
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Tiefe und räumlich kontrollierte Volumenablationen mit einem Zwei-Photonen-Mikroskop in der Zebrafisch-Gastrula
In diesem Artikel
Zusammenfassung
Die Embryonalentwicklung erfordert eine groß angelegte Koordination der Zellbewegung. Die durch Zwei-Photonen-Anregung vermittelte Laserablation ermöglicht die räumlich kontrollierte 3-dimensionale Ablation großer Gruppen tiefer Zellen. Darüber hinaus kann diese Technik die Reaktion kollektiv wandernder Zellen in vivo auf Störungen in ihrer mechanischen Umgebung untersuchen.
Zusammenfassung
Die Morphogenese beinhaltet viele Zellbewegungen, um Zellen in Geweben und Organen zu organisieren. Für eine ordnungsgemäße Entwicklung müssen all diese Bewegungen eng koordiniert werden, und sich häufende Beweise deuten darauf hin, dass dies zumindest teilweise durch mechanische Wechselwirkungen erreicht wird. Dies im Embryo zu testen, erfordert direkte körperliche Störungen. Laserablationen sind eine zunehmend eingesetzte Option, die es ermöglicht, mechanische Einschränkungen zu lindern oder zwei Zellpopulationen physisch voneinander zu isolieren. Viele Ablationen werden jedoch mit einem ultravioletten (UV) Laser durchgeführt, der eine begrenzte axiale Auflösung und Gewebepenetration bietet. Hier wird eine Methode beschrieben, um tiefe, signifikante und räumlich wohldefinierte Volumina mit einem Zwei-Photonen-Mikroskop abzutragen. Ablationen werden in einer transgenen Zebrafischlinie demonstriert, die das grün fluoreszierende Protein im axialen Mesendoderm exprimiert und verwendet wird, um das axiale Mesendoderm zu durchtrennen, ohne das darüber liegende Ektoderm oder die darunter liegende Dotterzelle zu beeinflussen. Das Zellverhalten wird durch Live-Bildgebung vor und nach der Ablation überwacht. Das Ablationsprotokoll kann in verschiedenen Entwicklungsstadien, auf jedem Zelltyp oder Gewebe, in Skalen von wenigen Mikrometern bis zu mehr als hundert Mikrometern verwendet werden.
Einleitung
Zell-Zell-Interaktionen spielen eine entscheidende Rolle bei der Entwicklung. Zellen liefern Signale, die ihre direkten Nachbarn oder weiter entfernte Zellen wahrnehmen können, wodurch ihr Schicksal und/oder Verhalten beeinflusst wird. Viele dieser Signale sind chemischer Natur. Zum Beispiel produziert eine Zellgruppe in den gut charakterisierten Induktionsereignissen diffusible Moleküle, die das Schicksal einer anderen Zellpopulation beeinflussen1. Andere Signale sind jedoch mechanisch; Zellen üben Kräfte und Einschränkungen auf ihre Nachbarn aus, die die Nachbarn wahrnehmen und auf die sie reagieren2.
Eine Möglichkeit, die Bedeutung dieser Zell-Zell-Interaktionen in vivo zu untersuchen, besteht darin, einige Zellen zu eliminieren und die nachfolgende Entwicklung zu beobachten. Leider sind die verfügbaren Techniken zum Entfernen oder Zerstören von Zellen begrenzt. Zellen können chirurgisch3,4 mit Nadeln oder kleinen Drähten entfernt werden, aber solche Behandlungen sind invasiv, nicht sehr präzise und werden normalerweise unter einem Stereomikroskop durchgeführt, wodurch eine sofortige Bildgebung unter einem Mikroskop verhindert wird. Darüber hinaus bedeutet das Targeting tiefer Zellen, ein Loch in darüber liegendes Gewebe zu stechen, wodurch unerwünschte Störungen entstehen. Genetisch kodierte Photosensibilisatoren wie KillerRed wurden verwendet, um den Zelltod durch Lichtbeleuchtung zu induzieren5. Photosensibilisatoren sind Chromophore, die bei Lichteinstrahlung reaktive Sauerstoffspezies erzeugen. Ihre Haupteinschränkung besteht darin, dass sie lange Lichtbeleuchtungen (etwa 15 Minuten) erfordern, die schwierig zu erreichen sind, wenn sich Zellen bewegen, und dass sie den Zelltod durch Apoptose induzieren, was nicht sofort der Fall ist.
Schließlich wurden Laserablationen entwickelt und in den letzten 15 Jahren weit verbreitet6,7,8,9,10,11,12. Ein Laserstrahl wird auf die Zielzelle/das Zielgewebe fokussiert. Es induziert seine Ablation durch Erhitzen, Photoablation oder plasmainduzierte Ablation; der involvierte Prozess hängt von der Leistungsdichte und der Belichtungszeit ab13. Die meisten Ablationsprotokolle verwenden UV-Laser für ihre hohe Energie. UV-Licht wird jedoch von biologischem Gewebe sowohl absorbiert als auch gestreut. Daher erfordert das Targeting tiefer Zellen eine hohe Laserleistung, die dann Schäden in oberflächlicheren Geweben außerhalb der Ebene induziert. Dies beschränkt den Einsatz von UV-Lasern auf oberflächliche Strukturen und erklärt ihre relativ geringe axiale Auflösung. Die nichtlineare Optik (sogenannte Zwei-Photonen-Mikroskopie) nutzt nichtlineare Eigenschaften des Lichts, um ein Fluorophor mit zwei Photonen von etwa halber Energie im Infrarotbereich anzuregen. Bei der Anwendung auf Ablationen hat dies drei Hauptvorteile. Erstens wird das Infrarotlicht weniger gestreut und weniger als UV-Licht von biologischen Geweben14 absorbiert, so dass tiefere Strukturen erreicht werden können, ohne die erforderliche Laserleistung zu erhöhen. Zweitens sorgt die Verwendung eines gepulsten Femtosekundenlasers für sehr hohe Leistungsdichten, wodurch eine Ablation durch Plasmainduktion entsteht, die im Gegensatz zur Erwärmung räumlich nicht diffundiert15. Drittens wird die Leistungsdichte, die die Plasmabildung induziert, nur am Brennpunkt erreicht. Dank dieser Eigenschaften können Zwei-Photonen-Laserablationen verwendet werden, um tiefe Zellen präzise anzugreifen, ohne die umgebende Gewebeumgebung zu beeinträchtigen.
Kollektive Migrationen sind ein hervorragendes Beispiel für Entwicklungsprozesse, bei denen Zell-Zell-Interaktionen grundlegend sind. Kollektive Migrationen sind definiert als Zellmigrationen, bei denen benachbarte Zellen das Verhalten einer Zelle beeinflussen16. Die Art dieser Wechselwirkungen (chemisch oder mechanisch) und wie sie die Zellmigration beeinflussen, kann stark variieren und ist oft nicht vollständig verstanden. Die Fähigkeit, Zellen zu entfernen und zu beobachten, wie sich dies auf die anderen auswirkt, ist entscheidend für die weitere Entschlüsselung dieser kollektiven Prozesse. Vor einigen Jahren haben wir – mit chirurgischen Ansätzen – festgestellt, dass die Migration der Polster während der Zebrafischgastrulation eine kollektive Migration ist17. Die Polster ist eine Gruppe von Zellen, die die ersten internalisierenden Zellen auf der dorsalen Seite des Embryos bildet18. Diese Zellen, die in der transgenen Linie Tg(gsc:GFP) grün markiert sind, befinden sich tief im Embryo, unter mehreren Schichten von Epiblastenzellen. Während der Gastrulation führt diese Gruppe die Ausdehnung des axialen Mesoderms an und wandert vom embryonalen Organizer zum Tierischen Pol19,20,21,22,23 (Abbildung 1A). Wir haben festgestellt, dass Zellen Kontakt zu ihren Nachbarn benötigen, um ihre Migration in Richtung des Tierpols auszurichten. Um die zellulären und molekularen Grundlagen dieser kollektiven Migration besser zu verstehen, müssen jedoch einige Zellen entfernt werden, um zu sehen, wie sich dies auf die verbleibenden auswirkt. Wir haben daher Ablationen von großen und tiefen Volumina mit einem Zwei-Photonen-Mikroskopie-Setup entwickelt. Hier demonstrieren wir die Verwendung dieses Protokolls, um die Polster in ihrer Mitte zu durchtrennen und die Folgen für die Zellmigration zu beobachten, indem wir mit Histone2B-mCherry markierte Kerne verfolgen.
Protokoll
Alle Tierarbeiten wurden von der Ethikkommission N 59 und dem Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche unter dem Aktenzeichen APAFIS#15859-2018051710341011v3 genehmigt. Einige der unten beschriebenen Schritte sind spezifisch für unsere Ausrüstung und Software, können aber leicht an verschiedene Geräte angepasst werden.
1. Injektionsvorbereitung
- Bereiten Sie 75 ml 1% ige Agaroselösung in Embryo Medium (EM) vor.
- Legen Sie die Injektionsform in eine 90-mm-Petrischale und gießen Sie etwa 50 ml Agarose, genug, damit die Form schwimmt. Lassen Sie die Agarose erstarren und entfernen Sie die Injektionsform.
- Bereiten Sie eine mit Agarose überzogene Schale zu, indem Sie 1 ml Agarose in eine 30 mm Petrischale gießen.
- Bereiten Sie 4 μL 30 ng/μL Histone2B-mCherry mRNA-Lösung vor, indem Sie die Stammlösung in RNase-freiem Wasser verdünnen und auf Eis aufbewahren.
HINWEIS: Achten Sie darauf, handschuhe zu tragen, während Sie mRNA manipulieren, um RNase-vermittelten Abbau zu vermeiden. - Ziehen Sie mit dem Mikropipettenabzieher eine Injektionsnadel aus einer Kapillare.
2. Embryonalisierung
- Sobald Fische Eier gelegt haben, sammeln, spülen und ernten Sie in einer 90 mm Petrischale in EM. Legen Sie die Embryonen in einen 28,5 °C großen Inkubator.
- Warten Sie 20 Minuten, bis die erste Zelle sichtbar wird.
- Übertragen Sie 30 Embryonen auf die mit EM gefüllte Injektionsplatte. Drücken Sie die Embryonen mit einer leicht stumpfen Pinzette in die Rillen und richten Sie sie mit der Tierstange nach oben aus.
- Füllen Sie mit einer Mikroladerspitze eine Injektionsnadel mit 2 μL mRNA-Lösung. Führen Sie die Nadel in den Kapillarhalter ein, der sich in einem Mikromanipulator befindet, der mit einem Polytetrafluorethylen (PTFE)-Schlauch mit einem Luftinjektor verbunden ist.
- Unter dem Stereomikroskop die Nadelspitze vorsichtig brechen.
- Injizieren Sie die mRNA-Lösung in die Embryonen im 1-Zell-Stadium, indem Sie die Nadel in die Zelle einführen.
HINWEIS: Das injizierte Volumen beträgt etwa ein Drittel des Zellvolumens. - Legen Sie die injizierten Embryonen zurück in den 28,5 °C-Inkubator.
3. Vorbereitung des Zwei-Photonen-Mikroskops
HINWEIS: In diesem Protokoll werden zwei Laser verwendet. Einer wird verwendet, um GFP (bei 920 nm) abzubilden und Ablationen (bei 820 nm) durchzuführen. Es wird als grüner / Ablationslaser bezeichnet. Die andere wird bei 1160 nm verwendet, um mCherry abzubilden. Er wird als roter Laser bezeichnet.
- Stellen Sie den grünen/Ablationslaser auf 820 nm (Ablationswellenlänge) und den roten Laser auf 1160 nm (mCherry-Anregung) ein.
- Richten Sie mit beweglichen Spiegeln auf dem Optischen Pfad grüne/ablations- und rote Laserstrahlen sowohl am Ein- als auch am Ausgang des Scankopfes aus.
HINWEIS: Dies erhöht den Laserstrahlfokus und minimiert das Fokusvolumen für Anregung und Ablation. - Messen Sie die maximale Leistung des Grün-/Ablationslasers bei 820 nm unter dem Objektiv. Platzieren Sie dazu den Leistungsmesser unter dem Objektiv, schließen Sie die schwarze Kammer, stellen Sie die grüne / Ablationslaserleistung auf 100% ein und öffnen Sie die Rollläden. Berechnen Sie den Prozentsatz der Laserleistung, der benötigt wird, um 300 mW zu erreichen.
- Stellen Sie den grünen/ablationslaser auf 920 nm zurück (GFP-Anregung) und stellen Sie die Laserleistung auf 7% ein. Stellen Sie die rote Laserleistung auf 15% ein.
- aktivieren Sie die epi-PhotoMultiplier Tubes (PMT) Detektoren für grüne und rote Linien; Setzen Sie die PMT-Empfindlichkeit der grünen und roten Linie auf 65.
- Stellen Sie das Sichtfeld auf 400 x 400 μm, die Bildauflösung auf 512 x 512 Pixel und die Scanfrequenz auf 800 Hz ein.
- Wählen Sie den 3D-Zeitraffer-Imaging-Modus . Erstellen Sie dann einen Ordner und aktivieren Sie autosave für Daten nach jeder Erfassung.
- Montieren Sie die Heizkammer und stellen Sie sie auf 28 °C ein. Warten Sie mindestens 10 Minuten, bis sich die Kammer und das Ziel erwärmt haben.
4. Montage des Embryos
- Identifizieren Sie unter einem Fluoreszenz-Stereomikroskop Embryonen mit 70% Epiboly, die GFP exprimieren.
HINWEIS: Wählen Sie Embryonen mit einem hellen Signal im axialen Mesoderm und ohne Hintergrundfluoreszenz für eine bessere Abbildungsqualität. - Drei bis vier ausgewählte Embryonen in die mit Agarose beschichtete Schale (Schritt 1.3) mit einer Pasteurpipette aus Kunststoff übertragen und vorsichtig mit einer feinen Pinzette dekumentiert.
HINWEIS: Dekumentierte Embryonen sind sehr empfindlich und platzen bei Kontakt mit Luft oder Kunststoff. - Gießen Sie 1 ml 0,2% Agarose in 1x Penicillin-Streptomycin EM in eine kleine Glasfläschchen. Legen Sie die Durchstechflasche in eine vorgewärmte 42 °C trockene Blockheizung.
HINWEIS: Die folgenden Schritte müssen schnell ausgeführt werden, um die Embryonenorientierung zu ermöglichen, bevor Agarose aushärtet. - Übertragen Sie einen dehorionierten Embryo in das 0,2% ige Agaroseglasfläschchen mit einer feuerpolierten Glaspipette. Achten Sie darauf, nicht zu viel EM in die Agarose zu geben, um eine Verdünnung zu vermeiden. Verwerfen Sie das verbleibende EM aus der Pipette und aspirieren Sie den Embryo zusammen mit genügend Agarose zurück, um den Objektträger der Glasbodenschale zu bedecken, bevor der Embryo aus der Pipette fällt.
- Blasen Sie die Agarose und den Embryo auf den Glasträger der Schale. Achten Sie darauf, dass der Embryo nicht die Luft oder die Kunststoffseite der Schale berührt. Als nächstes füllen Sie die Kammer um den Glasobjektträger mit Agarose.
- Verwenden Sie eine Wimper, um den Embryo so auszurichten, dass sich die Zielregion oben befindet (Abbildung 1B).
HINWEIS: Achten Sie bei der Orientierung von Embryonen darauf, nur das Blastoderm zu berühren, nicht das sehr zerbrechliche Eigelb. Agarose setzt je nach Raumtemperatur in ca. 1 min aus. - Warten Sie ~ 5 Minuten, bis die Agarose vollständig aushärtet, und fügen Sie dann ein paar Tropfen Penicillin-Streptomycin EM hinzu.
5. Lokalisierung des Embryos und Bildgebung vor der Ablation
- Stellen Sie die Glasbodenschale unter das Objektiv in die beheizte Kammer. Tauchen Sie das Objektiv in Penicillin-Streptomycin EM ein und schließen Sie die beheizte Kammer.
- Bewegen Sie den Schieberegler, um den Lichtpfad auf das Okular festzulegen. Dann finden Sie mit Okularen, Leuchtstofflampen und Bühnensteuerung einen Embryo und setzen sie den Fokus auf die Oberfläche des Embryos.
- Schalten Sie die Leuchtstofflampe aus, stellen Sie den Lichtweg auf PMTs ein und schließen Sie die schwarze Kammer.
HINWEIS: Achten Sie darauf, alle Lichtquellen in der schwarzen Kammer auszuschalten, da dies die PMTs beschädigen könnte. - Starten Sie die Live-Bildgebung und lokalisieren Sie das axiale Mesoderm. Passen Sie die grünen/ablations- und roten Laserleistungen an, um ein gutes Signal zu erhalten (d. h. zwischen 1.000 und 20.000 Photonen pro Pixel für GFP-exprimierende Bereiche). Verwenden Sie den roten Kanal, um das Stadium ganz nach oben auf den Embryo zu bewegen und diese Position als Z = 0 festzulegen.
- Wählen Sie einen Zeitschritt von 1 min und einen Z-Schritt von 2 μm. Ein Z-Verlauf von 110 μm reicht aus, um die gesamte Polsterung zu erfassen und wird mit diesen Einstellungen in weniger als 1 min erworben. Stellen Sie die erste Schicht 15 μm über dem axialen Mesoderm (im oberflächlicheren Ektoderm).
HINWEIS: Das Polster bewegt sich entlang einer gekrümmten Linie, so dass die untere Schicht des Z-Stapels 30 μm tiefer als die tiefste Position der Polster eingestellt werden sollte, um ihre Bewegung während der Zeitrafferaufnahme aufzunehmen (Abbildung 1E). - Nehmen Sie 10-15 Minuten Des Films vor der Ablation auf.

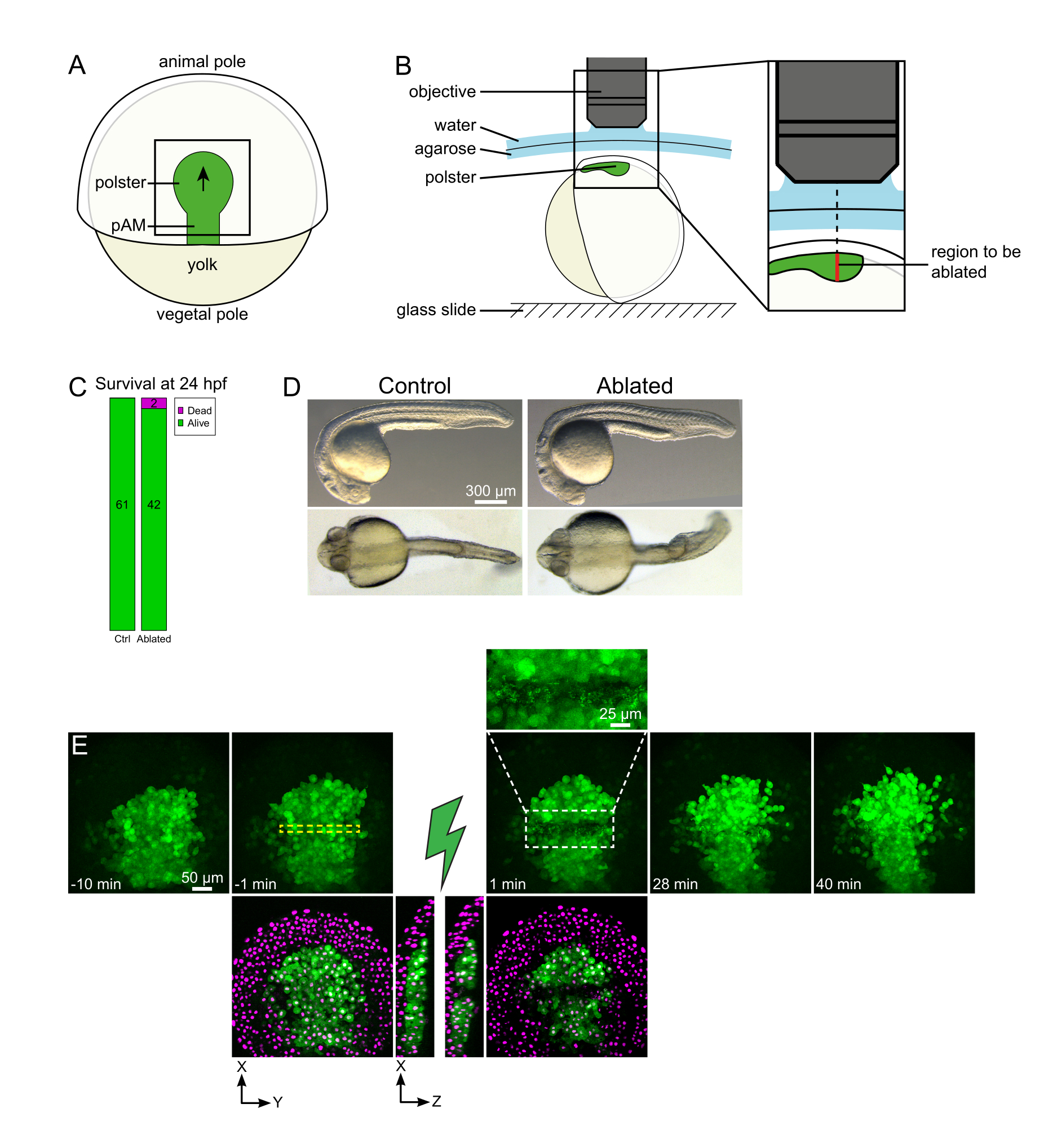
Abbildung 1: Erfolgreiches Ergebnis von Laserablationen. (A) Schema eines gastrulierenden Embryos bei 70% Epiboly in dorsaler Sicht; pAM: posteriores axiales Mesoderm; schwarzer Pfeil markiert die Richtung der Polsterwanderung; schwarzes Quadrat zeigt ein typisches Sichtfeld für Ablationen im Polster an. (B) Schema der Embryonenbefestigung zur Polsterabtrennung. Seitliche Ansicht. Der Embryo ist so montiert, dass die Ebene des Polsters senkrecht zur optischen Achse steht. (C) Überleben und (D) Morphologie der Kontroll- und abgetragenen Embryonen 24 Stunden nach der Befruchtung. Der Maßstabsbalken beträgt 300 μm. (E) Zeitsequenz aus der Laserablation in der Polsterung eines Tg(gsc:GFP) -Embryos, der Histone2B-mCherry exprimiert. Ansichten nur mit dem grünen Kanal sind maximale Projektionen. Die Nahaufnahme zeigt den abgetragenen Bereich, der Zelltrümmer enthält. Ansichten mit grünen und roten (als magentafarben dargestellten) Kanälen sind XY- und XZ-Scheiben vor und nach der Ablation (der grüne Blitz steht für ablation). XZ-Scheiben zeigen, dass die darüber liegenden Gewebe (magentafarbene Kerne ohne GFP-Expression) durch die Ablation der darunter liegenden Strukturen nicht beeinflusst wurden. Die gelb gestrichelte Box entspricht dem ROI, der für die Laserablationsbehandlung ausgewählt wurde. Der Maßstabsbalken beträgt 50 μm in großen Ansichten und 25 μm in der Nahaufnahme. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
6. Zielortung und Laserablation
- Suchen Sie die Polsterkontur auf Live-Aufnahmen und zeichnen Sie mit dem EOM-ROI-Werkzeug (Electro-Optic Modulator Region of Interest) ein 20 Pixel (15 μm) großes Rechteck, das sich über die Breite des Polsters erstreckt. Platzieren Sie dieses Rechteck in der Mitte der Polster (Abbildung 1E).
- Beachten Sie die axiale Position der höchsten und niedrigsten Ebene, die Polsterzellen enthalten. Ablationen werden alle 10 μm zwischen diesen beiden Ebenen durchgeführt. Achten Sie darauf, dass der ROI die Eigelbzelle auf keiner dieser Ebenen überlappt.
- Platzieren Sie die Bühne an der niedrigsten Z-Position des Intervalls. Ablationen müssen von unten nach oben durchgeführt werden, da Ablagerungen Licht absorbieren.
- Stellen Sie die grüne/Ablationslaserwellenlänge auf 820 nm und stellen Sie den Leistungsprozentsatz ein, um eine Austrittsleistung von 300 mW zu erhalten (Schritt 3.3).
- Stellen Sie die Abbildungsfrequenz auf 200 Hz ein.
- Stellen Sie green/ablation laser imaging EOM auf 0 und wählen Sie den ROI-Treat-Modus .
- Schalten Sie die EOM ein und stellen Sie die Behandlung so ein, dass sie sofort beginnt (nach 0 Frame).
- Stellen Sie den Imaging-Modus auf Zeitraffer und deaktivieren Sie das automatische Speichern.
- Stellen Sie den Zeitschritt auf den Schnellmodus ein.
- Legen Sie die Anzahl der Behandlungsrahmen und die Anzahl der Frames auf den Wert fest, der der Zieltiefe entspricht (Tabelle 1).
| Tiefe (μm) | Behandlungsrahmen |
| -30 | 1 |
| -35 | 1-2 |
| -40 | 1-2 |
| -45 | 2 |
| -50 | 2-3 |
| -55 | 3 |
| -60 | 3-4 |
| -65 | 4 |
| -70 | 4 |
| -75 | 4-5 |
| -80 | 4-5 |
| -85 | 5 |
| -90 | 5 |
| -95 | 5-6 |
| -100 | 6 |
| -105 | 6 |
Tabelle 1: Vorgeschlagene Anzahl von Laserbehandlungsrahmen in Abhängigkeit von der gezielten Zelltiefe im Embryo (0 ist die Oberfläche des Embryos).
- Starten Sie die Bildgebung. Die Erfassung ist schwarz, da sich der Verschluss für PMT während der EOM-Behandlung schließt.
- Bewegen Sie sich auf der Bühne an die nächste Z-Position der Liste (Schritt 6.2).
- Wiederholen Sie die Schritte 6.10 bis 6.12, bis die Spitze der Polster erreicht ist.
7. Verifizierung nach der Ablation und Bildgebung
- Stellen Sie den grünen/Ablationslaser auf 920 nm und 5 % Leistung ein. Stellen Sie die grüne/Ablations-Laserbildgebungs-EOM auf 100 und wählen Sie den Fullfield-Modus .
- Stellen Sie die Abbildungsfrequenz auf 800 Hz ein. Schalten Sie EOM aus.
- Gehen Sie den gesamten Stapel im Live-Modus durch, um zu überprüfen, ob jedes Flugzeug abgetragen wurde. Wenn dies nicht der Fall ist, kehren Sie zu Schritt 6.2 zurück.
HINWEIS: Die Ablation induziert manchmal eine vertikale Verschiebung benachbarter Gewebe, so dass der Z-Stack möglicherweise neu definiert werden muss. - Stellen Sie den Imaging-Modus auf 3D-Zeitraffer ein und aktivieren Sie das automatische Speichern erneut. Nehmen Sie 40-60 Minuten Des Films nach der Ablation auf.
- Überprüfen Sie im Film nach der Ablation, ob die Zielzellen effektiv abgetragen wurden. Die Fluoreszenzwiederherstellung oder Zielzellen, die Platz einnehmen und verhindern, dass sich Folgezellen durchbewegen, weisen darauf hin, dass zielgerichtete Zellen nur photogebleicht und nicht abgetragen wurden (Abbildung 1E und Abbildung 2A).

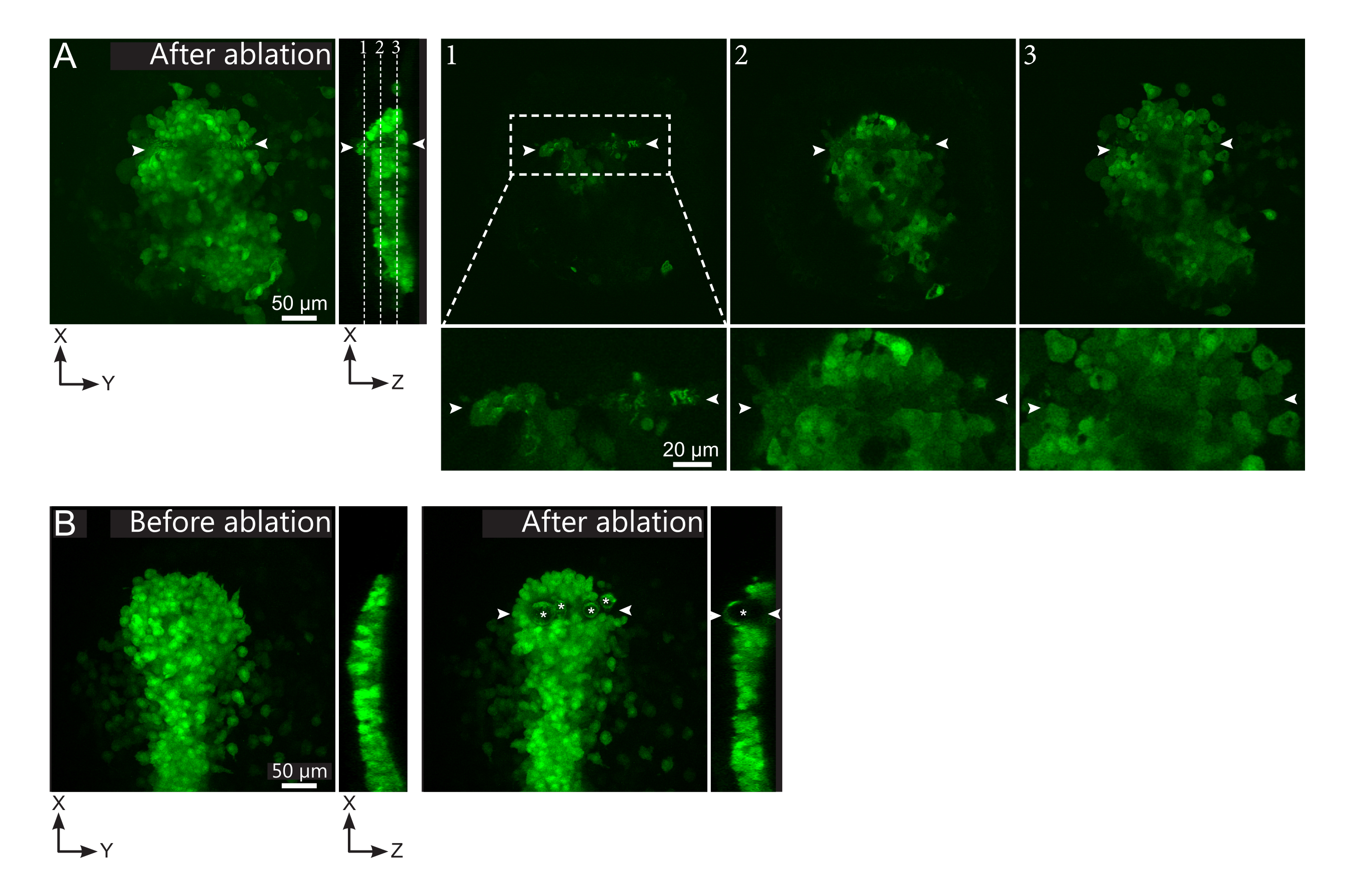
Abbildung 2: Negative Ergebnisse von Laserablationen. (A) Typische Beispiele für mögliche Fehler bei der Laserablation. Große XY-Ansichten sind maximale Projektionen, XZ-Ansicht ist ein rekonstruierter Ausschnitt. Der laserbehandelte Bereich befindet sich zwischen den beiden weißen Pfeilspitzen. Drei Fokusebenen sind im rekonstruierten Schnitt hervorgehoben und rechts dargestellt. Sie entsprechen drei verschiedenen Arten von Fehlern. Ebene 1 zeigt, dass Zellen oberhalb der Polster abgetragen wurden. Dies kann durch das Vorhandensein von autofluoreszierenden Trümmern auf dieser Brennebene (siehe Nahaufnahme) über der Polster identifiziert werden (siehe Position der Ebene 1 auf dem rekonstruierten Abschnitt). Dies resultiert wahrscheinlich aus einer falschen Definition der zu streichenden Region. Ebene 2 zeigt Zellen, die gebleicht, aber nicht abgetragen wurden. Sie können identifiziert werden, da das niedrige Fluoreszenzsignal noch intakte Zellkonturen offenbart (siehe Nahaufnahme). Ebene 3 zeigt intakte Zellen, die durch Laserbehandlung kaum gebleicht wurden. Dies könnte sich aus einer falschen Definition der zu löschenden Region oder aus einer schlechten Behandlung ergeben. In den in den Ebenen 2 und 3 dargestellten Situationen ist es möglich, die Ablationsbehandlung erneut auf die nicht abgetragenen Zielzellen anzuwenden. Die Maßstabsleiste beträgt 50 μm in großen Ansichten und 20 μm in Nahaufnahmen. (B) Ein typisches Beispiel für Blasen (gekennzeichnet durch weiße Sternchen), die durch Kavitation aufgrund einer zu intensiven Laserbehandlung gebildet werden. Solche Blasen sind nicht auf eine Z-Ebene beschränkt, die manchmal sogar die volle Höhe des Polsters überspannt und benachbarte Gewebe verformt. Der Maßstabsbalken beträgt 50 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
8. Datenanalyse
- Öffnen Sie Zeitrafferserien mit der Bildanalyse-Software und stellen Sie die richtige Pixelgröße ein.
- Stellen Sie in der Spot-Funktion die Objektgröße auf 10 μm ein, da dies die durchschnittliche Kerngröße während der Gastrulation ist. Führen Sie dann die Spot-Funktion aus, um die Kerne zu erkennen und zu verfolgen.
HINWEIS: Die Erkennung kann leicht verbessert werden, indem die niedrigere axiale Auflösung berücksichtigt wird, indem eine 12 μm lange ellipsoidische Form entlang der Z-Achse angepasst wird. - Verwenden Sie Filter, um Fehlalarme zu entfernen. In der Tg(Gsc:GFP) -Linie sind Zellen der Embryonalachse und einige endodermale Zellen grün markiert. Daher ermöglicht die Filterung nach Grünintensität eine schnelle Auswahl dieser Zellen (Abbildung 3A).
- Legen Sie den maximalen Abstand zwischen aufeinanderfolgenden Punkten auf einen Wert fest, der mit der Geschwindigkeit der Zellen kompatibel ist.
HINWEIS: Achten Sie darauf, das Zeitintervall zwischen zwei Frames zu berücksichtigen. Polsterzellen wandern mit 2,8 ± 0,8 μm/min. Wenn sie also eine maximale Verschiebung von 4 μm für einen Zeitschritt von 1 Minute zulassen, werden die meisten artefaktischen Spuren entfernt. - Das Zulassen von Lücken über ein oder zwei Zeitpunkte bietet längere kontinuierliche Spuren, kann jedoch zu Nachverfolgungsfehlern führen. Wenn ein Kern zu einem einmaligen Zeitpunkt nicht korrekt erkannt wird, sollten Sie eine erneute Ausführung der Punkterkennung mit anderen Parametern/Filtern in Betracht ziehen.
- Tracks visuell überprüfen und ggf. korrigieren.
- Exportieren Sie die Ergebnisse als .xlsx Datei. Verarbeiten Sie die Datei mit veröffentlichten Tabellenkalkulationsroutinen24 (Abbildung 3B) und benutzerdefinierten Routinen auf Datenanalysesoftware (auf Anfrage erhältlich).

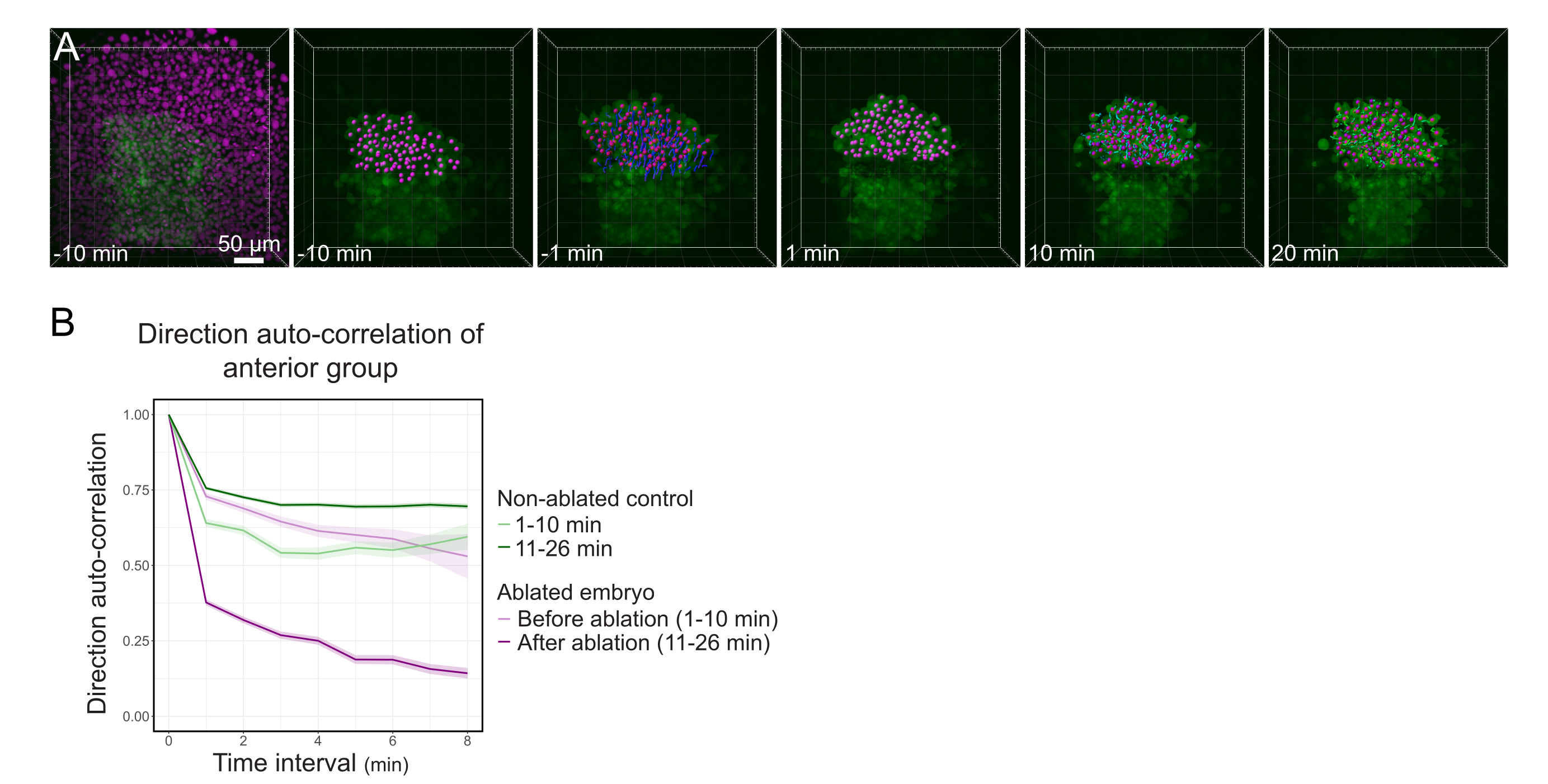
Abbildung 3: Die Isolierung der vorderen Hälfte des Polsters beeinflusst die Zelldirektionalität. (A) 3D-Rekonstruktion eines Tg(gsc:GFP) -Embryos, der Histone2B-mCherry exprimiert (dargestellt in Magenta), vor und nach einer Laserablation, die das Polster in seiner Mitte durchtrennt. Kerne, die zur vorderen Hälfte der Polster gehören, werden mit einem magentafarbenen Punkt markiert und vor und nach der Ablation über die Zeit verfolgt (siehe Film S1). Der Maßstabsbalken beträgt 50 μm. (B) Als Maß für die Migrationspersistenz gilt die Richtungsautokorrelation von Zellen, die vor und nach der Ablation zum vorderen Teil der Polsterung gehören. Zellen zeigen vor der Ablation eine kontinuierliche Bewegung, die nach der Ablation drastisch abnimmt, was auf einen Verlust der kollektivorientierten Migration hinweist. Die Richtungsautokorrelation wurde auch an Zellen gemessen, die die vordere Hälfte der Polster eines nicht abgetragenen Embryos bilden, als Kontrolle. Die Diagrammumschläge zeigen einen Standardfehler an. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Ergebnisse
Um das Polster in seiner Mitte zu durchtrennen, wurde ein Tg(gsc:GFP) -Embryo, injiziert mit Histone2B-mCherry mRNAs, im 70%igen Epiboly-Stadium montiert, wie in Schritt 4 beschrieben. Das Polster wurde durch GFP-Expression identifiziert, und der Embryo wurde so montiert, dass die Ebene des Polsters senkrecht zur optischen Achse steht (Abbildung 1B). Das Kippen des Embryos aus dieser Position erschwert das Verfahren. Das Licht muss durch mehr Gewebe gehen, um die Ablationsebenen zu ...
Diskussion
Hier beschreiben wir ein Protokoll, das nichtlineare Optik verwendet, um tiefe und räumlich gut definierte Volumenablationen durchzuführen. Der kritischste Schritt des Protokolls besteht darin, Behandlungsbedingungen zu finden, die genügend Energie liefern, um Ablationen zu ermöglichen, aber nicht zu viel Energie, um übermäßige Ablagerungen oder Kavitation zu vermeiden. Die Menge der am Zielort abgegebenen Energie hängt hauptsächlich von Folgendem ab: (1) der Laseraustrittsleistung, (2) der Qualität der Laserau...
Offenlegungen
Die Autoren erklären keine gegensätzlichen Interessen.
Danksagungen
Wir danken Emilie Menant für die Fischpflege, der Polytechnique Bioimaging Facility, insbesondere Pierre Mahou, für die Unterstützung bei der Live-Bildgebung auf ihren Geräten, die teilweise von der Région Ile-de-France (interDIM) und der Agence Nationale de la Recherche (ANR-11-EQPX-0029 Morphoscope2, ANR-10-INBS-04 France BioImaging) unterstützt werden. Diese Arbeit wurde durch die ANR-Zuschüsse 15-CE13-0016-1, 18-CE13-0024, 20-CE13-0016 und das Forschungs- und Innovationsprogramm Horizon 2020 der Europäischen Union im Rahmen der Marie-Skłodowska-Curie-Finanzhilfevereinbarung Nr. 840201, das Ministère de l'Enseignement Supérieur et de la Recherche und das Centre National de la Recherche Scientifique unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| 25x water immersion objective | Olympus | XLPLN25XWMP2 | |
| Agarose | PanReac AppliChem | A8963,0500 | |
| Data analysis software : Matlab | Math Works | ||
| Electro-optic modulator (EOM) | ConOptics | 350-80LA | |
| Embryo Medium (EM) solution | Westerfield, M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio), 5th Edition. University of Oregon Press, Eugene (Book). (2000). | ||
| Environmental chamber chamber | Okolab | H201-T-UNIT-BL | |
| EOM driver | ConOptics | 302RM | |
| Fluorescence source | Lumencor | SOLA | |
| Glass bottom dishes | MatTek | P35G-0-10-C | |
| Glass capillaries | Harvard Apparatus | 300085 | Outside diameter 1.0 mm, inside diameter 0.58 mm |
| Glass pipettes | Volac | D810 | Tip should be fire polished |
| Green/ablation laser | Spectra Physics | Mai Tai HP DeepSee | |
| Histone2B-mCherry mRNA | Synthesized from pCS2-H2B-mCherry plasmid (Dumortier& al. 2012) | ||
| Image analysis software: IMARIS | Bitplane | ||
| ImSpector software | Abberior Instruments Development Team | ||
| Injection mold | Adapative Science Tools | I-34 | |
| Microloader tips | Eppendorf | 5242956003 | |
| Micromanipulator | Narishige | MN-151 | |
| Micropipette puller | Sutter | P-1000 | |
| mMESSAGE mMACHINE SP6 Transcription Kit | Invitrogen | AM1340 | |
| Penicillin-Streptomycin | Thermofisher | 15140-122 | 10 000 units penicillin and 10 mgstreptomycin per ml |
| Photomultiplier tube (PMT) | Hammamatsu | H7422-40 | |
| PicoPump (Air injector) | World Precision Instrument | PV820 | |
| Red laser | Spectra Physics | OPO/Insight DeepSee | |
| RNAse free water for injection | Sigma | W3500 | |
| Spreadsheet software: Excel | Microsoft | ||
| Stereomicroscope | Nikon | SMZ18 | |
| Tg(gsc:GFP) zebrafish line | Doitsidou, M. et al. Guidance of primordial germ cell migration by the chemokine SDF-1. Cell. 111 (5), 647–59, doi: doi.org/10.1016/S0092-8674(02)01135-2 (2002). | ||
| TriM Scope II microscope | La Vision Biotech |
Referenzen
- Slack, J. M. W. Embryonic induction. Mechanisms of Development. 41 (2-3), 91-107 (1993).
- Fernandez-Sanchez, M. -. E., Brunet, T., Röper, J. -. C., Farge, E. Mechanotransduction's impact on animal development, evolution, and tumorigenesis. Annual Review of Cell and Developmental Biology. 31, 373-397 (2015).
- Shih, J., Fraser, S. E. Characterizing the zebrafish organizer: microsurgical analysis at the early-shield stage. Development. 122 (4), 1313-1322 (1996).
- Selleck, M. A. J. Culture and microsurgical manipulation of the early avian embryo. Methods in Cell Biology. 51 (51), 1-21 (1996).
- Bulina, M. E., et al. A genetically encoded photosensitizer. Nature Biotechnology. 24 (1), 95-99 (2006).
- Fang-Yen, C., Gabel, C. V., Samuel, A. D. T., Bargmann, C. I., Avery, L. Laser microsurgery in Caenorhabditis elegans. Methods in Cell Biology. 107, 177-206 (2012).
- Colombelli, J., Grill, S. W., Stelzer, E. H. K. Ultraviolet diffraction limited nanosurgery of live biological tissues. Review of Scientific Instruments. 75 (2), 472-478 (2004).
- Smutny, M., Behrndt, M., Campinho, P., Ruprecht, V., Heisenberg, C. -. P. UV laser ablation to measure cell and tissue-generated forces in the zebrafish embryo in vivo and ex vivo. Methods in Molecular Biology. 1189, 219-235 (2015).
- Behrndt, M., et al. Forces driving epithelial spreading in zebrafish gastrulation. Science. 338 (6104), 257-260 (2012).
- Volpe, B. A., Fotino, T. H., Steiner, A. B. Confocal microscope-based laser ablation and regeneration assay in zebrafish interneuromast cells. Journal of Visualized Experiments: JoVE. (159), (2020).
- Bonnet, I., et al. Mechanical state, material properties and continuous description of an epithelial tissue. Journal of the Royal Society, Interface. 9 (75), 2614-2623 (2012).
- Rauzi, M., Lenne, P. F., Lecuit, T. Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature. 468 (7327), 1110-1115 (2010).
- Niemz, M. H. . Laser-Tissue Interactions. Encyclopedia of Biomaterials and Biomedical Engineering, Second Edition - Four Volume Set. , (2019).
- Smith, A. M., Mancini, M. C., Nie, S. Bioimaging: second window for in vivo imaging. Nature Nanotechnology. 4 (11), 710-711 (2009).
- Rauzi, M., Lenne, P. -. F. Cortical forces in cell shape changes and tissue morphogenesis. Current Topics in Developmental Biology. 95, 93-144 (2011).
- Theveneau, E., David, N. B. Migrations cellulaires collectives. Medecine/Sciences. 30 (8-9), 751-757 (2014).
- Dumortier, J. G., Martin, S., Meyer, D., Rosa, F. M., David, N. B. Collective mesendoderm migration relies on an intrinsic directionality signal transmitted through cell contacts. Proceedings of the National Academy of Sciences of the United States of America. 109 (42), 16945-16950 (2012).
- Solnica-Krezel, L., Stemple, D. L., Driever, W. Transparent things: cell fates and cell movements during early embryogenesis of zebrafish. BioEssays. 17 (11), 931-939 (1995).
- Montero, J. -. A., Kilian, B., Chan, J., Bayliss, P. E., Heisenberg, C. -. P. Phosphoinositide 3-kinase is required for process outgrowth and cell polarization of gastrulating mesendodermal cells. Current Biology. 13 (15), 1279-1289 (2003).
- Ulrich, F., et al. Slb/Wnt11 controls hypoblast cell migration and morphogenesis at the onset of zebrafish gastrulation. Development. 130 (22), 5375-5384 (2003).
- Kai, M., Heisenberg, C. -. P., Tada, M. Sphingosine-1-phosphate receptors regulate individual cell behaviours underlying the directed migration of prechordal plate progenitor cells during zebrafish gastrulation. Development. 135 (18), 3043-3051 (2008).
- Smutny, M., et al. Friction forces position the neural anlage. Nature Cell Biology. 19 (4), 306-317 (2017).
- Johansson, M., Giger, F. A., Fielding, T., Houart, C. Dkk1 controls cell-cell interaction through regulation of non-nuclear β-Catenin pools. Developmental Cell. 51 (6), 775-786 (2019).
- Gorelik, R., Gautreau, A. Quantitative and unbiased analysis of directional persistence in cell migration. Nature Protocols. 9 (8), 1931-1943 (2014).
- Grill, S. W., Howard, J., Schäffer, E., Stelzer, E. H. K., Hyman, A. A. The distribution of active force generators controls mitotic spindle position. Science. 301 (5632), 518-521 (2003).
- Desprat, N., Supatto, W., Pouille, P. -. A. A., Beaurepaire, E., Farge, E. Tissue deformation modulates twist expression to determine anterior midgut differentiation in Drosophila embryos. Developmental Cell. 15 (3), 470-477 (2008).
- Farhadifar, R., Röper, J. -. C., Aigouy, B., Eaton, S., Jülicher, F. The influence of cell mechanics, cell-cell interactions, and proliferation on epithelial packing. Current Biology. 17 (24), 2095-2104 (2007).
- Willier, B. H., Oppenheimer, J. M. . Foundations of Experimental Embryology. , (1964).
- Ashby, W. J., Zijlstra, A. Established and novel methods of interrogating two-dimensional cell migration. Integrative Biology: Quantitative Biosciences from Nano to Macro. 4 (11), 1338-1350 (2012).
- Bosze, B., et al. Pcdh18a regulates endocytosis of E-cadherin during axial mesoderm development in zebrafish. Histochemistry and Cell Biology. 154 (5), 463-480 (2020).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten