
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Herstellung und Betrieb eines kontinuierlichen Durchfluss-Mikroelektroporationssystems mit Permeabilisierungsdetektion
In diesem Artikel
Zusammenfassung
Dieses Protokoll beschreibt die Mikrofabrikationstechniken, die erforderlich sind, um ein mikrofluidisches Lab-on-a-Chip-Elektroporationsgerät zu bauen. Der Versuchsaufbau führt kontrollierte Transfektionen auf Einzelzellebene in einem kontinuierlichen Fluss durch und kann mit populationsbasierter Kontrolle auf höhere Durchsätze erweitert werden. Es wird eine Analyse bereitgestellt, die die Fähigkeit zeigt, den Grad der Permeabilisierung der Zellmembran in Echtzeit elektrisch zu überwachen.
Zusammenfassung
Aktuelle therapeutische Innovationen, wie die CAR-T-Zelltherapie, sind stark von der viral-vermittelten Genabgabe abhängig. Obwohl diese Technik effizient ist, geht sie mit hohen Herstellungskosten einher, was zu einem Interesse an der Verwendung alternativer Methoden für die Genlieferung geführt hat. Die Elektroporation ist ein elektrophysikalischer, nicht-viraler Ansatz zur intrazellulären Abgabe von Genen und anderen exogenen Materialien. Bei Anlegen eines elektrischen Feldes ermöglicht die Zellmembran vorübergehend die molekulare Abgabe in die Zelle. Typischerweise wird die Elektroporation auf der Makroskala durchgeführt, um eine große Anzahl von Zellen zu verarbeiten. Dieser Ansatz erfordert jedoch eine umfangreiche empirische Protokollentwicklung, die bei der Arbeit mit primären und schwer zu transfizierenden Zelltypen kostspielig ist. Langwierige Protokollentwicklung, gepaart mit der Anforderung großer Spannungen, um ausreichende elektrische Feldstärken zur Permeabilisierung der Zellen zu erreichen, hat zur Entwicklung von Elektroporationsgeräten im Mikromaßstab geführt. Diese Mikroelektroporationsgeräte werden mit gängigen Mikrofabrikationstechniken hergestellt und ermöglichen eine bessere experimentelle Kontrolle mit dem Potenzial, hohe Durchsatzfähigkeiten aufrechtzuerhalten. Diese Arbeit baut auf einer mikrofluidischen Elektroporationstechnologie auf, die in der Lage ist, den Grad der Zellmembranpermeabilisierung auf Einzelzellebene unter kontinuierlichem Fluss zu detektieren. Diese Technologie war jedoch auf 4 pro Sekunde verarbeitete Zellen beschränkt, so dass hier ein neuer Ansatz zur Erhöhung des Systemdurchsatzes vorgeschlagen und vorgestellt wird. Diese neue Technik, die als zellpopulationsbasierte Rückkopplungskontrolle bezeichnet wird, berücksichtigt die Zellpermeabilisierungsantwort auf eine Vielzahl von Elektroporationspulsbedingungen und bestimmt die am besten geeigneten Elektroporationspulsbedingungen für den zu testenden Zelltyp. Anschließend wird ein Modus mit höherem Durchsatz verwendet, bei dem dieser "optimale" Impuls auf die Zellsuspension während des Transports angewendet wird. Die Schritte zur Herstellung des Geräts, zum Aufbau und zur Durchführung der mikrofluidischen Experimente und zur Analyse der Ergebnisse werden detailliert vorgestellt. Schließlich wird diese Mikroelektroporationstechnologie demonstriert, indem ein DNA-Plasmid, das für grün fluoreszierendes Protein (GFP) kodiert, in HEK293-Zellen eingebracht wird.
Einleitung
Aktuelle therapeutische Innovationen in der biomedizinischen Forschung, wie die CAR-T-Zelltherapie (Chimeric Antigen Receptor Engineered T cell) und die genetische Editierung mittels CRISPR (Clustered regularly interspaced short palindromic repeat DNA sequences)/Cas9, hängen stark von der Fähigkeit ab, exogenes Material sowohl erfolgreich als auch effizient in den intrazellulären Raum zu bringen1. In der CAR-T-Therapie ist der Goldstandard für die Durchführung des Genabgabeschritts bei der Zelltherapieherstellung die Verwendung viraler Vektoren2. Obwohl die viral vermittelte Genabgabe eine effiziente Verabreichungsmodalität ist, hat sie auch mehrere Nachteile. Dazu gehören Herstellungskosten, Zytotoxizität, Immunogenität, Mutagenese/Tumorgenesepotenzial und Größenbeschränkungen für das/die zu liefernde(n) Gen(e)3. Diese Einschränkungen haben zur Erforschung und Entwicklung alternativer, nicht-viraler Verabreichungstechnologien geführt.
Die Elektroporation, eine Alternative zur viral vermittelten Genabgabe, beruht auf der Anwendung einer optimalen elektrischen Pulswellenform, um DNA-, RNA- und Proteintransfektionen von Zellen durchzuführen. Nach dem Anlegen eines externen elektrischen Feldes wird die Zellmembran kurzzeitig kompromittiert, wodurch die Zelle anfällig für die intrazelluläre Abgabe von ansonsten undurchlässigen exogenen Materialienwird 4. Im Vergleich zur viralvermittelten Verabreichung ist die Elektroporation vorteilhaft, da sie im Allgemeinen sicher, einfach zu bedienen und mit niedrigen Betriebskosten verbunden ist. Die Elektroporation kann sowohl kleine als auch große molekulare Fracht liefern und kann bei der Transfizierung von Zellen unabhängig von der Linie5 effizient sein. Um wünschenswerte Ergebnisse nach der Elektroporation zu erzielen, d.h. gute Lebensfähigkeit und gute Elektrotransfektionseffizienz, müssen eine Vielzahl von experimentellen Parametern co-optimiert werden. Dazu gehören Zelltyp6, Zelldichte, Molekülkonzentration7, Elektroporationspuffereigenschaften (z. B. molekulare Zusammensetzung, Leitfähigkeit und Osmolarität)8, Elektrodengröße/-geometrie9 und elektrische Pulswellenform (Form, Polarität, Anzahl der Impulse)10 (siehe Abbildung 1 für eine Abbildung). Obwohl jeder dieser Parameter einen signifikanten Einfluss auf die Ergebnisse von Elektroporationsexperimenten haben kann, wurde die Pulswellenform besonders detailliert untersucht, da die elektrische Energie des angelegten Impulses (der angelegten Impulse) die Wurzel des intrinsischen Kompromisses zwischen der resultierenden Zelllebensfähigkeit und der Elektrotransfektionseffizienz ist8.
Typischerweise werden Elektroporationsexperimente auf der Makroskala durchgeführt, wo Zellen in 100 Mikrolitern Puffer zwischen einem Satz großer, paralleler Plattenelektroden innerhalb einer Elektroporationsküvette suspendiert werden. Die Elektroden werden üblicherweise aus Aluminium mit einem Elektrodenabstand von 1-4 mm hergestellt. Sobald die Zellen manuell per Pipette geladen sind, wird die Küvette elektrisch mit einem sperrigen, elektrischen Impulsgenerator verbunden, wo der Benutzer die Pulswellenformparameter einstellen und anwenden kann, um die Zellsuspension elektropolieren. Obwohl die Makro- oder Massenelektroporation Zelldichten >106 Zellen / ml verarbeiten kann, kann diese Funktion bei der Optimierung der Einstellungen der elektrischen Pulswellenform verschwenderisch sein. Dies ist besonders besorgniserregend, wenn primäre Zelltypen galoppieren, bei denen die Anzahl der Zellpopulationen begrenzt werden kann. Zusätzlich muss der Impulsgeber aufgrund des großen Abstands zwischen den Elektroden in der Lage sein, große Spannungen zu liefern, um elektrische Feldstärken >1kV/cm11 zu erreichen. Diese hohen Spannungen verursachen eine Widerstandsverlustleistung durch den Elektrolytpuffer, was zu einer Joule-Erwärmung führt, die sich nachteilig auf die resultierende Zelllebensfähigkeit auswirken kann12. Schließlich wird die Durchführung der Elektroporation an einer dichten Zellsuspension konsequent mit einer angeborenen Variabilität der resultierenden Elektrotransfektionseffizienz und Zelllebensfähigkeit belastet. Jede Zelle in Suspension könnte aufgrund der umgebenden Zellen eine andere elektrische Feldstärke erfahren. Je nachdem, ob die erlebte elektrische Feldstärke erhöht oder verringert wird, kann die resultierende Zelllebensfähigkeit oder die Elektrotransfektionseffizienz jeweils negativ beeinflusst werden11. Diese Nachteile der Elektroporation im Makromaßstab haben zur Verfolgung und Entwicklung alternativer Technologien geführt, die auf der Mikroskala arbeiten und eine bessere Kontrolle auf Einzelzellebene ermöglichen.
Das Gebiet der BioMEMS oder biomedizinischen mikroelektromechanischen Systeme stammt aus den technologischen Fortschritten in der Mikroelektronikindustrie. Insbesondere die Verwendung von Mikrofabrikationsprozessen zur Entwicklung von Mikrogeräten für die Förderung der biomedizinischen Forschung. Diese Fortschritte umfassen die Entwicklung von Mikroelektrodenarrays für die elektrische In-vivo-Überwachung 13, kapazitive Mikroelektroden für die In-situ-Elektroporation 14, miniaturisierte Organ-on-a-Chip-Geräte 15, mikrofluidische Point-of-Care-Diagnostik 16, Biosensoren 17 und Arzneimittelabgabesysteme18, einschließlich Nano- und Mikroelektroporationsvorrichtungen 19,20,21 . Aufgrund der Fähigkeit, Geräte im gleichen Größenmaßstab wie biologische Zellen zu entwerfen und herzustellen, sind Nano- und Mikroelektroporationstechnologien im Vergleich zu ihrem makroskaligen Gegenstück22,23 vorteilhaft. Diese Elektroporationsgeräte machen Hochspannungsimpulsanwendungen überflüssig, da typischerweise Elektrodensätze mit Abständen von 10s bis 100s Mikrometern integriert sind. Diese Eigenschaft reduziert den Strom durch den Elektrolyten drastisch, was wiederum die Ansammlung toxischer Elektrolyseprodukte und die Auswirkungen der Joule-Erwärmung in diesen Systemen reduziert. Die mikroskaligen Kanäle stellen auch sicher, dass während der Pulsapplikation zuverlässig ein viel gleichmäßigeres elektrisches Feld an die Zellen angelegt wird, was zu konsistenteren Ergebnissen führt24. Darüber hinaus ist es auch üblich, dass Mikroelektroporationsgeräte in eine mikrofluidische Plattform integriert werden, die sich für die zukünftige Integration in eine vollautomatische Technologie eignet, eine höchst wünschenswerte Fähigkeit bei der Herstellung von Zelltherapien25. Schließlich ermöglicht die Elektroporation im Mikromaßstab die elektrische Abfrage von Elektroporationsereignissen. Zum Beispiel kann der Grad der Zellmembranpermeabilisierung in Echtzeit auf Einzelzellebene überwacht werden26,27. Der Zweck dieser Methode besteht darin, die Mikrofabrikation, den Systembetrieb und die Analyse einer mikrofluidischen, einzelligen Mikroelektroporationsvorrichtung zu beschreiben, die in der Lage ist, den Grad der Zellmembranpermeabilisierung zur Optimierung von Elektroporationsprotokollen zu messen und gleichzeitig den Durchsatz gegenüber dem vorherigen Stand der Technik zu erhöhen.
Die Durchführung der Elektroporation auf Einzelzellebene ist keine neuartige Technik mehr, wie Rubinsky et al. erstmals 2001 mit der Entwicklung einer statischen Zellelektroporationstechnologiedemonstrierten 28. Ihr Mikrogerät war innovativ, da sie die ersten waren, die die Fähigkeit demonstrierten, das Ereignis der Elektroporation elektrisch zu überwachen. Dies hat weiter zur Entwicklung statischer, einzelliger Elektroporationstechnologien geführt, die in der Lage sind, den Grad der Permeabilisierung der Zellmembran parallel elektrisch zu erfassen, um den Durchsatz der Geräte zu erhöhen. Aber selbst bei Parallelisierung und Stapelverarbeitung fehlt diesen Geräten die Gesamtzahl der Zellen, die sie pro Zeiteinheit verarbeiten können,29,30. Diese Einschränkung hat zur Entwicklung von Durchflussvorrichtungen geführt, die in der Lage sind, Mikroelektroporation auf Einzelzellebene bei viel größeren Durchsätzen durchzuführen31. Dieser Geräteübergang von der statischen zur Durchflussumgebung begrenzt die Fähigkeit, den Grad der Permeabilisierung der Zellmembran nach der Anwendung des Elektroporationsimpulses elektrisch zu überwachen. Die in dieser Arbeit beschriebene Methode schließt die Lücke zwischen diesen beiden Technologien, eine Mikroelektroporationstechnologie, die in der Lage ist, den Grad der Zellmembranpermeabilisierung einzelner Zellen kontinuierlich seriell zu detektieren, zu pulsieren und zu überwachen.
Diese Technologie wurde kürzlich in Zheng et al. In dieser Arbeit wurden die Fähigkeiten dieser Technologie mit dem Abschluss einer parametrischen Studie eingeführt, bei der sowohl die Amplitude als auch die Dauer des Elektroporationsimpulses variiert wurden und das daraus resultierende elektrische Signal, das auf eine Permeabilisierung der Zellmembran hinweist, untersucht wurde32. Die Ergebnisse zeigten, dass eine Zunahme der Intensität des Elektroporationsimpulses (d.h. Zunahme des angelegten elektrischen Feldes oder Erhöhung der Pulsdauer) eine Erhöhung der gemessenen Zellmembranpermeabilisierung verursachte. Um das System weiter zu validieren, wurde der Zellsuspension ein üblicher Fluoreszenzindikator für eine erfolgreiche Elektroporation, Propidiumiodid33, zugegeben, und unmittelbar nach der Anwendung des elektrischen Impulses wurde ein Fluoreszenzbild aufgenommen. Das optische Signal, d.h. die Fluoreszenzintensität von Propidiumiodid in der Zelle, korrelierte stark mit der elektrischen Messung des Permeabilisierungsgrades der Zellmembran, was die Zuverlässigkeit dieser elektrischen Messung bestätigte. Diese Arbeit betrachtete jedoch nur die Abgabe des niedermolekularen Propidiumiodids, das wenig bis gar keine übersetzbare Bedeutung hat.
In dieser Arbeit wird eine neue Anwendung dieser Technologie vorgestellt, um den Durchsatz des Systems zu verbessern und gleichzeitig einen biologisch aktiven Plasmid-DNA-Vektor (pDNA) zu liefern und die Elektrotransfektionseffizienz von Zellen zu bewerten, die nach der Elektroporation neu plattiert und kultiviert werden. Obwohl die vorherige Arbeit bestehende Mikroelektroporationstechnologien übertrifft, die in der Lage sind, das Ereignis der Elektroporation elektrisch zu messen, erfordert der aktuelle Zustand des Geräts immer noch lange Zelllaufzeiten zwischen dem Elektrodensatz (~ 250 ms), um die Zellerkennung, die Pulsanwendung und die Permeabilisierungsmessung der Zellmembran durchzuführen. Bei einem einzigen Kanal begrenzt dies den Durchsatz auf 4 Zellen/s. Um diese Einschränkung zu bekämpfen, wird ein neues Konzept der zellpopulationsbasierten rückkopplungsgesteuerten Elektroporation eingeführt, um pDNA-Elektrotransfektion durchzuführen. Durch die Verwendung eines hypophysiologischen Leitfähigkeits-Elektroporationspuffers ermöglicht dieses System die elektrische Abfrage einzelner Zellen über eine Vielzahl von Elektroporationspulsanwendungen. Basierend auf der elektrischen Reaktion wird dann ein "optimaler" Elektroporationsimpuls bestimmt. Anschließend wird ein "Hochdurchsatz"-Modus implementiert, bei dem die Bestimmung der Permeabilisierung der Zellmembran aufgehoben, die Flussrate erhöht und der Tastverhältnis des Elektroporationsimpulses an die Zelllaufzeit angepasst wird, um einen Impuls pro Zelle im Transit zwischen den Elektroden sicherzustellen. Diese Arbeit wird umfangreiche Details zu den Mikrofabrikationsschritten für die Herstellung des Mikrogeräts, dem Material / der Ausrüstung und deren Aufbau liefern, die für die Durchführung des Experiments erforderlich sind, sowie dem Betrieb / der Analyse des Geräts und seiner Elektrotransfektionseffizienz (eTE).

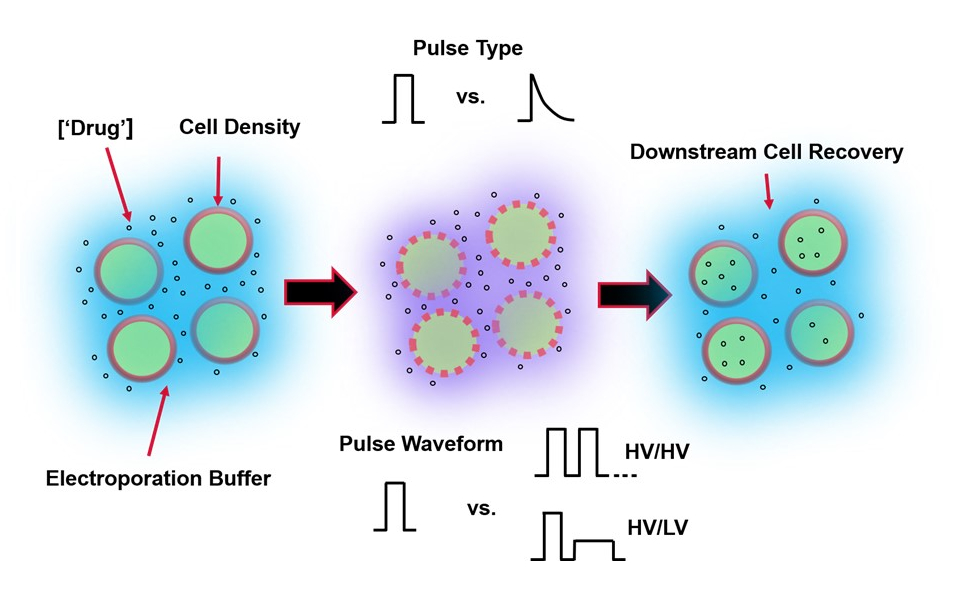
Abbildung 1: Experimentelle Faktoren, die die Ergebnisse der Elektroporation beeinflussen. (Links) Zellsuspension-Wichtige Faktoren, die vor Beginn der Elektroporation zu berücksichtigen sind, sind: Nutzlast (in diesem Fall pDNA), Konzentration, Zelldichte und Elektroporationspuffereigenschaften. Zu berücksichtigende Eigenschaften des Elektroporationspuffers sind Leitfähigkeit, Osmolarität und die genaue molekulare Zusammensetzung, die zu diesen Werten beiträgt. (Mitte) Pulsanwendung - Der genaue Pulstyp (Rechteckwelle vs. exponentieller Zerfall) und die Pulswellenform (Einzelpuls vs. Pulsfolge) müssen optimiert werden, um sowohl die resultierende Zelllebensfähigkeit als auch die Elektrotransfektionseffizienz zu maximieren. Übliche Pulsfolgen, die in Elektroporationsprozessen implementiert werden, bestehen typischerweise aus einer Reihe von Hochspannungsimpulsen (HV) oder einer Reihe von Impulsen, die zwischen HV- und Niederspannungsimpulsgrößen (LV) rotieren. (Rechts) Zellwiederherstellung-Nachgelagerte Verarbeitungsschritte, insbesondere die Rückgewinnungszellkulturmedien, auf die Zellen übertragen werden, sollten optimiert werden. Nicht vorgestellt (ganz links) können zusätzliche vorgeschaltete Zellverarbeitungsschritte zur Optimierung des gesamten Elektroporationsprozesses implementiert werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Protokoll
HINWEIS: Benutzer sollten alle Sicherheitsdatenblätter auf die in diesem Protokoll verwendeten Materialien und Verbrauchsmaterialien überprüfen. Bei jedem Schritt und jeder sterilen Technik, die während des Experimentierens verwendet wird, sollte eine geeignete PSA getragen werden. In den Abschnitten 1-7 wird die Geräteherstellung erläutert.
1. Geräteherstellung - Maskendesign
HINWEIS: In Abbildung 2 finden Sie eine Veranschaulichung des Mikrofabrikationsprozesses. Die Mikrofabrikationsschritte sollen in einer Reinraumumgebung durchgeführt werden. Zusätzliche PSA ist notwendig (Haarnetz, Gesichtshaarnetz, Maske, Reinraumanzug, Schuhüberzieher).
- Installieren Sie eine CAD-Software Ihrer Wahl, entwerfen Sie eine 2-dimensionale "Maske" sowohl des mikrofluidischen Kanals als auch der Elektroden und speichern Sie das Design im gewünschten Dateiformat (d. H. .dxf, .dwg).
HINWEIS: In der ergänzenden Abbildung 1 finden Sie ein Beispiel für einen 2-dimensionalen Maskenschema. - Senden Sie es an einen Lieferanten Ihrer Wahl, um gedruckt zu werden. Stellen Sie sicher, dass die Abmessungen der Designs innerhalb der Auflösungsmöglichkeiten des Lieferanten liegen.
2. Geräteherstellung - Photolithographie
HINWEIS: Die mitgelieferten Mikrofabrikationsrezepte sind den Empfehlungen des Fotolackherstellers entnommen und sollten nur als Ausgangspunkt34 verwendet werden. Genaue Werte für Backzeiten, Einwirkzeiten etc. müssen für jedes Fertigungsprotokoll optimiert werden. Es wird empfohlen, eine Waferpinzette für die Handhabung von Siliziumwafern und Glasobjektträgern zu verwenden.
- Mikrofluidische Kanalfertigung
- Reinigung von Siliziumwafern und Kalk-Natron-Glasobjektträgern: Führen Sie die Schritte 2.1.2-2.1.3 aus, um Siliziumwafer und 1" × 3" Kalk-Kalk-Kalk-Objektträgerreinigung (beide als "Substrat" bezeichnet) durchzuführen.
- Tauchen Sie die Substrate für jeweils 10 min in ein Acetonbad, ein Isopropanol (IPA)-Bad und ein entionisiertes Wasserbad. Führen Sie diese 3-stufige Wäsche seriell bei Raumtemperatur durch.
- Entfernen und trocknen Sie die Oberfläche mit einer unter Druck stehenden Stickstoff- oder gefilterten Luftgasquelle. Legen Sie die Substrate für mindestens 30 min in einen 150 °C heißen Ofen, damit die restliche Feuchtigkeit verdampfen kann.
- SU-8-Fotolithographie auf Siliziumwafer: Führen Sie eine Fotolithographie auf dem Siliziumwafer durch, indem Sie die Schritte 2.1.5-2.1.14 ausführen.
HINWEIS: Um eine mikrofluidische Kanalhöhe von 20 μm zu erreichen, wurde negativer Fotolack der SU-8 2000-Serie verwendet. Die genauen Spin-Raten variieren je nach Formulierung von SU-8 (z. B. 2010, 2015 usw.); Die folgenden Bedingungen gelten jedoch für die Formulierung SU-8 201035. - Nehmen Sie den Siliziumwafer aus dem 150 °C-Ofen und lassen Sie ihn auf Raumtemperatur (RT) abkühlen.
- Befestigen Sie den Wafer mit dem Vakuumsystem des Spin Coaters am Chuck des Wafer Spin Coaters. Programmieren Sie den Spinner. Schritt 1 - 500 U/min für 10 s bei einer Beschleunigung von 100 U/min/s, Schritt 2 - 1000 U/min für 30 s bei einer Beschleunigung von 300 U/min.
- Geben Sie 4 ml SU-8 2010 Fotolack auf die Mitte des Siliziumwafers. Führen Sie das Programm aus. Sobald das System zum Stillstand kommt, schalten Sie das Vakuum aus.
- Mit einer Pinzette den SU-8-beschichteten Siliziumwafer für 4-5 min auf eine Heizplatte bei 95 °C zum Weichbacken überführen. Nehmen Sie dann den Wafer von der Heizplatte und lassen Sie ihn auf RT abkühlen.
HINWEIS: Befolgen Sie das ordnungsgemäße Startverfahren für den laborspezifischen photolithografischen Masken-Aligner. - Befestigen Sie die Fotomaske mit den mikrofluidischen 2D-Kanaldesigns auf dem Maskenhalter. Setzen Sie den Siliziumwafer mit der SU-8-Beschichtung nach oben auf das Waferfutter ein.
- Stellen Sie die Belichtungseinstellungen auf 150 mJ/cm2 ein und starten Sie das Gerät.
VORSICHT: Schauen Sie nicht direkt auf die UV-Lichtquelle, um mögliche Augenschäden zu vermeiden. - Legen Sie den SU-8-beschichteten Siliziumwafer für 4-5 Minuten für den Backvorgang nach der Exposition auf eine Heizplatte bei 95 °C.
- Tauchen Sie den Siliziumwafer für 3-4 min in die SU-8-Entwicklerlösung (siehe Materialtabelle). Sanftes Rühren auftragen. Entfernen Sie den Wafer aus der Lösung und spülen Sie die Oberfläche mit IPA ab.
- Trocknen Sie die Oberfläche mit einer unter Druck stehenden Stickstoff- oder gefilterten Luftgasquelle. Untersuchen Sie die Merkmale unter dem Mikroskop mit einem UV-Filter und stellen Sie sicher, dass keine offensichtlichen Defekte in den mikrofluidischen Kanälen auftreten.
- Legen Sie den Siliziumwafer für mindestens 30 Minuten in einen 150 °C-Ofen für ein hartes Backen.
- Lassen Sie es auf RT abkühlen und verwenden Sie die Tasterprofilometrie, um die genaue Höhe und Neigung der Kanalseitenwände zu messen.
- Fotolithographie auf Glasobjektträgern
HINWEIS: Hexamethyldisilazan (HMDS) wird als Haftvermittler für den positiven Fotolack S181836 verwendet.- Nehmen Sie den Objektträger aus dem 150 °C heißen Ofen und lassen Sie ihn auf RT abkühlen.
- Befestigen Sie den Glasobjektträger mit Vakuum am Spannfutter des Spinners und programmieren Sie den Spinner. Schritt 1 - 500 U/min für 10 s bei einer Beschleunigung von 100 U/min/s. Schritt 2 - 3000 U/min für 30 saß eine Beschleunigung von 300 U/min/s.
- Geben Sie 3-4 Tröpfchen HMDS über die Oberfläche des Objektträgers. Führen Sie das Programm aus.
HINWEIS: Um eine Oberflächenbeschichtung von ~3 μm zu erreichen, sollte die positive Fotolackserie S1800 verwendet werden. Die genauen Schleuderraten variieren je nach Formulierung; Die folgenden Empfehlungen gelten für die S1818-Formulierung34. - Geben Sie 1 ml Fotolack auf die Oberfläche des Objektträgers. Stellen Sie sicher, dass Sie genügend Fläche abdecken.
- Führen Sie das Programm aus. Sobald das System zum Stillstand kommt, schalten Sie das Vakuum aus und entfernen Sie den Glasträger.
- Legen Sie den S1818 beschichteten Glasobjektträger für 4 min für einen weichen Backvorgang auf eine Heizplatte bei 120 °C. Entfernen und zu RT kommen lassen.
- Befestigen Sie die Fotomaske mit den 2D-Elektrodendesigns auf dem Maskenhalter.
- Setzen Sie den Objektträger mit der S1818-Beschichtung nach oben auf das Waferfutter ein und richten Sie es aus. Stellen Sie die Belichtungseinstellungen auf 250 mJ/cm2 ein und starten Sie das Gerät.
HINWEIS: Verschiedene Kontakt-Aligner-Modelle können mehr oder weniger für nicht kreisförmige, unterschiedlich dicke Substrate geeignet sein. - Tauchen Sie den Objektträger für 2 Minuten in die MF-319-Entwicklerlösung. Sanftes Rühren auftragen. Spülen Sie die Oberfläche des Objektträgers mit entionisiertem Wasser ab.
- Trocknen Sie die Oberfläche mit einer unter Druck stehenden Stickstoff- oder gefilterten Luftgasquelle und beobachten Sie die Merkmale unter einem Mikroskop mit einem UV-Filter. Stellen Sie sicher, dass die lithografischen Muster keine offensichtlichen Mängel aufweisen.
- Legen Sie den Glasobjektträger in den 150 °C-Ofen und stellen Sie sicher, dass die gewünschte Substratoberfläche nach oben zeigt, für mindestens 30 Minuten für einen harten Backvorgang. Aus dem Ofen nehmen und vor Licht geschützt aufbewahren.
3. Geräteherstellung: Flusssäure (HF) ätzen
ACHTUNG: Dieser Schritt beinhaltet die Handhabung und Entsorgung von Flusssäure (HF), die tiefe, schmerzhafte chemische Verbrennungen verursachen kann. Zum Schutz des Hundeführers sollte zusätzliche PSA verwendet werden (Gesichtsschutz, ellbogenlange, chemisch beständige Handschuhe, chemisch beständige Schürze mit Ärmeln). Calciumgluconatsäure-Neutralisator und Hautgel sollten in der Nähe des Labortisches aufbewahrt werden. Dieser Schritt sollte nicht alleine durchgeführt werden. HF sollte niemals in Glasbehältern gelagert oder in Glasbehältern abgegeben werden, da der Behälter von der Säure geätzt wird.
HINWEIS: Der HF ätzt das belichtete Glas (d. H. Das Elektrodendesign) gleichmäßig, um eine Aussparung im Glas zu bilden, was eine bessere Kantenauflösung des Elektrodenmusters nach der Metallabscheidung ermöglicht (Abschnitt 4).
- Tauchen Sie den Objektträger in 10:1 gepufferte HF-Lösung für 1 min in einen Polytetrafluorethylenbehälter. Übertragen und waschen Sie die Glasobjektträger in entionisiertem Wasser. Wiederholen Sie den Waschschritt 3 mal.
- Trocknen Sie die Oberfläche mit einer unter Druck stehenden Stickstoff- oder gefilterten Luftgasquelle. Glassubstrate über Nacht in einen 65 °C heißen Ofen geben, um die restliche Feuchtigkeit zu entfernen. Bedecken Sie die Substrate vor Licht.
4. Geräteherstellung: Physikalische Gasphasenabscheidung
HINWEIS: Dieser Schritt beinhaltet die Metallabscheidung auf den Glasträgersubstraten, um die Elektrodenmuster zu definieren. Häufig verwendete Metallelektroden sind Chrom/Gold und Titan/Platin. Gold und Platin haften nicht am Glassubstrat, so dass eine Samenhaftschicht aus Chrom bzw. Titan erforderlich ist, um die Adhäsionzu fördern 37.
- Befolgen Sie das reinraumspezifische Protokoll, um die hauseigene PVD-Anlage zu betreiben. Diese Arbeit verwendet ein DC-Sputtersystem und Sputtern mit 100 SCCM Argongas bei einem Druck von ~ 8 mTorr und 200 W Leistung.
- Titan 8 min mit einer Geschwindigkeit von ~100 Å/min sputtern. Platin für 10 min mit einer Rate von ~200 Å/min sputtern. Entfernen Sie die Substrate aus der PVD-Kammer.
5. Geräteherstellung: Fotolack-Abheben
HINWEIS: Dieser Schritt beinhaltet das Auflösen der Fotolackschicht in einem Acetonbad, wobei die aufgeklebten Platinelektroden auf den Glasobjektträgern gemustert bleiben.
- Tauchen Sie die metallbeschichteten Glasdias für ~10 min in ein Acetonbad.
- Beschallen Sie das Bad, um eine Bewegung einzuleiten, um den nicht haftenden Metallfilm aufzubrechen. Verwenden Sie ein mit Aceton getränktes Tuch, um bei Bedarf Rückstände zu entfernen.
- Sobald alle Fotolacke / Metall entfernt sind, waschen Sie die Elektrodenmuster mit entionisiertem Wasser und legen Sie sie über Nacht in einen 65 ° C Ofen, um die verbleibende Oberflächenfeuchtigkeit zu entfernen.
- Verwenden Sie die Tasterprofilometrie, um das Profil der strukturierten Elektroden zu messen.
6. Geräteherstellung: Softlithographie
HINWEIS: Dieser Schritt beinhaltet das Nachformen des mikrofluidischen Kanals auf die SU-8-Masterreliefstruktur unter Verwendung eines Elastomers, Polydimethylsiloxan (PDMS).
- Siliziumwafer-Silanisierung
HINWEIS: Dies ist ein optionaler Schritt. Es wird jedoch die Lebensdauer der SU-8-Reliefstruktur erhöhen, die in Unterabschnitt 2.1 hergestellt wurde. Dieser Schritt sollte in einem chemischen Abzug durchgeführt werden.- Befestigen Sie die Waffel am Boden einer Petrischale und legen Sie die Petrischale in einen Exsikkator.
- Umgeben Sie den Umfang des Siliciumwafers mit etwa 50 μL Trichlor(1H,1H,2H,2H-perfluorooctyl)silan. Vakuum anschließen (Vakuumpumpe oder Hausvakuumleitung) und 20 min laufen.
- PDMS-Replikatspritzguss
- Mischen Sie in einem Einwegbehälter (z. B. Wiegeboot, Plastikbecher) PDMS-Elastomerbasis mit Härter im Gewichtsverhältnis 10:1 auf einer elektronischen Waage. Gießen Sie die PDMS-Lösung über den Siliziumwafer und stellen Sie die Mischung unter Vakuum, um alle Luftblasen zu entfernen.
- Bei 65 °C für mindestens 4 h aushärten, damit das PDMS erstarren kann. Schneiden Sie mit der Spitze einer Rasierklinge das geformte PDMS aus und ziehen Sie es vom Siliziumwafer ab.
- Entfernen Sie PDMS mit einem geschärften Biopsiestempel aus dem Einlass / den Auslässen des Geräts. Für dieses Gerät wurden 0,75 mm bzw. 3 mm Biopsiestempel für die Einlässe bzw. die Auslässe verwendet.
HINWEIS: Der verwendete Biopsiestempel sollte einen etwas kleineren Durchmesser als der Außendurchmesser des Verbindungsschlauchs haben, um eine dichte Abdichtung der Schläuche in den Behältern zu gewährleisten.
- Beschallungsreinigung von PDMS
- Tauchen Sie die PDMS-Geräte in IPA ein und legen Sie sie für 30-45 Minuten in einen Ultraschallgerät, um PDMS-Ablagerungen aus dem Einlass / den Auslässen zu entfernen. PDMS kann in der IPA-Lösung anschwellen.
- Mit entionisiertem Wasser abspülen und über Nacht in einen 65 °C heißen Ofen stellen, damit das PDMS wieder auf die normale Größe anschwellen kann.
HINWEIS: Reste von Ablagerungen können das Gerät während des Experimentierens verstopfen. Große Schmutzteile können vor der Beschallung mit einem Stück Klebeband von der PDMS-Oberfläche entfernt werden.
7. Geräteherstellung: PDMS-Bonden / Drahtbefestigung
ANMERKUNG: Dieser Schritt beinhaltet die Behandlung der Oberfläche des PDMS und des Glassubstrats mit einem Sauerstoffplasma, um eine irreversible Verbindung zwischen dem PDMS und Glas38 zu bilden. Die mitgelieferte Rezeptur muss möglicherweise an das genaue System angepasst werden, das im Labor verwendet wird.
- Schneiden Sie die Geräte zu und stellen Sie sicher, dass die Oberfläche des PDMS-Geräts sauber ist. Wenn Sie nicht erneut reinigen, befolgen Sie die Schritte in Unterabschnitt 6.3.
- Programmieren Sie den Plasmagenerator. Leistung auf 70 W, Zeit auf 35 s, Druck auf 325 mTorr, Durchfluss des Sauerstoffgases auf 60 SCCM. Legen Sie PDMS und Elektrodenglasschieber mit den Funktionen nach oben in das System und führen Sie das Programm aus.
- Entfernen Sie die Geräte und richten Sie die Kanalmerkmale mithilfe eines Stereoskops schnell an den Elektroden aus. Üben Sie festen Druck von der Mitte des PDMS zu den Seiten aus, um unerwünschte Luftblasen an der Klebeschnittstelle zu entfernen.
- An einem heißen Ort bei 95 °C für mindestens 2 min aufbewahren, um den Klebevorgang abzuschließen, und das Gerät bei RT abkühlen lassen.
- Schneiden Sie 2 Stück 22-G-Volldraht bei ~ 6 "Längen und entfernen Sie den Isolator von beiden Enden.
- Verkleben Sie die Drähte mit silberleitfähigem Epoxidharz mit Elektrodenpads. Fertige Geräte über Nacht in einen 65 °C heißen Backofen stellen.

Abbildung 2: Herstellung von Mikrogeräten. (A) Mikrofluidische Kanalherstellung - Schlüsselschritte: Siliziumwaferreinigung (Schritte 2.1.1-2.1.3), Fotolackbeschichtung und weiches Backen (Schritte 2.1.7-2.1.8), UV-Exposition (Schritt 2.1.10), Entwicklung (Schritt 2.1.12) und PDMS-Gießen (Unterabschnitt 6.2). (B) Schlüsselschritte zur Elektrodenherstellung: Glasobjektträgerreinigung (Schritte 2.1.1-2.1.3), HMDS-Beschichtung und Fotolackbeschichtung (Schritte 2.2.3-2.2.4), UV-Exposition (Schritt 2.2.8), Entwicklung (Schritt 2.2.9), HF-Ätzen (Abschnitt 3), physikalische Gasphasenabscheidung (Abschnitt 4) und Fotolackabhebung (Abschnitt 5). (C) Schlüsselschritte zur Gerätefinalisierung: Zugang zum Einlass/Ausgang und Beschallung (Schritt 6.2.3 und Abschnitt 6.3), PDMS-Bonding und Drahtbefestigung (Abschnitt 7). Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
8. Zellkultur und Ernte
HINWEIS: Standard-Zellkultur- und sterile Handhabungsverfahren sollten verwendet werden. Befolgen Sie das zelltypspezifische Protokoll für die Zellkultur.
- Zellkultur
- Zellpassage: Kultur und Durchgang der Zellen nach den Schritten 8.1.2-8.1.5.
- Kultur von HEK293-Zellen in vollständiger DMEM-Lösung (88% DMEM, 10% hitzeinaktiviertes fetales Rinderserum, 1% L-Glutamin, 1% Penicillin-Streptomycin) in einem T25-Kolben in einem Inkubator bei 37 °C, 95% O2, 5% CO2. Durchgangszellen planmäßig, wenn ~80% Konfluenz erreicht wird.
- Die Medien werden entweder mit einer Pipette oder einem Vakuumsystem abgesaugt und die Zellen in 0,25% Trypsin-EDTA (2 ml-T25-Kolben) für 2 min bei 37 °C inkubiert. Neutralisieren Sie Trypsin mit dem doppelten Volumen von Kulturmedien.
- Die Zellsuspension in ein 15 mL konisches Röhrchen geben und HEK293-Zellen bei 770 x g für 2 min zentrifugieren. Absaugen des Überstands entweder mit einer Pipette oder einem Vakuumsystem
- Resuspendieren Sie HEK293-Zellen in 1 ml vorgewärmtem DMEM.
- Zellplattierung: Beschichten Sie die Zellen nach den Schritten 8.1.7-8.1.8
- Die Zellen werden in einer Verdünnung von 10:1 bis 20:1 in einem T25-Kolben (5 ml DMEM) beschichtet, um die Kultur fortzusetzen.
- Die Zellen werden in einer Verdünnung von 5:1 bis 20:1 in einer 6-Well-Platte (2 ml DMEM pro Well) für Elektroporationsexperimente geerntet.
HINWEIS: HEK293-Zellen wurden 24 Stunden vor Elektroporationsexperimenten plattiert, um ~70% Konfluenz bei der Zellernte zu erreichen (Unterabschnitt 8.3). Ein inkonsistenter Ernteplan kann zu Schwankungen in den Elektroporationsergebnissen führen.
- Elektroporationspuffer
- Elektroporationspuffer vorbereiten
HINWEIS: Einzelheiten zur Elektroporationspufferzubereitung8 finden Sie in Sherba et al. Die Pufferzusammensetzung betrug 285 mM Saccharose, 0,7 mMMgCl2, 1 mM KCl, 10 mM HEPES, 3 mM NaOH (pH: 7,4; Osmolalität: 310 mOsm, Leitfähigkeit: 500 μS/cm). Elektroporationspuffer sollte steril formuliert und bei 4 °C für eine Haltbarkeit von ~1 Monat gelagert werden. Die Formulierung des Elektroporationspuffers sollte pro Zelltyp optimiert werden.
- Elektroporationspuffer vorbereiten
- Zellernte und pDNA-Addition
- Führen Sie die gleichen Schritte wie bei der Zellpassage (8.1.2-8.1.4) aus.
- Waschen Sie die Zellen in sterilem 1x PBS, Transferzellsuspension in ein 15 ml konisches Röhrchen und zentrifugieren Sie die Zellen bei 770 x g für 2 min.
- HEK293 Zellpellet im Elektroporationspuffer waschen und bei 770 x g 2 min zentrifugieren. Resuspendieren Sie die Zellen im Elektroporationspuffer bei ~ 5 Millionen Zellen / ml.
HINWEIS: Die Zelldichte sollte pro Zelltyp optimiert werden. - Fügen Sie pDNA, die für grün fluoreszierendes Protein (GFP) kodiert, zu einer Endkonzentration von 20 μg / ml hinzu. Mischen Sie vorsichtig die pDNA/Zell-Suspension und geben Sie die Suspension zum Experimentieren in eine 1-cc-Spritze.
9. Hardware/Versuchsaufbau
HINWEIS: Stellen Sie vor der Ernte von Zellen für Experimente sicher, dass der Versuchsaufbau abgeschlossen ist, um die Zeit zu minimieren, in der die Zellen im Elektroporationspuffer suspendiert sind. Schalten Sie die Elektronik 20-30 Minuten vor dem Aufwärmen ein. Abbildung 3 zeigt einen schematischen Aufbau des Versuchsaufbaus für den Betrieb des Einzelzellen-Detektionsmoduls.
HINWEIS: Eine speziell angefertigte PA90-Operationsverstärkerschaltung wurde entwickelt, um sowohl die Empfindlichkeit zu berücksichtigen, die für die Einzelzellen-Füllstandserkennung mit dem Lock-in-Verstärker erforderlich ist, als auch die hohen Spannungen, die erforderlich sind, um ausreichend starke Elektroporationsimpulse zu verwenden. Spezifikationen der empfohlenen Schaltung39 finden Sie im PA90-Datenblatt.
- Initialisieren Sie den Lock-in-Verstärker mit aktuellen Vorverstärkereinstellungen und stellen Sie ihn über den Algorithmus ein. Einzelheiten zu den Lock-in-Einstellungen32 finden Sie in Zheng et al.
- Netzteile, Funktionsgenerator und Verstärker
- Stromversorgung 1: Stellen Sie auf -15 V ein, um das negative Ende des Stromkreises mit Strom zu versorgen.
- Stromversorgung 2 (Funktionsgenerator): Stellen Sie die Ausgabe des DC-Signals ein und stellen Sie die Amplitude auf 2 V ein.
- Programm Elektroporationsimpulsgenerator für die Rechteckwelle: Stellen Sie die gewünschte Pulsbreite (Tastverhältnis) und die gewünschte Pulsamplitude (Volt) ein.
- Stellen Sie den Ausgang auf den Triggermodus (1 Impuls). Verbinden Sie den Ausgang mit dem Eingang des 50x-Verstärkers.
HINWEIS: Beachten Sie die 50-fache Verstärkung bei der Programmierung der Impulsamplitude. D.h. um eine elektrische Feldstärke von 1 kV/cm zu erreichen, sind insgesamt 30 V erforderlich, 30 V/300 μm (Abstand zwischen den Elektroden), daher sollte die Leistung des Funktionsgenerators auf 30/50 oder 600 mV eingestellt werden. - Überprüfen Sie die Ausgänge des 50-fachen Verstärkers mit einem Oszilloskop. Ausgang 1-100 V aus Netzteil 2 (9.2.2). Ausgang 2-Variable Amplitude für den Elektroporationsimpuls (9.2.4).
- Schließen Sie eine 10x-Sonde an einen Oszilloskopkanal und an das fertige Mikrogerät (Device under Test, DUT) in Schritt 7.6 an, wo der Elektroporationsimpuls angelegt wird. Überwachen Sie das System während des Experimentierens, um sicherzustellen, dass Impulse angewendet werden.
- Stellen Sie sicher, dass der Lock-in-USB-Anschluss angeschlossen und registriert ist. Überprüfen Sie alle Lock-in-Einstellungen im Algorithmuscode (vor allem die Lock-in-Ausgangsfrequenz).
- Mikroskop/CCD-Kamera
- Legen Sie das Mikrogerät über einen Objektträgerhalter auf den Tisch des Mikroskops. Schalten Sie die CCD-Kamera ein und bringen Sie den mikrofluidischen Kanal in den Fokus. Verwenden Sie ein 4x- oder 10x-Objektiv.

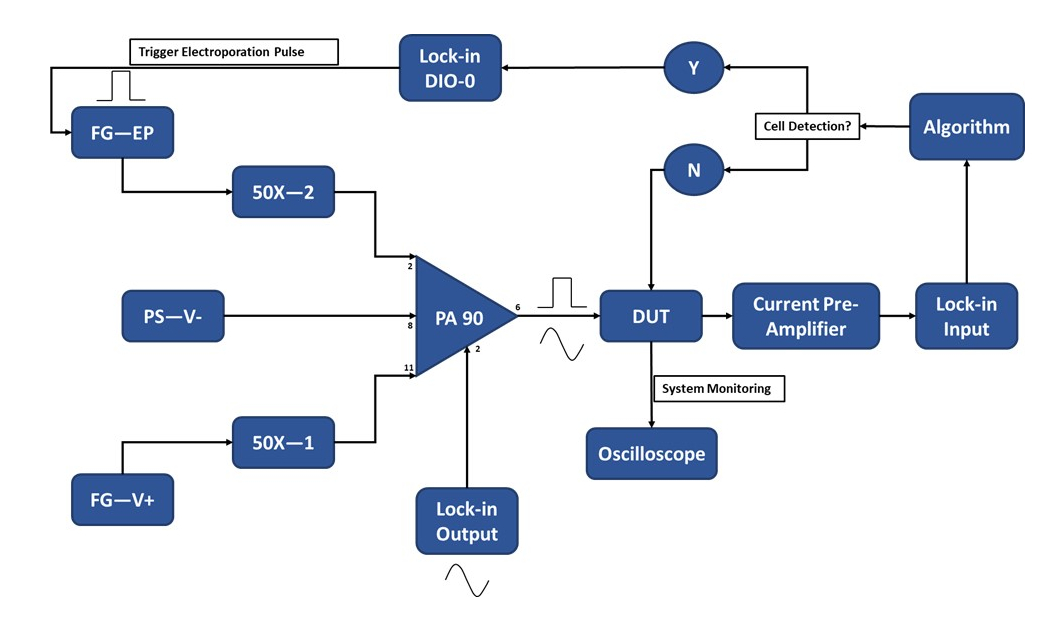
Abbildung 3: Schematische Darstellung des Versuchsaufbaus - Einzelzellenerkennung. Der Hochleistungs-Operationsverstärker (PA-90) ermöglicht die Überlagerung des Hochspannungs-Elektroporationsimpulses auf das Lock-in-Ausgangs-AC-Signal, das für die Einzelzellenerkennung erforderlich ist. Dieses Anregungssignal durchläuft das Mikroelektroporationsgerät (Device Under Test, DUT), wo der Strom dann durch den Stromvorverstärker verstärkt und in den Algorithmus eingespeist wird. Das System überwacht kontinuierlich das Zellerkennungsereignis. Beim Zelleintritt wird vom Lock-in-Verstärker ein digitales Signal erzeugt, um die Anwendung des Elektroporationsimpulses an die Zelle(n) während des Transports auszulösen. Legende: PA-90 (Hochleistungs-Operationsverstärker), DUT (Gerät unter Test), DIO (digitaler Ein-/Ausgang), FG-EP (Funktionsgenerator / Elektroporationsimpuls), 50X (50X Verstärker), PS-V- (Stromversorgung / negative Spannung für PA 90), FG-V+ (Funktionsgenerator, positive Spannung für PA 90). Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
10. Versuchsbetrieb
- Mikrofluidische Kanalgrundierung
- Entfernen Sie alle Luftblasen aus der zellbeladenen Spritze. Befestigen Sie eine 30 G Nadel an der zellbeladenen Spritze.
- Schieben Sie den Tygonschlauch mit einer Pinzette über die Länge der Nadel. Füllen Sie den Auslassbehälter mit Rückgewinnungsmedien vor (wie Schritt 8.1.2 ohne Antibiotika), ~40-50 μL.
- Üben Sie mit dem Daumen vorsichtigen Druck auf den Kolben aus, so dass die Flüssigkeit langsam das Ende der Schlauchleitung erreicht.
- Befestigen Sie die Spritze an der Spritzenpumpe. Schalten Sie die Spritzenpumpe ein und stellen Sie sicher, dass sie auf Durchblutung eingestellt ist.
- Programmieren Sie die Pumpe auf den richtigen Durchmesser der Spritze, um sicherzustellen, dass die Durchflussraten genau sind. Einzelheiten zu Spritzendurchmessern finden Sie im Pumpenhandbuch.
HINWEIS: Um zu verhindern, dass sich Zellen in der Spritze absetzen, befestigen Sie die Spritzenpumpe auf einem Klemmständer so, dass sie in vertikaler Position mit dem Spritzenende nach unten arbeiten kann. - Stellen Sie die Durchflussrate der Spritzenpumpe auf ~10-20 μL/min ein und lassen Sie die Pumpe laufen, bis die Flüssigkeit das Ende der Schlauchleitung erreicht. Befestigen Sie den Schlauch am mikrofluidischen Gerät.
- Senken Sie die Durchflussrate der Spritzenpumpe um ~ 5-10 μl / min und lassen Sie die Pumpe laufen, bis die gesamte Luft aus dem mikrofluidischen Gerät ausgestoßen ist und die Zellen zum Geräteauslass gelangen.
- Entfernen Sie die Zellen über die Pipettenaspiration aus dem Auslass. Füllen Sie den Auslassbehälter wieder mit Rückgewinnungsmedien (wie Schritt 8.1.2 ohne Antibiotika), ~40-50 μL.
- Single-Cell-Elektroporationszell-Membran-Permeabilisierungs-Mapping
HINWEIS: Siehe Abbildung 4 und Abbildung 5 für ein besseres Verständnis der elektrischen Daten, die auf die Permeabilisierung der Zellmembran und die Permeabilisierung der Zellmembran hinweisen.- Stellen Sie die Durchflussrate der Spritzenpumpe auf ~0,1-0,3 μl/min ein, um einen Fluss einzelner Zellen durch den Elektrodensatz zu gewährleisten. Die Zelllaufzeit zwischen den Elektroden sollte ~250 ms betragen.
- Starten Sie das Computerprogramm, indem Sie auf Ausführen klicken. Stellen Sie sicher, dass das System die elektrischen Daten speichert.
- Stellen Sie sicher, dass das System Zellen zuverlässig erkennt, um die computergesteuerten Impulsanwendungen auszulösen. Passen Sie den Erkennungsschwellenwert entsprechend an.
- Stellen Sie die Pulsparameter für den anfänglichen, stromsparenden Elektroporationsimpuls mit niedrigster elektrischer Energie ein. In Tabelle 1 finden Sie die pulsierenden Parameter der Elektroporation in dieser Studie.
- Schalten Sie den Ausgangskanal für den Elektroporationsimpulsgenerator ein (Schritt 9.2.3.).
- Folgen Sie einer vorgegebenen Anzahl von Zelldetektions-/Impulsanwendungen (n = 100). Am Ende jedes getesteten Zustands saugen Sie die Zellen aus dem Mikrogeräteauslass ab und füllen Sie den Auslass mit Wiederherstellungsmedien auf.
- Iterieren Sie zur nächsten Elektroporationsimpulsbedingung. Wiederholen Sie diesen Vorgang, bis alle Bedingungen des Elektroporationsimpulses getestet sind.
- Bestimmen Sie den Grad der Zellmembranpermeabilisierung für jede getestete Pulsanwendung. (Die Validierung nach dem Prozess wird in Unterabschnitt 11.1 beschrieben.) Generieren Sie die Permeabilisierungskarte der Zellmembran (Abbildung 5).
- Bestimmen Sie die Elektroporationspulsparameter für populationsbasiertes Feedback mit hohem Durchsatz.
- Schalten Sie die Spritzenpumpe aus, entfernen Sie Zellen aus dem Auslassbehälter und füllen Sie den Auslass mit Wiederherstellungsmedien auf.
- Populationsbasierte, rückkopplungsgesteuerte Elektroporation – hoher Durchsatz
HINWEIS: In Abbildung 6 finden Sie ein Schema, das den populationsbasierten Feedbackprozess veranschaulicht.- Stellen Sie die Durchflussrate der Spritzenpumpe auf ~1-3 μL/min ein, um einen Fluss einzelner Zellen durch den Elektrodensatz zu gewährleisten. Die Zelllaufzeit zwischen den Elektroden sollte ~25 ms betragen.
- Stellen Sie die Impulsamplitude auf den "optimierten" Zustand (10.2.9) ein, schalten Sie den Triggermodus aus und stellen Sie die Pulsbreite an die Sendezeit der Zelle an.
- Stellen Sie das Tastverhältnis so ein, dass die Einschaltzeit des Impulses der "optimierten" Bedingung entspricht. Siehe Tabelle 1.
- Stellen Sie den Ausgabekanal-Funktionsgenerator auf ON, schalten Sie die Spritzenpumpe ein und lassen Sie das System laufen, bis die gewünschte Anzahl von Zellen elektroporiert wurde.
- Wenn Sie fertig sind, schalten Sie sowohl die Spritzenpumpe als auch den Funktionsgenerator aus.
- Die Zellen werden aus dem Auslassbehälter in den mit vorgewärmten Rückgewinnungsmedien gefüllten Zellkulturkolben/die Zellkulturplatte überführt und in den Inkubator überführt.
11. Analyse
- Detektion der Membranpermeabilisierung auf Einzelzellebene
HINWEIS: Um sicherzustellen, dass der "optimale" Impuls während des Hochdurchsatzmoduls verwendet wurde, sollte nach dem Versuch eine Analyse durchgeführt werden, um die aus Unterabschnitt 10.2 exportierten elektrischen Daten zu überprüfen. In Abbildung 4 finden Sie eine grafische Darstellung des elektrischen Signals, das für die Permeabilisierung der Membran durch Elektroporation repräsentativ ist.- Laden Sie Daten in eine Analysesoftware (MATLAB, Python usw.). Generieren Sie für jede pulsierende Bedingung ein Diagramm von Strom und Zeit.
- Bestimmen Sie manuell den Grad der Permeabilisierung der Zellmembran (Δ IP / ΔIC). Siehe Abbildung 4. Generieren Sie die Permeabilisierungskarte der Zellmembran (Δ IP / ΔIC versus elektrische Energie, Abbildung 5) über alle getesteten Pulsbedingungen. Überprüfen Sie den "optimalen" pulsierenden Zustand.
- Elektro-Transfektionseffizienz (eTE)
- Nach der 24-stündigen Inkubationszeit die elektropolierten Zellen aus dem Inkubator entfernen.
- Führen Sie eine lebende Zellfärbung durch. DRAQ5 wird 1:1000 im Zellkulturgefäß auf eine Endkonzentration von 5 μM verdünnt. Die Zellen/Färbelösung vorsichtig mischen und bei 37 °C 5-30 min inkubieren.
HINWEIS: In diesem Schritt kann eine andere Färbung implementiert werden. Stellen Sie sicher, dass sich die fluoreszierenden Eigenschaften nicht mit dem Fluoreszenzmarker überlappen, der eine erfolgreiche Elektrotransfektion anzeigt (d. h. GFP ist in der grünen Wellenlänge und DRAQ5 ist die fernrote). - Schalten Sie ein Epifluoreszenzmikroskop, eine Lampe und Kameras ein (siehe Materialtabelle).
- Entfernen Sie die Zellen aus dem Inkubator und bringen Sie sie auf dem Mikroskop in den Fokus.
- Nehmen Sie ein Phasenkontrastbild (Hellfeld) des ausgewählten Felds auf.
- Nehmen Sie epifluoreszierende Bilder desselben Feldes mit FITC (GFP) und Far-Red (DRAQ5) Filtern auf. Analysieren Sie die Bildsets manuell oder über einen Algorithmus.
Hinweis: Repräsentative Bilder finden Sie in Abbildung 7 . - Zählen Sie die Gesamtzahl der GFP-positiven Zellen in allen Bildern. Zählen Sie die Gesamtzahl der DRAQ5-gefärbten Zellen in allen Bildern. Berechnen Sie eTE (Verhältnis von GFP-positiven Zellen zu DRAQ5-gefärbten Zellen).
Ergebnisse
Abbildung 4 zeigt die Funktionsprinzipien hinter der Einzelzell-Membranpermeabilisierungsdetektion für eine einzelne Pulsamplitude. Nach Beginn des Elektroporationsexperiments bestimmt der Zelldetektionsalgorithmus eine optimale Schwelle für die Zelldetektion über ein punktweises, steigungsbasiertes Detektionsverfahren. Das System überwacht dann kontinuierlich (1) auf eine signifikante negative Änderung des gemessenen elektrischen Stroms, was auf den Eintritt einer Zelle hinweist. Dies ...
Diskussion
Die in diesem Protokoll vorgestellte Methodik konzentriert sich in erster Linie auf die Mikrofabrikation eines mikrofluidischen Geräts, das dann in einen speziellen Elektroporationsversuchsaufbau integriert wird. Der Begriff "Rezept", der häufig zur Beschreibung der Besonderheiten des Mikrofabrikationsprozesses verwendet wird, deutet darauf hin, wie wichtig es ist, jeden Schritt zu befolgen / zu optimieren, um ein funktionierendes Gerät erfolgreich herzustellen. Bestimmte kritische Schritte innerhalb des Prozesses, we...
Offenlegungen
Die Autoren haben nichts offenzulegen.
Danksagungen
Die Autoren danken der National Science Foundation (NSF CBET 0967598, DBI IDBR 1353918) und der Graduate Training in Emerging Areas of Precision and Personalized Medicine (P200A150131) des US-Bildungsministeriums für die Finanzierung des Doktoranden J.J.S. auf Stipendium.
Materialien
| Name | Company | Catalog Number | Comments |
| 150-mm diameter petri dishes | VWR | 25384-326 | step 6.1.1 to secure wafer |
| 24-well tissue culture plates | VWR | 10062-896 | step 10.3.6 to plate electroporated cells |
| 33220A Waveform/Function generator | Agilent | step 9.2.3 electroporation pulse generator | |
| 4'' Si-wafers | University Wafer | subsection 2.1 for microfluidic channel fabrication | |
| 6-well tissue culture plates | VWR | 10062-892 | step 8.1.8 to plate cells |
| Acetone | Fisher Scientific | A18-4 | step 2.1.2 for cleaning and step 5.1 photoresist lift-off |
| Allegra X-22R Centrifuge | Beckman Coulter | steps 8.1.4 , 8.3.2. and 8.3.3. to spin down cells | |
| AutoCAD 2018 | Autodesk | subsection 1.1. to design transparency masks | |
| Buffered oxide etchant 10:1 | VWR | 901621-1L | subsection 3.1 for HF etching |
| CCD Monochrome microscope camera | Hamamatsu | Orca 285 C4742-96-12G04 | step 11.2.3. for imaging |
| CMOS camera- Sensicam QE 1.4MP | PCO | subsection 9.3 part of the experimental setup | |
| Conductive Epoxy | CircuitWorks | CW2400 | subsection 7.6. for wire attachement |
| Conical Centrifuge Tubes, 15 mL | Fisher Scientific | 14-959-70C | step 8.1.4. for cell centrifuging |
| Dektak 3ST Surface Profilometer | Veeco (Sloan/Dektak) | step 2.1.15 and 5.4 for surface profilometry | |
| Disposable biopsy punch, 0.75 mm | Robbins Instruments | RBP075 | step 6.2.3 for inlet access |
| Disposable biopsy punch, 3 mm | Robbins Instruments | RBP30P | step 6.2.3 for outlet access |
| DRAQ5 | abcam | ab108410 | step 11.2.2. for live cell staining |
| Dulbecco’s Modified Eagle’s Medium | ThermoFisher Scientific | 11885084 | step 8.1.2. part of media composition |
| E3631A Bipolar Triple DC power supply | Agilent | step 9.2.1.-9.2.2.part of the experimental setup | |
| Eclipse TE2000-U Inverted Microscope | Nikon | subsection 9.3. part of the experimental setup | |
| EVG620 UV Lithography System | EVG | step 2.1.9. and 2.2.7. for UV Exposure | |
| Fetal Bovine Serum | Neuromics | FBS001 | step 8.1.2. part of media composition |
| FS20 Ultrasonic Cleaner | Fisher Scientific | subsection 5.1. for photoresist lift-off | |
| Glass Media Bottle with Cap, 100mL | Fisher Scientific | FB800100 | step 8.2.1. for buffer storage |
| Glass Media Bottle with Cap, 500mL | Fisher Scientific | FB800500 | step 8.1.2.for media storage |
| HEK-293 cell line | ATCC | CRL-1573 | subsection 8.1 for cell culturing |
| HEPES buffer solution | Sigma Aldrich | 83264-100ML-F | step 8.2.1 part of electroporation buffer composition |
| Hexamethyldisilazane | Sigma Aldrich | 379212-25ML | step 2.2.3 adhesion promoter |
| HF2LI Lock-in Amplifier | Zurich Instruments | subsection 9.2 part of the experimental setup | |
| HF2TA Current amplifier | Zurich Instruments | subsection 9.2 part of the experimental setup | |
| Isopropyl Alcohol | Fisher Scientific | A459-1 | step 2.1.2 for cleaning, step 2.1.14 for rinsing wafer following SU-8 development, and step 6.3.1 for cleaning PDMS |
| IX81 fluorescence microscope | Olympus | step 11.2.3 for imaging | |
| L-Glutamine Solution | Sigma Aldrich | G7513-20ML | step 8.1.2. part of media composition |
| M16878/1BFA 22 gauge wire | AWC | B22-1 | subsection 7.5 for device fabrication |
| Magnesium chloride | Sigma Aldrich | 208337-100G | step 8.1.2 part of electroporation buffer composition |
| MF 319 Developer | Kayaku Advanced Materials | 10018042 | step 2.2.9. photoresist developer |
| Microposit S1818 photoresist | Kayaku Advanced Materials | 1136925 | step 2.2.4 positive photoresist for electrode patterning |
| Microscope slides, 75 x 25 mm | VWR | 16004-422 | step 2.2.1 electrode soda lime glass substrate |
| Model 2350 High voltage amplifier | TEGAM | 2350 | step 9.2.5. part of the experimental setup |
| National Instruments LabVIEW | National Instruments | data acquisition | |
| Needle, 30G x 1 in | BD Scientific | 305128 | step 10.1.1. part of the system priming |
| PA90 IC OPAMP Power circuit | Digi-key | 598-1330-ND | Part of the custom circuit |
| Penicillin-Streptomycin | Sigma Aldrich | P4458-20ML | step 8.1.2. part of media composition |
| Plasmid pMAX-GFP | Lonza | VCA-1003 | step 8.3.4. for intracellular delivery |
| Plastic tubing, 0.010'' x 0.030" | VWR | 89404-300 | step 10.1.2. for system priming |
| Platinum targets | Kurt J. Lesker | subsection 4.2. for physical vapor deposition | |
| Potassium chloride | Sigma Aldrich | P9333-500G | step 8.2.1. part of electroporation buffer composition |
| Pump 11 PicoPlus microfluidic syringe pump | Harvard Apparatus | MA1 70-2213 | step 10.1.4. for system priming |
| PVD75 Physical vapor deposition system | Kurt J. Lesker | subsection 4.1. for physical vapor deposition | |
| PWM32 Spinner System | Headway Research | steps 2.1.6 and 2.2.2. for substrate coating with photoresist | |
| PX-250 Plasma treatment system | March Instruments | subsection 7.2 for PDMS and glass substrate bonding | |
| SDG1025 Function/Waveform generator | Siglent | step 9.2.2. part of the experimental setup | |
| Sodium hydroxide | Sigma Aldrich | S8045-500G | step 8.2.1. part of electroporation buffer composition |
| SU-8 2010 negative photoresist | Kayaku Advanced Materials | Y111053 | step 2.1.7. for microfluidic channel patterning |
| SU-8 developer | Microchem | Y010200 | step 2.1.12. for photoresist developing |
| Sucrose | Sigma Aldrich | S7903-1KG | step 8.2.1. part of electroporation buffer composition |
| Sylgard 184 elastomer kit | Dow Corning | 3097358-1004 | step 6.2.1 10 : 1 mixture of PDMS polymer and hardening agent |
| Syringe, 1 ml | BD Scientific | 309628 | step 8.3.4. part of system priming |
| SZ61 Stereomicroscope System | Olympus | subsection 7.3. for channel and electrode alignment | |
| Tissue Culture Treated T25 Flasks | Falcon | 353108 | step 8.1.2 for cell culturing |
| Titanium targets | Kurt J. Lesker | subsection 4.2. for physical vapor deposition | |
| Transparency masks | CAD/ART Services | steps 2.1.9. and 2.2.7. for photolithography | |
| Trichloro(1H,1H,2H,2H-perfluorooctyl)silane | Sigma Aldrich | 448931-10G | step 6.1.2. for wafer silanization |
| Trypsin-EDTA solution | Sigma Aldrich | T4049-100ML | steps 8.1.3. and 8.3.1. for cell harvesting |
Referenzen
- Gao, Q. Q., et al. Therapeutic potential of CRISPR/Cas9 gene editing in engineered T-cell therapy. Cancer Medicine. 8 (9), 4254-4264 (2019).
- Aijaz, A., et al. Biomanufacturing for clinically advanced cell therapies. Nature Biomedical Engineering. 2 (6), 362-376 (2018).
- Milone, M. C., O'Doherty, U. Clinical use of lentiviral vectors. Leukemia. 32 (7), 1529-1541 (2018).
- Weaver, J. C., Chizmadzhev, Y. A. Theory of electroporation: A review. Bioelectrochemistry and Bioenergetics. 41 (2), 135-160 (1996).
- Kotnik, T., Rems, L., Tarek, M., Miklavcic, D. Membrane electroporation and electropermeabilization: mechanisms and models. Annual Review of Biophysics. 48, 63-91 (2019).
- Rosazza, C., Meglic, S. H., Zumbusch, A., Rols, M. P., Miklavcic, D. Gene electrotransfer: A mechanistic perspective. Current Gene Therapy. 16 (2), 98-129 (2016).
- Clauss, J., et al. Efficient non-viral T-cell engineering by sleeping beauty minicircles diminishing DNA toxicity and miRNAs silencing the endogenous T-cell receptors. Human Gene Therapy. 29 (5), 569-584 (2018).
- Sherba, J. J., et al. The effects of electroporation buffer composition on cell viability and electro-transfection efficiency. Scientific Reports. 10 (1), 3053 (2020).
- Lu, H., Schmidt, M. A., Jensen, K. F. A microfluidic electroporation device for cell lysis. Lab on a Chip. 5 (1), 23-29 (2005).
- Kar, S., et al. Single-cell electroporation: current trends, applications and future prospects. Journal of Micromechanics and Microengineering. 28 (12), (2018).
- Shi, J. F., et al. A review on electroporation-based intracellular delivery. Molecules. 23 (11), (2018).
- Wang, S. N., Zhang, X. L., Wang, W. X., Lee, L. J. Semicontinuous flow electroporation chip for high-throughput transfection on mammalian cells. Analytical Chemistry. 81 (11), 4414-4421 (2009).
- Wei, W. J., et al. An implantable microelectrode array for simultaneous L-glutamate and electrophysiological recordings in vivo. Microsystems & Nanoengineering. 1, (2015).
- Maschietto, M., Dal Maschio, M., Girardi, S., Vassanelli, S. In situ electroporation of mammalian cells through SiO2 thin film capacitive microelectrodes. Scientific Reports. 11 (1), (2021).
- Wu, Q. R., et al. Organ-on-a-chip: recent breakthroughs and future prospects. Biomedical Engineering Online. 19 (1), (2020).
- Pandey, C. M., et al. Microfluidics Based Point-of-Care Diagnostics. Biotechnology Journal. 13 (1), (2018).
- Vigneshvar, S., Sudhakumari, C. C., Senthilkumaran, B., Prakash, H. Recent advances in biosensor technology for potential applications - An overview. Frontiers in Bioengineering and Biotechnology. 4, (2016).
- Nuxoll, E. BioMEMS in drug delivery. Advanced Drug Delivery Reviews. 65 (11-12), 1611-1625 (2013).
- Kang, S., Kim, K. H., Kim, Y. C. A novel electroporation system for efficient molecular delivery into Chlamydomonas reinhardtii with a 3-dimensional microelectrode. Scientific Reports. 5, (2015).
- Zheng, M. D., Shan, J. W., Lin, H., Shreiber, D. I., Zahn, J. D. Hydrodynamically controlled cell rotation in an electroporation microchip to circumferentially deliver molecules into single cells. Microfluidics and Nanofluidics. 20 (1), (2016).
- Santra, T. S., Kar, S., Chang, H. Y., Tseng, F. G. Nano-localized single-cell nano-electroporation. Lab on a Chip. 20 (22), 4194-4204 (2020).
- Lee, W. G., Demirci, U., Khademhosseini, A. Microscale electroporation: challenges and perspectives for clinical applications. Integrative Biology. 1 (3), 242-251 (2009).
- Santra, T. S., Chang, H. Y., Wang, P. C., Tseng, F. G. Impact of pulse duration on localized single-cell nano-electroporation. Analyst. 139 (23), 6249-6258 (2014).
- Geng, T., Lu, C. Microfluidic electroporation for cellular analysis and delivery. Lab on a Chip. 13 (19), 3803-3821 (2013).
- Hsi, P., et al. Acoustophoretic rapid media exchange and continuous-flow electrotransfection of primary human T cells for applications in automated cellular therapy manufacturing. Lab on a Chip. 19 (18), 2978-2992 (2019).
- Khine, M., Ionescu-Zanetti, C., Blatz, A., Wang, L. P., Lee, L. P. Single-cell electroporation arrays with real-time monitoring and feedback control. Lab on a Chip. 7 (4), 457-462 (2007).
- Ye, Y. F., et al. Single-cell electroporation and real-time electrical monitoring on a microfluidic chip. 2020 33rd Ieee International Conference on Micro Electro Mechanical Systems (Mems 2020). , 1040-1043 (2020).
- Huang, Y., Rubinsky, B. Microfabricated electroporation chip for single cell membrane permeabilization. Sensors and Actuators a-Physical. 89 (3), 242-249 (2001).
- Guo, X. L., Zhu, R. Controllable in-situ cell electroporation with cell positioning and impedance monitoring using micro electrode array. Scientific Reports. 6, (2016).
- Punjiya, M., Nejad, H. R., Mathews, J., Levin, M., Sonkusale, S. A flow through device for simultaneous dielectrophoretic cell trapping and AC electroporation. Scientific Reports. 9, (2019).
- Wang, H. Y., Lu, C. Microfluidic electroporation for delivery of small molecules and genes into cells using a common DC power supply. Biotechnology and Bioengineering. 100 (3), 579-586 (2008).
- Zheng, M. D., et al. Continuous-flow, electrically-triggered, single cell-level electroporation. Technology. 5 (1), 31-41 (2017).
- Batista Napotnik, T., Miklavcic, D. In vitro electroporation detection methods - An overview. Bioelectrochemistry. 120, 166-182 (2018).
- MICROPOSIT™ S1800® G2 Series Photoresists. KAYAKU Available from: https://kayakuam.com/wp-content/uploads/2019/09/S1800-G2.pdf (2021)
- SU-8 2000 Permanent Negative Epoxy Photoresist. KAYAKU Available from: https://kayakuam.com/wp-content/uploads/2020/08/KAM-SU-8-2000-2000.5-2015-Datasheet-8.13.20-final.pdf (2001)
- Substrate Preparation. MicroChemicals Available from: https://www.microchemicals.com/technical_information/subtrate_cleaning_adhesion_photoresist.pdf (2021)
- Lisinenkova, M., Hahn, L., Schulz, J. . 4M 2006 - Second International Conference on Multi-Material Micro Manufacture. , 91-94 (2006).
- Beh, C. W., Zhou, W. Z., Wang, T. H. PDMS-glass bonding using grafted polymeric adhesive - alternative process flow for compatibility with patterned biological molecules. Lab on a Chip. 12 (20), 4120-4127 (2012).
- PA90 High Voltage Power Operational Amplifiers. APEX Available from: https://www.apexanalog.com/resources/products/pa90u.pdf (2021)
- Lissandrello, C. A., et al. High-throughput continuous-flow microfluidic electroporation of mRNA into primary human T cells for applications in cellular therapy manufacturing. Scientific Reports. 10 (1), 18045 (2020).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten