
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Einsatz des GAL4-UAS-Systems für die funktionelle Genetik bei Anopheles gambiae
In diesem Artikel
Zusammenfassung
Das bipartite GAL4-UAS-System ist ein vielseitiges Werkzeug zur Modifikation der Genexpression in kontrolliert raumzeitlicher Weise, das eine funktionelle genetische Analyse in Anopheles gambiae ermöglicht. Die beschriebenen Verfahren für die Verwendung dieses Systems sind eine semi-standardisierte Klonierungsstrategie, Sexing und Screening von Puppen auf fluoreszierende Proteinmarker und Embryofixierung.
Zusammenfassung
Das zweigliedrige GAL4-UAS-System ist ein vielseitiges und leistungsfähiges Werkzeug für die funktionelle genetische Analyse. Das Wesen des Systems besteht darin, transgene "Treiber" -Linien zu kreuzen, die den Hefe-Transkriptionsfaktor GAL4 auf gewebespezifische Weise exprimieren, wobei transgene "Responder" -Linien ein Kandidatengen / RNA-Interferenzkonstrukt tragen, dessen Expression durch Upstream Activation Sequences (UAS) gesteuert wird, die GAL4 binden. In den nachfolgenden Nachkommen wird das Gen oder Stummschaltungskonstrukt somit in einer vorgeschriebenen raumzeitlichen Weise exprimiert, so dass die resultierenden Phänotypen untersucht und die Genfunktion abgeleitet werden können. Das binäre System ermöglicht Flexibilität in experimentellen Ansätzen, um Phänotypen zu screenen, die durch transgene Expression in mehreren gewebespezifischen Mustern erzeugt werden, selbst wenn schwere Fitnesskosten induziert werden. Wir haben dieses System für Anopheles gambiae, den wichtigsten Malaria-Vektor in Afrika, angepasst.
In diesem Artikel stellen wir einige der gängigen Verfahren vor, die während der GAL4-UAS-Analyse verwendet werden. Wir beschreiben die bereits erzeugten GAL4-UAS-Linien von An. gambiae sowie das Klonen neuer Responder-Konstrukte für Upregulation und RNAi-Knockdown. Wir spezifizieren eine Schritt-für-Schritt-Anleitung für das Geschlecht von Mückenpuppen, um genetische Kreuzungen zu etablieren, die auch das Screening von Nachkommen umfasst, um die Vererbung von fluoreszierenden Genmarkern zu verfolgen, die den Treiber und die Responder-Insertionen markieren. Wir stellen auch ein Protokoll zur Klärung von An. gambiae-Embryonen vor, um die Embryonalentwicklung zu untersuchen. Schließlich stellen wir mögliche Anpassungen der Methode zur Erzeugung von Treiberlinien durch CRISPR/Cas9-Insertion von GAL4 stromabwärts von Zielgenen vor.
Einleitung
Das zweigliedrige GAL4-UAS-System ist das Arbeitspferd der funktionellen Charakterisierung von Genen im Insektenmodellorganismus Drosophila melanogaster1,2,3. Um das GAL4-UAS-System zu verwenden, werden transgene Treiberlinien, die den Hefetranskriptionsfaktor GAL4 unter Kontrolle einer regulatorischen Sequenz exprimieren, mit Responderlinien gekreuzt, die ein Gen von Interesse oder ein RNAi-Konstrukt (RNA Interference) tragen, das von einer von GAL4 erkannten Upstream Activation Sequence (UAS) gesteuert wird. Die Nachkommen dieses Kreuzes drücken das interessierende Transgen in einem raumzeitlichen Muster aus, das vom Promotor diktiert wird, der die GAL4-Expression kontrolliert (Abbildung 1). Phänotypen, die von Nachkommen von Driver-Responder-Kreuzungen gezeigt werden, können bewertet werden, um die Funktion von Kandidatengenen aufzuklären. Obwohl D. melanogaster verwendet wurde, um Gene aus anderen Organismen zu untersuchen4,5,6,7, wurde das GAL4-UAS-System nun für den Einsatz bei Insekten von medizinischer und landwirtschaftlicher Bedeutung angepasst, um eine direkte Analyse der interessierenden Arten zu ermöglichen 8,9,10,11,12,13,14.
Bei der afrikanischen Malariamücke Anopheles gambiae wurde das GAL4-UAS-System zunächst mittels Zelllinien-Co-Transfektion9 getestet. Mehrere Konstrukte wurden auf Effizienz in verschiedenen paarweisen Kombinationen untersucht und fanden heraus, dass 14 tandemly wiederholte UAS, ergänzt mit einem kleinen künstlichen Intron (UAS-14i), das breiteste Aktivierungspotenzial aufwiesen, wenn sie mit einem Panel von GAL4-Treibern verwendet wurden. Um die In-vivo-Funktionalität zu demonstrieren, wurden diese Konstrukte dann verwendet, um zwei separate transgene An. gambiae-Linien durch PiggyBac-Transformation8 zu erzeugen: eine Treiberlinie mit GAL4, die von einem Mitteldarm-spezifischen Promotor angetrieben wird, und eine Responder-Linie, die sowohl die Luciferase- als auch die eYFP-Gene (Enhanced Yellow Fluorescent Protein) unter Regulation von UAS-Sequenzen enthält. Darmspezifische Luciferase-Aktivität und Fluoreszenz in den Nachkommen zeigten, dass das System in Anopheles effizient war. Seitdem wurden Treiberlinien erstellt, die Transgene in anderen Geweben exprimieren, die für die Vektorkapazität und insektizidresistenz wichtig sind, einschließlich Derozyten15 und Derhämozyten16, und in einem nahezu allgegenwärtigen Muster10. Zahlreiche UAS-Linien wurden auch erzeugt, um Gene zu untersuchen, von denen angenommen wird, dass sie an stoffwechsel- und sequestrierungsvermittelter Insektizidresistenz, kutikulärer Kohlenwasserstoffsynthese beteiligt sind, und um verschiedene Zell- und Gewebetypen fluoreszierend zu markieren (Tabelle 1). Für die Responder-Linien wird nun die ortsgerichtete Integration des Transgens durch ΦC31 katalysierten Rekombinationskassettenaustausch17,18 durchgeführt, um den genomischen Kontext der UAS-regulierten Gene zu fixieren. Auf diese Weise wird die Transgenexpression in Bezug auf die position der genomischen Insertion normalisiert, was einen genaueren Vergleich der phänotypischen Effekte verschiedener Kandidatengene ermöglicht.
Die bisher erstellten Responder-Linien sind so konzipiert, dass sie das Transgen entweder in erhöhten Konzentrationen exprimieren oder die Genexpression durch RNA-Interferenz (RNAi) reduzieren. Normalerweise werden cDNA-Klone mit der UAS-Sequenz fusioniert, um geeignete Expressionsplasmide zu erzeugen, aber auch vollständige genomische Sequenzen sind möglich, vorausgesetzt, sie sind nicht zu groß für das Klonen. Um Silencing-Konstrukte zu erzeugen, haben wir drei verschiedene Methoden verwendet, um geeignete tandeminvertierte Sequenzen zu erhalten, die Hairpin dsRNA bilden, die RNAi stimuliert. Dazu gehörten Fusions-PCR, asymmetrische PCR und kommerzielle Synthese von Haarnadelkonstrukten. Allen Methoden gemeinsam ist die Aufnahme einer Intronsequenz zwischen die invertierten Sequenzen, um die Klonstabilität zu gewährleisten. Es wurden Responder-Plasmide entwickelt, in die ein Gen von Interesse/RNAi-Konstrukt eingefügt werden kann15. Diese Plasmide tragen auch die erforderlichen ΦC31 attB-Stellen für RMCE (beschrieben in Adolfi begleitendem JoVE-Papier, das die RCME-Technik im Detail beschreibt). Protokolle, die die wichtigen Schritte abdecken, die bei der Auswahl der Sequenz für die Insertion in eines dieser Plasmide zur Überexpression erforderlich sind, sind in diesem Manuskript enthalten. Zusätzlich werden zwei Protokolle für die Erstellung von RNAi-Haarnadelkonstrukten beschrieben und veranschaulicht.
Bei der Schaffung neuer Linien ist die Identifizierung seltener transgener Individuen entscheidend für die Zucht, um transgene Kolonien zu etablieren und zu erhalten. Am wichtigsten für das GAL4-UAS-System ist, dass die Responder- und Treiberlinien unterschieden werden müssen, um Kreuze zu ermitteln und einzelne Nachkommen zu identifizieren, die beide Transgene tragen. Dies wird durch die Verwendung verschiedener dominanter selektorierbarer Markergene erreicht, die mit den Treiber- und Responder-Kassetten verknüpft sind. Am häufigsten handelt es sich dabei um fluoreszierende Markergene, die durch optische Filter (z.B. eYFP, eCFP, dsRed) deutlich unterscheidbar sind. Es ist wichtig, dass Marker in einem bekannten und zuverlässigen raumzeitlichen Muster ausgedrückt werden, da dies die Identifizierung von Anomalien und Kontaminationen erleichtert. Die Expression des fluoreszierenden Markergens wird routinemäßig durch den synthetischen 3xP3-Promotor reguliert, der in allen Stadien der Entwicklung von An. gambiae eine augen- und ventrale Ganglien-spezifische Expression verursacht19. Fluoreszierende Marker, die von 3xP3 kontrolliert werden, sind in allen in diesem Artikel beschriebenen Transformationsplasmiden enthalten. Ein Protokoll, das die gängigen Methoden zum Screening von fluoreszierenden An. gambiae Puppen GAL4-UAS-Linien beschreibt, ist hier enthalten.
Eines der Schlüsselelemente des GAL4-UAS-Systems ist die Notwendigkeit, die differentiell markierten Treiber- und Responder-Linien zu überqueren. Um dies zu tun, müssen Männchen und Weibchen aus jeder Linie vor der Paarung getrennt werden. Erwachsene sind durch das Sehen leicht unterscheidbar, aber für die Feststellung genetischer Kreuzungen ist es sinnvoll, die Geschlechter vor dem Auftauchen des Erwachsenen zu trennen, um sicherzustellen, dass keine Paarung stattgefunden hat. Der allgemeine Größenunterschied zwischen männlichen und weiblichen An. gambiae-Puppen ist zu variabel, um eine effiziente und zuverlässige Methode der Geschlechtsbestimmung zu sein20. Stattdessen liefern deutliche morphologische Unterschiede in den äußeren Genitalien eine verlässliche Grundlage für das Sexing bei An. gambiae. In diesem Artikel beschreiben wir eine zuverlässige Methode zum Sexing von An. gambiae Puppen, um geeignete Kreuzungen aufzustellen.

Abbildung 1 - Schematische Darstellung des Prozesses zur Verwendung des zweigliedrigen GAL4-UAS-Systems in Anopheles gambiae. (A) Die Hauptkomponenten eines Beispielvektors (pSL-attB-UAS14-gyp[3xp3-eYFP]) werden dargestellt, wobei die verfügbaren Restriktionsstellen (EcoRI, NheI, XhoI und NcoI) innerhalb der multiplen Klonierungsstellen aufgeführt sind, die für die Verwendung geeignet sind, das Haarnadelkonstrukt oder die kodierende Sequenz für das interessierende Gen einzufügen. Der Aufbau der Andocklinie ist ebenfalls dargestellt. (B) Der Kreuzungsschritt ist dargestellt und zeigt die Verwendung von Männchen aus der Fahrerlinie (mit GAL4-Fahrer durch einen Promoter von Interesse und eCFP, der vom 3xP3-Promoter gesteuert wird) und Weibchen aus der Responder-Linie (mit dem von einem UAS-Promotor kontrollierten Gen oder Haarnadelkonstrukt und einem eYFP-Marker, der vom 3xP3-Promotor kontrolliert wird). (C) Eine schematische Darstellung der GAL4-treibenden Expression des Gens, das für die Nachkommen des Kreuzes von Interesse ist, in B und eine Liste einiger der typischen Phänotypen, die bewertet werden. Abkürzungen: Multiple Cloning Site (MCS), Recombinase mediated cassette exchange (RMCE), Upstream Activator Sequence (UAS), enhanced yellow fluorescent protein (eYFP), enhanced cyan fluorescent protein (eCFP). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
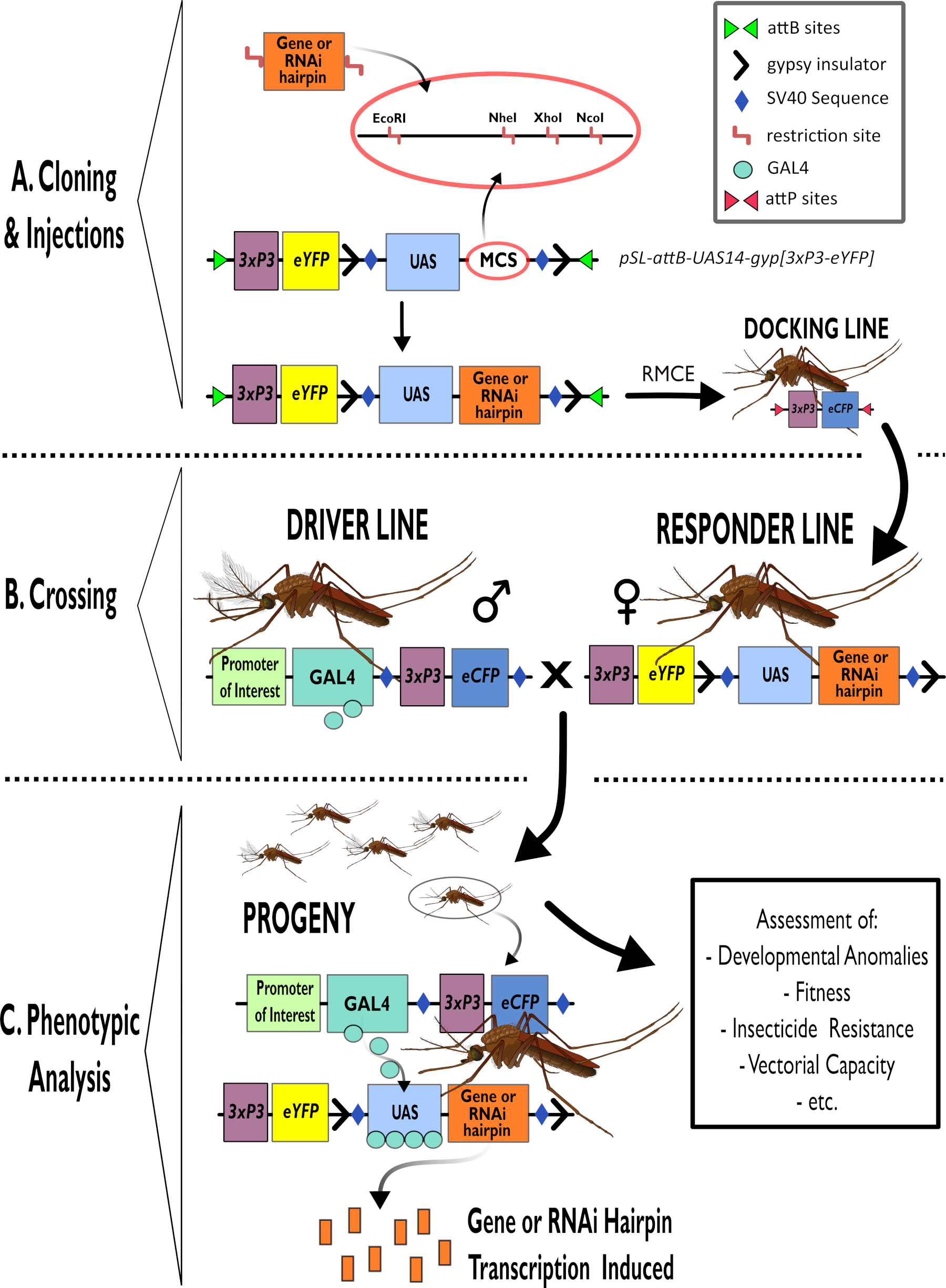{kind=link}
Es ist die Verwendung von Kreuzen, die die zweigliedrige Natur des GAL4-UAS-Systems bietet, die gegenüber lineareren Ansätzen deutliche Vorteile bietet. Beispielsweise können viel mehr Kombinationen von Treiber- und Responder-Linien bewertet werden, als es möglich wäre, wenn für jede Promotor-Gen-Kombination eine neue transgene Linie erzeugt und aufrechterhalten werden müsste. Noch wichtiger ist, dass es die Analyse von Genen ermöglicht, die tödliche oder sterile Phänotypen produzieren, wenn ihre Expression gestört ist, die in einem linearen System schwer zu erzeugen / aufrechtzuerhalten sind. Solche tödlichen Phänotypen können sich in allen Entwicklungsstadien manifestieren, abhängig von der Genfunktion und der raumzeitlichen Expression, werden aber am häufigsten während der Embryonalentwicklung beobachtet. Die Visualisierung der Entwicklung von Mückenembryonen erfordert die Reinigung des undurchsichtigen Chorions, das die Eier bedeckt. Nach Methoden, die in Trpiš (1970) 21 und Kaiser et al. (2014) 22 beschrieben sind, beschreiben wir die Protokolle, die wir verwenden, um Embryonen unter Beibehaltung der strukturellen Integrität zu fixieren, und Bleichen, um die Endochorion zu beseitigen, die eine mikroskopische Visualisierung und Bildgebung ermöglicht.
Protokoll
1. Planung und Konstruktion von UAS-Konstrukten
- Design und Montage von Vektoren für die Kandidaten-Genexpression
- Bestimmen Sie die Sequenz, die für die Hochregulierung von Kandidatengenen verwendet werden soll.
- Sequenzieren Sie die cDNA/gDNA aus dem interessierenden Stamm und vergleichen Sie sie mit der veröffentlichten Sequenz, um ihre Identität zu überprüfen und potenzielle SNPs und Einschränkungsstellen für diagnostische Digests zu identifizieren.
- Stellen Sie sicher, dass der für die Genamplifikation verwendete Vorwärtsprimer die native Kozak-Sequenz abdeckt und gegebenenfalls Codon beginnt. Ein Primer mit ~10 bp Bindung vor dem Startcodon umfasst die Kozak-Sequenz.
- Schließen Sie das Stopcodon in dem Fragment ein, das in den meisten Fällen aus der umgekehrten Grundierung verstärkt wird. Verwenden Sie 3'-Terminierungssequenzen, die in den beschriebenen Plasmidvektoren enthalten sind, oder amplifizieren Sie aus Genomsequenzen von Kandidatengen.
- Bestellen Sie auf Wunsch kommerzielle Sequenzen mit spezifischem Codon-Bias.
- Verwenden Sie Standard-Subklonierungsverfahren, um Genkassetten in UAS-Plasmidvektoren einzufügen, z. B. pSL-attB-UAS14-gyp[3xP3-eYFP]15 (Abbildung 1) sowohl für Hochregulierungs- als auch für RNAi-Konstrukte.
- Produzieren Sie transgene Moskitos, die mit ΦC31-Rekombinationsvermitteltem Kassettenaustausch hergestellt wurden10,17,18,23.
- Bestimmen Sie die Sequenz, die für die Hochregulierung von Kandidatengenen verwendet werden soll.
- Erstellung von RNAi-Haarnadelkonstrukten: einstufige Amplifikation mittels asymmetrischer PCR15,24
- Extrahieren Sie genomische DNA (gDNA) aus erwachsenen weiblichen An. gambiae, die das gewünschte Kandidatengen trägt, mit der Livak-Methode25.
- Entwerfen Sie die vordere Grundierung so, dass sie an das Ziel-Exon an den 5 'des gewünschten Fragments bindet, das auf das benachbarte Intron gerichtet ist. Entwerfen Sie das 3'-Ende einer Brückengrundierung so, dass es sich an das Ende des vorhergehenden Exons bindet, um das Intron zu verstärken. Das 5'-Ende ist komplementär zu einem kleinen Fragment des Ziel-Exons unmittelbar nach dem Intron.
- Führen Sie eine asymmetrische PCR-Reaktion durch, wie in Xiao (2006)24 (Abbildung 2) beschrieben.
- Klonen Sie das gereinigte PCR-Produkt in einen geeigneten Vektor, der den UAS-Promotor trägt (z. B. pSL-attB-UAS14-gyp[3xP3-eYFP]15).
HINWEIS: Enzyme innerhalb der mehrfachen Klonierungsstelle, die für das Klonen von pSL-attB-UAS14-gyp[3xP3-eYFP]15 geeignet sind, und die nächsten erforderlichen Schritte sind in Abbildung 1 dargestellt. Ein einzelner Enzymverdau ist wichtig, da nur eine Restriktionsstelle hinzugefügt wird. Die Dephosphorylierung des Plasmids verbessert die Klonierungseffizienz.
- Extrahieren Sie genomische DNA (gDNA) aus erwachsenen weiblichen An. gambiae, die das gewünschte Kandidatengen trägt, mit der Livak-Methode25.
- Aufbau von RNAi Haarnadelkonstrukten: Fusion PCR von cDNA und gDNA15
- Extrahieren Sie genomische DNA (gDNA) aus erwachsenen weiblichen An. gambiae, die das gewünschte Kandidatengen trägt, mit der Livak-Methode25.
- Fügen Sie gDNA in eine PCR-Reaktion ein, um den Zielbereich der Exon- und Intronsequenzen zusammen zu amplifizieren (Abbildung 2).
- Entwerfen Sie das 3'-Ende des Vorwärtsprimers so, dass es an die Reverse-Target-Exon-Sequenz bindet, um sich in Richtung der Ziel-Intronsequenz zu verstärken, und das 5'-Ende, um eine Restriktionsstelle zu tragen, um das Klonen zu erleichtern.
- Entwerfen Sie den umgekehrten Primer (1), um an das 5'-Ende des Introns zu binden, und der 5'-Endüberhang trägt die ersten Basen der Vorwärtssequenz des benachbarten Exons. Dieser Überhang wird bei der Fusions-PCR genutzt.
- Reinigen Sie das gewünschte Reaktionsprodukt.
- Extrahieren Sie RNA, entfernen Sie DNA mit DNas's und bereiten Sie cDNA aus erwachsenen weiblichen An. gambiae vor, die das gewünschte Kandidatengen gemäß den Protokollen des Herstellers tragen.
- Verwenden Sie cDNA in einer PCR-Reaktion, um nur den Zielbereich des Exons zu verstärken (Abbildung 2).
- Entwerfen Sie den Vorwärtsprimer (2), so dass das 3'-Ende am 3'-Ende der komplementären Ziel-Exon-Sequenz bindet und das 5'-Ende des Primers eine Restriktionsstelle für die Verwendung beim Klonen trägt.
HINWEIS: Der Vorwärtsprimer von 1.3.1.2 kann in dieser zweiten Reaktion erneut verwendet werden. Dies bedeutet jedoch, dass ein einziger Enzymverdaustand unerlässlich ist. Die Verwendung eines zweiten Vorwärtsprimers mit einer anderen Restriktionsstelle ermöglicht eine doppelte Verdauung, was die Kloneffizienz erhöhen kann. - Design Reverse Primer (2) - das 3'-Ende bindet an das 5'-Ende des benachbarten Exons und verstärkt das Ziel-Exon. Das 5'-Ende bindet an das 3'-Ende des Introns-Vorwärtsstrangs. Dieser Überhang wird bei der Fusions-PCR genutzt.
- Reinigen Sie das gewünschte Reaktionsprodukt.
- Entwerfen Sie den Vorwärtsprimer (2), so dass das 3'-Ende am 3'-Ende der komplementären Ziel-Exon-Sequenz bindet und das 5'-Ende des Primers eine Restriktionsstelle für die Verwendung beim Klonen trägt.
- Die Produkte der Schritte 1.3.1 und 1.3.2 sind als Schablonen für eine Fusions-PCR-Reaktion unter Verwendung von Standardkonzentrationen mit den Forward-Primern 1 und 2 enthalten. Reinigen Sie das gewünschte Produkt.
- Verdauen Sie das gereinigte Produkt, um die Überhänge für das Klonen zu erzeugen. Klonen Sie in einen geeigneten Vektor nach dem UAS-Promotor. Geeignete Enzyme für das pSL-attB-UAS14-gyp[3xP3-eYFP]-Klonen15 und die nächsten erforderlichen Schritte sind in Abbildung 1 dargestellt.
- Extrahieren Sie genomische DNA (gDNA) aus erwachsenen weiblichen An. gambiae, die das gewünschte Kandidatengen trägt, mit der Livak-Methode25.

Abbildung 2 - Schematische Darstellung der Erstellung von RNAi-Konstrukten zur Insertion in pSL-attB-UAS14-gyp[3xP3-eYFP] durch zwei Methoden: (A) Einstufige asymmetrische PCR (angepasst von Xiao. Y H et al (2006) und (B) mehrstufige Fusion PCR. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
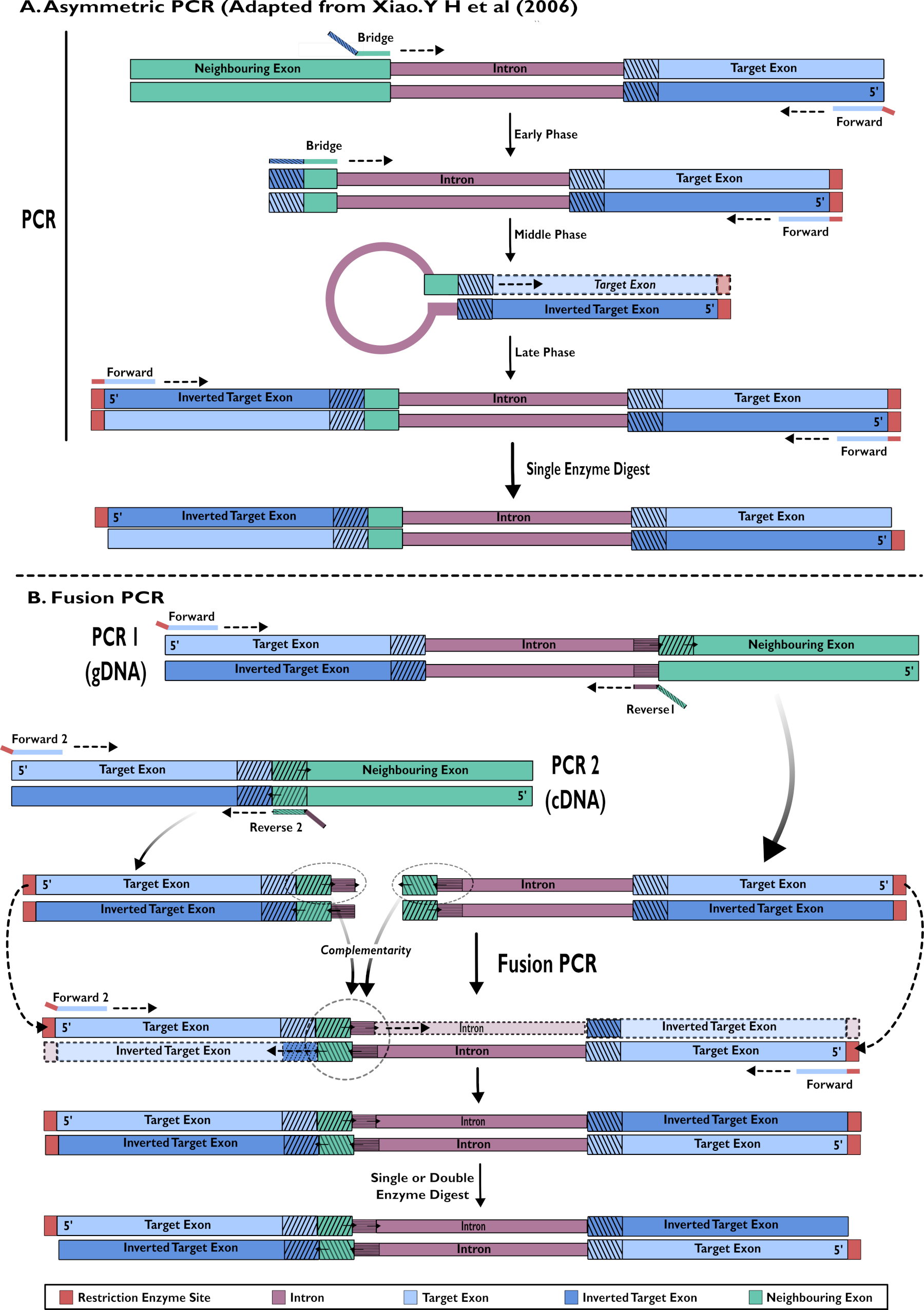{kind=link}
2. An. gambiae Puppenscreening
- Sammlung von Puppen zur mikroskopischen Charakterisierung
HINWEIS: In diesen Protokollen bezieht sich Wasser auf destilliertes Wasser, das mit 0,01% Teichsalz ergänzt wird.- Hintere An. gambiae Moskitos unter Verwendung von Standardprotokollen (z. B. MR426) bis zum Puppenstadium.
VORSICHT: Achten Sie darauf, puppen während dieses Prozesses nicht zu verletzen. - Sammeln Sie Puppen auf einer durchsichtigen flachen Schale, die für die Verwendung mit einem Stereomikroskop geeignet ist (z. B. eine 100 x 15 mm große Petrischale aus Kunststoff, wobei die Kanten vermieden werden).
HINWEIS: Um Puppen zu sammeln, verwenden wir eine 3 ml Kunststoff-Pasteurpipette mit etwa 10 mm Schnitt vom Ende, um das Ende zu verbreitern und Verletzungen der Moskitos zu vermeiden. Screening und Sexing können bei Einzelpersonen abgeschlossen werden, dies ist jedoch sehr langsam. Es wird empfohlen, Screening und Sexing an Gruppen von 50-200 Puppen durchzuführen (die Größe der möglichen Gruppe ist durch die Größe des verwendeten Gerichts begrenzt und unterliegt den persönlichen Vorlieben). Wenn eine große Anzahl gescreent wird, kann die Effizienz erhöht werden, indem zuerst Puppen etwa 4 bis 5 tief in Linien ausgerichtet und die Zielpuppen aus dieser Linie bewegt werden. - Entfernen Sie mit einer Pasteur-Pipette vorsichtig fast das gesamte Wasser um die Puppen herum. Lassen Sie gerade genug Wasser um die Puppen herum, damit sie effektiv unbeweglich sind, aber leicht mit einer feinen Bürste bewegt werden können. Wenn sie schwer zu bewegen sind, fügen Sie mehr Wasser hinzu.
HINWEIS: Wenn genügend Wasser entfernt wird, liegen die Puppen auf der Seite, was die Visualisierung der Augen zur Fluoreszenzerkennung und Identifizierung dimorpher Genitalien ermöglicht (Abbildung 4DE).
VORSICHT: Stellen Sie sicher, dass die Puppen nicht austrocknen. Wenn nur noch ein sehr kleines Wasservolumen übrig ist, kann es mit der Wärme aus der Lampe des Mikroskops und beim Aufteilen zwischen Puppenbecken weiter abnehmen. Zusätzliches Wasser muss manchmal während des Prozesses mit einer 3 ml Pasteurpipette zu den gewünschten Gruppen hinzugefügt werden.
- Hintere An. gambiae Moskitos unter Verwendung von Standardprotokollen (z. B. MR426) bis zum Puppenstadium.
- Identifizierung von Fluoreszenzmarkern in Puppen
HINWEIS: Die Verwendung eines Stereoskops mit geringer Vergrößerung ermöglicht ein Weitfeld-Screening, die Sortierung kann auf einem inversen Verbindungsmikroskop erfolgen, muss jedoch einzeln erfolgen.- Beim Screening nach einem fluoreszierenden Marker ist es zunächst entscheidend, die erwarteten Expressions- und Vererbungsmuster zu kennen. Berücksichtigen Sie Folgendes:
- Farbe(n): Bestimmen Sie, welche Filter den Ausdruck visualisieren sollen.
- Raumzeitliches Ausdrucksmuster: Verstehen Sie, wo und in welcher Lebensphase Sie Ausdruck erwarten.
- Verhältnis verschiedener Phänotypen: Legen Sie fest, welcher Prozentsatz der Bevölkerung die Marker von Interesse tragen sollte.
- Führen Sie bei Dunkelheit ein Fluoreszenzscreening durch, da selbst schwaches Licht die Auflösung der Fluoreszenz beeinträchtigen kann. Verwenden Sie jedoch eine Lampe neben dem Stereoskop, wenn Licht für andere Manipulationen benötigt wird.
VORSICHT: Stellen Sie sicher, dass der Arbeitsbereich um das fluoreszierende Stereoskop frei ist, bevor Sie das Licht ausschalten. - Schalten Sie die Leuchtstofflampe ein und lassen Sie sie für den vom Hersteller empfohlenen Zeitraum warm (normalerweise 10-15 min). Wählen Sie den gewünschten Filter am fluoreszierenden Stereoskop aus und prüfen Sie, ob ein farbiger Lichtstrahl sichtbar ist, der auf die Mitte der Bühnenplatte gerichtet ist. Wenn dies nicht sichtbar oder sehr schwach ist, hat sich die Leuchtstofflampe möglicherweise nicht vollständig erwärmt, der Verschluss ist geschlossen oder die Mikroskopoptik ist nicht gut ausgerichtet.
- Mit weißem Licht die Puppen im Sichtfeld zentrieren und in den Fokus rücken. Diese Vergrößerung muss je nach Fluoreszenzintensität möglicherweise geändert werden, wenn zwischen verschiedenen Filtern gewechselt wird.
- Achten Sie mit einem fein detaillierten Pinsel darauf, dass sich die untersuchten Puppen nicht überlappen.
- Schalten Sie das weiße Licht des Stereoskops aus und nutzen Sie den Feinfokus, um den Bereich der Puppen, der den interessierenden Phänotyp trägt, in den Fokus zu rücken. Das fluoreszierende Muster sollte sichtbar sein. Beispiele für 3xP3-Promotor-gesteuerte Fluoreszenz sind in Abbildung 3 dargestellt.
- Verwenden Sie die niedrigste Vergrößerung, bei der der erwartete fluoreszierende Phänotyp zuverlässig von Individuen ohne Fluoreszenz unterschieden werden kann.
- Für Dehnungen mit heller Fluoreszenz verwenden Sie während des Screenings auch ein Hellfeldlicht mit geringer Intensität, wenn das Fluoreszenzsignal noch eindeutig identifizierbar ist.
- Scannen Sie nach Abschluss des primären Screenings die Populationen schnell unter anderen Filtern, um eine mögliche Kontamination zu erkennen.
VORSICHT: Stellen Sie sicher, dass ein klarer Abstand zwischen den Gruppen sortierter Puppen besteht, um eine Kontamination durch Puppenbewegungen zu verhindern. Seien Sie sich bewusst, dass sich die Größe der Gruppen ändert, wenn Puppen geschlechtlich sind und dass Entfernungen größer erscheinen können, wenn Sie unter Vergrößerung schauen. Seien Sie besonders vorsichtig, wenn sich die Pools nicht im Sichtfeld befinden.
- Beim Screening nach einem fluoreszierenden Marker ist es zunächst entscheidend, die erwarteten Expressions- und Vererbungsmuster zu kennen. Berücksichtigen Sie Folgendes:

Abbildung 3 - Anopheles gambiae Puppen, die fluoreszierende Marker exprimieren, die vom 3xP3-Promotor (A) eYFP, (B) dsRed und (C) eCFP angetrieben werden. Vergrößerung: A = 16X, B, C = 20X.
-
Sexing Puppen
- Sammeln Sie Puppen. Überschüssiges Wasser entfernen, aber ausreichend bereitstellen, damit sich die Analpaddel leicht von den Genitalien trennen, um die Visualisierung und morphologische Charakterisierung zu erleichtern (Abbildung 4D,E).
- Wenn sich keine Puppen auf der Seite befinden, verwenden Sie einen feinen Detailpinsel, um die Puppe vorsichtig zu drehen und die Analpaddel so zu bewegen, dass äußere Genitalien identifiziert werden können.
- Separate Puppen basierend auf markanten äußeren Genitalien; Männchen haben eine lange Röhre, die aus dem letzten rückenförmigen Segment etwa halb so lang wie die Analpaddel austritt (Abbildung 4B). Die äußeren Genitalien der weiblichen Puppen sind deutlich kürzer und verzweigen sich (Abbildung 4A).
HINWEIS: Gelegentlich, wenn das Larven-Exoskelett des 4. Stadiums befestigt bleibt oder die äußeren Genitalien beschädigt sind (Abbildung 4C), ist die sichere Identifizierung des Geschlechts schwieriger. Wenn das Geschlecht einer Puppe nicht klar ist, ist es am besten, sie zu verwerfen. Wenn das Individuum gehalten werden soll, sollte die Puppe isoliert auftauchen und ihr Geschlecht anhand erwachsener morphologischer Merkmale bestimmt werden. Es ist wahrscheinlich, dass, wenn seine Genitalien beschädigt sind, das Individuum sich möglicherweise nicht erfolgreich paart. - Machen Sie einen Pool für jedes Geschlecht am gegenüberliegenden Ende des Gerichts zum ungeschlechtlichen Pool und bewegen Sie identifizierte Puppen mit einem feinen Detailpinsel über das Gericht. Beschriften Sie die Unterseite des Gerichts, auf der die beiden Pools gesammelt werden, um sie später zu identifizieren.
- Wenn sowohl ein Geschlechts- als auch ein Fluoreszenzscreening erforderlich ist, führen Sie zuerst ein Fluoreszenzscreening durch, da dies der schnellere Prozess der beiden ist.

Abbildung 4 - Sexing Anopheles gambiae Puppen. Einzelne Puppen, die die äußeren Genitalien von (A) einem Weibchen (B) einem Männchen und (C) einem Individuum anzeigen, das aufgrund einer unvollständigen Ablösung des Larven-Exoskeletts nicht ohne weiteres identifiziert werden kann. Vergrößerte Bilder unten, die die äußeren Genitalien hervorheben. Puppen mit ♀ (weiblich) und ♂ (männlich), die die äußeren Genitalien von Puppen mit (D) ~ 50% der Puppe in Wasser getaucht und mit (E) entferntem Wasser anzeigen, was den Unterschied in der Leichtigkeit der Visualisierung der äußeren Genitalien hervorhebt. Vergrößerung: A,B,C=40x, D,E=30x. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
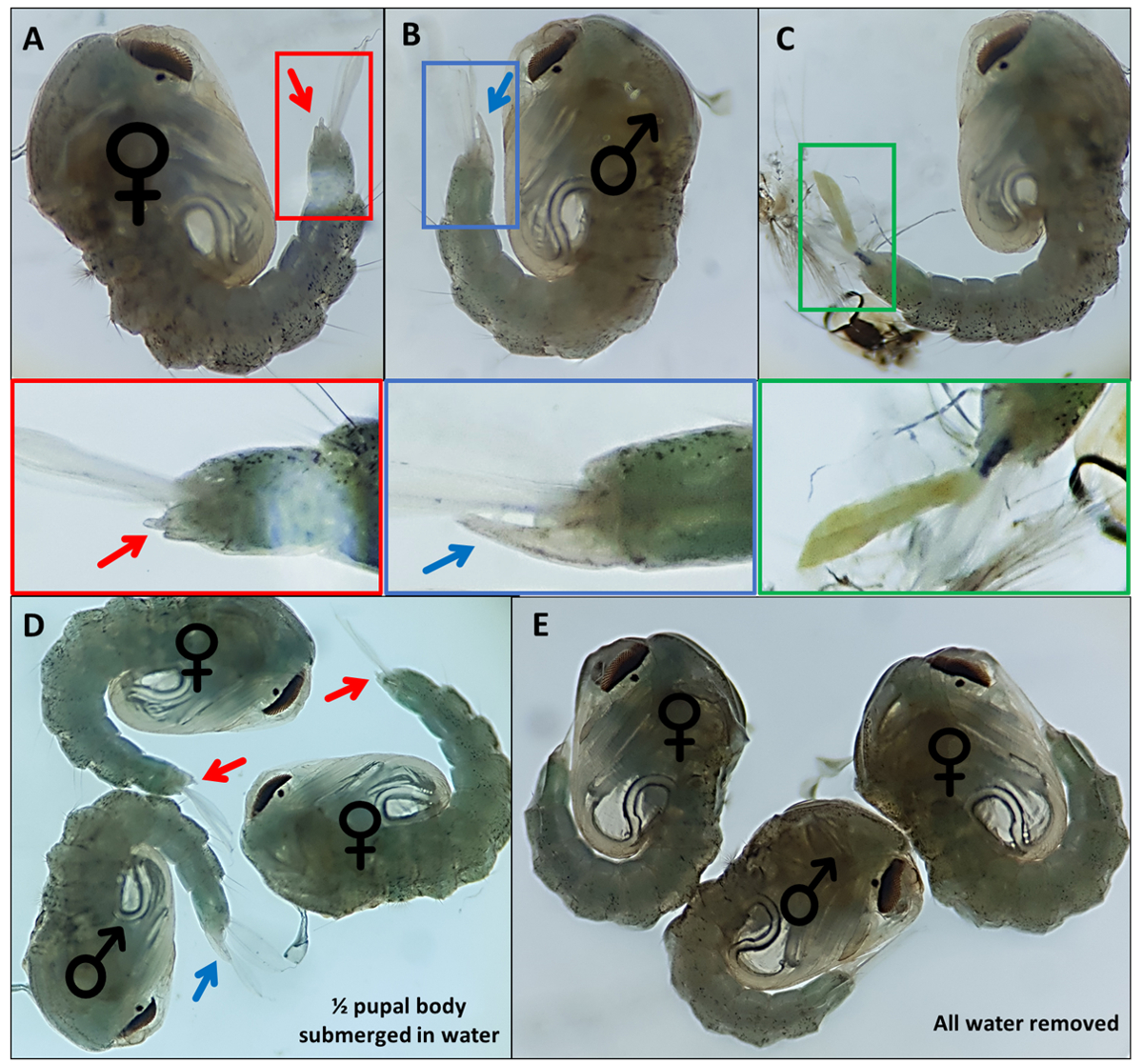{kind=link}
- Geschlechtsbestätigung als Erwachsene
- Bis eine sehr geringe Fehlerquote nachgewiesen wurde, bestätigen Sie das Puppen-Sexing durch die Morphologie des Erwachsenen nach dem Auftreten. Trennen Sie die geschlechtsspezifischen Puppen in Gruppen von 10 oder weniger in einem klaren 20-ml-Röhrchen mit einigen ml Wasser, versiegeln Sie es mit einem Wattebausch, beschriftet mit dem erwarteten Geschlecht und lassen Sie es über Nacht entstehen.
HINWEIS: Da Erwachsene am nächsten Morgen verlegt werden, ist es nicht notwendig, aufstrebende Erwachsene mit Nahrung zu versorgen. - Bestätigen Sie das Geschlecht der aufgetauchten Erwachsenen mit morphologischen Merkmalen am nächsten Tag.
- Wenn Männchen in den weiblichen Sammlungen vorhanden sind, entsorgen Sie die Weibchen, falls die Paarung bereits stattgefunden hat.
- Wenn Weibchen in der männlichen Sammlung vorhanden sind, entfernen Sie die Weibchen und bewahren Sie die Männchen zum Überqueren auf.
- Bis eine sehr geringe Fehlerquote nachgewiesen wurde, bestätigen Sie das Puppen-Sexing durch die Morphologie des Erwachsenen nach dem Auftreten. Trennen Sie die geschlechtsspezifischen Puppen in Gruppen von 10 oder weniger in einem klaren 20-ml-Röhrchen mit einigen ml Wasser, versiegeln Sie es mit einem Wattebausch, beschriftet mit dem erwarteten Geschlecht und lassen Sie es über Nacht entstehen.
- Einrichten des GAL4-UAS-Systems kreuzt
- Die gewünschte Anzahl männlicher und weiblicher Erwachsener aus den Schläuchen in Schritt 2.4 wird in einen Käfig oder einen kleinen Eimer abgesaugt, der in der für die An. gambiae-Aufzucht üblichen Weise aufgestellt ist.
HINWEIS: Achten Sie darauf, die Erwachsenen während dieses Transfers nicht zu beschädigen. - Verwenden Sie ungefähr 50 Weibchen mit einer gleichen Anzahl von Männchen, wenn ~ 2000 Erwachsene von den Nachkommen benötigt werden.
HINWEIS: Wenn ein Kreuz mehrmals gefüttert werden soll, um mehrere Chargen zu erzeugen, können bis zu 200 von jedem Geschlecht in 30 cm x 30 cm x 30 cm großen Käfigen aufgestellt werden. Wenn nur eine kleine Anzahl von Weibchen (<20) für die Kreuzung zur Verfügung steht, addieren wir ~4x die Anzahl der Männchen, um die Wahrscheinlichkeit einer erfolgreichen Paarung zu erhöhen. - Blutfütterung der gekreuzten Weibchen und der hinteren Nachkommen in ein geeignetes Stadium gemäß den Standardprotokollen26, um eine phänotypische Bewertung durchzuführen (z. B. Insektizidresistenz, vektorielle Kapazität und Eignungskostentests).
- Wenn eine maternale Wirkung der Transgenexpression wahrscheinlich ist, richten Sie reziproke Kreuze der Treiber- und Responderlinien ein und untersuchen Sie den erwarteten Phänotyp.
HINWEIS: Kreuzungen mit "heterozygoten" oder gemischten Populationen von Treiber- und Responderlinien erzeugen Nachkommen mit jedem der 4 möglichen Genotypen. Dies bietet Wild-Type-, UAS- und GAL4-Only-Kontrollen sowie die Transheterozygotes GAL4-UAS, mit denen der Phänotyp analysiert werden kann. Wenn homozygote Populationen gekreuzt werden, richten Sie zusätzliche Kreuzungen ein, um geeignete Kontrollen für den Vergleich von Phänotypen bereitzustellen. Die Nachkommen sollten wie oben beschrieben gescreent werden, wobei nachkommend getrennt werden sollte, die beide oder nur einen Marker sowie negative Marker zur phänotypischen Beurteilung tragen.
- Die gewünschte Anzahl männlicher und weiblicher Erwachsener aus den Schläuchen in Schritt 2.4 wird in einen Käfig oder einen kleinen Eimer abgesaugt, der in der für die An. gambiae-Aufzucht üblichen Weise aufgestellt ist.
- Etablierung homozygoter Populationen aus Linien, die durch RCME mit alternativen fluoreszierenden Markern erzeugt werden
HINWEIS: Es ist wichtig, dass der fluoreszierende Marker beider Linien an derselben genomischen Stelle vorhanden ist und sie vollständig unterscheidbar sind.- Richten Sie nach dem Screening ein Elternkreuz von etwa 200 Erwachsenen mit der gleichen Anzahl von unterschiedlich markierten Männchen einer Linie und Weibchen der anderen Linie ein, um Personen auszuwählen, die die korrekte Fluoreszenz und das richtige Geschlecht aufweisen, wie oben beschrieben. Etwa eine Woche später füttert das Blut das Kreuz nach etablierten Protokollen26.
- Ziehen Sie die F1-Nachkommen mit Standardprotokollen zu Puppen und sammeln Sie Puppen wie zuvor beschrieben.
- Screening auf Fluoreszenz, der diejenigen auswählt, die beide elterlichen Marker (transheterozygot) tragen. Richten Sie mit diesen Puppen ein F1-Intercross ein.
- Eine Woche später füttert das Blut die F1-Weibchen und die hinteren Nachkommen nach Standardprotokollen in das Puppenstadium.
- Überprüfen Sie die F2-Puppen und wählen Sie diejenigen aus, die NUR einen der Marker anzeigen. Diese werden homozygot für die Insertion sein. Nur 25% der Nachkommen werden für jede Insertion homozygot sein, also stellen Sie sicher, dass genügend Nachkommen aufgezogen werden, um einen Lagerkäfig (400-500) bereitzustellen.
HINWEIS: Die Auswahl der transheterozygoten Nachkommen muss völlig streng sein, sonst wird der Prozess kontaminiert und eine vollständige Homozygotie kann nicht erreicht werden. Überprüfen Sie alle Nachkommen, die für das F1-Intercross ausgewählt wurden.
- Richten Sie nach dem Screening ein Elternkreuz von etwa 200 Erwachsenen mit der gleichen Anzahl von unterschiedlich markierten Männchen einer Linie und Weibchen der anderen Linie ein, um Personen auszuwählen, die die korrekte Fluoreszenz und das richtige Geschlecht aufweisen, wie oben beschrieben. Etwa eine Woche später füttert das Blut das Kreuz nach etablierten Protokollen26.
3. An. gambiae Embryo Clearing Protokoll
-
Bluternährung und -pflege
- Hintere An. gambiae-Mücken für Erwachsene nach Standardprotokollen (z. B. MR4).
- Blut füttern 5-7 Tage alte weibliche Erwachsene, um sicherzustellen, dass die meisten vollständig gefüllt sind.
VORSICHT: Während dieses Protokolls ist es wichtig, schnell zu arbeiten, um sicherzustellen, dass Eier nicht austrocknen dürfen.
-
Induzierte Eiablage
- 3 Tage nach der Blutfütterung Eier durch induziertes Legen sammeln.
- Montieren Sie die Eiablagekammer.
- Füllen Sie den Eiablagetopf mit Wasser bis zu einer Tiefe von ca. 5 mm. Befestigen Sie den Topf an einem Ende eines 50-ml-Polypropylenrohrs, das zuvor mit einer Bügelsäge geschnitten wurde, so dass beide Enden offen sind. (Wir verwenden eine Kunststoffscheibe für einen Topf (Abbildung 5); stattdessen kann jedoch der Originaldeckel des Röhrchens verwendet werden).
- Bedecken Sie das andere Ende des geschnittenen Polypropylenrohrs mit Material (Schlauch /Strumpfhose) oder Teilen des Latexhandschuhs, die mit einem elastischen Band gesichert sind, so dass Erwachsene eingeführt werden können, aber nicht entkommen können (Abbildung 5). Andere alternative Eiablagekammer-Designs existieren und können verwendet werden26.
- Führen Sie vorsichtig 10-15 Weibchen (Blut, das in Schritt 3.1.2 gefüttert wird) in die Eiablagekammer ein. Decken Sie die Eiablagekammer ab, um Dunkelheit zu erzeugen, und lassen Sie sie 20 Minuten einwirken.
VORSICHT: Vermeiden Sie es, den Eiablagetopf zu bewegen, sobald die Eier gelegt wurden, um das Stranden und Austrocknen der Eier zu verhindern. - Lösen Sie vorsichtig das 50 ml Polypropylenrohr aus dem Eiablagetopf, während Sie sicherstellen, dass die Moskitos nicht freigesetzt werden. Weiße Eier sollten sichtbar sein. Überprüfen Sie, ob genügend für den verbotenen Zweck ausgelegt wurden. Wiederholen Sie dies bei Bedarf.
- Decken Sie den Topf ab (zum Staubschutz) und lassen Sie die Eier bis zum Entwicklungsstadium von Interesse reifen.
- Verwenden Sie einen feinen Detailpinsel, um Eier aus dem Topf aufzunehmen und sie in einem 40 mm2 großen ausgegrabenen Glasblock auf Wasser zu legen.

Abbildung 5 - Beispiel einer Eiablagekammer (A), die zerlegt wurde, um die Komponenten hervorzuheben, und (B) montiert wurde. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
-
Embryo Fixierung
ACHTUNG: Führen Sie alle Fixierschritte (Schritt 3.3) aufgrund der Verwendung von Formaldehyd in einem Abzug durch.- Bereiten Sie die FAA-Lösung vor, wie in Kaiser et al. (2014) 22 beschrieben. FAA besteht aus 3,6 M Formaldehyd, 0,87 M Essigsäure und 8,5 M absolutem Ethanol, das mit destilliertem Wasser (dH2O) auf Volumen gebracht wird.
- Für 10 ml FAA kombinieren Sie 2,68 ml 13,42 m Formaldehyd, 4,96 ml 17,14 m Ethanol und 0,5 ml 17,4 m Essigsäure mit 1,86 ml destilliertem H2O. Fixiermittel kann mindestens 3 Monate in einem dicht verschlossenen Glasbehälter aufbewahrt werden, der in einem dafür vorgesehenen Chemikalienschrank aufbewahrt wird.
- Entfernen Sie vorsichtig das Wasser mit einer Mikropipette aus dem Glasblock und bedecken Sie die Eier mit 500 μL FAA und oszillieren Sie sanft (~ 25 U / min) auf einem Orbitalschüttler bei Raumtemperatur für 30 Minuten. An dieser Stelle ist keine Farbänderung sichtbar.
- Eier gründlich mit destilliertem Wasser abspülen. Spülen Sie 15 Mal, um alle Spuren von Formaldehyd zu entfernen. Fügen Sie mit einer 1000 μL-Mikropipette 1 ml dH2O hinzu und entfernen Sie sie anschließend, um sicherzustellen, dass die Eier dabei nicht beschädigt werden.
- Lagern Sie Abwasser aus Spülungen in einem dafür vorgesehenen Formaldehyd-Entsorgungsbehälter zur Entsorgung gemäß den Sicherheitsrichtlinien.
- Zu diesem Zeitpunkt können festsitzende Eier bei 4 ° C über Nacht in Wasser gelagert werden, um sie hydratisiert zu halten.
- Bereiten Sie die FAA-Lösung vor, wie in Kaiser et al. (2014) 22 beschrieben. FAA besteht aus 3,6 M Formaldehyd, 0,87 M Essigsäure und 8,5 M absolutem Ethanol, das mit destilliertem Wasser (dH2O) auf Volumen gebracht wird.
-
Embryo-Bleiche
VORSICHT: Führen Sie alle Bleichschritte (Schritt 4) in einem Abzug aufgrund der möglichen Freisetzung von Chlorgas durch, wenn Natriumhypochlorit und Essigsäure kombiniert werden.- Bleichlösung herstellen (Trpiš-Lösung - beschrieben in Trpiš (1970)21 und modifiziert nach Kaiser et al. (2014)22). Trpiš-Lösung ist 0,59 M Natriumhypochlorit und 0,35 M Essigsäure, gelöst in destilliertem H2O.
- Für ein 10 mL Ovolum Trpiš-Lösung werden 2,68 ml 2,2 M Natriumhypochlorit und 0,2 ml 17,4 M Essigsäure mit 7,12 ml destilliertem H2O kombiniert.
HINWEIS:Trpiš-Lösung kann mindestens 3 Monate in einem dicht verschlossenen Glasbehälter gelagert und in einem sicheren Chemikalienschrank aufbewahrt werden. Die Lösung muss nach der Lagerung möglicherweise verwirbelt werden und sollte im Falle der Freisetzung von Chlorgas immer in einem Abzug geöffnet werden.
- Für ein 10 mL Ovolum Trpiš-Lösung werden 2,68 ml 2,2 M Natriumhypochlorit und 0,2 ml 17,4 M Essigsäure mit 7,12 ml destilliertem H2O kombiniert.
- Feste Eier mit 1 ml Trpiš-Lösung abdecken und 30 Minuten bei Raumtemperatur inkubieren. Eier beginnen nach etwa 5 Minuten Inkubation blasse Flecken zu entwickeln und erreichen schließlich eine milchig-weiße Farbe, sobald sie geklärt sind.
- Spülen Sie die Eier wie in Schritt 3.3.3, um trpiš Lösung zu entfernen.
- Lagern Sie das Abwasser in einem dafür vorgesehenen Abfallbehälter und entsorgen Sie überschüssiges Wasser in den Abfluss.
- Bleichlösung herstellen (Trpiš-Lösung - beschrieben in Trpiš (1970)21 und modifiziert nach Kaiser et al. (2014)22). Trpiš-Lösung ist 0,59 M Natriumhypochlorit und 0,35 M Essigsäure, gelöst in destilliertem H2O.
-
Lagerung
- In 500 μL dH2O lagern und einige Tage zwischen 2-8 °C aufbewahren. Entfernen Sie den größten Teil des Wassers vorsichtig vor der Betrachtung und Bildgebung auf Masse, aber vermeiden Sie die Austrocknung der Eier, indem Sie eine kleine Menge Wasser im Uhrglas lassen. Dies wird das Fotografieren der Eier nicht stören. Einzelne Eier können für eine bildgebung mit höherer Vergrößerung auf ein Objektträgerobjektiv gelegt werden.
Ergebnisse
Die 3xP3-Expression von eYFP, dsRed und eCFP ermöglicht eine zuverlässige, leicht unterscheidbare Identifizierung von Individuen, die die Markergene besitzen, die die Expression in Augen und ventralen Ganglien von An. gambiae puppen erzeugen (Abbildung 3). Die Differentielle Morphologie, die in männlichen und weiblichen äußeren Genitalien beobachtet wurde, die für die Geschlechtsbestimmung verwendet wurden, und ein Beispiel für eine nicht identifizierbare Puppe sind ...
Diskussion
Das Verständnis der Mückengenfunktion ist entscheidend, um neue Ansätze zur Kontrolle von Anopheles und zur Beeinflussung der Malariaübertragung zu entwickeln. Das beschriebene GAL4-UAS-System ist ein vielseitiges und leistungsfähiges System zur funktionellen Analyse von Kandidatengenen, und bisher haben wir das System verwendet, um die genetischen Grundlagen der Insektizidresistenz17 und der kutikulären Kohlenwasserstoffproduktion15,23 zu untersuchen sowie verschiedene Mückenzellpopulationen fluoresziere...
Offenlegungen
Die Autoren haben nichts preiszugeben.
Danksagungen
Wir danken ihnen für die Finanzierung durch LSTM und IVCC (Adriana Adolfi), BBSRC (New Investigator Award (AL), MRC (PhD studentship to BCP:MR/P016197/1), Wellcome (Sir Henry Wellcome Postdoctoral fellowship to LG: 215894/Z/19/Z), die die Gal4UAS-Analyse in die Vorschläge aufgenommen haben.
Materialien
| Name | Company | Catalog Number | Comments |
| 100 x 15 mm plastic Petri dish | SLS | 2175546 | Pack of 10 |
| 1000 µL Gilson Pipette | Gilson | F144059P | |
| 20/25 mL Universal Tubes | Starlab | E1412-3020 | Pack of 400 |
| 3 mL Pasteur Pipettes | SLS | G612398 | Greiner Pasteur pipette 3 mL sterile individually wrapped |
| 50 mL Falcon Tubes | Fisher Scientific | 11512303 | |
| Absolute Ethanol | Fisher Scientific | BP2818-500 | 500 mL |
| Acetic Acid | SLS | 45726-1L-F | 1 L |
| Cages | SLS | E6099 | 30x30x30 with screen port |
| Fine Paint Brushes | Amazon | UKDPB66 | KOLAMOON 9 Pieces Detail Painting Brush Set Miniture Brushes for Watercolor, Acrylic Painting, Oil Painting (Wine Red) |
| Fish food | Amazon | Tetra Min Fish Food, Complete Food for All Tropical Fish for Health, Colour and Vitality, 10 L | |
| Formaldehyde Solution | Sigma Aldrich | F8775 | |
| Mouth Aspirator | John Hock | 612 | |
| Pond Salt | Amazon | Blagdon Guardian Pond Tonic Salt, for Fish Health, Water Quality, General Tonic, pH Buffer, 9.08 kg, treats 9,092 L | |
| Pupae Pots | Cater4you | SP8OZ | 250 pots with lids |
| Small Plastic Buckets | Amazon | 2.5 L White Plastic Pail Complete with White Lid (Pack of 10) | |
| Sodium Hypochlorite | Fisher Scientific | S25552 |
Referenzen
- Brand, A. H., Perimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 118 (2), 401-415 (1993).
- Duffy, J. B. GAL4 system in drosophila: A fly geneticist's swiss army knife. Journal of Genetics and Development. 34 (1-2), 1-15 (2002).
- Dow, J. A. . ELS. , (2012).
- Edi, C. V., et al. CYP6 P450 Enzymes and ACE-1 Duplication Produce Extreme and Multiple Insecticide Resistance in the Malaria Mosquito Anopheles gambiae. PLoS Genetics. 10 (3), 1004236 (2014).
- Daborn, P. J., et al. Using Drosophila melanogaster to validate metabolism-based insecticide resistance from insect pests. Insect Biochemistry and Molecular Biology. 42 (12), 918-924 (2012).
- Riveron, J. M., et al. Genome-wide transcription and functional analyses reveal heterogeneous molecular mechanisms driving pyrethroids resistance in the major malaria vector Anopheles funestus across Africa. Genes Genomes Genetics. 7 (6), 1819-1832 (2017).
- Riveron, J. M., et al. A single mutation in the GSTe2 gene allows tracking of metabolically based insecticide resistance in a major malaria vector. Genome Biology. 15 (2), (2014).
- Lynd, A., Lycett, G. J. Development of the Bi-Partite Gal4-UAS System in the African Malaria Mosquito, Anopheles gambiae. PLoS ONE. 7 (2), 31552 (2012).
- Lynd, A., Lycett, G. J. Optimization of the Gal4-UAS system in an Anopheles gambiae cell line. Insect Molecular Biology. 20 (5), 599-608 (2011).
- Adolfi, A., Pondeville, E., Lynd, A., Bourgouin, C., Lycett, G. J. Multi-tissue GAL4-mediated gene expression in all Anopheles gambiae life stages using an endogenous polyubiquitin promoter. Insect Biochemistry and Molecular Biology. 96, 1-9 (2018).
- Kokoza, V. A., Raikhel, A. A. Targeted gene expression in the transgenic Aedes aegypti using the binary Gal4-UAS system. Insect Biochemistry and Molecular Biology. 41, 637-644 (2011).
- O'Brochta, D. A., Pilitt, K. L., Harrell, R. A., Aluvihare, C., Alford, R. T. Gal4-based Enhancer-Trapping in the Malaria Mosquito Anopheles stephensi. Genes Genomes Genetics. 2, 21305-21315 (2012).
- Zhao, B., et al. Regulation of the Gut-Specific Carboxypeptidase: A Study Using the Binary Gal4/UAS System in the Mosquito Aedes Aegypti. Insect Biochemistry and Molecular Biology. 54, 1-10 (2014).
- Imamura, M., et al. Targeted Gene Expression Using the GAL4/UAS System in the Silkworm Bombyx mori. Genetics. 165 (3), 1329-1340 (2003).
- Lynd, A., et al. Development of a functional genetic tool for Anopheles gambiae oenocyte characterisation: application to cuticular hydrocarbon synthesis. bioRxiv. , (2019).
- Pondeville, E., et al. Hemocyte-targeted gene expression in the female malaria mosquito using the hemolectin promoter from Drosophila. Insect Biochemistry and Molecular Biology. 120, 103339 (2020).
- Adolfi, A., et al. Functional genetic validation of key genes conferring insecticide resistance in the major African malaria vector, Anopheles gambiae. Proceedings of the National Academy of Sciences of the United States of America. 116 (51), 25764-25772 (2019).
- Pondeville, E., et al. Efficient integrase-mediated site-specific germline transformation of Anopheles gambiae. Nature Protocols. 9 (7), 1698-1712 (2014).
- Horn, C., Schmid, B. G. M., Pogoda, F. S., Wimmer, E. A. Fluorescent transformation markers for insect transgenesis. Insect Biochemistry and Molecular Biology. 32, 1221-1235 (2002).
- Clements, A. . A. Biology of Mosquitoes, Volume 1: Development, Nutrition and Reproduction. 1, (1992).
- Trpiš, M. A new bleaching and decalcifying method for general use in zoology. Canadian Journal of Zoology. 48, 892-893 (1970).
- Kaiser, M. L., Duncan, F. D., Brooke, B. D. Embryonic Development and Rates of Metabolic Activity in Early and Late Hatching Eggs of the Major Malaria Vector Anopheles gambiae. PLoS ONE. 9 (12), 114381 (2014).
- Grigoraki, L., Grau-Bové, X., Yates, H. C., Lycett, G. J., Ranson, H. Isolation and transcriptomic analysis of Anopheles gambiae oenocytes enables the delineation of hydrocarbon biosynthesis. eLife. 9, 58019 (2020).
- Xiao, Y. -. H., Yin, M. -. H., Hou, L., Pei, Y. Direct amplification of intron-containing hairpin RNA construct from genomic DNA. BioTechniques. 41 (5), 548-552 (2006).
- Livak, K. J. Organization and Mapping of a Sequence on the Drosophila melanogaster X and Y Chromosomes That Is Transcribed during Spermatogenesis. Genetics. 107 (4), 611-634 (1984).
- MR4, CDC, NEI & beiResources. . The MR4 Methods in Anopheles Research Laboratory Manual. 5th Edition. , (2015).
- Sik Lee, Y., Carthew, R. W. Making a better RNAi vector for Drosophila: use of intron spacers. Methods. 30 (4), 322-329 (2003).
- Cha-aim, K., Hoshida, H., Fukunaga, T., Akada, R., Peccoud, J. . Gene Synthesis: Methods and Protocols. , 97-110 (2012).
- Cavener, D. R. Comparison of the consensus sequence flanking translational start sites in Drosophila and vertebrates. Nucleic Acids Research. 15 (4), 1353-1361 (1987).
- Wang, Y., Wang, F., Wang, R., Zhao, P., Xia, Q. 2A self-cleaving peptide-based multi-gene expression system in the silkworm Bombyx mori. Scientific Reports. 5, (2015).
- Galizi, R., et al. A synthetic sex ratio distortion system for the control of the human malaria mosquito. Nature Communications. 5, 3977 (2014).
- Kondo, S., et al. Neurochemical organisation of the Drosophila Brain Visualised by Endogenously Tagged Neurotransmitter Receptors. Cell Reports. 30 (1), 284-297 (2020).
- Lee, P. -. T., et al. A gene-specific T2A-GAL4 library for Drosophila. eLife. 7, 35574 (2018).
- Marois, E., et al. High-throughput sorting of mosquito larvae for laboratory studies and for future vector control interventions. Malaria Journal. 11, 302 (2012).
- Crawford, J. E., et al. Efficient production of male Wolbachia-infected Aedes aegypti mosquitoes enables large-scale suppression of wild populations. Nature Biotechnology. 38 (4), 482-492 (2020).
- Goltsev, Y., et al. Developmental and evolutionary basis for drought tolerance of the Anopheles gambiae embryo. Developmental Biology. 330 (2), 462-470 (2009).
- Rezende, G. L., et al. Embryonic desiccation resistance in Aedes aegypti: presumptive role of the chitinized Serosal Cuticle. BMC Developmental Biology. 8 (1), 82 (2008).
- Vargas, H. C. M., Farnesi, L. C., Martins, A. J., Valle, D., Rezende, G. L. Serosal cuticle formation and distinct degrees of desiccation resistance in embryos of the mosquito vectors Aedes aegypti, Anopheles aquasalis and Culex quinquefasciatus. Journal of Insect Physiology. 62, 54-60 (2014).
- Chang, C. -. H., et al. The non-canonical Notch signaling is essential for the control of fertility in Aedes aegypti. PLOS Neglected Tropical Diseases. 12 (3), 0006307 (2018).
- Clemons, A., Flannery, E., Kast, K., Severson, D., Duman-Scheel, M. Immunohistochemical Analysis of Protein Expression during Aedes aegypti Development. Spring Harbor Protocols. 10, 1-4 (2010).
- Juhn, J., James, A. A. Hybridization in situ of Salivary Glands, Ovaries and Embryos of Vector Mosquitoes. Journal of Visualized Experiments. , e3709 (2012).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten