
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Linienformanalyse dynamischer NMR-Spektren zur Charakterisierung von Koordinationskugelumlagerungen an einem chiralen Rheniumpolyhydridkomplex
In diesem Artikel
Zusammenfassung
Die Linienformanalyse von NMR-Spektren, die über einen Temperaturbereich gesammelt wurden, dient als Leitfaden für die Umlagerung von Atomen der inneren Koordinationskugel an einem chiralen Rhenium(V)-Polyhydridkomplex mit acht Koordinaten,ReH5(PPh3)2(sec-Butylamin). Die Linienformanalyse wird auch verwendet, um die Aktivierungsparameter ΔH‡, ΔS‡ und ΔG‡ für diese Atomumlagerungen zu bestimmen.
Zusammenfassung
Die Dynamische Lösung Kernspinresonanz (NMR) Spektroskopie ist die typische Methode zur Charakterisierung der dynamischen Umlagerungen von Atomen innerhalb der Koordinationssphäre für Übergangsmetallpolyhydridkomplexe. Die Anpassung der Linienform der dynamischen NMR-Spektren kann zu Schätzungen der Aktivierungsparameter der dynamischen Umlagerungsprozesse führen. Eine Kombination aus dynamischer 31 P-{1 H} NMR-Spektroskopie metallgebundener Phosphoratome mit dynamischer 1H-{31P} NMR-Spektroskopie von Hydridliganden kann Hydridligandenumlagerungen identifizieren, die in Verbindung mit einer Phosphoratomumlagerung auftreten. Für Moleküle, die ein solches gekoppeltes Paar von Umlagerungen aufweisen, kann die dynamische NMR-Spektroskopie verwendet werden, um theoretische Modelle für die Ligandenumlagerungen zu testen. Dynamische 1H-{31P} NMR-Spektroskopie und Linienformanpassung können auch das Vorhandensein eines Austauschprozesses identifizieren, der einen bestimmten Hydridliganden über die innere Koordinationssphäre des Metalls hinaus durch einen Protonenaustausch mit einem Lösungsmittelmolekül wie zufälligem Wasser bewegt. Die Herstellung einer neuen Verbindung,ReH5(PPh 3)2(sec-Butylamin), die mehrere dynamische Umlagerungsprozesse veranschaulicht, wird zusammen mit der Linienformanpassung dynamischer NMR-Spektren des Komplexes vorgestellt. Die Ergebnisse der Linienanpassung können durch die Eyring-Gleichung analysiert werden, um die Aktivierungsparameter für die identifizierten dynamischen Prozesse abzuschätzen.
Einleitung
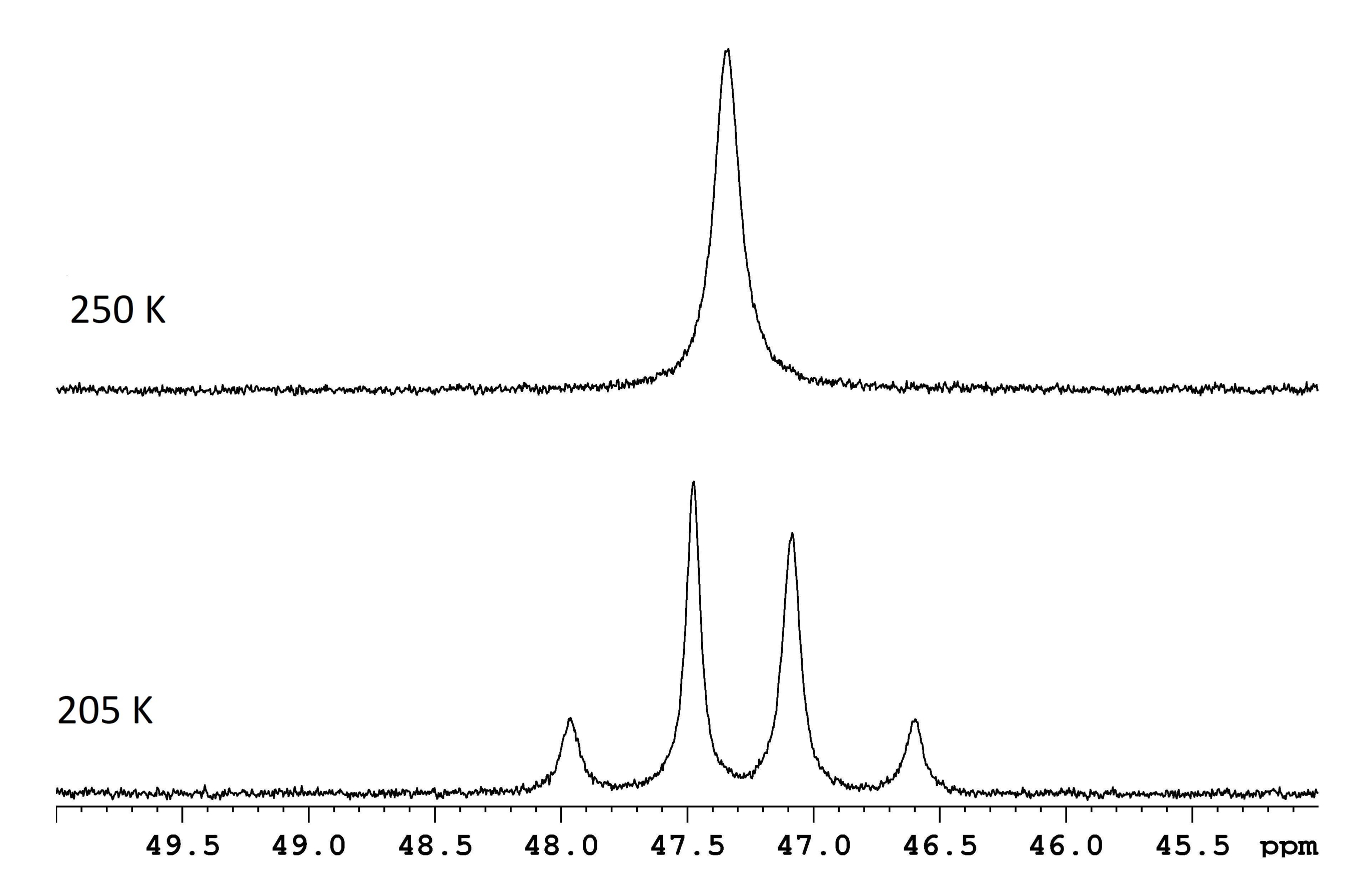
NMR-Spektroskopie wird häufig verwendet, um dynamische Prozesse zu charakterisieren, die innerhalb oder zwischen Molekülen ablaufen. Für viele einfache intramolekulare Umlagerungen ist die Schätzung von ΔG‡ so einfach wie die Messung der Frequenzdifferenz Δν zwischen zwei Resonanzen an der Grenze des langsamen Austauschs und die Bestimmung der Koaleszenztemperatur für dieselben Resonanzen (Abbildung 1)1. Die Beziehung,
ΔG‡ = 4,575 x 10-3 kcal/mol x T c [9,972 + log (Tc/Δν)]
wobei Tc die Koaleszenztemperatur für ein Paar von Resonanzen ist, die die langsame Austauschform einer dynamischen Probe darstellen, kann verwendet werden, um die freie Aktivierungsenergie für eine solche dynamische Umlagerung zu lösen. Komplexere dynamische Systeme erfordern die Anpassung der Linienform dynamischer NMR-Spektren oder eine andere NMR-Technik wie die zweidimensionale Austauschspektroskopie (2D-EXSY) oder die zweidimensionale Overhauser-Effektspektroskopie (2D-ROESY), um Aktivierungsparameter abzuschätzen.

Abbildung 1: NMR-Spektren für eined-8-Toluol-Lösung vonReH5(PPh3)2(sec-Butylamin) bei zwei Temperaturen. Die Frequenzdifferenz zwischen den beiden langsamen Austauschdubletten (untere Spur, 117,8 Hz) und eine Koaleszenztemperatur von 250 K (obere Spur) entsprechen einer Energiebarriere (ΔG‡) von 11,8 kcal/mol. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Die Linienformanpassung dynamischer NMR-Spektren ist eine gängige Technik, die seit langem zur Abschätzung von Aktivierungsparametern verwendet wird, die dynamische Umlagerungen für Substanzen mit einer Aktivierungsenergie von etwa 5 bis 25 kcal/mol 2,3,4,5 beschreiben. Die Bestimmung der Energiebarrieren für den Protonenaustausch zwischen Wasser und Aminmolekülen6, die Energiebarriere gegen die Rotation um die C-N-Bindung in Dimethylformamid7 oder die allgemeine Größe organischer Einheiten8 sind nur einige Beispiele für die vielen Eigenschaften, die durch Linienformanpassung dynamischer NMR-Spektren bewertet wurden. Dieses Manuskript demonstriert die Verwendung der Linienformanpassung zur Charakterisierung der intermolekularen und intramolekularen dynamischen Prozesse, die für den KomplexReH5(PPh 3)2(sec-Butylamin) ablaufen. Die Ziele dieser und ähnlicher NMR-Experimente sind: 1) Charakterisierung aller NMR-beobachtbaren intramolekularen dynamischen Atomaustauschprozesse, falls vorhanden, 2) Identifizierung und Charakterisierung von NMR-beobachtbaren intramolekularen dynamischen Atomaustauschprozessen, falls vorhanden, 3) Identifizierung korrelierter intramolekularer Atomaustausch, der in diesem Beispiel sowohl für Wasserstoff- als auch für Phosphoratome auftritt, und 4) für das hier vorgestellte Beispiel, Vergleichen Sie zwei veröffentlichte Modelle für die dynamischen Prozesse, die im KomplexReH5(PPh3)2(sec-Butylamin) ablaufen.
Acht-Koordinaten-Rhenium(V)-Polyhydridsysteme sind komplexe dynamische Systeme, in denen die Liganden an mehreren dynamischen Prozessen teilnehmen und die Phosphoratome an einem einzigen dynamischen Prozess teilnehmen können, der ein zweiter Aspekt eines Hydridligandenaustauschprozesses ist 9,10,11,12,13,14,15,16,17,18 ,19,20,21,22,23,24,25,26,
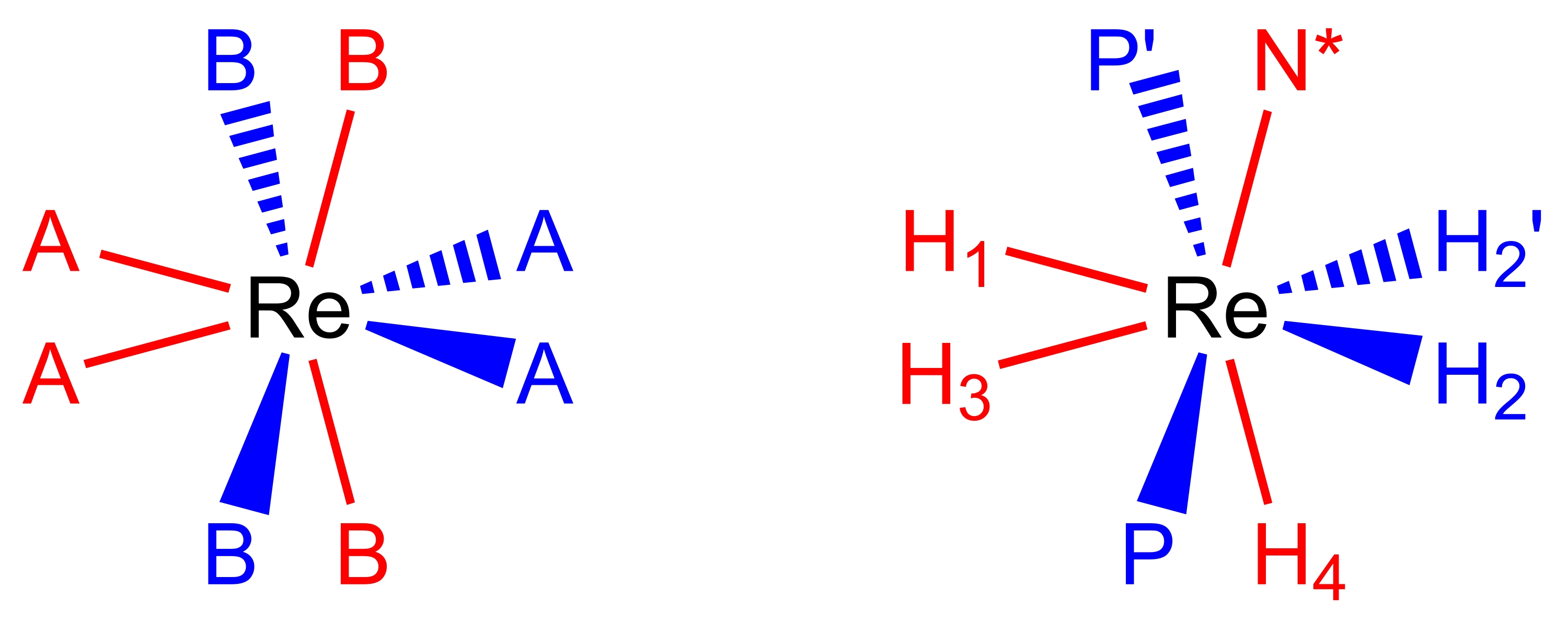
27,28,29. Achtkoordinaten, pseudododekaedrische Rhenium(V)-Polyhydridkomplexe nehmen eine molekulare Geometrie an (Abbildung 2), die als Paar orthogonaler Trapeze der Liganden17,26 beschrieben werden kann. Die Eckpunkte an den langen Kanten der Trapeze werden üblicherweise als B-Stellen markiert und sind in Rheniumpolyhydridkomplexen normalerweise die Stellen, die von neutralen Zwei-Elektronen-Donorliganden wie tertiären Phosphinen oder Aminliganden besetzt sind. Die Eckpunkte an den kurzen Rändern der Trapeze werden üblicherweise als A-Stellen markiert und sind typischerweise von anionischen Zwei-Elektronen-Donor-Hydrid-Liganden besetzt. Die Raumtemperatur-NMR-Spektren von Rhenium(V)-Polyhydridkomplexen sind aufgrund der verschiedenen dynamischen Prozesse, die in Raumtemperaturlösungen ablaufen, typischerweise täuschend einfach.

Abbildung 2: Ein dodekaedrischer Koordinationssatz (links) und der KomplexReH5(PPh3)2(sec-Butylamin) aus der gleichen Perspektive (rechts). Die rot gefärbten Stellen stellen Koordinationsstellen dar, die ein vertikales Trapez bilden, und die blau gefärbten Stellen stellen Koordinationsstellen dar, die ein horizontales Trapez bilden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Komplexe der FormReH5(PPh3)2(amin) sind die am gründlichsten untersuchte Klasse von Rheniumpolyhydridkomplexen in Bezug auf dynamische Prozesse 9,10,12,13,16,30,31. Drei dynamische Prozesse (Abbildung 3) wurden fürReH5(PPh3)2(amin)-Komplexe identifiziert: 1) ein Protonenaustausch zwischen dem einzigen B-Site-Hydrid-Liganden und einem Proton aus einem Wassermolekül (zufällig oder beabsichtigt)9,13, 2) ein Drehkreuzaustausch eines Paares von A-Sitehydrid-Liganden mit einem benachbarten B-Sitehydrid-Liganden 9, 11,13,30,31 und 3) eine sterische Inversion (oder Pseudorotation), die sich als paarweiser Austausch der A-Site-Hydridliganden und eine paarweise Bewegung der B-Site-Atome zur gegenüberliegenden Seite des Rheniumzentrums manifestiert (wie in Abbildung 4 dargestellt)4,5,6,8,26,27 . Die Bewegung von B-Site-Atomen zur gegenüberliegenden Seite von Rhenium ist durch dynamische NMR-Spektroskopie wie folgt beobachtbar: 1) ein Prozess, der die inäquivalenten 3 und 5 Protonen von N = Pyridinäquivalent bei Raumtemperatur10,30,31 erzeugt, 2) ein Prozess, der bewirkt, dass die E- und Z-Isomere von N = unsymmetrisch substituierte aromatische Aminliganden bei Raumtemperatur schnell ausgetauschtwerden 9, 10,13,30,31 oder 3) ein Prozess, der einen schnellen Austausch der sterischen Perspektiven eines diastereotopen Paares von Phosphoratomen in Bezug auf ein chirales Zentrum auf dem Aminliganden9,30,31 bewirkt. Der bisher nicht berichtete chirale KomplexReH5(PPh 3)2(sec-Butylamin) bietet die Möglichkeit, die Methoden, mit denen die dynamischen Umlagerungen von Rheniumpolyhydridkomplexen identifiziert und charakterisiert werden können, allgemein zu beschreiben.

Abbildung 3: Darstellungen der dynamischen Prozesse, die mittels NMR-Spektroskopie für Lösungen vonReH5(PPh 3)2(sec-Butylamin) beobachtet werden. Darstellung A zeigt den Austausch eines einzelnen Protons von Adventivwasser gegen den einzigartigen B-Site-Hydridliganden. Darstellung B zeigt den Drehkreuzaustausch von drei benachbarten Hydridliganden, von denen sich zwei an der A-Stelle befinden, während der dritte der einzigartige B-Standorthydrid-Ligand ist. Darstellung C zeigt sowohl den paarweisen Austausch von A-Sitehydrid-Liganden als auch die sterische Inversion der Phosphoratome in Bezug auf den chiralen Aminliganden (N*). Es sollte beachtet werden, dass der paarweise Austausch von A-Sitehydrid-Liganden keine Verschiebung der A-Sitehydrid-Liganden auf die gegenüberliegende Seite des Rheniumzentrums erfordert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Für chemische Systeme wie Rheniumpolyhydridkomplexe, die einen komplexen Satz dynamischer Prozesse aufweisen, ist die Linienformanpassung dynamischer NMR-Spektren die am häufigsten verwendete NMR-Technik zur Charakterisierung der Prozesse 9,11,13,16,21,29. Zweidimensionale EXSY 9,32 oder 2D-ROESY11 sind alternative dynamische NMR-Techniken, mit denen auch die dynamischen Prozesse quantitativ charakterisiert werden können. Zweidimensionale EXSY-Spektren werden typischerweise im Bereich der langsamen Austauschtemperatur gemessen; zweidimensionale ROESY-Spektren werden typischerweise im Bereich der schnellen Austauschtemperatur gemessen. Beide zweidimensionalen Techniken können im Spektrometer viel Zeit für die Datenerfassung benötigen, da jede der Techniken bei einer bestimmten Temperatur einen viel größeren Datensatz erfasst als die eindimensionalen Datensätze, die für die Linienformanpassungsanalyse benötigt werden. Einfache dynamische Prozesse, die gut verstanden werden, wie der dynamische Austausch der beiden Methylgruppen von Dimethylformamid, können leicht durch jede der drei NMR-Techniken charakterisiert werden. Komplexere Systeme wieReH5(PPh3)2(sec-Butylamin), in denen einzelne Hydridliganden an mehreren dynamischen Prozessen beteiligt sind, oder Systeme, die nicht unbedingt gut verstanden werden, wie ein neuartiger Übergangsmetallpolyhydridkomplex, der Protonen zwischen einem Hydridliganden und Adventivwasser austauschen kann oder nicht, werden durch die linienförmige NMR-Methode leichter quantitativ charakterisiert als durch die zweidimensionalen NMR-Methoden. Im Gegensatz zu den zweidimensionalen NMR-Methoden bietet die Linienformanpassungsmethode eine leicht interpretierbare Visualisierung der Übereinstimmung zwischen einem getesteten Modell und den experimentellen Daten sowie einen visuellen Nachweis eines Austauschs, der einen Hydridliganden über die innere Koordinationssphäre von Rhenium hinaus bewegt. Basierend auf Peakhöhen und Peakformen in langsamen Austauschspektren kann selbst ein komplexes dynamisches System wie ReH5(PPh3)2(sec-Butylamin) zu einem leicht zu testenden ersten Satz von Austauschmodellen führen. Wenn mehrere theoretische Modelle für eine molekulare Transformation berichtet wurden, kann die Anpassung der Linienform dynamischer NMR-Spektren einen visuellen Vergleich jedes Modells mit beobachteten Spektren ermöglichen.
Über die drei oben genannten NMR-Techniken hinaus wurden Isotopensubstitutions-NMR-Experimente mitD2Ooder HD verwendet, um den intermolekularen Austausch von Atomen für komplexe Rheniumpolyhydridsysteme qualitativ nachzuweisen, wurden jedoch nicht für quantitative Charakterisierungen 9,33,34,35 verwendet. Theoretische Berechnungen stellen eine zusätzliche Methode zur Charakterisierung der dynamischen Prozesse komplexer dynamischer Systemedar 30,31,36. Theoretische Berechnungen haben gegenüber der Linienformanpassung den Vorteil, dass sie zur Unterscheidung von Möglichkeiten verwendet werden können, die durch die Linienformanpassungsanalyse nicht unterschieden werden können. Zum Beispiel wurden theoretische Berechnungen verwendet, um einen Austausch, der drei benachbarte Hydridliganden auf bestimmten Rhenium(V)-Komplexen umfasst, als Drehkreuzaustausch aller drei Hydridliganden zu beschreiben, anstatt ein alternierendes Paar paarweisen Austauschs mit jedem paarweisen Austausch, einschließlich eines einzigartigen Hydridliganden und eines von zwei chemisch äquivalenten Hydridliganden30, 31. Die Ergebnisse theoretischer Berechnungen werden typischerweise mit experimentell beobachteten quantitativen Charakterisierungen aus einer der drei oben genannten NMR-Techniken verglichen, um die Gültigkeit der berechneten Ergebnisse zu überprüfen.
Die Linienformanpassung dynamischer NMR-Spektren nutzt die Veränderung des Erscheinungsbilds von NMR-Spektren, die auftritt, wenn sich NMR-aktive Kerne während einer NMR-Messung zwischen verschiedenen chemischen Umgebungen bewegen. NMR-Spektren mit langsamem Austausch (Spektren mit unabhängigen Lorentzschen Resonanzen für jeden Satz austauschbarer Kerne) treten bei Temperaturen auf, bei denen die Frequenzdifferenz zwischen den Resonanzen für Kerne, die ausgetauscht werden, im Vergleich zur Austauschrate der Kerne37 groß ist. Schneller Austausch NMR-Spektren (Spektren mit einer einzigen Lorentzschen Resonanz zum Austausch von Kernen) treten bei Temperaturen auf, bei denen die Austauschrate der Kerne viel größer ist als die Frequenzdifferenz zwischen den langsamen Austauschresonanzen37. Zwischenwechselraten treten für Temperaturen zwischen dem langsamen Austauschtemperaturbereich und dem schnellen Austauschtemperaturbereich37 auf. Wenn die grundlegenden Parameter der Larmorfrequenz, der chemischen Verschiebung der Austauschkerne, der Kopplungskonstanten (falls vorhanden) für die austauschenden Kerne und der relativen Populationen jedes Kerntyps bekannt sind, können Ratenkonstanten für den mutmaßlichen Austausch zwischen Kernen durch Vergleich simulierter Spektren mit beobachteten Spektren bei mehreren Zwischentemperaturen bestimmt werden. Gute Anpassungen für Simulationen bei mehreren Temperaturen führen zu temperatur- und ratenkonstanten Daten, die mit der Eyring-Gleichung verwendet werden können, um Aktivierungsparameter für den mutmaßlichen Austausch zu schätzen. Die Ergebnisse der Methode haben sich sowohl als genau als auch reproduzierbar erwiesen.
Protokoll
1. Probenvorbereitung
- Herstellung von ReH7(PPh 3)235
- 0,15 g Natriumborhydrid und 0,41 g ReOCl 3(PPh 3)2 werden in einem zwei- oder dreihalsigen 100-ml-Rundkolben mit Gummischeidewand und Gasanschluss oder einem 100-ml-Kjeldahlkolben (mit Seitenarmgasanschluss) mit einem Gummiseptum kombiniert (ergänzende Abbildung 1).
- Fügen Sie dem Reaktionsgefäß einen Spin-Stab hinzu.
- Verwenden Sie in einem Abzug einen Gummidruckschlauch, um den Gasanschluss des Reaktionsgefäßes mit einem der Absperrhähne eines doppelten Glasverteilers für Vakuum- und Stickstoffgas zu verbinden. Verbinden Sie den Glasvakuumverteiler mit einer Vakuumpumpe mit Gummidruckschlauch und verbinden Sie den Glasstickstoffverteiler mit einer geregelten Stickstoffgasflasche.
- Verbinden Sie das Austrittsgas aus dem Stickstoffgasverteiler mit einem Absperrhahn, mit dem das entlüftete Gas entweder durch eine 2 cm lange Mineralölsäule oder eine 2 cm lange Quecksilbersäule geleitet werden kann.
- Öffnen Sie den Wasserhahn an der Stickstoffflasche und stellen Sie den Druck auf das strömende Gas auf 34 Pfund pro Quadratzoll ein. Entlüften Sie den Stickstoffgasstrom durch den Quecksilbersprudler.
- Evakuieren Sie das Gas im Reaktionsgefäß, indem Sie den Absperrhahn am Glasverteiler einstellen, um den Behälter mit dem Vakuumverteiler zu verbinden. Füllen Sie das Reaktionsgefäß mit Stickstoffgas, indem Sie den Glaskrümmerhahn so wechseln, dass er den Gasverteiler mit dem Reaktionsgefäß verbindet.
- Wiederholen Sie die Schritte 1.1.5 und 1.1.6 noch zweimal, um die Luft im Reaktionsgefäß vollständig durch Stickstoffgas zu ersetzen. Kühlen Sie den Kolben und seinen Inhalt in einem Eisbad.
- Fügen Sie 8 mL sauerstoffarmes Wasser und 8 ml sauerstoffarmes Tetrahydrofuran über eine Spritze zu den Feststoffen im Reaktionsgefäß hinzu. Schalten Sie den gasentlüftenden Absperrhahn so, dass das Gas durch den Mineralölsprudler austritt. Die Suspension 15 min im Eisbad mild umrühren. Entfernen Sie das Reaktionsgefäß nach den ersten 15 Minuten Rühren aus dem Eisbad.
- Lassen Sie die Mischung weitere 45 Minuten unter Rühren ziehen. Beachten Sie die Farbe des Reaktionsgemisches als Indikator dafür, wann die Reaktion abgeschlossen ist. Eine braune bis orange Reaktionsmischungsfarbe (ergänzende Abbildung 1) zeigt an, dass die Reaktion ihren Endpunkt erreicht hat.
- Wenn Sie eine orange bis braune Farbe für das Reaktionsgemisch erreicht haben, filtern Sie die Mischung durch einen 30-ml-mittleren gesinterten Glastrichter. Waschen Sie den zurückgewonnenen Feststoff dreimal mit jeweils 15 ml Portionen Wasser, Methanol und Ethylether. Trocknen Sie den Feststoff unter Vakuum, um adsorbiertes Lösungsmittel zu entfernen.
HINWEIS: Die Reaktion erzeugt im Allgemeinen zwischen 0,20 g und 0,25 g Produkt.
- Herstellung vonReH5(PPh3)2(sec-Butylamin)
- 0,070 g ReH7(PPh3)2 werden eingewogen und in einen 50-ml-Einhalskolben mit rundem Boden überführt, der einen Spinnbalken enthält. Bringen Sie den Kolben an einen Kondensator an, der mit einem Gasanschluss ausgestattet ist. Das Reaktionsgefäß wird mit der Pump- und Füllmethode aus den Schritten 1.1.3 bis 1.1.7 mit Sauerstoff versetzt.
- Über eine Spritze wird ein Volumen von 8 ml sauerstoffarmem Tetrahydrofuran in das Reaktionsgefäß gegeben, indem die Verbindung zwischen dem Rundkolben und dem Kondensator geknackt wird. Fügen Sie auf ähnliche Weise ein Volumen von 0,2 ml sec-Butylamin hinzu. Schalten Sie den gasentlüftenden Absperrhahn so, dass die Gasentlüftungen zum Mineralölsprudler wechseln.
- Das Reaktionsgemisch wird auf 65 °C mit einem Heizmantel erhitzt, der mit einem variablen Wechselstromtransformator verbunden ist, der auf einer Skala von 0 bis 140 für 40 min auf 40 eingestellt ist. Kühlen Sie das Reaktionsgemisch auf eine Temperatur ab, die eine bequeme Handhabung des Kolbens ermöglicht.
- Gießen Sie das Reaktionsgemisch in 25 mL Methanol in einen 125 mL Erlenmeyerkolben. Die Mischung 5 min kräftig umrühren. Fügen Sie 5 ml Wasser hinzu, um die Bildung eines flockigen gelben Niederschlags zu induzieren.
- Sammeln Sie den gelben Niederschlag durch Vakuumfiltration in einem gesinterten Glastrichter. Waschen Sie den Feststoff mit 15 ml Methanol. Trocknen Sie den Feststoff unter Vakuum. Nach diesem Prozess beträgt die typische Produktausbeute 0,035 g.
2. Erfassung und Analyse von NMR-Spektren
- Messung dynamischer NMR-Spektren
- Bereiten Sie eine NMR-Probe mit etwa 8 mg des KomplexesReH5(PPh3)2(sec-Butylamin) in etwa 0,8 mld8-Toluol vor. Setzen Sie die Probe in das Gerät ein.
- Klicken Sie auf die Registerkarte Datei , und wählen Sie Neu aus den angezeigten Optionen aus, um ein Dialogfeld zu öffnen, das zum Erstellen eines NMR-Experiments verwendet wird.
- Erstellen Sie ein 1-H-Experiment, indem Sie die folgenden Schritte ausführen.
- Weisen Sie dem neuen Experiment einen Ordnernamen zu, indem Sie das Eingabefeld Name mit einem eindeutigen Dateinamen ausfüllen. Weisen Sie dem 1-H-Experiment im Feld EXPNO eine Testnummer zu, z. B. 1.
- Weisen Sie dem Experiment in der Box PROCNO die Prozessnummer 1 zu. Weisen Sie den Ordner mithilfe der Dropdown-Liste für DIR einem Verzeichnis zu. Identifizieren Sie das Lösungsmittel, das das Gerät sperrt, aus der Dropdown-Liste Lösungsmittelauswahl .
- Wählen Sie das Verzeichnis, das die Parameter für das 1-H-Experiment enthält, aus der Dropdown-Liste der Verzeichnisse in den Testverzeichnissen aus. Wählen Sie das Proton-Experiment aus den Auswahlmöglichkeiten in der Dropdown-Liste Experiment aus, und fügen Sie (optional) einen Titel für die Daten im Feld Titel aus.
- Geben Sie einen Eda-Befehl in die Befehlszeile ein und passen Sie die Parameter nach Bedarf an, um den Beschreibungen des Experiments im zweiten Absatz des Abschnitts Diskussion unten zu entsprechen.
- Klicken Sie auf die Registerkarte Fenster, wählen Sie Neues Fenster aus der Liste aus, und wiederholen Sie die Schritte 2.1.3.1-2.1.3.8, um ein 1H-{31P}-Experiment mit einem EXPNO-Wert von 2 vorzubereiten, um das Experiment von dem zuvor erstellten 1H-Experimentzu unterscheiden.
- Klicken Sie auf die Registerkarte Fenster, wählen Sie Neues Fenster aus der Liste aus, und wiederholen Sie die Schritte 2.1.3.1-2.1.3.8, um ein 31 P-{1 H}-Experiment mit einem EXPNO-Wert von 3 vorzubereiten, um das Experiment von den zuvor erstellten Experimenten1 H und 1 H-{31P} zu unterscheiden (detaillierte Parameterinformationen finden Sie in der Zusatztabelle 1).
- Geben Sie einen Lock-Befehl in die Befehlszeile ein und wählen Sie die Option d 8-Toluol aus der Liste aus. Klicken Sie auf OK, um die Lösungsmittelauswahl zu übernehmen. Geben Sie einen Atma-Befehl in die Befehlszeile ein, falls erforderlich, wegen einer variablen Kern-X-Band-Sonde, um die reflektierte Energie bei den Larmor-Frequenzen für 1H und 31P auf dem Instrument zu minimieren.
- Geben Sie in der Befehlszeile einen Ro-Befehl ein, geben Sie den Wert 20 in das Feld ein und klicken Sie auf die Schaltfläche Rotation starten . Geben Sie in der Befehlszeile einen Shim-Befehl ein. Wählen Sie eine geeignete Autoshim-Routine wie Topshim aus der Liste der Shimroutinen aus, und klicken Sie auf die Schaltfläche Start .
- Geben Sie in der Befehlszeile einen Rga-Befehl ein. Wählen Sie die Auswahl Automatische Empfängeranpassung und klicken Sie auf OK. Messen Sie wiederum die drei Spektren der Probe bei Raumtemperatur mit 64 Scans für jedes Spektrum mit einem Go-Befehl in der Befehlszeile.
- Transformieren Sie die Daten aus einem Experiment in ein Spektrum mit einem Efp-Befehl , der in die Befehlszeile eingegeben wird.
- Passen Sie die Phase des Spektrums mit den folgenden Befehlen an.
- Klicken Sie auf die Registerkarte Phase und anschließend auf die Registerkarte Phase anpassen. Bewegen Sie den Mauszeiger über die Schaltfläche 0 in der Phasing-Symbolleiste und halten Sie die linke Maustaste gedrückt, sodass die Schaltfläche 0 grün wird.
- Halten Sie die linke Maustaste gedrückt, indem Sie die Maus vorwärts oder rückwärts drehen, bis die Grundlinie über das gesamte Spektrum flach ist und alle Resonanzen als Absorptionswerte angezeigt werden (Spitzen steigen über die Basislinie).
- Wenn die Grundlinie nicht nur mit der Schaltfläche 0 flach gemacht werden kann, passen Sie die Schaltfläche 1 wie in den Schritten 2.1.10.1 und 2.1.10.2 beschrieben sowie die Schaltfläche 0 an, bis die Grundlinie für das gesamte Spektralfenster flach ist.
- Speichern Sie die Phasenanpassung mit den Daten, indem Sie in der Phasensymbolleiste auf die Schaltfläche Speichern und Zurück klicken.
- Passen Sie die Anzahl der Scans für jede Messung nach Bedarf basierend auf dem Signal-Rausch-Verhältnis im Spektrum an, wobei zu beachten ist, dass das Signal-Rausch-Verhältnis bei niedrigeren Temperaturen aufgrund der Dekoaleszenz der Signale zu einzelnen Resonanzen typischerweise abnimmt (Abbildung 4).
- Bereiten Sie das Spektrometer gemäß den Anweisungen des Herstellers für die Temperaturregelung vor. Geben Sie einen Durchfluss von 200 L/h für das Kühlgas und eine Zieltemperatur von 290 K für die Sonde ein. Lassen Sie das Spektrometer 2 Minuten lang bei der Zieltemperatur stabilisieren. Erhöhen Sie den Kühlgasdurchsatz bei Bedarf auf 210 oder 220 l/h, um die Temperatur zu stabilisieren.
- Schneiden Sie die Probe wie in Schritt 2.1.7 bei 290 K ab. Ändern Sie den Dateinamen für jedes der zuvor gemessenen Spektren, indem Sie die Temperatur am Ende des Dateinamens hinzufügen (Schritte 2.1.2 und 2.1.3.1) und einen Satz von drei Spektren bei 290 K erfassen.
- Erhöhen Sie den Kühlgasdurchsatz um ≥ 30 l/h, je nach Bedarf, um sich bei der nächsten Temperatur zu stabilisieren, und verringern Sie die Zieltemperatur um 10 K. Lassen Sie das Spektrometer bei der nächsten Temperatur für 2 min stabilisieren und legen Sie dann die Probe wie in Schritt 2.1.7 vor. Messen Sie den Satz von drei Spektren.
- Wiederholen Sie die Schritte 2.1.13 und 2.1.14 nach Bedarf, um Spektren bis zur niedrigsten gewünschten Temperatur zu erfassen.
HINWEIS: Eine Temperatur von 200 K ist in der Regel ausreichend für einen vollständigen Datensatz, der zur Bestimmung der Aktivierungsparameter für die dynamischen Prozesse der Probe geeignet ist. - Die Probe wird in Schritten von 10 K wieder auf Raumtemperatur erwärmt. Stabilisieren Sie die Temperatur für 2 min bei jeder Temperatur, bevor Sie die Probe erneut erwärmen, um Schäden an der Glasauskleidung der Sonde zu vermeiden.
- Linienformanalyse der gemessenen Spektren
- Klicken Sie im NMR-Programm auf die Befehlsleiste oben links im Fenster und wählen Sie Öffnen aus dem Dropdown-Menü. Wählen Sie Im Standardformat gespeicherte NMR-Daten öffnen aus. Klicken Sie auf OK , um das Datei-Explorer-Fenster für das Programm zu öffnen.
- Navigieren Sie zu dem Ordner, in dem die Daten analysiert werden sollen, indem Sie die Linienform anpassen. Wählen Sie die Dateinummer aus, die dem zu analysierenden Spektrum entspricht, und klicken Sie auf die Schaltfläche Anzeige . Das Spektrum (falls zuvor verarbeitet) oder die FID-Kurve (Free Induction Decay) wird in der NMR-Software angezeigt.
- Verarbeiten Sie den FID gegebenenfalls, indem Sie einen Efp-Befehl (Exponentialmultiplikation, Fouriertransformation und Phasenkorrektur) in die Befehlszeile eingeben. Stellen Sie die Phase des Spektrums ein (Schritt 2.1.10).
- Passen Sie die Grundlinie des Spektrums an; Wenn es nicht über das gesamte Spektrum flach ist, dann gleichen Sie mit der 0-Intensitätslinie wie folgt aus.
- Klicken Sie auf die Registerkarte Prozess und dann auf die Registerkarte Baseline . Bewegen Sie den Mauszeiger über die Schaltfläche A . Drücken Sie die linke Maustaste und rollen Sie die Maus vorwärts oder rückwärts, um die rote Einstelllinie mit dem linken (unten) Ende des Spektrums auszugleichen.
- Wenn die Grundlinie immer noch nicht auf Höhe der roten Anpassungslinie liegt, wiederholen Sie den Vorgang mit den verbleibenden Buchstabenschaltflächen, bis die rote Anpassungslinie zur Grundlinie des Spektrums passt. Verwenden Sie die Schaltfläche Speichern und Return , um die Anpassung zu speichern, wenn die rote angepasste Basislinie mit der tatsächlichen Basislinie übereinstimmt.
- Wählen Sie die Registerkarte Analysieren in der NMR-Software. Wählen Sie in den Analyseoptionen die Option Linienformen und anschließend die Option Dynamische NMR-Modelle anpassen aus.
- Das Spektrum wird nun im Fenster des linienförmigen Moduls angezeigt. Verwenden Sie die Symbolleisten über dem Spektrum, um die Anzeige des Spektrums anzupassen. Das Fenster links neben dem Spektrum behandelt die Anpassung der Linienform an das Spektrum.
- Stellen Sie die Spektrumanzeige mit dem Smooth Zoom Tool so ein, dass der Teil des Spektrums, der angepasst werden soll, im Spektrumfenster angezeigt wird. Verwenden Sie die Symbolleistenschaltfläche Spektrum nach links und rechts verschieben , um einen Teil des Spektrums im Anzeigefenster zu zentrieren.
- Greifen Sie auf das chemische Verschiebungsfenster für die Anpassung der Linienform zu, indem Sie im Fenster für die Anpassung an die Linienform die Registerkarte Spektrum auswählen.
- Klicken Sie auf die Schaltfläche Bereich bearbeiten . Geben Sie die oberen und unteren chemischen Verschiebungen für die Anpassung der Linienform ein, und klicken Sie auf die Schaltfläche OK , um diese Grenzwerte zu akzeptieren.
- Starten Sie ein Modell für die Anpassung der Linienform, indem Sie im Fenster für die Anpassung der Linienform auf die Registerkarte System drehen klicken. Klicken Sie auf die Schaltfläche Hinzufügen , um den Aufbau eines Modell-Spin-Systems zu ermöglichen.
- Deaktivieren Sie LB (für Linienverbreiterung) und geben Sie den Wert für die Linienverbreiterung manuell mit der Maus und der Schaltfläche LB auf der Symbolleiste für die Anpassung an die Linienform ein.
- Fügen Sie den ersten Kern zum Modell hinzu, indem Sie auf die Registerkarte Kern und anschließend auf die Schaltfläche Hinzufügen klicken. Ein Satz von Standardwerten wird für Nucleus 1 angezeigt. Passen Sie die chemische Verschiebung für Kern 1 an, indem Sie einen Wert für die chemische Verschiebung in das Feld Nu(iso) eingeben oder mit dem chemischen Verschiebungswerkzeug auf der Werkzeugleiste für die Anpassung an die Linienform verwenden.
HINWEIS: Wenn das Auswahlfeld in der angekreuzten Form belassen wird, wird die chemische Verschiebung dieses Kerns variiert, um die beste Anpassung zu erzielen. Ungeprüfte Variablen werden im Linienanpassungsprozess nicht variiert. - Verwenden Sie das Feld Pseudospin für Kern 1, um die Anzahl der äquivalenten Kerne für Kern 1 einzugeben, wobei jeder Spin 1/2 Kern 0,5 entspricht. Geben Sie die Summe der Spins in das Pseudospin-Feld ein, um alle äquivalenten Kerne zu berücksichtigen.
- Verwenden Sie das Feld In Molekül, um Modelle aufzunehmen, die mehr als ein einzelnes Molekül benötigen, um an einem dynamischen Prozess teilzunehmen. Weisen Sie Resonanzen, die von verschiedenen Molekülen entstehen, separaten Molekülen zu, indem Sie Bezeichnungen wie 1, 2 usw. für verschiedene Moleküle verwenden. Für Resonanzen, die von einem einzelnen Molekül ausgehen, weisen Sie 1 für alle In-Molecule-Werte zu.
- Fügen Sie den zweiten und alle nachfolgenden Kerne zum Modell hinzu, indem Sie auf die Registerkarte Kern und anschließend auf die Schaltfläche Hinzufügen klicken. Fügen Sie die Spin-Spin-Kopplung zwischen Kernen hinzu, indem Sie entweder die Kopplung in das entsprechende JN-Feld eingeben (wobei N der Kern ist, mit dem der hinzugefügte Kern gekoppelt ist, N = 1, 2, ...) oder indem Sie die Schaltfläche Skalare Kopplung auf der Werkzeugleiste für die Anpassung an die Linienform anpassen.
- Beginnen Sie mit der Beschreibung des Atomaustauschs, indem Sie auf die Registerkarte Reaktion klicken. Aktivieren Sie das Kontrollkästchen , wenn die Kurskonstante für den Umtausch in der Linienformanpassung variiert werden soll. Geben Sie die Anzahl der auszutauschenden Kerne (Anzahl in Bezug auf ihre Identifikationsregisterkarten wie Kern 1 und Kern 2) in das Feld Austausch für den ersten Austausch im Modell ein.
- Beschreiben Sie die zu testenden Börsen in den Feldern unter dem Feld Austausch . Definieren Sie den Austausch zwischen Nucleus-Registerkarten in den folgenden Feldern. Ein Zwei-Kern-Austausch würde als Kern 1 zu Kern 2 und Kern 2 zu Kern 1 eingegeben werden. Stellen Sie sicher, dass der Austausch zyklisch ist, denn wenn ein Kern von Kern 1 verschoben wird, muss ein anderer Kern in Kern 1 verschoben werden.
- Verwenden Sie die Schaltfläche Geschwindigkeit austauschen auf der Symbolleiste für die Anpassung an die Linienform, um den Anfangswert von k zu ändern, um den Wert von k iterativ anzupassen, auch wenn das Kontrollkästchen für die Ratenkonstante aktiviert ist.
- Fügen Sie dem Modell weitere Austauschvorgänge hinzu, indem Sie auf die Registerkarte Reaktion und anschließend auf die Schaltfläche Hinzufügen klicken. Fügen Sie dem Modell nach Bedarf Austauschvorgänge hinzu. Verwenden Sie die Werkzeuge auf der Werkzeugleiste für die Anpassung an die Linienform, um die Anfangsvariablen, einschließlich der Spektrumintensität, so anzupassen, dass sie gut an das zu passende Spektrum angepasst sind.
- Beginnen Sie mit der iterativen Anpassung der Linienform, indem Sie auf der Werkzeugleiste für die Linienformanpassung auf die Schaltfläche Spektrumanpassung starten klicken. Setzen Sie die iterative Anpassung fort, bis keine Änderung in der besten Überlappung zwischen Spektrum und Modell gefunden wird oder bis 1000 Iterationen erreicht sind. Wenn die Anpassung bei 1000 Iterationen aufhört, setzen Sie weitere Iterationen mit der Schaltfläche Start the Spectrum Fit fort. Das Modellspektrum wird zum Vergleich mit dem tatsächlichen Spektrum angezeigt.
- Notieren Sie die Werte für die optimale Anpassung auf den entsprechenden Registerkarten. Speichern Sie das am besten angepasste Spektrum, indem Sie im Fenster zur Anpassung der Linienform auf die Registerkarte Spektrum klicken und anschließend auf die Schaltfläche Speichern klicken.
HINWEIS: Das am besten geeignete Spektrum wird in demselben Ordner gespeichert, in dem die Daten gesammelt wurden. Das Best-Fit-Spektrum wird von den Originaldaten unterschieden, indem es mit einer anderen Verarbeitungsnummer gespeichert wird, die beim Speichern eingegeben wird. - Speichern Sie das Modell, das für die Anpassung der Linienform verwendet wurde, indem Sie auf die Registerkarte Main und anschließend auf die Schaltfläche Speichern klicken. Geben Sie einen Namen für das Modell ein.

Abbildung 4: Ein Vergleich von 31P-{1H}-Signalintensitäten für eine einzelne Probe vonReH5(PPh3)2(sec-Butylamin) in d8-Toluol. Eine repräsentative Demonstration des Unterschieds in den Signalintensitäten zwischen einer schnellen Austausch-Einzelphosphorresonanz und einem Paar von Phosphorresonanzen in der Nähe der Koaleszenztemperatur für diese Resonanzen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
3. Bestimmung der Aktivierungsparameter aus einem Eyring-Diagramm 1
- Geben Sie Daten aus der Linienformanpassung für einen modellierten dynamischen Prozess in eine Tabelle ein, wobei die unabhängige Variable als 1/T und die abhängige Variable als ln(k/T) eingegeben wird.
- Fügen Sie ein Punktdiagramm der Daten in die Tabelle ein. Fügen Sie eine Trendlinie durch die Daten hinzu. Verwenden Sie die Steigung und den Schnittpunkt der Trendlinie, um für ΔH‡ und ΔS‡ zu lösen. Die Steigung der Trendlinie beträgt -ΔH‡/R, während der Schnittpunkt der Trendlinie ΔS‡/R + 23,76 beträgt.
- Lösen Sie für ΔG‡ bei einer gegebenen Temperatur unter Verwendung der Beziehung
ΔG‡(T) = ΔH‡ - TΔS‡.
HINWEIS: Für einen einfachen Austausch von zwei Kernen mit koaleszierenden Resonanzen kann eine Überprüfung der Werte von ΔH‡ und ΔS‡ durchgeführt werden, indem ΔG‡, berechnet bei der Koaleszenztemperatur, mit dem Wert von ΔG‡ verglichen wird, der sich aus der langsamen Austauschfrequenzdifferenz zwischen Resonanzen und der Koaleszenztemperatur ergibt.
Ergebnisse
Die Charakterisierungen der beiden in diesem Manuskript beschriebenen Rheniumpolyhydridprodukte lassen sich am besten durch 1 H-{31 P} und 31P-{1H} NMR-Spektroskopie erreichen. In einer d-6-Benzollösung bei Raumtemperatur erscheint die Hydridligandenresonanz von ReH7(PPh3)2 als Binomialtriplett bei δ = -4,2 ppm mit 2 JPH = 18 Hz bei 1H NMR-Spektroskopie (ergänzende Abbildung 2). Di...
Diskussion
Es gibt vier Punkte bei der Herstellung von ReH7 (PPh 3) 2, die die Menge und Reinheit des hergestellten Materials beeinflussen können. Erstens ist die Verwendung eines Eisbades während der ersten 15 Minuten der Reaktion wichtig, um Wärme aus der Reaktion zu entfernen, die zwischen Natriumborhydrid und Wasser auftritt. Höhere Anfangstemperaturen führen zu einer verminderten Ausbeute des ProduktsReH7(PPh3)2 durch Bildung des thermischen Zersetzungsprod...
Offenlegungen
Die Autoren haben keine Interessenkonflikte offenzulegen.
Danksagungen
Die Autoren danken dem Department of Chemistry and Physics und dem Creativity and Research Grant Program (Naik, Moehring) der Monmouth University für die finanzielle Unterstützung dieser Arbeit.
Materialien
| Name | Company | Catalog Number | Comments |
| Bruker Avance II 400 MHz NMR spectrometer | Bruker Biospin | The instrument includes a two channel probe (1H and X) with the X channel tunable from 162 MHz to 10 Mhz. The instrument is also VT capable with a dewar and heat exchanger for VT work. | |
| d8-toluene | MilliporeSigma | 434388 | |
| Powerstat variable transformer | Powerstat | ||
| sec-butyl amine | MilliporeSigma | B89000 | |
| Sodium borohydride | MilliporeSigma | 452882 | |
| Tetrahydrofuran | MilliporeSigma | 186562 | |
| Thermowell C3AM 100 mL | Thermowell | ||
| Topspin 3.0 or 4.1.4 with dNMR | Bruker Biospin | Data was acquired with Topspin version 3.0 and data handling was performed on a second computer that was running Topspin version 4.1.4.. | |
| Trichlorooxobis(triphenylphosphine) rhenium(V) | MilliporeSigma | 370193 | |
| Vacuubrand PC3000 vacuum pump with a CVC 3000 controller | Vacuubrand |
Referenzen
- Zimmer, K. D., Shoemaker, R., Ruminski, R. R. Synthesis and characterization of a fluxional Re(I) carbonyl complex fac-[Re(CO)3(dpop')Cl] with the nominally tri-dentate ligand dipyrido(2,3-α:3',2'-j)phenazine (dpop). Inorganica Chimica Acta. 359 (5), 1478-1484 (2006).
- McGlinchey, M. J. Symmetry breaking in NMR spectroscopy: the elucidation of hidden molecular rearrangement processes. Symmetry. 6 (3), 622-654 (2014).
- Casarini, D., Luazzi, L., Mazzanti, A. Recent advances in stereodynamics and conformational analysis by dynamic NMR and theoretical calculations. European Journal of Organic Chemistry. 2010 (11), 2035 (2010).
- Palmer, A. G., Williams, J., McDermott, A. Nuclear magnetic resonance studies of biopolymer dynamics. Journal of Physical Chemistry. 100 (31), 13293-13310 (1996).
- Kern, D., Kern, G., Scherer, G., Fischer, G., Drakenberg, T. Kinetic analysis of cyclophilin-catalyzed prolyl cis/trans isomerization by dynamic NMR spectroscopy. Biochemistry. 34 (41), 13594-13602 (1995).
- Menger, F. M., Lynn, J. L. Fast proton transfer at a micelle surface. Journal of the American Chemical Society. 97 (4), 948-949 (1975).
- Pines, A., Rabinovitz, M. A nuclear magnetic resonance total line-shape treatment of internal rotation in dimethylformamide. Tetrahedron Letters. 9 (31), 3529-3532 (1968).
- Mancinelli, M., Bencivenni, G., Pecorari, D., Mazzanti, A. Stereochemistry and recent applications of axially chiral organic molecules. European Journal of Organic Chemistry. 2020 (27), 4070-4086 (2020).
- Streisel, D. J., et al. Fluxionality, substitution, and hydrogen exchange at eight-coordinate rhenium(V) polyhydride centers. Inorganica Chimica Acta. 496 (1), 119028 (2019).
- Jimenez, Y., Strepka, A. M., Borgohain, M. D., Hinojosa, P. A., Moehring, G. A. Ortho-metalation, rotational isomerization, and hydride-hydride coupling at rhenium(V) polyhydride complexes stabilized by aromatic amine ligands. Inorganica Chimica Acta. 362 (9), 3259-3266 (2009).
- Lee, J. C., Yao, W., Crabtree, R. H., Ruegger, H. Fluxionality in [ReH5(PPh3)2(pyridine)]. Inorganic Chemistry. 35 (3), 695-699 (1996).
- Patel, B. P., Kavallieratos, K., Crabtree, R. H. Effects of dihydrogen bonding on fluxionality in ReH5(PPh3)2L. Journal of Organometallic Chemistry. 528 (1), 205-207 (1997).
- Geetha, B., et al. Chiral amine ligands at rhenium(V) pentahydride complexes allow for characterization of an energetically accessible and reversible steric inversion of diastereotopic phosphorus atoms. Inorganica Chimica Acta. 531 (1), 120741 (2022).
- Paulo, A., Ascenso, J., Domingos, A., Galvao, A., Santos, I. Rhenium-(III) and -(V) hydride complexes with modified poly(pyrazolyl)borates. Journal of the Chemical Society, Dalton Transactions. 1999 (8), 1293-1300 (1999).
- Bianchini, C., et al. Synthesis and characterization of rhenium polyhydrides stabilized by the tripodal ligand MeC(CH2PPh2)3. Journal of Organometallic Chemistry. 451 (1), 97-106 (1993).
- Scorzelli, A. G., Macalush, B. E., Naik, D. V., Moehring, G. A. Comparative study of fluxional processes at two different classes of eight-coordinate rhenium(V) polyhydride complexes. Inorganica Chimica Acta. 516 (1), 120120 (2021).
- Luo, X. -. L., Crabtree, R. H. Synthesis and spectroscopic characterization of rhenium complexes ReH5(triphos)] and [ReH6(triphos)]+ [triphos = PPh(CH2CH2PPh2)2]. Journal of the Chemical Society. 1991 (5), 587-590 (1991).
- Kim, Y., Deng, H., Gallucci, J. C., Wojcicki, A. Rhenium polyhydride complexes containing PhP(CH2CH2CH2PCy2)2 (Cyttp): protonation, insertion, and ligand substitution reactions of ReH5(Cyttp) and structural characterization of ReH5(Cyttp) and [ReH4(η2-H2)(Cyttp)]SbF6. Inorganic Chemistry. 35 (24), 7166-7173 (1996).
- Bolano, S., et al. Synthesis, characterization, protonation studies and X-ray crystal structure of ReH5(PPh3)2(PTA) (PTA = 1,3,5-triaza-7-phosphaadamantane). Journal of Organometallic Chemistry. 691 (4), 629-637 (2006).
- Ginsberg, A. P., Abrahams, S. C., Jamieson, P. B. Nonrigid stereochemistry in eight-coordinate pentahydridorhenium complexes. Journal of the American Chemical Society. 95 (14), 4751-4752 (1973).
- Bolano, S., Bravo, J., Garcia-Fontan, S. Mono- and dinuclear rhenium polyhydride complexes bearing the chelating ligand 1,2-bis(dicyclohexylphosphinanyloxy)ethane. European Journal of Inorganic Chemistry. 2004 (24), 4812-4819 (2004).
- Leeaphon, M., Rohl, K., Thomas, R. J., Fanwick, P. E., Walton, R. A. Reactions of the polyhydride complex ReH7(PPh3)2 with quinoline, 2-hydroxyquinoline, and 2-mercaptoquinoline. The preparation and characterization of hydrido complexes of rhenium(V) and chloro complexes of rhenium(III). Inorganic Chemistry. 32 (24), 5562-5568 (1993).
- Mejia, E., Togni, A. Rhenium complexes containing the chiral tridentate ferrocenyl ligand pigiphos. Organometallics. 30 (17), 4765-4770 (2011).
- Moehring, G. A., Walton, R. A. Reactions of heptahydrobis(triphenylphosphine)rhenium with bidentate aromatic heterocycles. Inorganic Chemistry. 26 (17), 2910-2912 (1987).
- Kosanovich, A. J., Reibenspies, J. H., Ozerov, A. V. Complexes of high-valent rhenium supported by the PCP pincer. Organometallics. 35 (4), 513-519 (2016).
- Emge, T. J., Koetzle, T. F., Bruno, J. W., Caulton, K. G. Pentahydridorhenium: crystal and molecular structure of ReH5(PMePh2)3. Inorganic Chemistry. 23 (24), 4012-4017 (1984).
- Costello, M. T., Fanwick, P. E., Green, M. A., Walton, R. A. Reactions of Heptahydridobis(triphenylphosphine)rhenium with 1-(diphenylphosphino)-2-(diphenylarsino)ethane (arphos) and 1,2-bis(diphenylarsino)ethane (dpae). Structural characterization of ReH5(PPh3)2(arphos-As) and ReH5(PPh3)2(dpae-As). Inorganic Chemistry. 30 (4), 861-864 (1991).
- Alvarez, D., Lundquist, E. G., Ziller, J. W., Evans, W. J., Caulton, K. G. Synthesis, structure and applications of transition-metal polyhydride anions. Journal of the American Chemical Society. 111 (22), 8392-8398 (1989).
- Albinati, A., et al. Synthesis, characterization, and interconversion of the rhenium polyhydrides ReH3(η4-NP3)] and [ReH4(η4-NP3)]+ {NP3 = tris[2-(diphenylphosphanyl)ethyl]amine}. European Journal of Inorganic Chemistry. 2002 (6), 1530-1539 (2002).
- Bosque, R., et al. Site preference energetics, fluxionality, and intramolecular M−H···H−N hydrogen bonding in a dodecahedral transition metal polyhydride. Inorganic Chemistry. 36 (24), 5505-5511 (1997).
- Tao, Y., Sou, W., Luo, G. -. G., Kraka, E. Describing polytopal rearrangement processes of octacoordinate structures. I. renewed insights into fluxionality of the rhenium polyhydride complex ReH5(PPh3)2(Pyridine). Inorganic Chemistry. 60 (4), 2492-2502 (2021).
- Beringhelli, T., D'Alfonso, G., Minoja, A. P. Rhenium-platinum mixed metal clusters. Characterization in solution and dynamic behavior of the isomers of [Re3Pt(µ-H3)(CO)14]. An example of a labile metal fragment that undergoes intermolecular exchange. Organometallics. 13 (2), 663-668 (1994).
- Grieco, G., Blacque, O. Solution and solid-state structure of the first NHC-substituted rhenium heptahydrides. European Journal of Inorganic Chemistry. 2019 (34), 3810-3819 (2019).
- Wazio, J. A., Jimenez, V., Soparawalla, S., John, S., Moehring, G. A. Hydrogen exchange of rhenium(VII) heptahydridobis(triphenylphosphine) with water, aniline, methanol, and itself. Inorganica Chimica Acta. 362 (1), 159-165 (2009).
- Chatt, J., Coffey, R. S. Hydrido-complexes of rhenium-containing tertiary phosphines. Journal of the Chemical Society, A. 1969, 1963-1972 (1969).
- Tao, Y., Wang, X., Zou, W., Luo, G. -. G., Kraka, E. Unusual intramolecular motion of ReH92- in K2ReH9 crystal: circle dance and three-arm turnstile mechanisms revealed by computational study. Inorganic Chemistry. 61 (2), 1041-1050 (2022).
- Berger, X., Braun, S. . 200 and More NMR Experiments a Practical Course. , (2004).
- He, G., Chen, J., Sung, H. H. -. Y., Williams, I. D., Jia, G. Substituent effect on reactions of ReH5(PMe2Ph)3 with propargyl alcohols. Inorganica Chimica Acta. 518 (1), 120239 (2021).
- Donnelly, L. J., Parsons, S., Morrison, C. A., Thomas, S. P., Love, J. B. Synthesis and structures of anionic rhenium polyhydride complexes of boron-hydride ligands and their application in catalysis. Chemical Science. 11 (9), 9994-9999 (2020).
- Donnelly, L. J., et al. C-H borylation catalysis of heteroaromatics by a rhenium boryl polyhydride. ACS Catalysis. 11 (12), 7394-7400 (2021).
- Jin, H., et al. CO-enabled rhenium hydride catalyst for directed C(sp2)-H bond alkylation with olefins. Organic Chemistry Frontiers. 2 (4), 378-382 (2015).
- Takaya, H., Ito, M., Murahashi, S. -. I. Rhenium-catalyzed addition of carbonyl compounds to the carbon−nitrogen triple bonds of nitriles: α-C−H activation of carbonyl compounds. Journal of the American Chemical Society. 131 (31), 10824-10825 (2009).
- Carr, S. W., Fowles, E. H., Fontaine, X. L. R., Shaw, B. L. Multihydride complexes of rhenium, osmium or iridium containing monodentate ditertiary phosphine ligands: selective hydrogen-deuterium exchanges of the rhenium multihydrides. Journal of the Chemical Society, Dalton Transactions. 1990 (2), 573-579 (1990).
- Jin, H., et al. Rhenium-catalyzed acceptorless dehydrogenative coupling via dual activation of alcohols and carbonyl compounds. ACS Catalysis. 3 (10), 2195-2198 (2013).
- Loza, M. L., de Gala, S., Crabtree, R. H. Steric crowding in a rhenium polyhydride induced by a chelating disilyl ligand: synthesis, characterization, and reactivity of ReH5(disil)(PPh3)2 (disil = 1,2-Bis(dimethylsilyl)benzene and 1,2-Bis(dimethylsilyl)ethane). Inorganic Chemistry. 33 (22), 5073-5078 (1994).
- Lin, Y., Zhu, X., Xiang, M. Transition metal polyhydrides-catalyzed addition of activated nitriles to aldehydes and ketones via Knoevenagel condensation. Journal of Organometallic Chemistry. 448 (1-2), 215-218 (1993).
- Abdukader, A., Jin, H., Cheng, Y., Zhu, C. Rhenium-catalyzed amination of alcohols by hydrogen transfer process. Tetrahedron Letters. 55 (30), 4172-4174 (2014).
- Lin, Y., Zhou, Y. Selective transfer hydrogenation catalyzed by transition metal polyhydrides. Fenzi Cuihua. 5 (2), 119-124 (1991).
- Green, M. A., Huffman, J. C., Caulton, K. G., Rybak, W. K., Ziolkowski, J. J. Ligand scavenging and catalytic utilization of the phototransient ReH5(PMe2Ph)2. Journal of Organometallic Chemistry. 218 (2), 39-43 (1981).
- Komiya, S., Chigira, T., Suzuki, T., Hirano, M. Polymerization of alkyl methacrylate catalyzed by hydridorhenium complexes. Chemistry Letters. 4 (4), 347-348 (1999).
- Michos, D., Luo, X. L., Faller, J. W., Crabtree, R. H. Tungsten(VI) hexahydride complexes supported by chelating triphosphine ligands: protonation to give η2-dihydrogen complexes and catalytic dehydrogenation of cyclooctane to cyclooctene. Inorganic Chemistry. 32 (8), 1370-1375 (1993).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten