
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Herstellung von enantiopuren nicht-aktivierten Aziridinen und Synthese von Biemamid B, D und Epiallo-Isomuscarin
In diesem Artikel
Zusammenfassung
In dieser Studie stellen wir sowohl Enantiomere von Aziridin-2-carboxylat her, die bei der asymmetrischen Synthese von Alkaloiden, einschließlich Biemamid B und D, als auch (-)-Epiallo-Isomuscarin verwendet werden.
Zusammenfassung
Stickstoffhaltige heterocyclische Aziridine sind synthetisch sehr wertvoll für die Herstellung von azacyclischen und acyclischen Molekülen. Es ist jedoch sehr schwierig und mühsam, Aziridine in optisch reiner Form in großem Maßstab herzustellen, um eine asymmetrische Synthese von Azaverbindungen anzuwenden. Glücklicherweise gelang es uns, sowohl Enantiomere (2R)- als auch (2S)-aziridin-2-carboxylate mit der elektronenspendenden α-Methylbenzylgruppe am Ringstickstoff als nicht aktivierte Aziridine zu erreichen. Diese Startaziridine haben zwei verschiedene funktionelle Gruppen - hochreaktiver dreigliedriger Ring und vielseitiges Carboxylat. Sie sind in der Ringöffnung oder Ringtransformation mit Aziridin und in der funktionellen Gruppentransformation zu anderen aus Carboxylat anwendbar. Beide Enantiomere wurden bei der Herstellung von biologisch wichtigen aminoacyclischen und/oder aza-heterocyclischen Verbindungen in asymmetrischer Weise verwendet. Insbesondere beschreibt dieser Bericht die erste zweckmäßige asymmetrische Synthese beider Enantiomere der 5 ,6-Dihydrouracil-marinen Naturstoffe Biemamid B und D als potenzielle TGF-β-Inhibitoren. Diese Synthese bestand aus der Regio- und der stereoselektiven Ringöffnungsreaktion von Aziridin-2-carboxylat und der anschließenden Bildung von 4-Aminoteterahydropyrimidin-2,4-dion. Ein weiteres Beispiel in diesem Protokoll befasste sich mit einer hochstereoselektiven Mukaiyama-Reaktion von Aziridin-2-Carboxylat und Silylenolether nach intramolekularer Aziridinringöffnung, um einen einfachen und einfachen Zugang zu (-)-Epiallo-Isomuscarin zu ermöglichen.
Einleitung
Kleine Ringe, die aus Cyclopropanen, Oxiranen und Aziridinen bestehen, finden sich in verschiedenen Verbindungen wie Naturstoffen und Medikamenten 1,2. Sie werden vor allem als Ausgangsstoffe für die Verwertung ihrer Ringsorte verwendet. Unter den Drei-Ring-Verbindungen wurde Aziridin aufgrund seiner Instabilität und unkontrollierbaren Reaktivität weniger umfassend untersucht3. Wie in den elektrostatischen Potentialkarten gezeigt (Abbildung 1), macht eine Gruppe, die an den Aziridin-Ring-Stickstoff gebunden ist, ob elektronenspendend oder elektronenanziehend, die Basizität von Stickstoff unterschiedlich. Dieser Unterschied bildet einen markanten Kontrast zur Reaktivität und Selektivität der entsprechenden Aziridine.

Abbildung 1: Chemische Strukturen von "aktivierten" und "nicht aktivierten" Aziridinen und elektrostatische Potentialkarten ihrer repräsentativen Beispiele N-Methylaziridin und N-Acetylaziridin4. Diese Figur wurde mit Genehmigung von Ranjith et al.4 modifiziert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
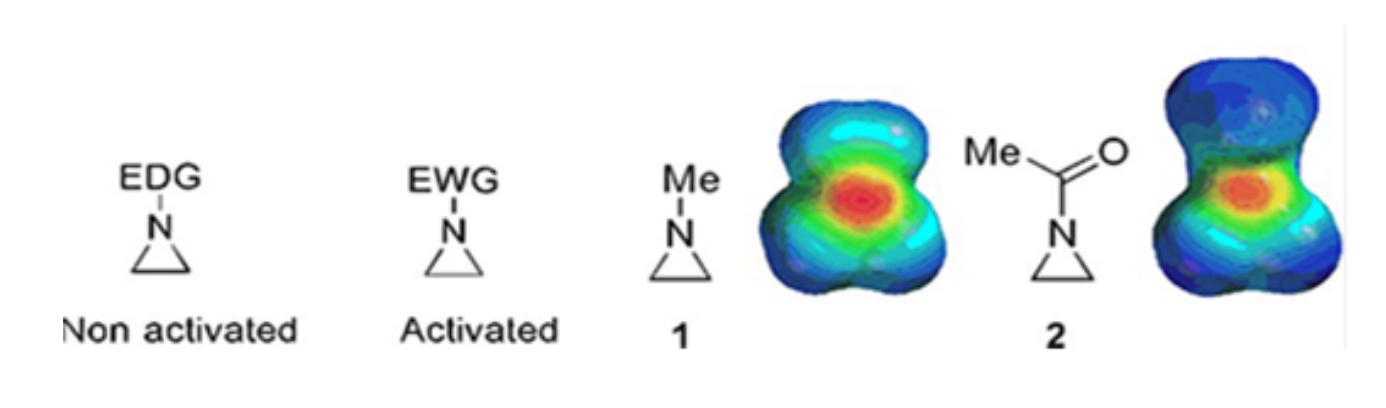{kind=link}
Wenn der Ringstickstoff eine elektronenentziehende Gruppe wie Sulfonat, Phosphonat und Carbamat hat, nennen wir ihn "aktiviertes" Aziridin. Dies ist bei Nucleophilen leicht reaktiv, um seine Instabilität mit einem begrenzten Umfang der Regiochemie auszugleichen. Diese aktivierten Aziridine werden durch verschiedene katalytische Methoden hergestellt und als Ausgangsmaterial verwendet. Ein Großteil der neueren Aziridinchemie hat sich mit diesen aktivierten Aziridinen befasst. Aktivierte Aziridine leiden jedoch unter bestimmten Einschränkungen, die sich aus ihrer Instabilität und dem begrenzten Reaktionsumfang der Ringöffnung ergeben. Auf der anderen Seite sind Aziridine, die elektronenspendende Substituenten wie Alkyl oder substituierte Alkylgruppen tragen, am Ring Stickstoff, der als "nicht aktiviert" bezeichnet wird4, unter den meisten Umständen relativ stabil und können lange Zeit ohne signifikante Zersetzung auf der Bank belassen werden. Die nukleophilen Ringöffnungsreaktionen von nicht aktiviertem Aziridin treten über die Bildung von Aziridinium-Ionen auf. Die meisten Reaktionen der Aziridin-Ringöffnung und Ringtransformationen verlaufen in einer stark regiochemischen Weise. Allerdings wird in nur sehr wenigen Literaturberichten die Herstellung von optisch reinen nicht aktivierten Aziriden mit Substituenten an den C2- oder C3-Positionen 5,6 diskutiert.
Diese Arbeit zeigt die erfolgreiche Herstellung von α-Methylbenzylgruppen-haltigen chiralen Aziridin-2-carboxylat-Derivaten, insbesondere (-)-Mentholyl(1 R)-phenylethylaziridin-2-carboxylates als diastereomeres Gemisch, aus der Reaktion von 2,3-Dibrompropionat und (1R)-Phenylethylamin. Aus diesem diastereomeren Gemisch wurden enantiopure (1 R)-phenylethyl-(2R)- und (2S)-aziridin-2-carboxylate als ihre (-)-Mentholylester in optisch reiner Form durch selektive Rekristallisation aus MeOH und n-Pentan auf Mehrhundert-Kilo-Skalen erhalten (Abbildung 1)7. Diese (-)-Mentholylester können durch Umesterung in Gegenwart von Magnesium oder Kaliumcarbonat7 leicht in ihre Ethyl- oder Methylester umgewandelt werden. Diese Verbindungen können auch leicht im Labormaßstab aus den Reaktionen von Alkyl-2,3-Dibrompropionaten oder der Vinyltriflate von α-Ketoester mit chiralem 2-Phenylethylamin hergestellt werden, gefolgt von einer Trennung des diastereomeren Gemisches mittels einfacher Blitzsäulenchromatographie 8.
Sobald wir enantioreines chirales Aziridin-2-carboxylat haben, können wir verschiedene zyklische und acyclische stickstoffhaltige biologisch wichtige Zielmoleküle synthetisieren, die auf funktionellen Gruppentransformationen von Carboxylat und hochregio- und stereoselektiven Aziridin-Ringöffnungsreaktionen basieren 6,9,10. Die erste zweckmäßige asymmetrische Synthese wurde sowohl für Enantiomere der 5,6-Dihydrouracil-marinen Naturstoffe Biemamid B und D als potentielle TGF-β-Inhibitoren11,12 angewendet. Zweitens wurde die diastereoselektive Synthese von β-(Aziridin-2-yl)-β-hydroxyketonen durch Mukaiyama-Aldol-Reaktion von optisch reinem 1-(1-phenylethyl)-aziridin-2-carboxaldehyd und verschiedenen Enolsilanen in Gegenwart von ZnCl2 in hoher Ausbeute (>82%) mit nahezu perfekter Stereoselektivität (98:2 dr) über einen chelatgesteuerten Übergangszustand erreicht. Diese wurden für die asymmetrische Synthese der Epiallo-Isomuscarin-Alkaloide13,14,15 verwendet.
Protokoll
1. Synthese der diastereomeren Mischung aus chiralem Aziridin(-)-Mentholylesterderivat (1)
- 2,3-Dibrompropan (-)-Mentholester 1a (5,0 g, 13,58 mM, 1,0 equiv) und eine magnetische Rührstange in einen ofengetrockneten 250 ml Zweihals-Rundbodenkolben unter Stickstoffatmosphäre (N2) geben.
- Wasserfreies Acetonitril (60 ml) mit einer luftdichten Spritze in den Reaktionskolben geben.
- Dann das Reaktionsgemisch mit einem Eisbad bei 0 °C abkühlen und das Reaktionsgemisch 5 min rühren.
- Kaliumcarbonat (5,6 g, 40,74 mM, 3,0 equiv) bei gleicher Temperatur in das Reaktionsgemisch geben und 30 min rühren lassen.
- (2R)-Phenylethylamin (2,0 ml, 16,29 nM, 1,2 equiv) tropfenweise bei Raumtemperatur (RT) zugeben und das Reaktionsgemisch 12 h lang rühren lassen.
- Überwachen Sie den Fortschritt der Reaktion mittels Dünnschichtchromatographie unter Verwendung von 9:1 v/v Hexan:Ethylacetat (EtOAc; Rf = 0,4) als Elutionsmittel.
- Nachdem die Reaktion abgeschlossen ist, filtrieren Sie die Mischung über Filterpapier (Porengröße 70 mm).
- Dann fügen Sie Wasser (30 ml) in das organische Filtrat hinzu und extrahieren Sie die organische Schicht mit Et 2 O (2x 50 ml) zweimal mit einem Trenntrichter.
- Die kombinierten organischen Extrakte werden über 7,5 g wasserfreiesNa2SO4 getrocknet und im Vakuum (<15 mbar) mit einem Rotationsverdampfer konzentriert.
HINWEIS: Nun wird eine rohe Mischung aus diastereomerem chiralem Aziridin, die beide Isomere von (R)-(1 R,2 S,5 R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridin-2-carboxylat und (S)-(1 R,2 S,5 R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridin-2-carboxylat (4,1 g,90%) erhalten wird. - Isolierung von chiralem Aziridin (R)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl 1-((R)-1-phenylethyl)aziridin-2-carboxylat (2) durch ein selektives Kristallisationsverfahren
- 8,7 g des Rohölgemisches chirales Aziridin (-)-Mentholester 1-Derivat zugeben und in 70 ml Methanol in einem ofengetrockneten 250 ml-Einhalskolben mit rundem Boden lösen.
- Nun das Reaktionsgemisch mit einem Heißwasserbad auf 70 °C erwärmen, dann das Reaktionsgemisch bei -10 °C abkühlen, bis sich fester Kristall bildet.
- Filtern Sie die feste Verbindung über einem Filterpapier (Porengröße 70 mm), um 2,2 g (R)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl 1-((R)-1-phenylethyl)aziridin-2-carboxylat(2)-ester zu erhalten.
- Die Filtratlösung wird mit einem Rotationsverdampfer erneut im Vakuum (<15 mbar) konzentriert, 50 ml Ethanol im verbleibenden Reaktionsgemisch gelöst und bei -10 °C zu 1,2 g (R)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl-1-((R)-1-phenylethyl)aziridin-2-carboxylat (2) umkristallisiert.
- Verwenden Sie zu diesem Zeitpunkt das andere Alkoholethanol auf die gleiche Weise wie Methanol.
- Nach der Rekristallisation erneut über ein Filterpapier (Porengröße 70 mm) filtrieren, die restlichen rohen 5,3 g Filtratlösung mit einem Rotationsverdampfer vollständig im Vakuum (<15 mbar) konzentrieren und 50 ml eines Pentankohlenwasserstofflösungsmittels hinzufügen.
- Halten Sie die verbleibende Reaktionslösung bei -15 °C.
HINWEIS: Nun wird eine feste Verbindung von fast 1,9 g (S)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridin-2-carboxylatester(3) erhalten. - Nachdem Sie Kristalle erhalten haben, konzentrieren Sie die Lösung erneut im Vakuum (<15 mbar) mit einem Rotationsverdampfer und lösen Sie sie in 30 ml Pentankohlenwasserstofflösungsmittel auf.
- Bei -15 °C erneut rekristallisieren, erhält man 0,8 g (S)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridin-2-carboxylate ester (2').
- Gewinnung von (R)-ethyl-1-(R)-1-phenylethyl)aziridin-2-carboxylat (3)
- (R)-(1R,2 S,5 R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridin-2-carboxylat (2) (0,167 g, 0,57 mM) und einen magnetischen Rührstab in einen ofengetrockneten 25-ml-Zweihalskolben mit rundem Boden unter Stickstoff (N2)-Atmosphäre geben.
- Mit einer luftdichten Spritze 1,8 ml Ethanol in den Reaktionskolben geben und bei RT umrühren.
- Dann fügen Sie Kaliumcarbonat (0,40 g, 20,28 mmol, 4,0 equiv) hinzu und rühren Sie es bei RT für 2 Tage um.
- Überwachen Sie den Fortschritt der Reaktion mittels Dünnschichtchromatographie (Eluent, 8:2 v/v, Hexan:Ethylacetat (EtOAc), Rf = 0,6).
- Nach Abschluss der Reaktion die Mischung über Filterpapier (Porengröße 70 mm) filtrieren, dann Wasser (5 ml) in das organische Filtrat geben und die organische Schicht mit CH 2 Cl 2 (2x 15 ml) zweimal mit einem Trenntrichter extrahieren.
- Trocknen Sie die kombinierten organischen Extrakte über 3,0 g wasserfreiesNa2 SO4 und konzentrieren Sie sie im Vakuum (<15 mbar) mit einem Rotationsverdampfer.
- Reinigen Sie das Rohprodukt durch normale Phasensäulenchromatographie auf Kieselgel (70-230 mesh), um ein reines Produkt (R)-ethyl 1-((R)-1-phenylethyl)aziridin-2-carboxylat (3) (950 mg, 88%) zu erhalten. Rf (30% EtOAc/Hexan = 0,50).
2. Regio und stereoselektive Aziridinringöffnung durch Azidnucleophil zur Totalsynthese von Biemamid B und Biemamid D
- Synthese von (R)-ethyl-2-azido-3-(((R)-1-phenylethyl)amino)propanoat (5)
- Transfer (500 mg, 2,20 mM, 1,0 Äquiv) von chiralem (S)-ethyl 1-((R)-1-phenylethyl)aziridin-2-carboxylat (4) und einem magnetischen Rührstab in einen ofengetrockneten 50 ml-Zweihalskolben mit rundem Boden unter offener Atmosphäre.
- 50% wässriges Ethanol (15 ml) in das Reaktionsgemisch geben.
- Das Reaktionsgemisch bei 0 °C abkühlen lassen und konzentrierte Schwefelsäure (36 N) tropfenweise hinzufügen, um einen pH-Wert von nahezu 4,0 zu halten, und 5 min umrühren.
- Natriumazid (370 mg, 5,70 mM, 2,5 equiv) bei 0 °C zugeben und das Reaktionsgemisch 10 min bei gleicher Temperatur rühren und dann bei RT erwärmen lassen.
- Dann AlCl 3∙6H2O (55 mg, 0,22 mM, 0,1 equiv) als Katalysator bei derselben RT zugeben und weitere3 h rühren lassen.
- Überwachen Sie den Fortschritt der Reaktion mittels Dünnschichtchromatographie (Eluent, 6:4 v/v, Hexan:Ethylacetat (EtOAc), Rf = 0,2).
- Das Reaktionsgemisch mit zwei Portionen 20 mL gesättigtemNaHCO 3 abschrecken.
- Anschließend die Rohmischung über einem Celitpad mit Ethanol (2 x 10 ml) filtrieren.
- Konzentrieren Sie das Reaktionsgemisch im Vakuum (<15 mbar) mit einem Rotationsverdampfer.
- Die organische Schicht mit CH 2 Cl 2 (2x 50 ml) zweimal mit einem Trenntrichter extrahieren.
- Dann trocknen Sie die kombinierte organische Schicht über 5,0 g wasserfreiesNa2 SO4 für 5 min.
- Konzentrieren Sie die rohe organische Schicht im Vakuum (<15 mbar) mit einem Rotationsverdampfer, um ein Rohazidprodukt zu erhalten.
- Reinigen Sie das Rohprodukt mit normaler Phasensäulenchromatographie auf Kieselgel (70-230 mesh), indem Sie mit 40% EtOAc/Hexan (R f = 0,20) eluieren, um 490 mg (90% Ausbeute) (R)-ethyl 2-azido-3-(((R)-1-phenylethyl)amino)propanoat (5) als viskose Flüssigkeit zu erhalten.
- Synthese von (9H-Fluoren-9-yl)methyl(3-((R)-3-methyl-2,4-dioxo-1-((R)-1-phenylethyl)hexahydropyrimidin-5-yl)amino)-3-oxopropyl)carbamat (7)
- 150 mg chirales (R)-5-amino-3-methyl-1-((R)-1-phenylethyl)-dihydropyrimidin-2,4(1H,3H)-dion (6) (150 mg, 0,60 mM, 1,0 equiv) und eine magnetische Rührstange in einen ofengetrockneten 25-ml-Zweihalskolben mit rundem Boden unterN2-Atmosphäre.
- Trockenes CH 2Cl2 (15,0 ml) mit einer luftdichten Spritze in den Reaktionskolben geben.
- Dann das Reaktionsgemisch mit einem Eisbad bei 0 °C abkühlen und das Reaktionsgemisch 5 min rühren.
- Fmoc-beta-Alanin (377 mg, 1,20 mM, 2,0 equiv) und DIPEA (0,67 ml, 3,64 mM, 6,0 equiv) bei 0 °C zugeben und 5 min umrühren.
- EDCI (347 mg, 1,82 mM, 3,0 equiv) und HOBt (165 mg, 1,21 mM, 2,0 equiv) in das Reaktionsgemisch bei 0 °C geben und 10 min bei gleicher Temperatur rühren lassen.
- Halten Sie das Reaktionsgemisch bei RT und lassen Sie es weitere 8 h rühren.
- Überwachen Sie den Fortschritt der Reaktion mittels Dünnschichtchromatographie mit (2:8 v/v, Hexan:Ethylacetat (EtOAc), Rf = 0,4) als Elutionsmittel.
- Das Reaktionsgemisch mit Wasser (10 ml) abschrecken.
- Waschen Sie die kombinierte organische Schicht mit Sole (15 ml) und extrahieren Sie dann die organische Schicht mit CH 2 Cl2 (2 x 20 ml) zweimal mit einem Trenntrichter.
- Dann trocknen Sie die kombinierte organische Schicht über 5,0 g wasserfreiesNa2 SO4 für 5 min und konzentrieren Sie sie im Vakuum (<15 mbar) mit einem Rotationsverdampfer.
- Reinigen Sie das Rohprodukt mit normaler Phasensäulenchromatographie auf Kieselgel (70-230 mesh), indem Sie mit 80% EtOAc/Hexan (Rf = 0,40) eluieren, um 295 mg (7) (90% Ausbeute) zu erhalten.
3. Stereoselektive Mukaiyama-Aldol-Reaktion mit chiralem Aziridin-2-carboxaldehyd und dessen Regio und stereoselektiver Aziridin-Ringöffnung durch internes Hydroxynucleophil zur Totalsynthese von (-)-Epiallo-Somuscin (17)
- Synthese von (S)-4-hydroxy-4-((R)-1-((R)-1-phenylethyl)aziridin-2-yl)butan-2-on (12)
- (R)-1-((R)-1-phenylethyl)aziridin-2-carbaldehyd(10) (140 mg, 0,8 mM, 1,0 equiv) und einen magnetischen Rührstab in einen ofengetrockneten 25 ml Zweihals-Rundbodenkolben unterN2-Atmosphäre.
- Trockener CH3CN (4,0 ml) mit einer luftdichten Spritze in den Reaktionskolben geben.
- Dann das Reaktionsgemisch mit einem Eis-Aceton-Bad bei -20 °C abkühlen und das Reaktionsgemisch 5 min rühren.
- ZnCl2 wasserfrei (108 mg, 0,8 mM, 1,0 equiv) in das Reaktionsgemisch bei -20 °C geben und 5 min rühren lassen.
- Dann wird Trimethyl(prop-1-en-2-yloxy)silan (11) (104 mg, 0,8 mM, 1,0 equiv), gelöst in trockenem CH 3 CN (3,0ml), zu dem Reaktionsgemisch bei -20 °C tropfenweise gegeben und das Reaktionsgemisch 1 h bei gleicher Temperatur rühren lassen.
- Überwachen Sie den Fortschritt der Reaktion mittels Dünnschichtchromatographie mit 8:2 v/v Hexan:Ethylacetat (EtOAc), Rf = 0,2) als Elutionsmittel.
- Das Reaktionsgemisch mit gesättigtemNaHCO 3 (4 ml) abschrecken.
- Extrahieren Sie die organische Schicht mit EtOAc (2 x 15 ml) zweimal mit einem Trenntrichter.
- Trocknen Sie die kombinierte organische Schicht mit 3,0 g wasserfreiemNa2 SO4 und konzentrieren Sie sie im Vakuum (<15 mbar) mit einem Rotationsverdampfer.
- Reinigen Sie das Rohprodukt mit normaler Phasensäulenchromatographie auf Kieselgel (70-230 mesh), indem Sie mit 80% EtOAc/Hexan (R f = 0,20) eluieren, um 58 mg (S)-4-hydroxy-4-((R)-1-(R)-1-phenylethyl)aziridin-2-yl)butan-2-on) (12) (85% Ausbeute) zu erhalten.
- Synthese von (R)-N-((2 R,3S,5 R)-3-((tert-butyldimethylsilyl)oxy)-5-methyltetrahydrofuran-2-yl)methyl)-1-phenylethanamin(15)
- (S)-4-((tert-butyldimethylsilyl)oxy)-4-((R)-1-(R)-1-phenylethyl)-aziridin-2-yl)butan-2-on (13) (400 mg, 1,15 mM, 1,0 equiv) und eine magnetische Rührstange in einen ofengetrockneten 25 ml Zweihals-Rundbodenkolben unterN2-Atmosphäre.
- Wasserfreies THF (50 ml) mit einer luftdichten Spritze in den Reaktionskolben geben.
- Das Reaktionsgemisch mit einem Trockeneis-Aceton-Bad bei -78 °C abkühlen lassen und 5 min rühren lassen.
- Dann wird Lithium-tri-sec-butylborohydrid (L-Selectrid) (1 M Lösung in THF) (2,3 ml, 2,0 equiv) tropfenweise in das Reaktionsgemisch bei -78 °C gegeben und weitere 25 min rühren lassen.
- Die Reaktionsmischung auf RT erwärmen und 8 h rühren lassen.
- Überwachen Sie den Fortschritt der Reaktion mittels TLC unter Verwendung von 30% EtOAc/Hexan (Rf = 0,40) als Elutionsmittel.
- Nach dem vollständigen Verbrauch von Verbindung 13 wird das Reaktionsgemisch mit 0,1 M NaOH (5 ml) abgeschreckt.
- Die organische Schicht mit EtOAc (3 x 15 ml) extrahieren und dann mit Salzlake (15 ml) waschen.
- Die organische Schicht über 3,0 g wasserfreiesNa2 SO4 trocknen und im Vakuum (<15 mbar) mit einem Rotationsverdampfer konzentrieren.
- Das Rohprodukt wird mitnormaler Phasensäulenchromatographie auf Kieselgel (70-230 mesh, Eluent, 8:2 v/v, Hexan:Ethylacetat (EtOAc), R f = 0,2) gereinigt, um eine reine Verbindung (15) (382 mg, 95% Ausbeute) zu erhalten.
- Synthese von (-)-Epiallo-Isomuscariniodid (17)
- Transferverbindung 16 (20 mg, 0,15 mM, 1,0 equiv) und eine magnetische Rührstange in einen ofengetrockneten 10 ml Rundkolben unterN2-Atmosphäre .
- Fügen Sie 3 ml EtOAc-Reaktionskolben mit einer luftdichten Spritze hinzu.
- Anschließend wird dem Reaktionsgemisch bei RT Methyliodid (0,4 mL, 3,0 mM) zugegeben.
- 1,2,2,6,6-Pentamethylpiperidin (PMP) (0,05 mL, 0,3 mM, 2,0 Equiv) in das Reaktionsgemisch bei 0 °C geben.
- Halten Sie dann das Reaktionsgemisch bei RT und lassen Sie es für weitere 16 h rühren.
- Verdampfen Sie das Lösungsmittel im Vakuum (<15 mbar) mit einem Rotationsverdampfer, um das Rohprodukt zu erhalten.
- Dreimal waschen, indem (3 x 5 ml) 10% MeOH in EtOAc in die rohe Reaktionsmischung gegeben werden.
- Dann die Rohmischung mit n-Pentan (5 ml) waschen und unter Vakuum (<15 mbar) mit einem Rotationsverdampfer konzentrieren, um reines (-)-Epiallo-Isomuscariniodid (17) (32 mg, 68%) zu erhalten.
4. Charakterisierung aller Produkte
- Charakterisieren Sie alle neuen Verbindungen durch 1H, 13 C NMR-Spektroskopie und hochauflösende Massenspektrometrie (HRMS) 7,8,11.
Ergebnisse
Hier berichten wir über die Synthese von enantiopuren Aziridin-2-carboxylaten. Die diastereomeren Gemische aus (R)-(1 R,2 S,5 R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridin-2-carboxylat(2) und (S)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridin-2-carboxylat (3) (4,1 g,90 %) wurden in quantitativer Ausbeute aus 2,3-Dibrompropan(-)-mentholylester und (1 R)-pheny...
Diskussion
Aziridine als stickstoffhaltige dreigliedrige Heterocyclen haben ein enormes Potenzial für synthetische Ausgangsmartiale oder Zwischenprodukte, um stickstoffreiche organische Moleküle herzustellen. Basierend auf der Gruppe, die am Ring Stickstoff lagert, werden sie als "aktivierte" und "nicht aktivierte" Aziridine klassifiziert, deren chemische Reaktivität und Selektivität unterschiedlich sind. Es stehen jedoch nur sehr begrenzte Methoden zur Verfügung, um dieses wertvolle Aziridin in optisch aktiver Form herzustell...
Offenlegungen
Die Autoren erklären, dass an dieser Studie kein Interessenkonflikt bestand.
Danksagungen
Diese Forschung wurde von der National Research Foundation of Korea (NRF-2020R1A2C1007102 und 2021R1A5A6002803) mit dem Center for New Directions in Organic Synthesis und einem HUFS Grant 2022 unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| (2R)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester, 98% | Sigma-Aldrich | 57054-0 | |
| (2S)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester | Sigma-Aldrich | 57051-6 | |
| 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride | TCI | 424331-25 g | CAS No: 25952-53-8 |
| 1,4-Dioxane | SAMCHUN | D0654-1 kg | CAS No: 123-91-1 |
| 1-Hydroxybenzotriazole hydrate | Aldrich | 219-989-7-50 g | CAS No: 123333-53-9 |
| 2,6-Lutidine | Alfa Aesar | A10478-AP, 500 mL | CAS No: 108-48-5 |
| Acetonitrile | SAMCHUN | A0127-18 L | CAS No: 75-05-8 |
| Acetonitrile-d3 | Cambridge Isotope Laboratories, | 15G-744-25 g | CAS No: 2206-26-0 |
| Aluminum chloride hexahydrate | Aldrich | 231-208-1, 500 g | CAS No : 7784-13-6 |
| Bruker AVANCE III HD (400 MHz) spectrometer | Bruker | NA | |
| Chloroform-d | Cambridge Isotope Laboratories, | 100 g | CAS No: 865-49-6 |
| Dichloromethane | SAMCHUN | M0822-18 L | CAS No: 75-09-2 |
| Dimethyl sulfoxide-d6 | Cambridge Isotope Laboratories, | 25 g | CAS No: 2206-27-1 |
| Ethanol | EMSURE | 1009831000,1L | CAS No: 64-17-5 |
| Ethyl acetate | SAMCHUN | E0191-18 L | CAS No: 141-78-6 |
| High resolution mass spectra/MALDI-TOF/TOF Mass Spectrometry | AB SCIEX | 4800 Plus | High resolution mass spectra |
| JASCO P-2000 | JASCO | P-2000 | For optical rotation |
| Lithium aluminum hydride | TCI | L0203-100 g | CAS No: 16853-85-3 |
| L-Selectride, 1 M solution in THF | Acros | 176451000, 100 mL | CAS No: 38721-52-7 |
| Methanol | SAMCHUN | M0585-18 L | CAS No: 67-56-1 |
| N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-β-alanine | TCI | F08825G-5 g | CAS No: 35737-10-1 |
| N-Ethyldiisopropylamine | Aldrich | 230-392-0, 100 mL | CAS No: 7087-68-5 |
| n-Hexane | SAMCHUN | H0114-18 L | CAS No: 110-54-3 |
| Ninhydrin | Alfa Aesar | A10409-250 g | CAS No: 485-47-2 |
| p-Anisaldehyde | aldrich | A88107-5 g | CAS No: 123-11-5 |
| Phosphomolybdic acid hydrate | TCI | P1910-100 g | CAS No: 51429-74-4 |
| Sodium azide | D.S.P | 703301-500 g | CAS No: 26628-22-8 |
| Sodium Hydride 60% dispersion in mineral oil | Sigma-Aldrich | 452912-100 G | CAS No: 7646-69-7 |
| Sodium hydroxide | DUKSAN | A31226-1 kg | CAS No: 1310-73-2 |
| Sodium sulfate | SAMCHUN | S1011-1 kg | CAS No: 7757-82-6 |
| Thin Layer Chromatography (TLC) | Merck | 100390 | |
| Tert-Butyldimethylsilyl trifluoromethanesulfonate, 98% | Aldrich | 274-102-0, 25 g | CAS NO: 69739-34-0 |
| Tetrahydrofuran | SAMCHUN | T0148-18 L | CAS No: 109-99-9 |
| Triethylethylamine | DAEJUNG | 8556-4400-1 L | CAS No: 121-44-8 |
| UV light | Korea Ace Sci | TN-4C | 254 nm |
| Zinc chloride, anhydrous, 98+% | Alfa Aesar | A16281-22100 g | CAS No : 7646-85-7 |
Referenzen
- Pitzer, K. S. Strain energies of cyclic hydrocarbons. Science. 101 (2635), 672 (1945).
- Dudev, T., Lim, C. Ring strain energies from ab initio calculations. Journal of the American Chemical Society. 120 (18), 4450-4458 (1998).
- D'hooghe, M., Ha, H. -. J. . Synthesis of 4- to 7-Membered Heterocycles by Ring Expansion. , (2016).
- Ranjith, J., Ha, H. -. J. Synthetic applications of aziridinium ions. Molecules. 26 (6), 1744 (2021).
- Sweeney, J. B. Aziridines: epoxides' ugly cousins. Chemical Society Reviews. 31 (5), 247-258 (2002).
- Stankovic, S., et al. Regioselectivity in the ring opening of non-activated aziridines. Chemical Society Reviews. 41 (2), 643-665 (2012).
- Lee, W. K., Ha, H. -. J. Highlights of the chemistry of enantiomerically pure aziridine-2-carboxylates. Aldrichimica Acta. 36 (2), 57-63 (2003).
- Tranchant, M. J., Dalla, V., Jabin, I., Decroix, B. Reaction of vinyl triflates of α-keto esters with primary amines: efficient synthesis of aziridine carboxylates. Tetrahedron. 58 (42), 8425-8432 (2002).
- Ha, H. -. J., Jung, J. -. H., Lee, W. K. Application of regio- and stereoselective functional group transformation of chiral aziridine-2-carboxylate. Asian Journal of Organic Chemistry. 3 (10), 1020-1035 (2014).
- Kim, Y., et al. Preparation of 2,3-diaminopropionate from ring opening of aziridine-2-carboxylate. Tetrahedron Letters. 46 (25), 4407-4409 (2005).
- Srivastava, N., Macha, L., Ha, H. -. J. Total synthesis and stereochemical revision of biemamides B and D. Organic Letters. 21 (22), 8992-8996 (2019).
- Zhang, F., et al. Biemamides A-E, inhibitors of the TGF-β pathway that block the epithelial to mesenchymal transition. Organic Letters. 20 (18), 5529-5532 (2018).
- Srivastava, N., Ha, H. -. J. Highly efficient and stereoselective Mukaiyama Aldol reaction with chiral aziridine-2-carboxaldehyde and its synthetic applications. Asian Journal of Organic Chemistry. 11 (1), 2021005671 (2021).
- Kempter, I., et al. Synthesis and structural characterization of the isomuscarines. Tetrahedron. 70 (10), 1918-1927 (2014).
- Pirrrung, M. C., DeAmicis, C. V. Total synthesis of the muscarines. Tetrahedron Letters. 29 (2), 159-162 (1988).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten