
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Verwendung der postnatalen In-vivo-Elektroporation zur Untersuchung der Morphologie von Kleinhirngranula-Neuronen und der Synapsenentwicklung
In diesem Artikel
Erratum Notice
Zusammenfassung
Hier beschreiben wir eine Methode zur Visualisierung der Synaptogenese von Körnerneuronen im Kleinhirn der Maus über den zeitlichen Verlauf der postnatalen Gehirnentwicklung, wenn diese Zellen ihre synaptischen Strukturen verfeinern und Synapsen bilden, um sich in den gesamten Gehirnkreislauf zu integrieren.
Zusammenfassung
Neuronen verändern sich während der Entwicklung des Gehirns dynamisch in ihrer Struktur und Funktion, um geeignete Verbindungen zu anderen Zellen zu bilden. Das Kleinhirn von Nagetieren ist ein ideales System, um die Entwicklung und Morphogenese eines einzelnen Zelltyps, des Kleinhirngranula-Neurons (CGN), im Laufe der Zeit zu verfolgen. Hier wurde die In-vivo-Elektroporation von Körnerneuronen-Vorläuferzellen im sich entwickelnden Kleinhirn der Maus eingesetzt, um Zellen für nachfolgende morphologische Analysen spärlich zu markieren. Die Wirksamkeit dieser Technik zeigt sich in ihrer Fähigkeit, wichtige Entwicklungsstadien der CGN-Reifung aufzuzeigen, mit besonderem Schwerpunkt auf der Bildung von dendritischen Klauen, bei denen es sich um spezialisierte Strukturen handelt, in denen diese Zellen den Großteil ihrer synaptischen Inputs erhalten. Diese Technik liefert nicht nur Momentaufnahmen von CGN-Synapsenstrukturen während der gesamten Kleinhirnentwicklung, sondern kann auch angepasst werden, um Körnerneuronen zellautonom genetisch zu manipulieren, um die Rolle eines Gens von Interesse und seine Auswirkungen auf die CGN-Morphologie, die Klauenentwicklung und die Synaptogenese zu untersuchen.
Einleitung
Die Entwicklung des Gehirns ist ein langwieriger Prozess, der sich von der Embryogenese bis ins postnatale Leben erstreckt. Während dieser Zeit integriert das Gehirn eine Kombination aus intrinsischen und extrinsischen Reizen, die die Verdrahtung von Synapsen zwischen Dendriten und Axonen formen, um letztendlich das Verhalten zu steuern. Das Kleinhirn von Nagetieren ist ein ideales Modellsystem, um zu untersuchen, wie sich Synapsen entwickeln, da die Entwicklung eines einzelnen Neuronentyps, des Kleinhirn-Körner-Neurons (CGN), beim Übergang von einer Vorläuferzelle zu einem reifen Neuron verfolgt werden kann. Dies ist zum Teil auf die Tatsache zurückzuführen, dass sich ein Großteil der Kleinhirnrinde postnatal entwickelt, was eine einfache genetische Manipulation und Zellmarkierung nach der Geburt ermöglicht1.
Bei Säugetieren beginnt die CGN-Differenzierung am Ende der Embryonalentwicklung, wenn eine Untergruppe proliferativer Zellen im Hinterhirn über die rhombische Lippe wandert, um eine sekundäre Keimzone auf der Oberfläche des Kleinhirns zu bilden 2,3,4. Obwohl sie vollständig an eine Granule Neuron Progenitor (GNP)-Identität gebunden sind, vermehren sich diese Zellen bis zum postnatalen Tag 14 (P14) im äußeren Teil der äußeren Körnerschicht (EGL) weiter. Die Proliferation dieser Schicht führt zu einer massiven Ausdehnung des Kleinhirns, da diese Zellen ausschließlich CGNs5 hervorbringen. Sobald neugeborene CGNs den Zellzyklus in der EGL verlassen, wandern sie nach innen in Richtung der inneren Körnerschicht (IGL) und hinterlassen ein Axon, das sich in der molekularen Schicht des Kleinhirns gabelt und wandert und parallele Fasern bildet, die auf Purkinje-Zellen synapsieren6. Die Position dieser Fasern innerhalb der Molekülschicht hängt vom Zeitpunkt des Zellzyklusaustritts ab.
CGNs, die sich zuerst differenzieren, verlassen ihre parallelen Fasern zum unteren Rand der Molekülschicht, während die Axone von CGNs, die sich später differenzieren, oben 7,8 gruppiert sind. Sobald die CGN-Zellkörper das IGL erreichen, beginnen sie, Dendriten zu entwickeln und Synapsen mit nahe gelegenen inhibitorischen und exzitatorischen Neuronen zu bilden. Der ausgereifte dendritische Baum eines CGN weist eine stereotype Architektur mit vier Hauptprozessen auf. Im Laufe der CGN-Reifung bilden die Strukturen am Ende dieser Dendriten eine Klaue, die mit postsynaptischen Proteinen angereichert wird 9,10. Diese spezialisierten Strukturen, dendritische Klauen genannt, enthalten die Mehrheit der Synapsen auf Körnerneuronen und sind wichtig, um sowohl exzitatorische Inputs von moosigen Faserinnervationen aus den Pons als auch inhibitorische Inputs von lokalen Golgi-Zellen zu erhalten. Einmal vollständig konfiguriert, ermöglichen die synaptischen Verbindungen von CGNs diesen Zellen, Eingaben von präzerebellären Kernen an Purkinje-Zellen weiterzuleiten, die aus der Kleinhirnrinde in die tiefen Kleinhirnkerne projizieren.
Die postnatale In-vivo-Elektroporation von GNPs ist vorteilhaft gegenüber anderen markierungsbasierten Methoden, wie z. B. Virusinfektion und Erzeugung transgener Mauslinien, da die Expression gewünschter Konstrukte auf einer schnellen Zeitachse erreicht werden kann und die Methode auf eine kleine Population von Zellen abzielt, was bei der Untersuchung zellautonomer Effekte nützlich ist. Diese Methode wurde in früheren Studien verwendet, um die morphologische Entwicklung von CGNs zu untersuchen. Diese Studien konzentrierten sich jedoch entweder auf einen einzelnen Zeitpunkt oder ein kurzes Zeitfenster 9,10,11,12,13. Diese Markierungsmethode wurde mit einer Bildanalyse gepaart, um die Veränderungen in der CGN-Morphologie zu dokumentieren, die über den gesamten zeitlichen Verlauf der CGN-Differenzierung in den ersten drei Wochen des postnatalen Lebens auftreten. Diese Daten zeigen die Dynamik der Entwicklung von CGN-Dendriten, die dem Aufbau von Kleinhirnschaltkreisen zugrunde liegt.
Protokoll
HINWEIS: Alle Verfahren wurden gemäß Protokollen durchgeführt, die vom Duke University Institutional Animal Care and Use Committee (IACUC) genehmigt wurden.
1. DNA-Vorbereitung für die In-vivo-Elektroporation oder IVE (1 Tag vor der Operation)
- Sammeln Sie die folgenden Materialien: gereinigte DNA (0,5-25 μg pro Tier), 3 M Natriumacetat, Ethanol, Fast Green-Farbstoff, hochreines destilliertes Wasser, Phosphatpufferlösung (PBS) (siehe Materialtabelle).
ANMERKUNG: Für DNA wurde ein Konstrukt, das grün fluoreszierendes Protein (GFP) unter einem humanen Ubiquitin-Promotor exprimiert, aus Addgene (FUGW, https://www.addgene.org/14883/) erhalten. Jedes Konstrukt, das GFP oder ein anderes fluoreszierendes Protein unter der Kontrolle eines ubiquitären Promotors exprimiert, sollte funktionieren. Die CGN-spezifische Markierung bei dieser Technik ist nicht vom Konstrukt abhängig, sondern von der Elektroporation. - Bereiten Sie die DNA für die Elektroporation vor, indem Sie die gewünschte Menge DNA, 10 Vol.-% 3 M Natriumacetat und 250 Vol.-% 100 % eiskaltes Ethanol mischen. Beachten Sie, dass die DNA sofort aus der Lösung ausfällt.
- Fällen Sie das DNA-Gemisch über Nacht bei -20 °C oder eine Stunde lang bei -80 °C aus.
- Pellet fällte DNA in einer Tischzentrifuge bei >16.000 × g aus und wusch zweimal mit 70% Ethanol.
- Lassen Sie das DNA-Pellet vollständig trocknen und rekonstituieren Sie es in einer 1x PBS + 0,02% Fast Green-Lösung.

Abbildung 1: Begrenzung der Einspritztiefe auf 1,5 mm mit einem Abstandshalter. (A) Ein 11,2-mm-Segment wird mit einer Rasierklinge von einer Ladepipette abgeschnitten. (B) Der Abstandshalter wird auf die Spitze der Hamilton-Spritze aufgesetzt (Gesamtlänge beträgt 1,27 cm oder 0,5 Zoll) und entweder mit Klebstoff oder Parafolie gesichert. Die freiliegende Spitze sollte 1,5 mm lang sein. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
2. In-vivo-Elektroporation von Körnerneuronen-Vorläuferzellen in sieben Tage alten postnatalen Mäusen
HINWEIS: Alle Elektroporationsoperationen wurden in einem sterilen und hochbelüfteten Operationssaal durchgeführt, und das gesamte Personal trug eine komplette persönliche Schutzausrüstung wie Handschuhe, Gesichtsmaske, Haarhaube, Kittel und Überschuhe. Alternativ können die Operationen in einer belüfteten und sterilen Haube durchgeführt werden.
- Sammeln Sie die folgenden Materialien: DNA für die Elektroporation, kleine chirurgische Schere, kleine chirurgische Pinzette, maßgeschneiderte Hamilton-Spritze, Applikator mit Wattestäbchen, Heizkissen, Betadin, 70% Ethanol, 1x PBS, Parafilm, Gewebekleber (n-Butylestercyanacrylat), Isofluran, Elektroporator und Pinzettenelektroden (siehe Materialtabelle).
- Schneiden Sie einen Abstandshalter von einer sterilisierten Ladespitze ab, der über die Hamilton-Spritze passt, um die Injektionstiefe auf 1,5 mm zu begrenzen (Abbildung 1A, B). Befestigen Sie den Abstandshalter mit Klebstoff oder Parafolie.
- Anästhesieren Sie den P7-Welpen in einer Isoflurankammer mit einer Abgaberate von 0,8 l/min. Bestätigen Sie die Vollnarkose, indem Sie das Tier auf verminderte Atmung und das Fehlen einer Zehen- oder Schwanzquetschreaktion überwachen (Abbildung 2A).
- Sobald das Tier vollständig betäubt ist, legen Sie die Welpen auf einen Sockel, der mit einem Nasenkegel ausgestattet ist, der konstant 4% Isofluran bei einer Abgaberate von 0,8 l/min abgibt. Reinigen Sie die Oberseite des Kopfes des Welpen 3 Mal mit einem sterilen Tupfer Betadin und dann 70% Ethanol, abwechselnd zwischen den beiden, um die Stelle vorzubereiten. Lassen Sie die Lösung trocknen, bevor Sie fortfahren.
- Machen Sie mit einer sterilisierten Schere einen kleinen Einschnitt mit einem Schnitt, der den Abstand von der Oberseite bis zur Basis der Ohren überspannt, um das Hinterhirn freizulegen (Abbildung 2B).
- Lokalisieren Sie das Kleinhirn (Abbildung 2C), führen Sie die freiliegende Spitze der Hamilton-Spritze senkrecht zum Gehirn durch den Schädel ein und injizieren Sie 1,5 μl DNA-Gemisch in das Kleinhirnarenchym, indem Sie den hinteren Kolben der Spritze langsam drücken. Ziehen Sie nach der Abgabe der DNA-Mischung die Nadel langsam zurück, um ein Verschütten des Rückens zu verhindern, und lassen Sie die DNA-Lösung 30 Sekunden lang diffundieren.
- Schalten Sie das Isofluran aus und legen Sie den Welpen auf ein 37 °C heißes Heizkissen. Bereiten Sie die Pinzettenelektrode für die Elektroporation vor, indem Sie beide Enden in steriles 1x PBS tauchen.
HINWEIS: Das Benetzen der Pinzettenelektrode verhindert Kontaktverbrennungen auf der Haut des Welpen während der Verabreichung der elektrischen Impulse. - Richten Sie die Pinzettenelektrode über der Injektionsstelle aus, wobei das Plusende nach unten und das negative Ende über dem Kopf des Tieres zeigt (Abbildung 2D). Verabreichen Sie fünf elektrische Impulse vom Elektroporator mit den folgenden Einstellungen: 50 ms, 130 V und 950 ms Intervall zwischen den Impulsen.
HINWEIS: Führen Sie bei Bedarf eine Testinjektion durch, um sicherzustellen, dass sich die Injektionsstelle auf dem Kleinhirnwurm befindet (Abbildung 2E). - Drücken Sie den Einschnitt zu und versiegeln Sie die Wunde mit einem ungiftigen n-Butylester-Cyanacrylat-Gewebekleber. Reinigen Sie die Wunde mit 70% Ethanol, da jede Spur von Blut die Wahrscheinlichkeit von elterlichem Kindsmord und Kannibalismus erhöht.
- Lassen Sie das Tier sich auf einem 37 °C warmen Heizkissen erholen, bevor Sie den Welpen zum Muttertier zurückbringen. Überwachen Sie den/die Welpen alle 30 Minuten für mindestens 2 Stunden nach der Operation, um eine vollständige Genesung sicherzustellen.
HINWEIS: Kindstötung durch einen Elternteil ist weit verbreitet. Um Kannibalismus vorzubeugen, bringen Sie den Vater in einem anderen Käfig unter, bevor Sie mit der Elektroporation beginnen, und bringen Sie die gereinigten und geborgenen Welpen (d. h. ohne Blutflecken , voll beweglich) immer in den ursprünglichen Käfig auf der ursprünglichen Einstreu zurück. Welpen können auch mit Kot aus dem ursprünglichen Käfig abgewischt werden, um den Blutgeruch zu minimieren. Die Verwendung einer Ersatzmutter kann erforderlich sein, wenn die ursprüngliche Mutter ihre Welpen weiterhin kannibalisiert.

Abbildung 2: In vivo zerebelläre Elektroporation von granulären Neuronen-Vorläuferzellen in P7-Wildtyp-Mauswelpen. (A) Welpen werden mit 4% Isofluran anästhesiert, das mit einer Geschwindigkeit von 0,8 l / min verabreicht wird, um die Anästhesie während der gesamten Injektion der DNA-Lösung sicherzustellen. Isofluran wird mit einer Geschwindigkeit von 0,8 l/min abgegeben. (B) Nach 3-maliger Sterilisation der Maus mit Betadin und 70% Ethanol wird ein Einschnitt vorgenommen, der sich über den Abstand der Ohren erstreckt und das Hinterhirn freilegt. (C) Ein vergrößertes Bild einer weißen Abgrenzung auf dem Schädel, einem Orientierungspunkt für die Injektionsstelle. Das DNA-Konstrukt sollte innerhalb von 1 mm über der Markierung injiziert werden. Gepunktete Linien umreißen die Abgrenzung und ein schwarzer Pfeil kennzeichnet die Injektionsstelle. Die Kämme des Kleinhirnwurms können sichtbar sein und für das Auffinden der Injektionsstelle nützlich sein. (D) Pinzetten-Elektrodenorientierung für eine effiziente Elektroporation. Das Plus-Ende (+) muss nach unten ausgerichtet sein, um negativ geladene DNA vor der Verabreichung elektrischer Impulse in das Kleinhirnarenchym zu ziehen. (E) Die Testinjektion von 1 μl eines 0,02%igen Fast Green-Farbstoffs zeigt, dass die Injektion in der Mitte des Kleinhirnwurms zwischen den Läppchen 5-7 lokalisiert ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
3. Immunhistochemie elektroporierter CGNs
- Sammeln Sie die folgenden Materialien: Isofluran, 1x PBS, 4% Paraformaldehyd (PFA), 30% Saccharose, normales Ziegenserum, nichtionisches Reinigungsmittel, Objektträger, Glasdeckgläser, Nagellack, Eindeckmittel, Hoechst-Kernfarbstoff und geeignete Primär- und Sekundärantikörper (siehe Materialtabelle).
- Betäuben Sie das Versuchstier mit Isofluran und bestätigen Sie die Vollnarkose mit einer Zehen- und Schwanzklemme.
- Führen Sie eine transkardiale Perfusion durch, indem Sie langsam 1x PBS und 4% PFA in den linken Ventrikel des Herzens des Tieres injizieren. Lassen Sie das Blut aus dem Tier abfließen, indem Sie die Hohlvene abschneiden.
- Fixieren Sie das Gehirn über Nacht, indem Sie es in 4% PFA bei 4 °C tauchen. Spülen Sie das Gehirn am nächsten Tag schnell mit 1x PBS aus und übertragen Sie das Gehirn in 30% Saccharose in 1x PBS zur Kryoprotektion für mindestens 24 Stunden.
- Falls erforderlich, schneiden Sie das Gehirn entlang der rostral-kaudalen Achse in zwei Hälften und bestätigen Sie die Expression des transfizierten Reporterkonstrukts mit einem aufrechten fluoreszierenden Präpariermikroskop.
HINWEIS: Halten Sie das Gehirn in 1x PBS in einer kleinen Schüssel untergetaucht, um ein Austrocknen zu verhindern. - Montieren Sie das Gehirn auf einem gefrierenden Mikrotom, schneiden Sie 25 μm sagittale Schnitte und lassen Sie die Schnitte in einer 1:1-Mischung aus 1x PBS und Glycerin entfalten.
HINWEIS: Schnitte können in dieser Kryoprotektivlösung bei -20 °C zur Langzeitlagerung gelagert werden. - Waschen Sie die Schnitte dreimal in 1x PBS für jeweils 10 Minuten, um das Kryoprotektivum zu entfernen, und blockieren Sie das Gewebe in 1x PBS + 10% normalem Ziegenserum + 0,2% nichtionischem Reinigungsmittel auf einem Orbitalschüttler bei Raumtemperatur für 1 h.
- Bereiten Sie eine primäre Antikörperlösung vor: 1x PBS, 10% normales Ziegenserum, 0,2% nichtionisches Detergens und Anti-GFP-Antikörper und zentrifugieren Sie die Lösung 5 Minuten lang bei >16.000 × g. Schnitte in der Antikörperlösung werden bei 4 °C auf einem Orbitalschüttler für 48 h inkubiert.
- Waschen Sie die primäre Antikörperlösung fünfmal 15 Minuten lang mit 1x PBS + 0,2% nichtionischem Reinigungsmittel ab.
- Sekundäre Antikörperlösung vorbereiten: 1x PBS, 10% normales Ziegenserum, 0,2% nichtionisches Detergens und ein geeigneter sekundärer Antikörper zum Nachweis von GFP; Zentrifugieren Sie die Lösung bei >16.000 × g. Inkubationsabschnitte in der Antikörperlösung auf einem Orbitalschüttler bei Raumtemperatur für 2-3 h. Schützen Sie die Abschnitte vor Lichteinwirkung, um ein Ausbleichen zu vermeiden.
- Waschen Sie die sekundäre Antikörperlösung dreimal mit 1x PBS + 0,2% nichtionischem Reinigungsmittel für jeweils 15 Minuten ab. Inkubieren Sie Schnitte in 1x PBS + Hoechst für 5 Minuten, um Kerne zu färben.
- Hoechst-Lösung mit 1x PBS + 0,2% nichtionischem Reinigungsmittel abwaschen und auf Objektträger montieren. Decken Sie die Abschnitte mit Montagemedien ab, decken Sie die Objektträger ab und versiegeln Sie den Objektträger mit Nagellack, um Verdunstung zu verhindern.
4. Morphologische Analysen von CGNs - dreidimensionale (3D) Rekonstruktion und Oberfläche und zelluläres Volumen
- Bilden Sie einzelne elektroporierte CGNs auf einem konfokalen Mikroskop mit einem 63-fachen Objektiv mit einem 2-fachen Zoom ab und nehmen Sie Z-Stapel-Bilder mit 0,5 μm pro Stapel auf. Bilden Sie eine Zelle pro Bildfenster ab, um eine einfache Bildanalyse und -rekonstruktion zu ermöglichen.
- Installieren Sie das Simple Neurite Tracer Plug-in für FIDSCHI über den folgenden Link (https://imagej.net/Simple_Neurite_Tracer:_Basic_Instructions), um die Struktur von elektroporierten CGNs im dreidimensionalen (3D) Raum einfach und effizient zu verfolgen.
HINWEIS: Es gibt eine aktualisierte Version des Plug-Ins (https://imagej.net/SNT). - Analysieren Sie die Neuritenlänge und die dendritische Klauenbildung verblindet mit Simple Neurite Tracer. Laden Sie einkanalige Z-Stack-Bilder von elektroporierten CGNs auf FIDSCHI hoch und klicken Sie auf Plugins | Segmentierung | Einfacher Neuriten-Tracer (Abbildung 3D).
- Rufen Sie das Dropdown-Menü auf, und wählen Sie Neuen 3D-Viewer erstellen aus (Abbildung 3D).
- Scrollen Sie zur Basis eines Dendriten, wo er sich mit dem Zellsoma verbindet, und starten Sie einen Pfad, indem Sie auf die Kreuzung klicken. Verfolgen Sie den Pfad manuell, indem Sie durch die Abschnitte klicken, in denen das Zellenfüllsignal am hellsten ist, und [y] drücken, um die Spur beizubehalten. Verfolgen Sie bis zum Ende des Dendriten, wenn er keine Klaue enthält, oder bis zur Basis der Klaue und bestätigen Sie den Pfad durch Drücken von [f] (Abbildung 4D).
- Verfolgen Sie als Nächstes die Klaue, indem Sie einen Pfad an der Basis der Struktur beginnen und bis zum Ende des längsten Neuriten verfolgen. Verfolgen Sie sekundäre und tertiäre Zweige, indem Sie unter Windows [Strg] oder unter Mac OS [alt] gedrückt halten und auf den Pfad klicken. Bestätigen Sie den Pfad, indem Sie [f] drücken.
- Beachten Sie, dass die Messungen für die Spuren in einem separaten Fenster sichtbar sind. Addieren Sie alle Maße der Klauenäste (primär, sekundär, tertiär), um die Gesamtlänge für jede Klaue zu erhalten.
- Für die Analyse der Oberfläche und des zellulären Volumens von elektroporierten CGNs laden Sie die Imaris-Zellanalysesoftware herunter (https://imaris.oxinst.com/).
HINWEIS: FIDSCHI kann auch verwendet werden, um Zellen in 3D aus Z-Stack-Bildern zu rekonstruieren, indem leicht verfügbare und kostenlose Plug-Ins verwendet werden. Darüber hinaus gibt es eine volumetrische Rendering-Funktion in Simple Neurite Tracer, aber Imaris wurde aus den unten aufgeführten Gründen verwendet. - Laden Sie das Z-Stack-Bild eines elektroporierten CGN auf Imaris hoch. Greifen Sie auf das 3D-Rekonstruktions-Toolkit zu, indem Sie auf Übernehmen drücken.
- Um das CGN zu rekonstruieren, drücken Sie Flächen, und wählen Sie einen gewünschten Bereich aus, der die gesamte Zelle im Bildfenster umfasst. Wenn Sie fertig sind, drücken Sie den blauen Vorwärtspfeil in der unteren rechten Ecke unter Erstellen.
- Wenn das Bild mehrere Kanäle für unterschiedliche Signale enthält, wählen Sie den Kanal aus, der das elektroporierte CGN enthält, und drücken Sie den blauen Vorwärtspfeil.
- Stellen Sie mit dem Schieberegler einen gewünschten Schwellenwert ein, der am genauesten zum Signal der elektroporierten Zelle passt. Zoomen Sie näher an die Oberfläche der Zelle, um den Schwellenwert genau zu bestimmen. Wenn Sie fertig sind, drücken Sie den doppelten grünen Pfeil, um die Zelle zu rekonstruieren und die Oberfläche und die Volumegröße aus den Metadaten abzurufen.

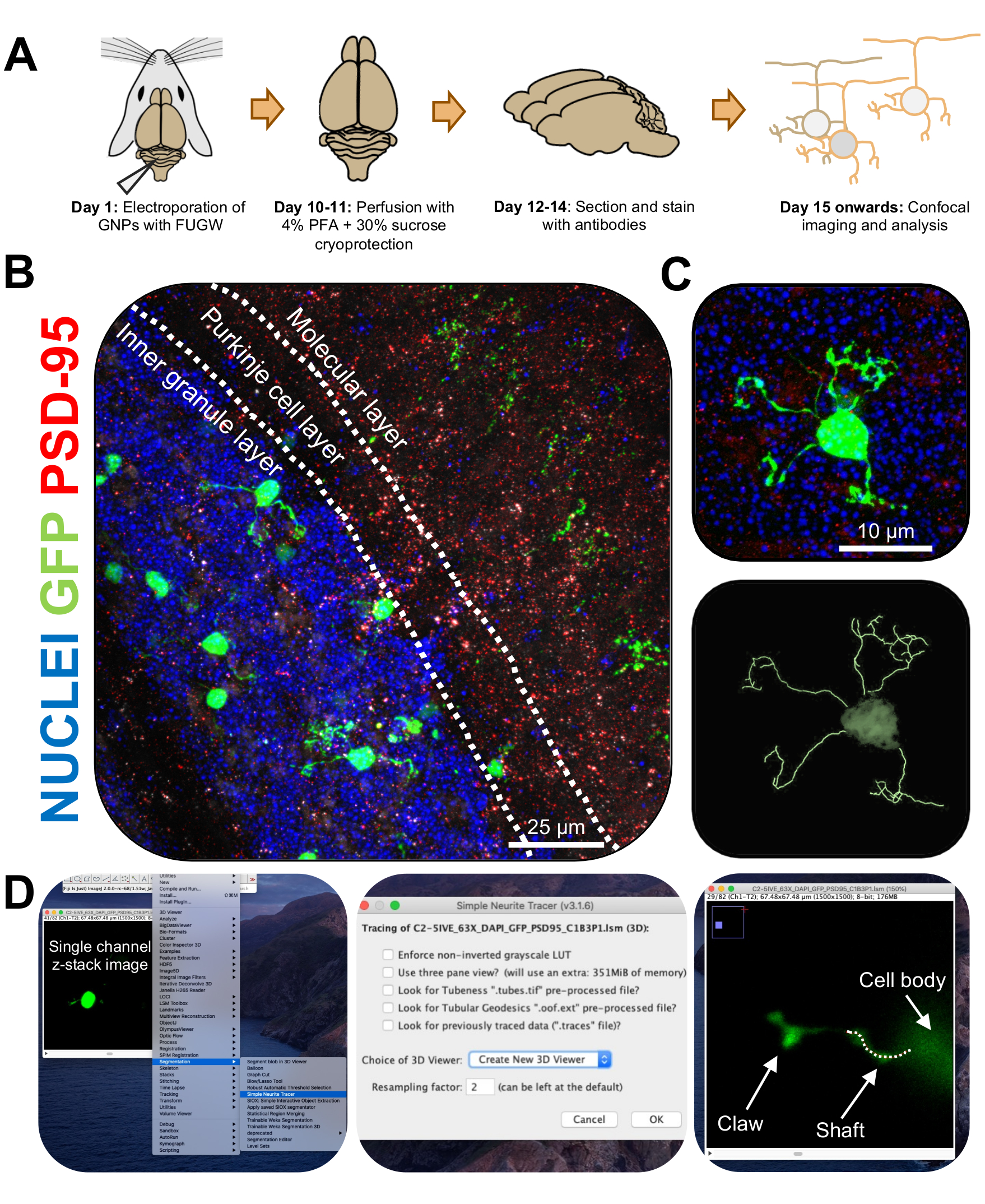
Abbildung 3: Immunhistochemische Analyse und dreidimensionale Rekonstruktion von elektroporierten Körnerneuronen. P7 CD-1-Mäuse wurden mit einem GFP-exprimierenden Konstrukt elektroporiert. Die Gehirne wurden gesammelt und einer Immunhistochemie, konfokalen Mikroskopie und 3D-Rekonstruktion für die morphologische Analyse unterzogen. (A) Zeitleiste von der Elektroporation bis zur Bildverarbeitung einer 10-DPI-Maus. (B) Maximales Projektionsbild eines sagittalen Querschnitts des elektroporierten Kleinhirns 10-DPI; Weiße Linien grenzen Kleinhirnschichten ab, und der Maßstabsbalken beträgt 25 μm. (C) Maximales Projektionsbild eines einzelnen elektroporierten Körnerneurons 10-DPI und der entsprechenden 3D-Spur, der Maßstabsbalken beträgt 10 μm. (D) 3D-Rekonstruktionen wurden mit dem FIDSCHI-Plugin Simple Neurite Tracer erstellt. Alle Messungen wurden durch den z-Stapel verfolgt, wobei dem Cell-Fill-Signal gefolgt wurde. Schaft- und Klauenmessungen wurden für jeden Dendriten separat verfolgt; Die gepunktete Linie bezeichnet einen Teil des Dendriten innerhalb der aktuellen Ebene. Abkürzungen: 3D = dreidimensional; GFP = grün fluoreszierendes Protein; DPI = Tage nach der Injektion; PSD-95 = postsynaptisches Dichteprotein 95; BSPs = Vorläuferzellen von Körnerneuronen; PFA = Paraformaldehyd. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Ergebnisse

Abbildung 4: Analyse der Morphologie von Körnerneuronen während der Kleinhirnentwicklung. (A) Maximale Projektionsbilder von elektroporierten CGNs von 3-DPI bis 14-DPI (postnatales Alter P10 bis P21), Kernen (blau) und GFP (grün); Pfeilspitzen zeigen einzelne Dendriten an, und der Maßstabsbalken beträgt 10 μm. (B) Durchschnittliche Anzahl der Dendrite...
Diskussion
Kleinhirngranula-Neuronen sind die am häufigsten vorkommenden Neuronen im Säugetiergehirn und machen fast 60-70% der gesamten Neuronenpopulation im Nagetiergehirnaus 1,14. Das Kleinhirn wurde ausgiebig genutzt, um Mechanismen der zellulären Proliferation, Migration, Dendritenbildung und Synapsenentwicklung aufzuklären 6,9,10,11,15,16,17,18,19,20
Offenlegungen
Die Autoren erklären keine Interessenkonflikte.
Danksagungen
Die Arbeit wurde durch die NIH-Zuschüsse R01NS098804 (A.E.W.), F31NS113394 (U.C.) und das Summer Neuroscience Program (D.G.) der Duke University unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| Betadine | Purdue Production | 67618-150-17 | |
| Cemented 10 µL needle | Hamilton | 1701SN (80008) | 33 gauge, 1.27 cm (0.5 in), 4 point style |
| Chicken anti-GFP | Millipore Sigma | AB16901 | Our lab uses this antibody at a 1:1000 concentration |
| Cotton-tip applicator | |||
| Donkey anti-chicken Cy2 | Jackson ImmunoResearch | 703-225-155 | Our lab uses this antibody at a 1:500 concentration |
| Ethanol (200 proof) | Koptec | V1016 | |
| Electroporator ECM 830 | BTX Harvard Apparatus | 45-0052 | |
| Fast Green FCF | Sigma | F7252-5G | |
| FUGW plasmid | Addgene | 14883 | |
| Glass slides | VWR | 48311-703 | Superfrost plus |
| Glycerol | Sigma-Aldrich | G5516 | |
| Heating pad | Softheat | ||
| Hoescht 33342 fluorescent dye | Invitrogen | 62249 | |
| Imaris | Bitplane | ||
| Isoflurane | Patterson Veterinary | 07-893-1389 | |
| Micro cover glass | VWR | 48382-138 | |
| Nail polish | Sally Hansen | Color 109 | |
| Normal goat serum | Gibco | 16210064 | |
| O.C.T. embedding compound | Tissue-Tek | 4583 | |
| Olympus MVX10 Dissecting Scope | Olympus | MVX10 | |
| P200 pipette reach tip | Fisherbrand | 02-707-138 | Used for needle spacer |
| Parafilm | Bemis | PM-996 | |
| PBS pH 7.4 (10x) | Gibco | 70011-044 | |
| Simple Neurite Tracer | FIJI | https://imagej.net/Simple_Neurite_Tracer:_Basic_ Instructions | |
| Sucrose | Sigma | S0389 | |
| Surgical tools | RWD Life Science | Small scissors and tweezers | |
| Triton X-100 | Roche | 11332481001 | non-ionic detergent |
| Tweezertrodes | BTX Harvard Apparatus | 45-0489 | 5 mm, platinum plated tweezer-type electrodes |
| Ultrapure distilled water | Invitrogen | 10977-015 | |
| Vectashield mounting media | Vectashield | H1000 | |
| Vetbond tissue adhesive | 3M | 1469SB | |
| Zeiss 780 Upright Confocal | Zeiss | 780 |
Referenzen
- Altman, J., Bayer, S. A. . Development of the cerebellar system : in relation to its evolution, structure, and functions. , (1997).
- Rahimi-Balaei, M., Bergen, H., Kong, J., Marzban, H. Neuronal migration during development of the cerebellum. Frontiers in Cellular Neuroscience. 12, 484 (2018).
- Alder, J., Cho, N. K., Hatten, M. E. Embryonic precursor cells from the rhombic lip are specified to a cerebellar granule neuron identity. Neuron. 17 (3), 389-399 (1996).
- Hatten, M. E., Heintz, N. Mechanisms of neural patterning and specification in the developing cerebellum. Annual Review of Neuroscience. 18, 385-408 (1995).
- Ben-Arie, N., et al. Math1 is essential for genesis of cerebellar granule neurons. Nature. 390 (6656), 169-172 (1997).
- Borghesani, P. R., et al. BDNF stimulates migration of cerebellar granule cells. Development. 129 (6), 1435-1442 (2002).
- Espinosa, J. S., Luo, L. Timing neurogenesis and differentiation: insights from quantitative clonal analyses of cerebellar granule cells. Journal of Neuroscience. 28 (10), 2301-2312 (2008).
- Markwalter, K. H., Yang, Y., Holy, T. E., Bonni, A. Sensorimotor coding of vermal granule neurons in the developing mammalian cerebellum. Journal of Neuroscience. 39 (34), 6626-6643 (2019).
- Shalizi, A., et al. PIASx is a MEF2 SUMO E3 ligase that promotes postsynaptic dendritic morphogenesis. Journal of Neuroscience. 27 (37), 10037-10046 (2007).
- Shalizi, A., et al. A Calcium-regulated MEF2 sumoylation switch controls poststynaptic differentiation. Science. 311 (5763), 1012-1017 (2006).
- Konishi, Y., Stegmuller, J., Matsuda, T., Bonni, S., Bonni, A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 303 (5660), 1026-1030 (2004).
- Holubowska, A., Mukherjee, C., Vadhvani, M., Stegmuller, J. Genetic manipulation of cerebellar granule neurons in vitro and in vivo to study neuronal morphology and migration. Journal of Visualized Experiments: JoVE. (85), e51070 (2014).
- Yang, Y., et al. Chromatin remodeling inactivates activity genes and regulates neural coding. Science. 353 (6296), 300-305 (2016).
- Herculano-Houzel, S. Coordinated scaling of cortical and cerebellar numbers of neurons. Frontiers in Neuroanatomy. 4, 12 (2010).
- Wilson, P. M., Fryer, R. H., Fang, Y., Hatten, M. E. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. Journal of Neuroscience. 30 (25), 8529-8540 (2010).
- Kokubo, M., et al. BDNF-mediated cerebellar granule cell development is impaired in mice null for CaMKK2 or CaMKIV. Journal of Neuroscience. 29 (28), 8901-8913 (2009).
- Schwartz, P. M., Borghesani, P. R., Levy, R. L., Pomeroy, S. L., Segal, R. A. Abnormal cerebellar development and foliation in BDNF-/- mice reveals a role for neurotrophins in CNS patterning. Neuron. 19 (2), 269-281 (1997).
- Segal, R. A., Pomeroy, S. L., Stiles, C. D. Axonal growth and fasciculation linked to differential expression of BDNF and NT3 receptors in developing cerebellar granule cells. Journal of Neuroscience. 15 (7), 4970-4981 (1995).
- Zhou, P., et al. Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron. 55 (1), 53-68 (2007).
- Dhar, M., Hantman, A. W., Nishiyama, H. Developmental pattern and structural factors of dendritic survival in cerebellar granule cells in vivo. Scientific Reports. 8 (1), 17561 (2018).
- Ito, M. Synaptic plasticity in the cerebellar cortex and its role in motor learning. Canadian Journal of Neurological Sciences. 20, 70-74 (1993).
- Jorntell, H., Hansel, C. Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron. 52 (2), 227-238 (2006).
- Nakanishi, S. Genetic manipulation study of information processing in the cerebellum. Neuroscience. 162 (3), 723-731 (2009).
- Chang, C. H., et al. Atoh1 controls primary cilia formation to allow for SHH-triggered granule neuron progenitor proliferation. Developmental Cell. 48 (2), 184-199 (2019).
Erratum
Formal Correction: Erratum: Utilizing In Vivo Postnatal Electroporation to Study Cerebellar Granule Neuron Morphology and Synapse Development
Posted by JoVE Editors on 4/06/2023. Citeable Link.
An erratum was issued for: Utilizing In Vivo Postnatal Electroporation to Study Cerebellar Granule Neuron Morphology and Synapse Development. A figure was updated.
Figure 2 was updated from:
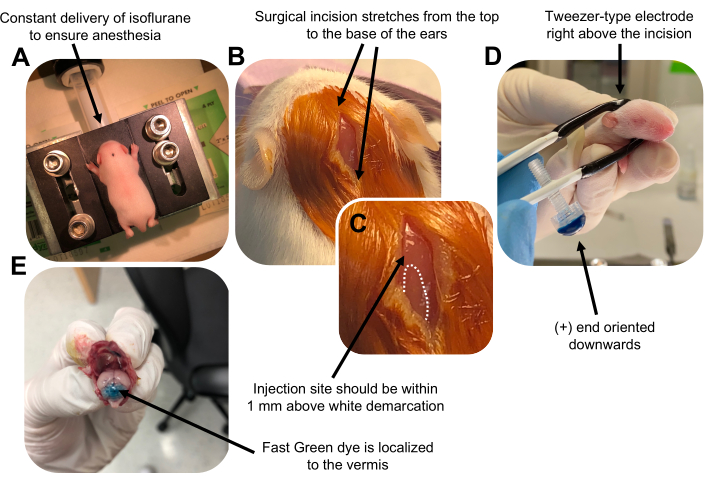
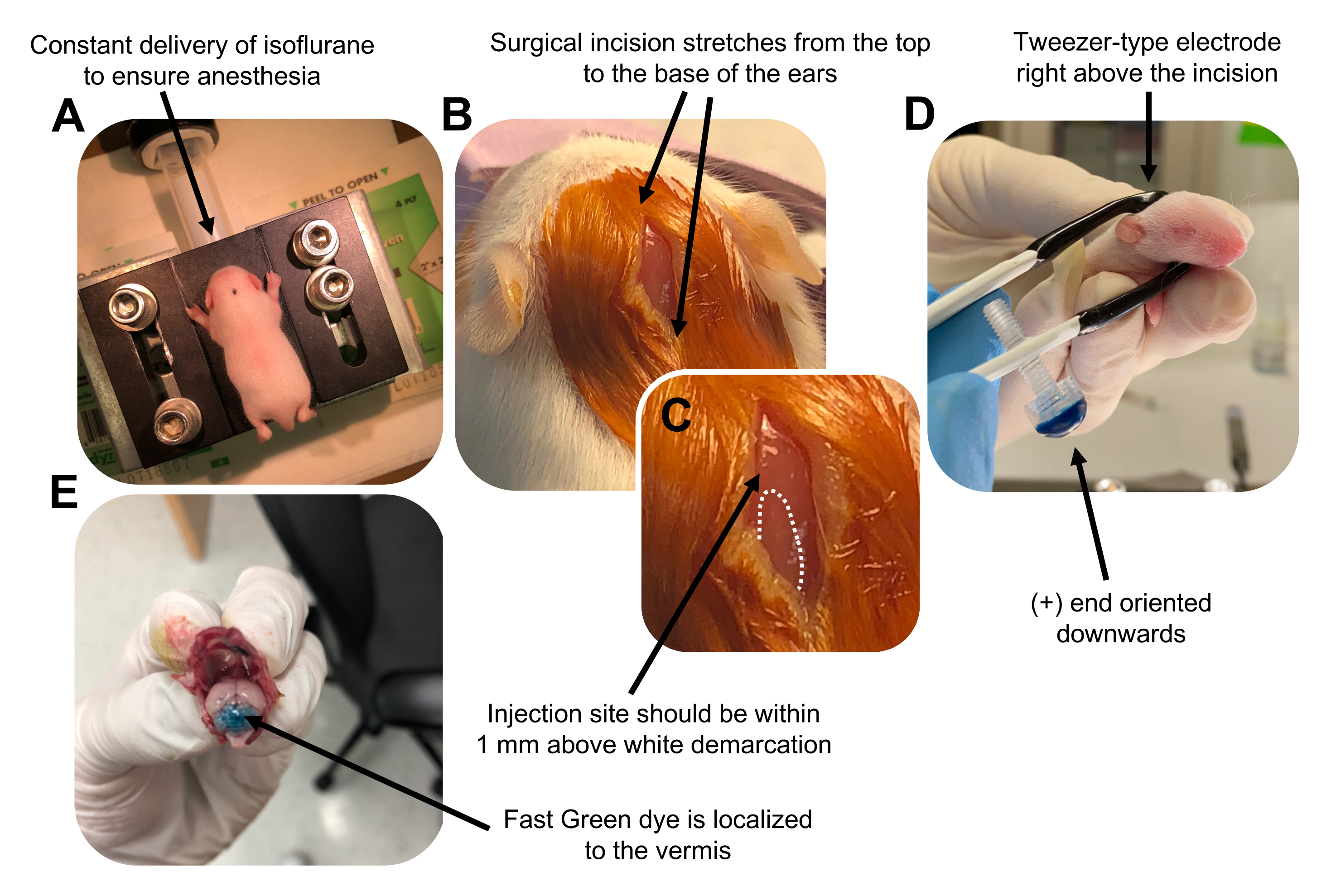
Figure 2: In vivo cerebellar electroporation of granule neuron progenitors in P7 wildtype mouse pups. (A) Pups are anesthetized with 4% isoflurane delivered at a rate of 0.8L/min to ensure anesthesia throughout the injection of the DNA solution. Isoflurane is delivered at a rate of 0.8 L/min. (B) After sterilizing the mouse 3 times with betadine and 70% ethanol, an incision is made that spans the distance of the ears, revealing the hindbrain. (C) A magnified image of a white demarcation on the cranium, a landmark for the injection site. DNA construct should be injected within 1 mm above the mark; dotted lines outline the demarcation, and black arrow denotes the injection site. The ridges of the cerebellar vermis may be visible and can be useful for finding the injection site. (D) Tweezer-type electrode orientation for efficient electroporation. Plus (+) end must be oriented downwards to pull negatively charged DNA into the cerebellar parenchyma prior to administration of electrical pulses. (E) Test injection of 1 µL of a 0.02% Fast Green dye shows injection is localized to the middle of the cerebellar vermis between lobules 5-7. Please click here to view a larger version of this figure.
{kind=link}
to:
Figure 2: In vivo cerebellar electroporation of granule neuron progenitors in P7 wildtype mouse pups. (A) Pups are anesthetized with 4% isoflurane delivered at a rate of 0.8L/min to ensure anesthesia throughout the injection of the DNA solution. Isoflurane is delivered at a rate of 0.8 L/min. (B) After sterilizing the mouse 3 times with betadine and 70% ethanol, an incision is made that spans the distance of the ears, revealing the hindbrain. (C) A magnified image of a white demarcation on the cranium, a landmark for the injection site. DNA construct should be injected within 1 mm above the mark; dotted lines outline the demarcation, and black arrow denotes the injection site. The ridges of the cerebellar vermis may be visible and can be useful for finding the injection site. (D) Tweezer-type electrode orientation for efficient electroporation. Plus (+) end must be oriented downwards to pull negatively charged DNA into the cerebellar parenchyma prior to administration of electrical pulses. (E) Test injection of 1 µL of a 0.02% Fast Green dye shows injection is localized to the middle of the cerebellar vermis between lobules 5-7. Please click here to view a larger version of this figure.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten