H. Anzahl der Vertiefungen + 10%).
- Fügen Sie das vorbereitete Reagenz in Vertiefungen hinzu. Für das 96-Well-Format fügen Sie 20 μL 6x Reagenz zu jeder Vertiefung hinzu, die bereits ein Volumen von 100 μL enthält. Für das 384-Well-Format fügen Sie 8 μL 6x Reagenz zu jeder Vertiefung hinzu, die 40 μL Zellen enthält. Mischen Sie kurz auf einem Mikrotiterplatten-Wirbelmischer, dann inkubieren Sie die Platte für 30 min in einem 37 °C Inkubator.
- Messen Sie am gewünschten Messendpunkt (d. h. 6 oder 24 Stunden nach der Behandlung, Schritt 1.2.3) die Fluoreszenz auf einem Instrument, das Fluoreszenz (380-400nmEx/505nmEm) im 96- oder 384-Well-Format lesen kann.
- Bereiten Sie 2x lytisches Detektionsreagenz durch Zugabe von 20 μL lytischem Substrat und 10 μL LgBiT-Protein pro 1 ml lytischem Puffer vor. Bereiten Sie genügend 2x-Detektionsreagenz für die Anzahl der zu untersuchenden Vertiefungen vor, einschließlich eines zusätzlichen Volumens zur Berücksichtigung von Pipettierfehlern (z. B. Anzahl der Vertiefungen + 10%).
- Fügen Sie das vorbereitete Reagenz für lytische Detektion in Bohrlöcher hinzu. Für das 96-Well-Format fügen Sie 120 μL 2x lytisches Detektionsreagenz zu jeder Vertiefung hinzu, die bereits ein Volumen von 120 μL enthält. Für das 384-Well-Format fügen Sie 48 μL 2x lytisches Detektionsreagenz zu jeder Vertiefung hinzu, die bereits ein Volumen von 48 μL enthält. Mischen Sie die Platte auf einem Mikrotiterplatten-Wirbelmischer für 10-20 min.
- Messen Sie die Lumineszenz auf einem Luminometer, das die Lumineszenz in 96- oder 384-Well-Platten lesen kann.
- Quantifizierung des Abbaus und der Zelllebensfähigkeit
- Mittelwert der relativen Lichteinheiten (RLU) aus dem DMSO-Steuerelement zum gemessenen Zeitpunkt. Verwenden Sie diesen Wert als Ausgangsproteingehalt des Ziels, um den fraktionierten Abbau zu berechnen, indem alle anderen zu diesem Zeitpunkt getesteten Behandlungen auf diesen Wert verweisen. Wenn beispielsweise die durchschnittliche RLU für die DMSO-Kontrollbohrungen bei 6 h 10.000 und die RLU für eine gegebene Verbindungsbehandlung bei 6 h 5.000 beträgt, würde der fraktionierte Abbau als 5.000 ÷ 10.000 = 0,5 berechnet (Gleichung 1).
Gleichung 1:
- Bestimmen Sie die prozentuale Degradation aus fraktionierter RLU:
Gleichung 2:
- Zeichnen Sie fraktionierten RLU- oder %-Abbau zu bestimmten Zeitpunkten auf, um die Aktivität von Verbindungen zu bewerten.
- Optional können Sie RFU-Daten (Relative Fluoresecence Unit) für die Messung des Zelllebensfähigkeitstests analysieren, indem Sie die Werte aller Behandlungen mit der DMSO-Kontrolle vergleichen. Wenn ein signifikanter Rückgang der RFU für eine Behandlung im Vergleich zur DMSO-Kontrolle beobachtet wird, können die Abbaudaten zusätzlich zu den Zelllebensfähigkeitstestdaten normalisiert werden, um Änderungen des Proteinspiegels relativ zu Verlusten in der Zelllebensfähigkeit zu bestimmen.
- Mittelwert der relativen Lichteinheiten (RLU) aus dem DMSO-Steuerelement zum gemessenen Zeitpunkt. Verwenden Sie diesen Wert als Ausgangsproteingehalt des Ziels, um den fraktionierten Abbau zu berechnen, indem alle anderen zu diesem Zeitpunkt getesteten Behandlungen auf diesen Wert verweisen. Wenn beispielsweise die durchschnittliche RLU für die DMSO-Kontrollbohrungen bei 6 h 10.000 und die RLU für eine gegebene Verbindungsbehandlung bei 6 h 5.000 beträgt, würde der fraktionierte Abbau als 5.000 ÷ 10.000 = 0,5 berechnet (Gleichung 1).
2. Kinetischer Abbau von HiBiT-CRISPR-Zielproteinen in Echtzeit und optionaler Zelllebensfähigkeits-Lumineszenz-Assay
ANMERKUNG: Die Fähigkeit, kinetisches Screening und Abbau durchzuführen, erfordert eine LgBiT-Protein-Coexpression in der Zelle, die zuvorbeschrieben wurde 18,19,63. Dies kann durch transiente Transfektion eines LgBiT-Vektors, Verwendung von BacMam LgBiT oder durch HiBiT-CRISPR-Insertion in eine LgBiT-stabile Zelllinie erreicht werden.
- Plattierung von adhärenten Zelllinien.
- Entfernen Sie das Medium aus dem Zellkolben durch Aspiration, waschen Sie die Zellen mit DPBS, dissoziieren Sie die Zellen mit 0,05% Trypsin-EDTA und lassen Sie die Zellen vom Kolbenboden dissoziieren. Für Suspensionszelllinien fahren Sie mit Abschnitt 2.2 fort.
- Trypsin mit serumhaltigem Zellkulturmedium neutralisieren, Mischen zum Sammeln und Resuspendieren von Zellen und Übertragen der Zellsuspension in ein konisches Röhrchen.
- Zellen mit 125 x g für 5 min herunterdrehen. Das Zellkulturmedium wird verworfen und in gleicher Menge frisches Zellkulturmedium resuspendiert.
- Plattenzellen in Assayplatten mit einem Minimum an dreifachen Vertiefungen pro Versuchs- und Kontrollbedingung. Für die 96-Well-Formatzählung zur Schätzung der Zelldichte stellen Sie die Dichte auf 2 x 105 Zellen/ml im Assay-Medium ein und dosieren Sie 100 μL (20.000 Zellen) pro Vertiefung in eine 96-Well-Platte. Für die 384-Well-Formatzählung zur Schätzung der Zelldichte passen Sie die Dichte auf 4,44 x 105 Zellen/ml im Assay-Medium an und dosieren Sie 18 μL (8.000 Zellen) pro Vertiefung.
- Platten bei 37 °C, 5% CO2 über Nacht oder unter für ihr Wachstum optimalen Bedingungen inkubieren.
- Beschichtung von Suspensionszellen
- Die Zelldichte wird auf 2,22 x 105 Zellen/ml in CO 2-unabhängigem Medium eingestellt, ergänzt mit 10% FBS und 1x Endrazin (1:100 Verdünnung des Stammreagens).
- Plattenzellen in Assayplatten mit mindestens 3 Vertiefungen pro Versuchs- und Kontrollbedingung. Für das 96-Well-Format 90 μL (20.000 Zellen) pro Well. Für das 384-Well-Format 36 μL (8.000 Zellen) pro Well.
HINWEIS: Für Suspensionszelllinien mit geringer Signal-zu-Hintergrund-Lumineszenz (S:B), z. B. bei der Arbeit mit CRISPR-Pools anstelle von Klonen, ist es möglich, die Lumineszenz zu erhöhen, indem die Anzahl der beschichteten Zellen auf bis zu 100.000 Zellen / Vertiefung im 96-Well-Format oder 40.000 Zellen / Welle im 384-Well-Format erhöht wird.
- Kinetische Abbauassays mit HiBiT-CRISPR-Zellen, die LgBiT exprimieren
- Bei Suspensionszellen, die bereits Endurazin enthalten, das im Beschichtungsschritt in 2.2. enthalten war, fahren Sie direkt mit Schritt 2.3.3 fort. Für adhärente Zelllinien Nano-Glo Endurazine Lösung herstellen. Für das 96-Well-Format wird eine 1x-Lösung von Endurazin hergestellt, indem das Stammreagenz 1:100 in CO 2-unabhängiges Medium verdünnt wird, das mit 10% FBS ergänzt wird. Für das 384-Well-Format wird eine 2x-Lösung von Endurazin hergestellt, indem das Stammreagenz 1:50 in ein CO 2-unabhängiges Medium verdünnt wird, das mit 10% FBS ergänzt wird.
- Fügen Sie Endurazin-Lösung zu jeder Vertiefung der adhärenten Zellen hinzu. Für 96-Well-Format aspirieren Medium und fügen Sie 90 μL 1x Endurazine Lösung hinzu. Für das 384-Well-Format fügen Sie 18 μL 2x Endurazin-Lösung zu 18 μL Zellen hinzu. Das Medium darf nicht abgesaugt werden, da der Abbautest in einer 50:50-Mischung aus Kulturmedium und CO2-unabhängigem Medium im 384-Well-Format durchgeführt wird.
- Inkubieren Sie Suspensions- oder anhaftende Zellplatten, die Endurazin enthalten, für 2,5 h in einem Inkubator bei 37 °C und 5%CO2 , damit die Lumineszenz ausgeglichen werden kann.
- Bereiten Sie eine 10-fache Konzentration der Test-PROTAC-Titration in CO 2-unabhängigem Medium vor und geben Sie 10 μL zu jeder Vertiefung der 96-Well-Platte oder 4 μL für 384-Well-Platte. Für Verbindungen mit unbekannter Wirksamkeit wird eine Endkonzentration von 1-10 μM am höchsten Punkt als Ausgangspunkt empfohlen.
- Sammeln Sie kinetische Messungen der Lumineszenz in einem auf 37 °C voräquilibrierten Luminometer für einen Zeitraum zwischen 0-48 h. Die Zeitschritte der Messung können für jedes Experiment angepasst werden, aber ein empfohlenes Anfangsexperiment wären Lumineszenzmessungen alle 5-15 Minuten für 24 Stunden oder die gewünschte Zeitspanne.
- Optionale Zelllebensfähigkeits-Multiplexanalyse nach abschließender kinetischer Messung
HINWEIS: Dieser Assay wird mit einem handelsüblichen CellTiter-Glo (CTG)-Kit durchgeführt (siehe Materialtabelle).- CTG-Reagenz auf Raumtemperatur ausgleichen.
- Nach der Abbaumessung zum letzten Zeitpunkt der kinetischen Analyse werden 100 μL (96-Well-Platte) oder 40 μL (384-Well-Platte) des Reagenzes pro Vertiefung der Platte hinzugefügt und auf einem Plattenschüttler bei 500-700 U/min (96-Well-Platte) oder einem Mikroplatten-Wirbelmischer (384-Well-Platte) für 5 min gemischt.
- Inkubieren Sie die Platte bei Raumtemperatur für 30 Minuten, um die Zelllyse und das Löschen des HiBiT-Signals zu ermöglichen.
- Messen Sie die Gesamtlumineszenz auf einem Luminometer gemäß der Empfehlung des Herstellers.
- Quantifizierung kinetischer Abbauprofile
- Normalisieren Sie unter Verwendung der gesammelten kinetischen Lumineszenzmessungen die Roh-RLUs für jede PROTAC-Konzentration auf die replizierte gemittelte DMSO-Bedingung zu jedem Zeitpunkt, um Änderungen der freien Furimazinkonzentration im Laufe der Zeit zu berücksichtigen. Berechnen Sie die fraktionierte RLU mithilfe von Gleichung 1.
Gleichung 1: - Passen Sie aus den Degradationskurven ein einkomponentiges exponentielles Zerfallsmodell unter Verwendung von Gleichung 2 an den anfänglichen Degradationsabschnitt jeder Kurve bis zu dem Punkt an, an dem die Daten ein Plateau erreichen.
HINWEIS: Es kann hilfreich sein, die ersten Datenpunkte von der Anpassung auszuschließen, da es eine kurze Verzögerung geben kann, bevor eine Verschlechterung beobachtet wird.
Gleichung 2:
- Bestimmen Sie aus Gleichung 2 den Parameter ƛ, der die Abbaugeschwindigkeitskonstante darstellt, und das Plateau, das die geringste verbleibende Proteinmenge darstellt.
- Berechnen Sie Dmax, die die maximale Teilmenge an abgebautem Protein ist und als 1-Plateau berechnet wird.
- Zeichnen Sie Dmax für jede Konzentration von PROTAC auf, um eine zeitunabhängige Abbaupotenzkurve zu bestimmen.
- Bestimmen Sie den Dmax-50-Wert für das Diagramm in 2.3.5, um die Wirksamkeit von Verbindungen zu analysieren.
HINWEIS: Um einen DC50 zu einem bestimmten Zeitpunkt zu bestimmen, zeichnen Sie die berechnete prozentuale Degradation für jede Konzentration zum gewählten Zeitpunkt auf. Dies kann als DC 50 t=4 h oder DC50 t=12 h angegeben werden.
- Normalisieren Sie unter Verwendung der gesammelten kinetischen Lumineszenzmessungen die Roh-RLUs für jede PROTAC-Konzentration auf die replizierte gemittelte DMSO-Bedingung zu jedem Zeitpunkt, um Änderungen der freien Furimazinkonzentration im Laufe der Zeit zu berücksichtigen. Berechnen Sie die fraktionierte RLU mithilfe von Gleichung 1.
Ergebnisse
Um die Analyse des lytischen Abbaus mit einem Endpunkt zu demonstrieren, mehrere CDK-Zielproteine; CDK2, CDK4, CDK7 und CDK10 wurden endogen mit HiBiT an ihrem C-Terminus in HEK293-Zellen markiert und mit einer 1 μM-Konzentration des Pan-Kinase-Cereblon-basierten PROTAC, TL12-18654 behandelt (Abbildung 1A). Der Gehalt an CDK-Protein wurde zu verschiedenen Zeitpunkten gemessen und die fraktionierte RLU relativ zur DMSO-Kontrolle bestimmt (Abbildung 1A). Jedes CDK-Protein zeigte unterschiedliche Abbaugrade als Reaktion auf die Behandlung der Verbindung und die verschiedenen Zeitpunkte (Abbildung 1A). Um zu verstehen, wie CDK-Proteine in Bezug auf den Proteinverlust direkt miteinander verglichen werden, wurden die fraktionierten RLUs in Abbildung 1A als Gesamtabbau in % berechnet und für jeden Zeitpunkt in Abbildung 1B aufgetragen. Dies zeigt, dass selbst zu frühen Zeitpunkten, 2 oder 4 h, einige Mitglieder der CDK-Familie einen hohen Abbau aufweisen, der im Laufe der Zeit weiter nach oben tendiert (Abbildung 1B).
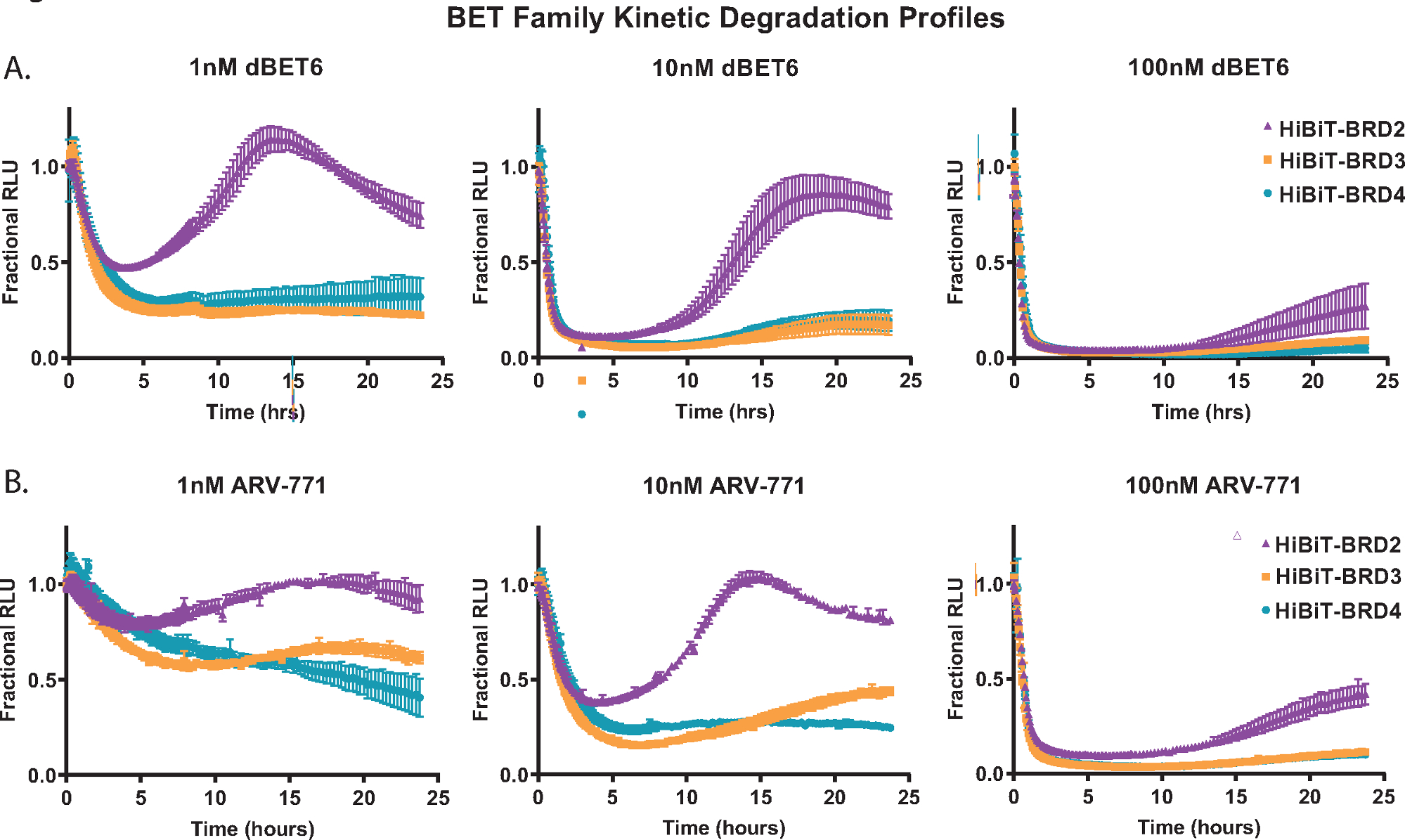
Um die kinetische Abbauanalyse zu demonstrieren, jedes der Mitgliedsproteine der BET-Familie; BRD2, BRD3 und BRD4 wurden endogen mit HiBiT an ihrem N-Terminus in HEK293-Zellen markiert, die das LgBiT-Protein18 stabil exprimieren. Diese wurden dann mit drei verschiedenen Konzentrationen der pan-BET PROTACs behandelt; das Cereblon-basierte dBET650 (Abbildung 2A) und das VHL-basierte ARV-77141 (Abbildung 2B). Kinetische Messungen wurden über einen Zeitraum von 24 Stunden gesammelt, und aus den Profilen bei jeder Konzentration sind die Unterschiede in der Reaktion der Mitglieder der BET-Familie leicht ersichtlich. Die Fähigkeit von BRD2, eine schnellere Wiederfindungsreaktion nach der Behandlung von Abbauverbindungen einzuleiten (Abbildung 2A,B), wurde zuvor bei anderen pan-BET-PROTACs beobachtet und ist wahrscheinlich auf eine transkriptionelle Rückkopplungsreaktion zurückzuführen, die mit dem Abbauprozess konkurriertist 18.
Sowohl die Endpunkt- als auch die kinetische Analyse können mit vollständigen Dosis-Wirkungs-Behandlungen durchgeführt werden. In Abbildung 3 sind kinetische Dosis-Wirkungs-Abbauprofile der Behandlung von Ikaros/IKZF1-HiBiT CRISPR Jurkat-Zellen gezeigt, die das LgBiT-Protein stabil mit vier verschiedenen molekularen Klebstoffverbindungenexprimieren 2,26,55,57; Lenalidomid (Abbildung 3A), Iberdomid (CC-220) (Abbildung 3B), Thalidomid (Abbildung 3C) und Pomalidomid (Abbildung 3D). Diese Degrader zeigen signifikante Unterschiede in der Abbaureaktion zwischen den Verbindungen sowie über die Konzentrationsreihen hinweg (Abbildung 3).
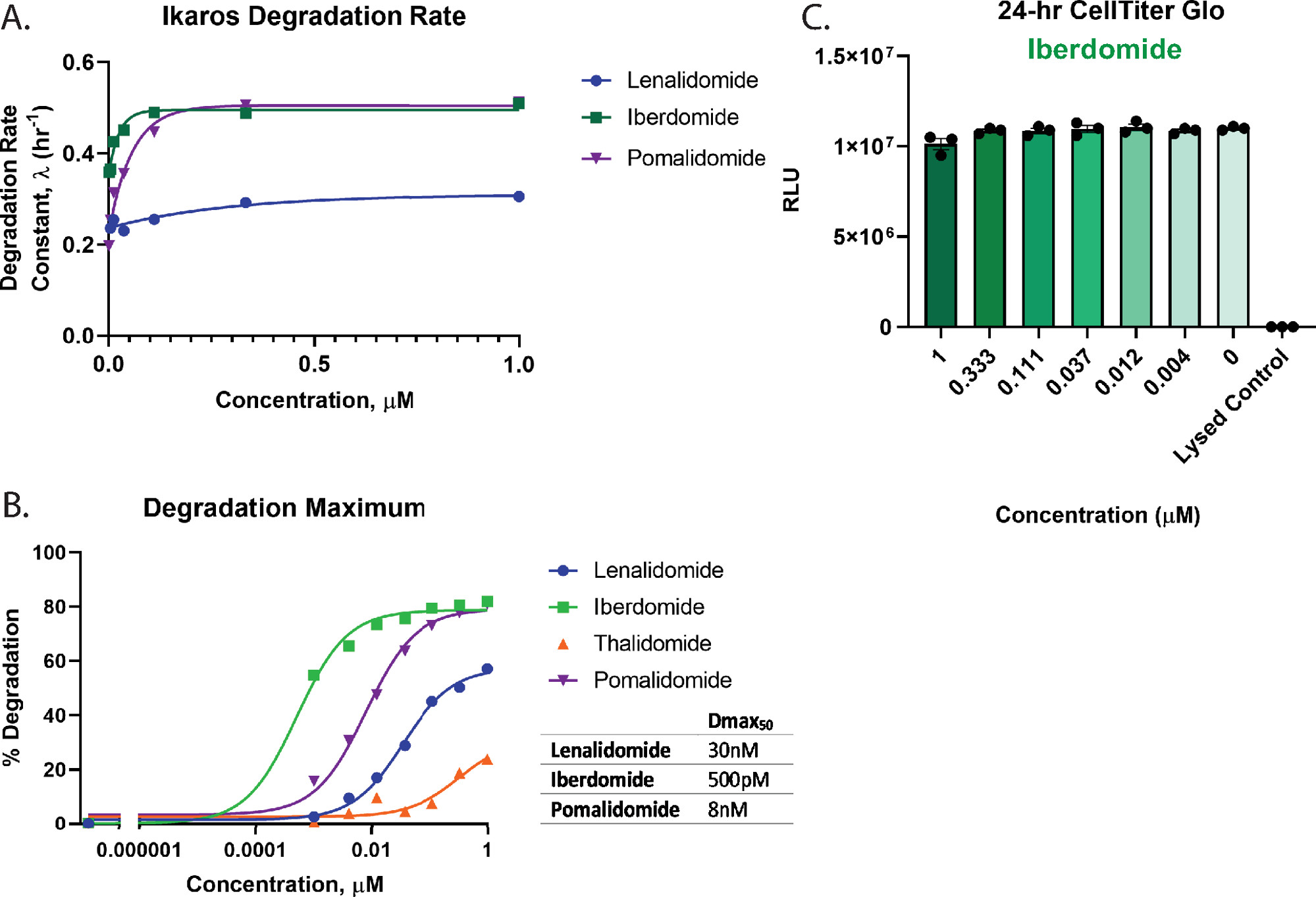
Um den Abbau und die Rangfolge der Verbindungen in Abbildung 3 quantitativ zu bewerten, wurden die Dosis-Wirkungs-Profile verwendet, um wichtige Abbauparameter einschließlich der Abbaurate (Abbildung 4A), Dmax (Abbildung 4B) und Dmax50-Werte (Abbildung 4B) zu berechnen. Diese Analysen zeigen, dass Iberdomid (CC-220) und Pomalidomid sehr ähnliche schnelle anfängliche Abbauraten aufweisen (Abbildung 4A), aber Iberdomid (CC-220) hat die höchste Potenz, wie zuvor in orthogonalen Studien beobachtet wurde55,57 (Abbildung 4B). Da Iberdomid eine so hohe Wirksamkeit aufweist und alle getesteten Konzentrationen einen Abbau von mehr als 50% aufweisen, stellt der für Iberdomid erhaltene Dmax50-Wert eine Schätzung dar, die auf der Einschränkung bei der genauen Anpassung der Daten basiert. Aus den Grafiken in Abbildung 3C,D und Abbildung 4B geht hervor, dass weder Lenalidomid noch Thalidomid das Ikaros/IKZF1-Ziel bei den höchsten getesteten Konzentrationen bis zur Vollendung abbauen. Aufgrund des sehr geringen Abbaus mit Thalidomid konnten die Abbauspuren nicht genau an ein exponentielles Zerfallsmodell angepasst werden, daher wurde die Abbaurate für diese Behandlung nicht quantifiziert. Für den stärksten Degrader, Iberdomid (CC-220)55,57 (Abbildung 4B). Zelllebensfähigkeits-Multiplex-Assays zeigten keinen Verlust an Zelllebensfähigkeit für die getesteten Konzentrationen (Abbildung 4C).

Abbildung 1: CDK-Endpunktabbau und Toxizität mit Pan-Kinase PROTAC, TL12-18654. (A) Auswahl eines Panels endogener CDK-Zielproteine, die mit HiBiT am C-Terminus über CRISPR/Cas9 fusioniert und mit 1 μM TL12-186 PROTAC 54 nach 2 h, 4 h, 8 h und24 h Behandlung auf Abbau untersucht wurden. Die Werte werden als Fractional RLU relativ zu einem DMSO-Steuerelement dargestellt, das zu jedem Zeitpunkt gemessen wird. Fehlerbalken stellen SD des Mittelwerts von 3 technischen Replikaten dar. (B) Prozentualer Abbau des Panels von CDK-Zielproteinen, berechnet aus (A), der die Menge des Abbaus jedes Familienmitglieds darstellt, die zu 2-, 4-, 8- und 24-Stunden-Zeitpunkten beobachtet wurde. Fehlerbalken stellen SD des Mittelwerts von 3 technischen Replikaten dar. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 2: Profiling der kinetischen Abbauselektivität von Mitgliedern der BET-Familie mit BET-Degradern, dBET650 und ARV-77141. Kinetische Abbauprofile von endogenen Mitgliedern der BET-Familie, BRD2, BRD3 und BRD4, markiert mit HiBiT am N-Terminus über CRISPR/Cas9 mit Behandlung von Einzelkonzentrationen von 1 nM (links), 10 nM (Mitte) oder 100 nM (rechts) dBET650 (A) oder ARV-77141 (B) PROTACs. Die Werte werden als fraktionierte RLU dargestellt, die von einem DMSO-Steuerelement zu jedem kinetischen Zeitpunkt berechnet werden. Fehlerbalken stellen SD des Mittelwerts von 4 technischen Replikaten dar. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 3: Dosis-Wirkungs-Profile des kinetischen Abbaus lebender Zellen von Ikaros/IKZF1-HiBiT mit einem molekularen Klebstoffpanel 2,26,55,57. Jurkat-Zellen, die das LgBiT-Protein stabil exprimieren, wurden mit CRISPR/Cas9 hergestellt, um den C-Terminus von Ikaros/IKZF1 mit dem HiBiT-Peptid zu markieren. Die Zellen wurden mit einer 8-Punkt-Dosis-Wirkungs-Konzentrationsreihe einschließlich DMSO von vier verschiedenen molekularen Klebstoffverbindungen 2,26,55,57 behandelt: (A) Lenalidomid, (B) Iberdomid (CC-220), (C) Thalidomid oder (D) Pomalidomid. Die Lumineszenz wurde alle 5 min für insgesamt 19,5 h gemessen. RLU-Daten (Relative Light Unit) von (A-D) wurden wie in Schritt 2.4.1 beschrieben in fraktionierte RLU konvertiert und als Funktion der Zeit grafisch dargestellt. Fehlerbalken stellen SD von 3 technischen Replikaten dar. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 4: Berechnung der Abbaurate und Dmax50 für Ikaros/IKZF1-HiBiT und Multiplexing-Zellgesundheitstests. Kinetische Abbaudaten aus Abbildung 3 wurden verwendet, um quantitative Abbauparameter zu berechnen. (A) Abbauraten und (B) Abbauhöchstwerte (Dmax) werden bei jeder Wirkstoffkonzentration für die angegebenen molekularen Klebstoffverbindungen 2,26,55,57 grafisch dargestellt. (B) Dmax50-Werte für jede Verbindung wurden unter Verwendung eines Dosis-Wirkungs-Modells mit eingeschränkter Hill-Steigung von 1 berechnet, das verwendet werden kann, um Ordnungsabbauverbindungen für ein Ziel zu bewerten. (C) Zelllebensfähigkeitstests mit der Abbaudosis-Wirkungs-Beziehung von Iberdomid (CC-220)55,57 aus Abbildung 3B wurden nach Abschluss der kinetischen Abbaumessungen als Endpunktmessung durchgeführt. Fehlerbalken stellen SD von 3 technischen Replikaten dar. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Diskussion
Wir stellen hier zwei Methoden zum Screening der Abbausubstanzaktivität entweder im Endpunkt-lytischen Format oder im kinetischen Lebendzellmodus vor. Diese Ansätze basieren auf den gleichen lumineszierenden Messprinzipien, bieten jedoch unterschiedliche Detail- und Verständnisebenen. Die Wahl beider Ansätze hängt wahrscheinlich von den Screening-Zielen und der Größe der Substanzbibliothek ab. Für große Compound-Screening-Decks oder Primär-Screens, um jede nachweisbare Degradation zu beobachten, bietet das Endpunkt-Lytic-Screening empfindliche und effiziente Hochdurchsatz-Kompatibilität, wo andere Endpunktansätze, wie Western Blot oder Massenspektrometrie, entweder unpraktisch oder schwer anzupassen sind14. Ein Ausgangspunkt für diese Bildschirme könnte mit einer begrenzten Anzahl von Konzentrationen und Zeitpunkten durchgeführt werden. Die empfohlenen Anfangskonzentrationen liegen im Bereich von 100 nM-10 μM, um anfängliche Degrader mit geringer Potenz, schlechter Permeabilität oder in einigen Fällen mit hochwirksamen Verbindungen das Vorhandensein eines Hakeneffekts zu berücksichtigen. Es wird ferner empfohlen, mindestens zwei verschiedene Zeitpunkte zu testen, um einen früh einsetzenden Abbau nach 4-6 h und einen latenten oder anhaltenden Abbau nach 18-24 h festzustellen. Verbindungen, die eine hohe Abbaupotenz und einen On-Target-Mechanismus aufweisen, werden innerhalb eines Zeitrahmens von 4-6 h leicht beobachtet, während der Abbau oder scheinbare Proteinverlust, der nur zu späteren Zeitpunkten beobachtet wird, auf eine Vielzahl von Mechanismen zurückzuführen sein könnte. Es wird dringend empfohlen, die Zelllebensfähigkeit sowohl zu frühen als auch zu späten Zeitpunkten zu überwachen, so dass der Proteinverlust vom Verlust aufgrund des Zelltods entkoppelt werden kann. Ähnlich wie bei jeder Art von Lumineszenz- oder Fluoreszenztest besteht die Möglichkeit, dass Verbindungen innerhalb von Bibliotheken das Signal stören oder hemmen, daher sind orthogonale Folgeexperimente mit Leitverbindungen unter Verwendung nicht verwandter Fusionen oder alternativer Ansätze zur Überwachung des Proteinspiegels wichtig, um zu beurteilen, ob der Verlust von RLU in diesen Assays direkt mit dem Abbau des Zielproteins verbunden ist.
Die Fähigkeit, über längere Zeiträume im kinetischen Format lebender Zellen zu screenen, hängt stark vom Assay-Signal zum Hintergrund (S:B) ab. Zu den Faktoren, die zu S:B beitragen, gehören das Expressionsniveau des Zielproteins selbst, das mehrere Größenordnungen umfassen kann, die Effizienz der LgBiT-Expression in der für die Peptidinsertion ausgewählten Zelllinie, und die Verfügbarkeit des markierten Ziels zur Ergänzung in seinen verschiedenen nativen Komplexen. Wir haben eine allgemeine Cutoff-Anforderung festgelegt, die aus einem S:B von 15 besteht, um den Abbau im kinetischen Modus mit Endurazin oder Vivazin erfolgreich zu messen. Der S:B wird bestimmt, indem das Baseline-Signal der HiBiT-editierten Zellen, die LgBiT co-exprimieren, relativ zu uneditierten Elternzellen, die LgBiT allein exprimieren, in Gegenwart von lebenden Endurazin- oder Vivazin-Zellsubstraten gemessen wird. Vivazin erzeugt ein höheres Lumineszenzsignal, zerfällt jedoch schneller als Endurazin und kann die Signalerfassung auf 24 Stunden oder weniger begrenzen. Darüber hinaus kann S:B auch stark davon abhängig sein, ob CRISPR-Pools oder Klone verwendet werden. Für Ziele in Zelllinien, die zugänglicher sind und eine hohe Effizienz für das CRISPR/Cas9-Engineering aufweisen, kann eine heterogene CRISPR-Poolpopulation editierter Zellen ausreichend S:B für die kinetische Analyse aufweisen. Für Ziele in schwierigeren Zelllinien, bei denen eine weniger effiziente genomische Integration über CRISPR zu Pools mit niedrigem S:B führt, könnte die Isolierung von CRISPR-Klonen erforderlich sein, um editierte Populationen anzureichern und einen ausreichend hohen S:B für die kinetische Analyse zu erreichen. Für jedes dieser Szenarien, wenn S:B mit Endurazin- oder Vivazin-Substraten kleiner als 15 ist, wird ein lytisches Endpunkt-Screening empfohlen.
Zum besseren Verständnis und zur Charakterisierung von Verbindungen, einschließlich der Bestimmung eines Abbauprofils mit quantitativen Parametern, ist die kinetische Echtzeitanalyse in lebenden Zellen der empfohlene Screening-Ansatz14,18. Wie bei der oben beschriebenen Endpunktanalyse kann das anfängliche kinetische Screening mit einer begrenzten Anzahl von Konzentrationen im Bereich von 100nM-10μM im Hochdurchsatz durchgeführt werden. Im 384-Well-Format können über 100 Verbindungen in dreifacher Ausführung in einer Konzentration auf einer einzigen Platte gesiebt werden. Die resultierenden Abbauprofile geben nicht nur Hinweise auf das Ausmaß des beobachteten Abbaus, sondern auch auf die Abbaugeschwindigkeit, die Dauer des Abbaus und die mögliche Wiederfindung des Proteins14,18 (Abbildung 2 und Abbildung 3). Auch die Formen des Abbauprofils liefern wertvolle Informationen. Spezifische und potente Degrader zeigen oft einen anfänglichen schnellen Verlust des Zielproteins auf ein Plateau innerhalb von Stunden18,53, während andere Mechanismen wie transkriptionelle Rückkopplung oder Verbindungstoxizität typischerweise zu einem lineareren Verlust des Proteins im Laufe der Zeit führen. Diese Details und Nuancen werden bei der lytischen Endpunktanalyse übersehen, und bei der Echtzeitanalyse über 24-48 Stunden muss man nicht die Zeit vorhersagen, um die wahre Dmax in Sätzen neuer oder unbekannter Verbindungen zu erfassen.
Die Echtzeit-Kinetik ermöglicht auch ein effizientes Dosis-Wirkungs-Screening, um die Wirksamkeit von Verbindungen besser zu verstehen, wie sich die Konzentration der Verbindung auf die anfängliche Abbaurate auswirkt, und bietet Möglichkeiten, Verbindungen basierend auf mehr als einem Parameter zu bewerten. Klassische Messungen der Abbaupotenz beinhalten DC50-Berechnungen zu einem bestimmten Zeitpunkt basierend auf scheinbaren Abbaumaxima. Im Gegensatz dazu beinhaltet unser kinetischer Ansatz zur Bewertung der Potenz das wahre Abbaumaximum bei jeder Konzentration, unabhängig davon, wann sie in Zeit18 auftritt. Wir nennen diese Messung der kinetischen Abbaupotenz Dmax5018. Die Analyse auf diese Weise berücksichtigt Verbindungen, die den Abbau bei niedrigeren Konzentrationen langsamer initiieren können und daher eine längere Zeit nach der Behandlung benötigen, um ihre Dmax zu erreichen. Es kann besonders informativ sein, Verbindungen sowohl nach Abbaurate als auch nach Dmax zu bewerten. Für die stärksten Degrader wird dies langsame, aber potente Degrader weiter von denen unterscheiden, die sowohl schnell als auch potent sind. Zusammen sind sowohl lytisches als auch lebendzellkinetisches Screening unter Verwendung von HiBiT-CRISPR-Zelllinien leistungsstarke Ansätze, die ein umfassenderes Bild des gezielten Proteinabbaus und der Verbindungsfunktion liefern und den Screening-Prozess von der anfänglichen Aktivitätsbewertung bis zur nachgeschalteten chemischen Optimierung durch Verbesserung der wichtigsten Abbauparameter ermöglichen.
Offenlegungen
Promega Corporation ist der kommerzielle Eigentümer durch die Übertragung von Patenten der HiBiT- und NanoLuc-Technologien und -Anwendungen.
Danksagungen
K.M.R, S.D.M, M.U. und D.L.D sind alle Mitarbeiter der Promega Corporation
Materialien
| Name | Company | Catalog Number | Comments |
| CellTiter-Glo 2.0 reagent | Promega | G9241 | Cell Viability luminescent assay |
| CellTiter-Fluor Cell Viability Assay | Promega | G6080 | Cell Viability fluorescent assay |
| CO2-independent medium | ThermoFisher | 18045-088 | Cell culture |
| DMSO | Sigma Aldrich | D2650 | For compound dilution and control |
| DPBS | Gibco | 14190 | Cell culture |
| Fetal Bovine Serum | Seradigm | 89510-194 | Cell culture |
| HEK293 LgBiT stable cell line | Promega | N2672 | For complementation with HiBiT to generate luminescence |
| HiBiT CRISPR mammalian cell line | Promega | https://www.promega.com/crispr-tpd | |
| Hygromycin B solution | Gibco | 10-687-010 | Cell culture |
| LgBiT BacMam | Promega | CS1956C01 | For complementation with HiBiT to generate luminescence |
| LgBiT Expression Vector | Promega | N2681 | For complementation with HiBiT to generate luminescence |
| Luminometer Plate Reader | Luminomenter capable of measuring luminescence and fluorescence (e.g. GloMax Discover System, Promega GM3000) | ||
| NanoGlo Endurazine live cell substrate | Promega | N2570 | Kinetic HiBiT reagent |
| NanoGlo Vivazine live cell substrate | Promega | N2580 | Kinetic HiBiT reagent |
| NanoGlo HiBiT Lytic Detection system | Promega | N3030 | Enpoint lytic HiBiT reagent |
| Opti-MEM Reduced Serum Medium, no phenol red (ThermoFisher) | ThermoFisher | 11058-021 | Cell culture |
| Tissue culture plates, white, 96 well plate | Costar | 3917 | Cell culture |
| Tissue culture plates, white, 384 well plate | Corning | 3570 | Cell culture |
| Trypsin/EDTA | Gibco | 25300 | Cell culture |
Referenzen
- Burslem, G. M., Crews, C. M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. 181 (1), 102-114 (2020).
- Chamberlain, P. P., Hamann, L. G. Development of targeted protein degradation therapeutics. Nature Chemical Biology. 15 (10), 937-944 (2019).
- Churcher, I. Protac-induced protein degradation in drug discovery: Breaking the rules or just making new ones. Journal of Medicinal Chemistry. 61 (2), 444-452 (2018).
- Ciulli, A., Farnaby, W. Protein degradation for drug discovery. Drug Discovery Today: Technologies. 31, 1-3 (2019).
- Crews, C. M. Inducing protein degradation as a therapeutic strategy. Journal of Medicinal Chemistry. 61 (2), 403-404 (2018).
- Cromm, P. M., Crews, C. M. Targeted protein degradation: from chemical biology to Drug Discovery. Cell Chemical Biology. 24 (9), 1181-1190 (2017).
- Deshaies, R. J. Protein degradation: Prime time for PROTACs. Nature Chemical Biology. 11 (9), 634-635 (2015).
- Lai, A. C., Crews, C. M. Induced protein degradation: an emerging drug discovery paradigm. Nature Reviews Drug Discovery. 16 (2), 101-114 (2017).
- Ottis, P., Crews, C. M. Proteolysis-targeting chimeras: Induced protein degradation as a therapeutic strategy. ACS Chemical Biology. 12 (4), 892-898 (2017).
- Wu, T., et al. Targeted protein degradation as a powerful research tool in basic biology and drug target discovery. Nature Structural and Molecular Biology. 27, 605-614 (2020).
- Hanan, E. J., et al. Monomeric targeted protein degraders. Journal of Medicinal Chemistry. , (2020).
- Collins, I., Wang, H., Caldwell, J. J., Chopra, R. Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway. Biochemical Journal. 474 (7), 1127-1147 (2017).
- Carmony, K. C., Kim, K. B. PROTAC-induced proteolytic targeting. Methods in Molecular Biology. 832, 627-638 (2012).
- Daniels, D. L., Riching, K. M., Urh, M. Monitoring and deciphering protein degradation pathways inside cells. Drug Discovery Today: Technologies. 31, 61-68 (2019).
- Gu, S., Cui, D., Chen, X., Xiong, X., Zhao, Y. PROTACs: An emerging targeting technique for protein degradation in drug discovery. Bioessays. 40 (4), 1700247 (2018).
- Neklesa, T. K., Winkler, J. D., Crews, C. M. Targeted protein degradation by PROTACs. Pharmacology and Therapy. 174, 138-144 (2017).
- Raina, K., Crews, C. M. Targeted protein knockdown using small molecule degraders. Current Opinion in Chemical Biology. 39, 46-53 (2017).
- Riching, K. M., et al. Quantitative live-cell kinetic degradation and mechanistic profiling of PROTAC mode of action. ACS Chemical Biology. 13 (9), 2758-2770 (2018).
- Schwinn, M. K., et al. CRISPR-mediated tagging of endogenous proteins with a luminescent peptide. ACS Chemical Biology. 13 (2), 467-474 (2018).
- Schwinn, M. K., Steffen, L. S., Zimmerman, K., Wood, K. V., Machleidt, T. A simple and scalable strategy for analysis of endogenous protein dynamics. Science Reports. 10 (1), 8953 (2020).
- Bensimon, A., et al. Targeted degradation of SLC transporters reveals amenability of multi-pass transmembrane proteins to ligand-induced proteolysis. Cell Chemical Biology. 27 (6), 728-739 (2020).
- Bondeson, D. P., et al. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chemical Biology. 25 (1), 78-87 (2018).
- Buckley, D. L., et al. HaloPROTACS: Use of small molecule PROTACs to induce degradation of HaloTag fusion proteins. ACS Chemical Biology. 10 (8), 1831-1837 (2015).
- Bulatov, E., Ciulli, A. Targeting Cullin-RING E3 ubiquitin ligases for drug discovery: structure, assembly and small-molecule modulation. Biochemical Journal. 467 (3), 365-386 (2015).
- Burslem, G. M., et al. The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chemical Biology. 25 (1), 67-77 (2018).
- Chamberlain, P. P., et al. Evolution of cereblon-mediated protein degradation as a therapeutic modality. ACS Medicinal Chemistry Letters. 10 (12), 1592-1602 (2019).
- Crew, A. P., et al. Identification and characterization of Von Hippel-Lindau-recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. Journal of Medicinal Chemistry. 61 (2), 583-598 (2018).
- DeMars, K. M., Yang, C., Castro-Rivera, C. I., Candelario-Jalil, E. Selective degradation of BET proteins with dBET1, a proteolysis-targeting chimera, potently reduces pro-inflammatory responses in lipopolysaccharide-activated microglia. Biochemical and Biophysical Research Communication. 497 (1), 410-415 (2018).
- Erb, M. A., et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature. 543 (7644), 270-274 (2017).
- Farnaby, W., et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nature Chemical Biology. 15 (7), 672-680 (2019).
- Gadd, M. S., et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nature Chemical Biology. 13 (5), 514-521 (2017).
- Gechijian, L. N., et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nature Chemical Biology. 14 (4), 405-412 (2018).
- Gustafson, J. L., et al. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angewandte Chemie International Edition England. 54 (33), 9659-9662 (2015).
- Kerres, N., et al. Chemically induced degradation of the oncogenic transcription factor BCL6. Cell Reports. 20 (12), 2860-2875 (2017).
- Lohbeck, J., Miller, A. K. Practical synthesis of a phthalimide-based Cereblon ligand to enable PROTAC development. Bioorganic and Medicinal Chemistry Letters. 26 (21), 5260-5262 (2016).
- Lu, J., et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chemical Biology. 22 (6), 755-763 (2015).
- Lu, M., et al. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eurupean Journal of Medicinal Chemistry. 146, 251-259 (2018).
- Nabet, B., et al. The dTAG system for immediate and target-specific protein degradation. Nature Chemical Biology. 14 (5), 431-441 (2018).
- Nowak, R. P., et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nature Chemical Biology. 14, 706-714 (2018).
- Powell, C. E., et al. Chemically induced degradation of Anaplastic Lymphoma Kinase (ALK). Journal of Medicinal Chemistry. 61 (9), 4249-4255 (2018).
- Raina, K., et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proceedings of National Academy of Science U. S. A. 113 (26), 7124-7129 (2016).
- Sakamoto, K. M., et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proceeding National Academy Science. 98 (15), 8554-8559 (2001).
- Sakamoto, K. M., et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Molecular Cell Proteomics. 2 (12), 1350-1358 (2003).
- Schiedel, M., et al. Chemically induced degradation of Sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). Journal of Medicinal Chemistry. 61 (2), 482-491 (2018).
- Slabicki, M., et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature. , (2020).
- Smith, B. E., et al. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nature Communications. 10 (1), 131 (2019).
- Sun, B., et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia. 32 (2), 343-352 (2018).
- Tong, B., et al. A Nimbolide-based kinase degrader preferentially degrades oncogenic BCR-ABL. ACS Chemical Biology. 15 (7), 1788-1794 (2020).
- Winter, G. E., et al. Drug Development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 348 (6241), 1376-1381 (2015).
- Winter, G. E., et al. BET Bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Molecular Cell. 67 (1), 5-18 (2017).
- Zengerle, M., Chan, K. H., Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chemical Biology. 10 (8), 1770-1777 (2015).
- Zhang, C., et al. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK). European Journal of Medicinal Chemistry. 151, 304-314 (2018).
- Zoppi, V., et al. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel-Lindau (VHL) based dual degrader probe of BRD9 and BRD7. Journal of Medicinal Chemistry. 62 (2), 699-726 (2019).
- Huang, H. T., et al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chemical Biology. 25 (1), 88-99 (2018).
- Bjorklund, C. C., et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia. 34 (4), 1197-1201 (2020).
- Matyskiela, M. E., et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nature Chemical Biology. 14 (10), 981-987 (2018).
- Matyskiela, M. E., et al. A cereblon modulator (CC-220) with improved degradation of Ikaros and Aiolos. Journal of Medicinal Chemistry. 61 (2), 535-542 (2018).
- Bussiere, D. E., et al. Structural basis of indisulam-mediated RBM39 recruitment to DCAF15 E3 ligase complex. Nature Chemical Biology. 16 (1), 15-23 (2020).
- Du, X., et al. Structural basis and kinetic pathway of RBM39 recruitment to DCAF15 by a sulfonamide molecular glue E7820. Structure. 27 (11), 1625-1633 (2019).
- Ting, T. C., et al. Aryl sulfonamides degrade RBM39 and RBM23 by recruitment to CRL4-DCAF15. Cell Reports. 29 (6), 1499-1510 (2019).
- Hughes, S. J., Ciulli, A. Molecular recognition of ternary complexes: a new dimension in the structure-guided design of chemical degraders. Essays in Biochemistry. 61 (5), 505-516 (2017).
- Schapira, M., Calabrese, M. F., Bullock, A. N., Crews, C. M. Targeted protein degradation: expanding the toolbox. Nature Review Drug Discovery. 18 (12), 949-963 (2019).
- Dixon, A. S., et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chemical Biology. 11 (2), 400-408 (2016).
- Gilan, O., et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immuno-inflammation. Science. 368 (6489), 387-394 (2020).
- Oh-Hashi, K., Furuta, E., Fujimura, K., Hirata, Y. Application of a novel HiBiT peptide tag for monitoring ATF4 protein expression in Neuro2a cells. Biochemical Biophysical Report. 12, 40-45 (2017).
- Ottis, P., et al. Cellular resistance mechanisms to targeted protein degradation converge toward impairment of the engaged ubiquitin transfer pathway. ACS Chemical Biology. 14 (10), 2215-2223 (2019).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten