
Method Article
Industrialisierte, von künstlicher Intelligenz gesteuerte Laser-Mikrodissektion zur mikroskaligen proteomischen Analyse der Tumormikroumgebung
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Dieses Protokoll beschreibt einen Hochdurchsatz-Workflow für die künstliche Intelligenz-gesteuerte Segmentierung von pathologiebestätigten Regionen von Interesse aus gefärbten, dünnen Gewebeschnittbildern zur Anreicherung von histologieaufgelösten Zellpopulationen mittels Laser-Mikrodissektion. Diese Strategie beinhaltet einen neuartigen Algorithmus, der die Übertragung von Demarkationen, die Zellpopulationen von Interesse bezeichnen, direkt auf Lasermikroskope ermöglicht.
Zusammenfassung
Die Tumormikroumgebung (TME) stellt ein komplexes Ökosystem dar, das aus Dutzenden verschiedener Zelltypen besteht, darunter Tumor-, Stroma- und Immunzellpopulationen. Um die Variation auf Proteomebene und die Tumorheterogenität auf Skala zu charakterisieren, sind Hochdurchsatzmethoden erforderlich, um diskrete Zellpopulationen selektiv in soliden Tumormalignitäten zu isolieren. Dieses Protokoll beschreibt einen Hochdurchsatz-Workflow, der durch künstliche Intelligenz (KI) ermöglicht wird und Bilder von Hämatoxylin und Eosin (H&E)-gefärbten, dünnen Gewebeschnitten in pathologiebestätigte Regionen von Interesse für die selektive Ernte histologieaufgelöster Zellpopulationen mittels Lasermikrodissektion (LMD) segmentiert. Diese Strategie beinhaltet einen neuartigen Algorithmus, der die Übertragung von Regionen, die mit digitaler Bildsoftware auf Zellpopulationen von Interesse hinweisen, direkt auf Lasermikroskope ermöglicht und so einfachere Sammlungen ermöglicht. Es wurde eine erfolgreiche Implementierung dieses Workflows durchgeführt, die den Nutzen dieser harmonisierten Methode zur selektiven Ernte von Tumorzellpopulationen aus dem TME für die quantitative, gemultiplexte Proteomanalyse durch hochauflösende Massenspektrometrie demonstrierte. Diese Strategie lässt sich vollständig in die routinemäßige histopathologische Überprüfung integrieren und nutzt die digitale Bildanalyse, um die Anreicherung von Zellpopulationen von Interesse zu unterstützen, und ist vollständig generalisierbar, was harmonisierte Ernten von Zellpopulationen aus dem TME für multiomische Analysen ermöglicht.
Einleitung
Die TME stellt ein komplexes Ökosystem dar, das von einer Vielzahl von Zelltypen wie Tumorzellen, Stromazellen, Immunzellen, Endothelzellen, anderen mesenchymalen Zelltypen und Adipozyten zusammen mit einer komplexen extrazellulären Matrix 1 bevölkertist. Dieses zelluläre Ökosystem variiert innerhalb und zwischen verschiedenen Krankheitsorganstellen, was zu einer komplexen Tumorheterogenität führt 2,3. Neuere Studien haben gezeigt, dass heterogene Tumoren und Tumoren mit geringer Tumorzellularität (geringe Reinheit) oft mit einer schlechten Krankheitsprognose korrelieren 2,3.
Um das molekulare Zusammenspiel zwischen Tumor- und Nicht-Tumorzellpopulationen innerhalb der TME auf Skala zu verstehen, sind standardisierte und Hochdurchsatzstrategien erforderlich, um selektiv verschiedene Zellpopulationen zu ernten, die für die nachgelagerte multiomische Analyse von Interesse sind. Die quantitative Proteomik stellt eine sich schnell entwickelnde und immer wichtiger werdende Technik dar, um das Verständnis der Krebsbiologie zu fördern. Bisher hat die überwiegende Mehrheit der Studien, die Proteomik verwenden, dies mit Proteinen getan, die aus Ganztumorgewebepräparaten extrahiert wurden (z. B. kryopulverisiert), was zu einem Mangel am Verständnis der Heterogenität auf Proteomebene im TME 4,5,6 führte.
Die Entwicklung von Probenentnahmestrategien, die sich nahtlos in klinische Pathologie-Workflows integrieren und Informationen aus klinischen Pathologie-Workflows nutzen, wird eine neue Generation von histologie-aufgelösten Proteomics ermöglichen, die die Goldstandard-Workflows der diagnostischen Pathologie in hohem Maße ergänzen. LMD ermöglicht die direkte und selektive Erfassung zellulärer Subpopulationen oder Regionen von Interesse (ROIs) durch mikroskopische Inspektion von histologisch gefärbten Gewebedünnschnitten7. Jüngste große Fortschritte in der digitalen Pathologie und KI-fähigen Analyse haben die Fähigkeit gezeigt, einzigartige Zusammensetzungsmerkmale und ROIs innerhalb der TME automatisiert zu identifizieren, von denen viele mit molekularen Veränderungen und klinischen Krankheitsmerkmalen wie Therapieresistenz und Krankheitsprognosekorrelieren 8.
Der in dem hier vorgestellten Protokoll beschriebene Workflow nutzt kommerzielle Softwarelösungen, um Tumor-ROIs selektiv in digitalen Histopathologiebildern zu kommentieren, und verwendet intern entwickelte Softwaretools, um diese Tumor-ROIs auf Lasermikroskope zu übertragen, um diskrete Zellpopulationen von Interesse zu erfassen, die sich nahtlos in nachgelagerte multiomische Analyse-Workflows integrieren lassen. Diese integrierte Strategie verringert die LMD-Bedienerzeit erheblich und minimiert die Dauer, für die Gewebe Umgebungstemperatur haben müssen. Die Integration von automatisierter Merkmalsauswahl und LMD-Ernte mit quantitativer Proteomik mit hohem Durchsatz wird durch eine differentielle Analyse der TME aus zwei repräsentativen histologischen Subtypen des epithelialen Ovarialkarzinoms, dem hochgradigen serösen Eierstockkrebs (HGSOC) und dem Ovarialklarzellkarzinom (OCCC), demonstriert.
Protokoll
Alle Studienprotokolle wurden für die Verwendung im Rahmen eines vom westlichen IRB genehmigten Protokolls "An Integrated Molecular Analysis of Endometrial and Ovarian Cancer to Identify and Validate Clinically Informative Biomarkers" zugelassen, das gemäß der US-Bundesverordnung 45 CFR 46.102 (f) als befreit gilt. Alle experimentellen Protokolle mit menschlichen Daten in dieser Studie stimmten mit der Erklärung von Helsinki überein. Die Einwilligung nach Aufklärung wurde von allen an der Studie beteiligten Probanden eingeholt.
VORSICHT: Die folgenden Reagenzien, die im gesamten Protokoll verwendet werden, sind bekannte oder vermutete Karzinogene und / oder enthalten gefährliche Stoffe: Ethanol, DEPC-Wasser, Mayers Hämatoxylinlösung, Eosin Y-Lösung, Methanol, Acetonitril und Ameisensäure. Eine ordnungsgemäße Handhabung, wie in den jeweiligen Sicherheitsdatenblättern (SDB) beschrieben, und die Verwendung geeigneter persönlicher Schutzausrüstung (PSA) ist obligatorisch.
1. Generieren der Standard-Shape-Listendatendatei (.sld), die Kalibrator-Fiducials enthält
HINWEIS: Die in diesem Abschnitt beschriebenen Protokollschritte sind spezifisch für die Verwendung mit einem inversen Lasermikroskop und der zugehörigen Software (siehe Materialtabelle). Die Erstellung einer Standard-SLD-Datei ist nur einmal pro Lasermikroskop erforderlich. Die resultierende Datei kann zum Schneiden von Treuhändern in alle danach verwendeten PEN-Objektträger verwendet werden. Ungefähre Zeit: 5 min (nur einmal).
- Öffnen Sie die LMD-Software und laden Sie den Membranträger aus Polyethylennaphthalat (PEN) mit der Vorderseite nach unten auf die LMD-Bühne, wobei das Etikett dem Benutzer am nächsten ist. Deaktivieren Sie das Kontrollkästchen Zeile(n) schließen auf der rechten Seite des Programmfensters.
- Verwenden Sie die PtoP-Funktion (Punkt zu Punkt) unter hoher Vergrößerung (63x), um drei "V" -Pfeile zu zeichnen, die als Kalibrierfiduciales dienen. Beginnend an einem externen Punkt auf dem V, zeichnen Sie eine Linie bis zur Mitte des V und klicken Sie mit einem einzigen Klick. Zeichnen Sie dann eine zweite Linie vom zentralen Punkt des V bis zum Ende des zweiten externen V-Punktes, und doppelklicken Sie, um eine einzelne, nicht geschlossene V-Form aus den beiden Linien zu erstellen.
HINWEIS: Diese Kalibrierfiduzierer sollten in drei Ecken des Objektträgers platziert werden: vorne rechts, hinten rechts, hinten links. - Wählen Sie die Option AF (Autofokus) vor dem Schneiden . Schneiden Sie das Dia in Position 1 ein, bewegen Sie es in jede der verbleibenden Gleitpositionen und zeichnen Sie es präzise über die Kalibrierungsschnitte.
- Speichern Sie die SLD-Datei und wählen Sie im Popup-Dialogfeld die Option Ohne Kalibrierung speichern , um zu vermeiden, dass die Kalibrierungsfiducials in die Membran geschnitten werden.
HINWEIS: Eine repräsentative .sld-Datei mit Standardkalibrator-Fiducials für vier Objektträgerpositionen finden Sie in der Zusatzdatei 1.
2. Vorbereitung der LMD-Folie(n)
HINWEIS: Die in diesem Abschnitt beschriebenen Protokollschritte sind spezifisch für die Verwendung mit einem inversen Lasermikroskop und der zugehörigen Software (siehe Materialtabelle). Ungefähre Zeit: 5 min.
- Stellen Sie sicher, dass der Objektträger vollständig trocken ist, bevor Sie die Referenzkalibrierungsfiducials schneiden. Öffnen Sie die LMD-Software und öffnen Sie die Standardkalibrierungsdatei .sld unter der Option Formen importieren.
- Wählen Sie die Option AF (Autofokus) vor dem Schneiden . Laden Sie die Schiene(n) mit dem Gewebe nach unten und die Etikettenfolie näher an den Bediener in den Objektträgerhalter auf der LMD-Bühne.
- Schneiden Sie mit dem Lasermikroskop und der Standardkalibrierungsdatei .sld Kalibrierfiducials in die PEN-Membran.
- OPTIONAL: Schneiden Sie die Kalibrierfiziermittel entweder vor oder nach dem Platzieren der Gewebeabschnitte auf dem Objektträger in die PEN-Membran. Wenn die Kalibrierfikatoren vor der Gewebeplatzierung geschnitten werden, stellen Sie sicher, dass sich das Gewebe und/oder Fixiermittel nicht mit den Kalibratoren überlappt, wenn das Gewebe in Schritt 2.5 auf den Objektträger gelegt wird. Wenn Kalibrierfiducials nach der Gewebeplatzierung geschnitten werden, beenden Sie sie nach Abschluss von Schritt 2.4 und fahren Sie mit Abschnitt 3 fort.
- Überprüfen Sie alle Kalibratoren einzeln, um sicherzustellen, dass jeder Schnitt vollständig und sichtbar ist.
HINWEIS: Verwenden Sie die Funktion "Verschieben und Schneiden ", um den Laser manuell über Kalibrierfiduzien zu richten, die die PEN-Membran nicht vollständig durchtrennt haben. - Legen Sie den gefrorenen oder formalinfixierten, paraffineingebetteten (FFPE) Gewebeabschnitt auf den Objektträger mit den Kalibrierfiduziermitteln.
3. Gewebefärbung
HINWEIS: Ungefähre Zeit: 30 min.
- Fixieren Sie die gefrorenen LMD-Gewebeobjektträger in 70% Ethanol (EtOH), das Phosphatase-Inhibitor-Cocktail-Reagenzien enthält, für 5 min.
- Waschen Sie die Objektträger in Diethylpyrocarbomat (DEPC) Wasser, das Phosphatase-Inhibitor-Cocktailreagenzien enthält, für 1 min.
- Waschen Sie die Rutschen in DEPC-Wasser für 1 min.
- Inkubieren Sie die Objektträger in Mayers Hämatoxylinlösung für 3 min.
- Spülen Sie die Rutschen in DEPC-Wasser für 3 min.
- Spülen Sie die Objektträger in einem frischen Austausch von DEPC-Wasser für 1 min.
- Inkubieren Sie die Objektträger in wässriger Eosin Y-Lösung für 1 s.
- Spülen Sie die Objektträger 2 x 5 s in 95% EtOH.
- Spülen Sie die Objektträger 3 x 10 s in 100% EtOH.
- Wischen Sie den überschüssigen EtOH von der Rückseite der Objektträger ab und lassen Sie die Objektträger an der Luft trocknen.
- Lagern Sie die Dias bei -80 °C, wenn LMD nicht sofort durchgeführt werden soll.
4. Folienbebilderung
HINWEIS: Die in diesem Abschnitt beschriebenen Protokollschritte gelten speziell für gescannte Dias (siehe Materialverzeichnis) und die resultierenden Bilder, die als SVS-Dateien gespeichert sind. Verwenden Sie einen beliebigen Scanner und die zugehörige Software, die Bilddateien in einem Format generieren, das die Bildanalysesoftware (siehe Materialtabelle) öffnen kann. Zu den unterstützten Dateitypen, die pyramidenförmige Tiffs verwenden, gehören JPG, TIF, MRXS, QPTIFF, COMPONENT TIFF, SVS, AFI, SCN, LIF, DCM, OME. TIFF, ND2, VSI, NDPI, NDPIS, CZI, BIF, KFB und ISYNTAX. Ungefähre Zeit: 5 min.
- Schalten Sie den Scanner ein und öffnen Sie die Diascanner-Software. Legen Sie das Dia mit dem Gewebe nach oben auf die einzelne Diastufe im Scanner. Stellen Sie sicher, dass der Objektträger vollständig trocken ist und legen Sie vorsichtig ein Deckglas über das Gewebe. Verwenden Sie kein Ethanol oder Tauchöl unter dem Deckglas.
- Erfassen Sie das Mikrobild mit Einstellungen, die kalibriert wurden, um sich an die PEN-Membran anstelle des Glashintergrunds anzupassen und die Färbung der Hintergrundmembran gemäß den Anweisungen des Herstellers zu ignorieren.
- Passen Sie den Bildgebungsbereich an, indem Sie den inneren grünen Umfang ziehen und in der Größe ändern, um den gesamten PEN-Membranbereich nach Bedarf zu erfassen. Fügen Sie dem Gewebe vier Fokuspunkte hinzu, indem Sie auf das Schnappschuss-Übersichtsbild doppelklicken, und drei Fokuspunkte auf der Membran in der Nähe der Kalibrierfiducials (ein Fokuspunkt pro der drei Kalibrierfiducials).
HINWEIS: Die vier Fokuspunkte können fast überall auf dem Gewebeabschnitt platziert werden, obwohl das Aufsetzen auf Gewebe, das zu dunkel gefärbt ist und schwarz erscheint, dazu führen kann, dass der Scan fehlschlägt. - Wählen Sie im Menü Ansicht die Option Videomonitor aus. Passen Sie den Fokus bei Bedarf manuell mit dem Fein- und/oder Makrofokusschieberegler für jeden Punkt um das LMD-Gewebe an. Erfassen Sie den Bildscan unter hoher Vergrößerung (20x). Vergewissern Sie sich, dass alle Kalibrierungsfidukte im gespeicherten Bild sichtbar und klar sind.
5. Automatisierte Feature-Auswahl mittels Bildanalyse-Software
- Für ganze Tumorsammlungen (ungefähre Zeit: 5 min; fallabhängig):
- Öffnen Sie die Bildanalysesoftware (siehe Materialverzeichnis). Wählen Sie Bilder öffnen und wählen Sie im Popup-Fenster die SVS-Bilddatei aus, die beim Scannen des Dias auf dem AT2-Scanner generiert wurde.
HINWEIS: Eine repräsentative SVS-Bilddatei wird in der Zusatzdatei 2 bereitgestellt. - Navigieren Sie zur Registerkarte Anmerkungen Wählen Sie in der Anmerkungssymbolleiste das Zeichenstiftwerkzeug aus, und zeichnen Sie eine Form um das Gewebe herum.
- Wählen Sie die Form aus und klicken Sie mit der rechten Maustaste auf das Bild. Wählen Sie im Dropdown-Menü Erweitert die Option Partitionierung (gekachelt) aus. Legen Sie die Kachelgröße und den Abstand zwischen 500 und 40 fest, und wählen Sie OK aus, um die Kacheln zu generieren. Wählen Sie die Umkreisform aus, die zum Generieren der Kacheln in Schritt 5.1.2 verwendet wurde, und löschen Sie sie.
- Wählen Sie das Dropdown-Menü Ebenenaktionen | Exportieren, um die gekachelten Anmerkungen als ANMERKUNGSDATEI zu speichern.
HINWEIS: Eine repräsentative .annotation-Datei für eine vollständige Tumorgewebeentnahme wird in der Ergänzungsdatei 3 bereitgestellt. - Erstellen Sie einen Ordner für die Sitzung oder das Projekt, und speichern Sie die ANMERKUNGSDATEI in einem Unterordner, der mit dem eindeutigen Bezeichner für die Folie gekennzeichnet ist.
- Navigieren Sie zur Registerkarte Anmerkungen Wählen Sie die Dropdown-Liste Ebenenaktionen | aus Löschen Sie alle Ebenen , um alle Anmerkungen aus dem Bild zu entfernen. Wählen Sie das Stiftwerkzeug aus und zeichnen Sie für jedes Kalibrierfiducial eine kurze Linie von der inneren Spitze der Pfeilspitze. Zeichnen Sie die Linien aus den Markierungen in der folgenden Reihenfolge: oben links, oben rechts, unten rechts.
- Wählen Sie das Dropdown-Menü Ebenenaktionen | Exportieren, um die Linienanmerkungen als ANMERKUNGSDATEI zu speichern. Fügen Sie dem Dateinamen _calib hinzu, und legen Sie die Datei in dem Unterordner ab, der die Koordinaten für die gekachelten Formen enthält.
HINWEIS: Eine repräsentative _calib.annotation-Datei wird in der Zusatzdatei 4 bereitgestellt. - Kopieren Sie die Adresse für das Hauptprojekt oder den Sitzungsordner. Öffnen Sie das XML-Import-generierende Skript "Malleator" (verfügbar über https://github.com/GYNCOE/Mitchell.et.al.2022) mit der integrierten IDLE-Entwicklungsumgebung und fügen Sie die Projektordneradresse zwischen die Anführungszeichen am Ende des Skripts ein.
- Wählen Sie das Dropdown-Menü Ausführen | Führen Sie Module aus , um das Skript auszuführen.
HINWEIS: Die .xml LMD-Importdatei wird in dem Unterordner generiert, der für das Bild / die Folie erstellt wurde. Eine repräsentative .xml Datei finden Sie in der Ergänzungsdatei 5.
- Öffnen Sie die Bildanalysesoftware (siehe Materialverzeichnis). Wählen Sie Bilder öffnen und wählen Sie im Popup-Fenster die SVS-Bilddatei aus, die beim Scannen des Dias auf dem AT2-Scanner generiert wurde.
- Nur für LMD-angereicherte Sammlungen (ungefähre Zeit: 15 min; fallabhängig):
- Öffnen Sie die Bildanalysesoftware (siehe Materialverzeichnis). Wählen Sie Bilder öffnen und wählen Sie im Popup-Fenster die SVS-Bilddatei aus, die beim Scannen der Folie generiert wurde.
- Navigieren Sie zur Registerkarte Anmerkungen Wählen Sie das Rechteck-Anmerkungswerkzeug aus, und verwenden Sie es, um ein Feld um das Gewebe zu zeichnen.
- Wählen Sie das Kontrollkästchen Anmerkung und klicken Sie mit der rechten Maustaste auf das Bild. Wählen Sie das Dropdown-Menü Erweitert | Partitionierungsoption (gekachelt). Legen Sie die Kachelgröße und den Abstand zwischen 500 und 40 fest, und wählen Sie OK aus, um die Kacheln zu generieren. Wählen Sie die Randfeldanmerkung aus, die zum Generieren der Kacheln in Schritt 5.2.2 verwendet wurde, und löschen Sie sie.
- Wählen Sie das Dropdown-Menü Ebenenaktionen | Exportieren , um die gekachelten Anmerkungen als ANMERKUNGSDATEI zu speichern.
- Legen Sie eine gespeicherte Kopie des Python-Algorithmus "Dapọ" (verfügbar über https://github.com/GYNCOE/Mitchell.et.al.2022), der zum Zusammenführen der KI-klassifizierten Anmerkungsebenen entwickelt wurde, im selben Ordner wie die gekachelte Anmerkungsdatei ab.
- Kopieren Sie den Namen der gekachelten Anmerkungsdatei. Öffnen Sie das Python-Programm mit der integrierten IDLE-Entwicklungsumgebung und fügen Sie den Namen der gekachelten Anmerkungsdatei zwischen den Zitaten am unteren Rand des Programms ein.
- Wählen Sie das Dropdown-Menü Ausführen | Führen Sie das Modul aus. Warten Sie, bis eine neue Datei generiert wurde, in der alle gekachelten Anmerkungen unter einer einzigen Ebene zusammengeführt werden.
- Öffnen Sie die Bildanalysesoftware und navigieren Sie zur Registerkarte Anmerkungen . Wählen Sie das Dropdown-Menü Ebenenaktionen | Löschen Sie alle Ebenen , um alle Anmerkungen aus dem Bild zu entfernen.
- Wählen Sie das Dropdown-Menü Ebenenaktionen | Importieren Sie die lokale Anmerkungsdatei. Wählen Sie im Popup-Fenster die zusammengeführte ANNOTATION-Datei aus, die vom Skript generiert wurde. Stellen Sie sicher, dass sich alle importierten Kacheln unter derselben Anmerkungsebene befinden.
- Navigieren Sie zur Registerkarte Klassifizierer, und befolgen Sie die Anweisungen des Herstellers, um Shapes für die ROIs zu generieren. Bevor Sie den Klassifikator ausführen, wählen Sie die gewünschten Anmerkungsschicht(en) (d. h. die Tumor- oder Stromaschicht) aus, indem Sie das ROI-Kontrollkästchen auf der Registerkarte Anmerkungen unter den erweiterten Klassifizierungsoptionen aktivieren. Verwenden Sie die Option Anmerkungsebene (Annotation Layer) aus dem Menü Aktionen (Classifier Actions), um den Classifier auszuführen.
- Nachdem die Klassifizierungsanalyse abgeschlossen ist, navigieren Sie zur Registerkarte Anmerkungen, und wählen Sie die aus der Analyse generierte Anmerkungsfolie aus. Wählen Sie das Dropdown-Menü Ebenenaktionen | Löschen Sie "Alle Ebenen, aber aktuell ", um alle anderen Anmerkungsebenen aus dem Bild zu entfernen.
- Wählen Sie das Dropdown-Menü Ebenenaktionen | Export, um die Anmerkungen als ANMERKUNGSDATEI zu speichern. Erstellen Sie einen Ordner für die Sitzung oder das Projekt, und speichern Sie die ANMERKUNGSDATEI in einem Unterordner, der mit dem eindeutigen Bezeichner für die Folie gekennzeichnet ist.
HINWEIS: Eine repräsentative .annotation-Datei für eine klassifizierte LMD-angereicherte Gewebeentnahme finden Sie in der Zusatzdatei 6. - Navigieren Sie zur Registerkarte Anmerkungen, wählen Sie die Dropdown-Liste Ebenenaktionen | Löschen Sie alle Ebenen, um alle Anmerkungen aus dem Bild zu entfernen. Wählen Sie das Stiftwerkzeug aus und zeichnen Sie eine kurze Linie von jedem Kalibrierfiducial. Zeichnen Sie Linien aus den Markierungen in der folgenden Reihenfolge: oben links, oben rechts, unten rechts.
- Wählen Sie das Dropdown-Menü Ebenenaktionen | Exportieren , um die Linienanmerkungen als ANMERKUNGSDATEI zu speichern. Fügen Sie dem Dateinamen _calib hinzu, und legen Sie die Datei in dem Unterordner ab, der die Koordinaten für die gekachelten Formen enthält.
- Kopieren Sie die Adresse für das Hauptprojekt oder den Sitzungsordner. Öffnen Sie das XML-Import-generierende Skript "Malleator" (verfügbar über https://github.com/GYNCOE/Mitchell.et.al.2022) mithilfe der integrierten IDLE-Entwicklungsumgebung und fügen Sie dann die Projektordneradresse zwischen den Anführungszeichen am Ende des Skripts ein.
- Wählen Sie das Dropdown-Menü Ausführen | Führen Sie Module aus, um das Skript auszuführen.
HINWEIS: Die .xml LMD-Importdatei wird in dem Unterordner generiert, der für das Bild / die Folie erstellt wurde.
6. Laser-Mikrodissektion
HINWEIS: Die in diesem Abschnitt beschriebenen Protokollschritte sind spezifisch für die Verwendung mit einem inversen Lasermikroskop und der zugehörigen Software (siehe Materialtabelle). Ungefähre Zeit: 2 h; fallabhängig.
- Laden Sie den markierten Membranträger (mit Kalibrierfiducials) mit dem Gewebe nach unten und der Etikettenseite näher am Bediener in den Objektträgerhalter auf dem Lasermikroskoptisch.
- Wählen Sie im Dropdown-Menü Datei die Option Formen importieren aus. Wählen Sie die .xml LMD-Importdatei aus, die für die Folie generiert wurde. Wählen Sie Nein im Popup-Fenster, um das Laden von Referenzpunkten aus der Datei zu vermeiden, und Nein im zweiten Popup-Fenster, um die Verwendung zuvor gespeicherter Referenzpunkte für die Kalibrierung zu vermeiden.
- Folgen Sie den Anweisungen der LMD-Anwendung und richten Sie das Kalibrierungskreuz an jedem der drei Kalibrierfiduzierer auf dem Objektträger aus. Suchen Sie in der Bildanalysesoftware nach Kalibrierungsfiduzierern, die oben links, oben rechts und unten rechts im Objektträgerbild angezeigt werden und den Referenzpunkten in der vorderen rechten, rechten und linken hinteren Ecke des invertierten LMD-Objektträgers auf der Mikroskopstufe entsprechen. Wechseln Sie zwischen der Verwendung des 5-fachen Objektivs zum Lokalisieren und des 63-fachen Objektivs, um jede Kalibrierungsfiducial auszurichten. Wählen Sie Nein im Popup-Fenster, um zu vermeiden, dass die Referenzpunkte in der Datei gespeichert werden, und OK im zweiten Popup-Fenster, um zu bestätigen , dass die Folie eingefügt wurde.
- Bewegen Sie das 5-fache Objektiv in Position und wählen Sie im Popup-Fenster Ja, um die tatsächliche Vergrößerung zu verwenden. Sobald die importierten Formen angezeigt werden, fokussieren Sie die Kamera auf das Gewebe.
- Markieren und markieren Sie alle Formen im Fenster Formenliste , ziehen Sie sie mit einer oder zwei Anmerkungen im Sichtfeld als Referenzen an ihren Platz, und richten Sie die vertikale Z-Achse zum Schneiden mit dem Laser aus.
- Überprüfen Sie die importierten Formen, und weisen Sie sie der entsprechenden Rohrposition zum Sammeln zu. Drücken Sie Start Cut , um den Laser zu starten.
HINWEIS: Importierte Formen in der .xml Datei werden automatisch der Position "Keine Kappe" im Fenster Formenliste zugewiesen. Um Gewebe zu ernten, müssen die importierten Formen einer Position zugewiesen werden, die ein beladenes Rohr enthält.
7. Proteinverdauung durch Druckzyklustechnologie (PCT)
HINWEIS: Ungefähre Zeit: 4 h (3 h ohne Vakuumzentrifugentrocknungszeit).
- 0,5 ml Röhrchen mit den gedeckelten PCT-Mikroröhrchen mit LMD-geerntetem Gewebe in 20 μL 100 mM TEAB/10% Acetonitril in einen Thermocycler geben und 30 min bei 99 °C erhitzen, dann 10 min auf 50 °C abkühlen.
- Drehen Sie die Röhrchen für 30 s bei 4.000 × g und entfernen Sie dann die MicroTubes aus den 0,5 ml Röhrchen. Entfernen und verwerfen Sie mit dem MicroCap-Tool die MicroCaps aus den PCT-MicroTubes. Trypsin (siehe Materialtabelle) im Verhältnis 1 μg pro 30 mm2 Gewebe hinzufügen und mit dem MicroCap-Werkzeug einen MicroPestle in den MicroTube einführen.
- Übertragen Sie die MicroTubes in eine Barocycler-Kartusche und montieren Sie die komplette Kartusche. Legen Sie die Kartusche in die Barocycler-Druckkammer und befestigen Sie den Deckel. Barocycle bei 45.000 psi für 50 s und atmosphärischem Druck für 10 s bei 50 °C für 60 Zyklen.
- Sobald das Barocycling abgeschlossen ist, geben Sie die Mikroröhrchen für 2 Minuten bei 4.000 × g in ein 0,5-ml-Mikrozentrifugenröhrchen und eine Zentrifuge.
- Entfernen Sie das MicroTube mit dem Kappenwerkzeug aus dem 0,5 ml Mikrozentrifugenröhrchen. Entfernen Sie den Stößel vorsichtig mit dem Kappenwerkzeug und spülen Sie die untere Hälfte des Stößels mit 20 μL flüssigem Chromatographie-Massenspektrometrie-Wasser (LC-MS) ab und sammeln Sie die Wäsche in ein sauberes 0,5 ml Mikrozentrifugenröhrchen.
- Klopfen Sie vorsichtig auf den MicroTube auf der Tischplatte, um die Flüssigkeit nach unten zu bewegen und die gesamte Lösung aus dem MicroTube in das 0,5-ml-Mikrozentrifugenröhrchen zu übertragen.
- Fügen Sie dem MicroTube 20 μL LC-MS-Wasser hinzu und klopfen Sie es vorsichtig auf die Tischplatte. Die Waschlösung in das 0,5 ml Tubus geben und diesen Waschschritt noch einmal wiederholen.
- Vakuumzentrifuge zum Trocknen der Proben auf ~ 2 μL und Zugabe von 100 μL von 100 mM TEAB, pH 8,0.
- Bestimmen Sie die Peptidkonzentration mit einem kolorimetrischen Assay (Bicchincinsäure (BCA)-Assay; siehe Materialtabelle) gemäß dem Protokoll des Herstellers.
8. Tandem-Mass Tag (TMT) Etikettierung und EasyPep Bereinigung
HINWEIS: Ungefähre Zeit: 7 h 20 min (2 h 20 min ohne Vakuumzentrifugentrocknungszeit).
- Bringen Sie die isobaren TMT-Markierungsreagenzien vor dem Öffnen auf Umgebungstemperatur. 500 μL 100% Acetonitril in jede TMT-Durchstechflasche (5 mg) geben. Inkubieren Sie für 10 Minuten mit gelegentlichen Wirbeln.
- 5 μg der Peptidprobe werden in 100 μL 100 mM TEAB, pH 8,0, gelöst und 10 μL eines gegebenen TMT-Reagenzes zugegeben. Erstellen und schließen Sie Referenzpools ein, die jede einzelne Probe im Experiment in jedem TMT-Multiplex-Satz von Proben darstellen, um die Quantifizierung von Proben über mehrere TMT-Multiplexe hinwegzu erleichtern 9. Inkubieren Sie Reaktionen für 1 h bei Umgebungstemperatur mit gelegentlichem Schütteln / Klopfen.
- Quen Sie die TMT-Markierungsreaktion durch Zugabe von 10 μL 5% Hydroxylamin und inkubieren Sie für 30 min bei Umgebungstemperatur mit gelegentlichem Klopfen. Nach dem Abschrecken die TMT-markierten Proben in einem Röhrchen kombinieren und auf ca. 200 μL trocknen.
- 1.800 μL 0,1% Ameisensäure hinzufügen. Überprüfen Sie den pH-Wert mit pH-Papier: Wenn pH ~ 3, fügen Sie 1 ml 0,1% Ameisensäure hinzu; Wenn der pH-Wert >3 beträgt, fügen Sie 10-20 μL 5% Ameisensäure bis zum pH-Wert ~ 3 hinzu. Fügen Sie 0,1% Ameisensäure hinzu, um ein Endvolumen von 3 ml zu erreichen.
- Entfernen Sie die Lasche am unteren Rand der Peptidbereinigungsspalte, entfernen Sie die Kappe und legen Sie sie in einen 15 ml konischen Schlauch. Übertragen Sie die TMT-gekennzeichnete Probe in die Spalte und fahren Sie mit der Bereinigung gemäß dem Protokoll des Herstellers fort.
- Vakuumzentrifuge zum Trocknen der eluierten Peptide auf ~ 20 μL, Transfer in eine LC-Durchstechflasche mit 25 mM Ammoniumbicarbonat für ein Endvolumen von 80 μL und Offline-Fraktionierung.
9. TMT-Multiplex-Probenfraktionierung und Pooling
HINWEIS: Ungefähre Zeit: 3 h 30 min (1 h 30 min ohne Vakuumzentrifugentrocknungszeit).
- Die TMT-markierten Peptidmultiplexe werden durch basische Umkehrphasenchromatographie in 96 Fraktionen fraktioniert, indem ein zunehmender linearer Gradient (0,69% min-1) der mobilen Phase B (Acetonitril) in die mobile Phase A (10 mM NH4HCO3, pH 8,0) entwickelt wird.
- Erzeugen Sie 36 verkettete Fraktionen durch Pooling von Stichprobenbohrungen. Vakuumzentrifuge zum Trocknen von Fraktionen bis ~2 μL und Resuspension in 25 mM Ammoniumbicarbonat (Endkonzentration 1,5 μg/10 μL), Zentrifuge bei 15.000 × g für 10-15 min und Transfer auf LC-Fläschchen zur MS-Analyse.
10. Flüssigkeitschromatographie Tandem-Massenspektrometrie (LC-MS/MS)
HINWEIS: Ungefähre Zeit: Instrumentenmethode und experimentelles Design abhängig.
- Kalibrieren Sie das Massenspektrometer gemäß den Anweisungen / Protokollen des Herstellers.
- Bereiten Sie frische mobile Phasen und Standards vor und führen Sie geeignete LC-Vorlaufvorbereitungen durch (einschließlich, aber nicht beschränkt auf Spüllösungsmittel, Spülluft und Dichtheitsprüfskripte für das referenzierte Instrument [siehe Materialtabelle]). Gleichgewichten Sie die prä- und analytischen Spalten und die Probenschleife vor Beginn der Analysen.
- Überprüfen Sie vor und zwischen seriellen TMT-Multiplex-Analysen, ob das LC-MS-System zuvor gebenchmarkte Leistungsmetriken erfüllt, indem Sie TTT-markierte Peptidfermente (z. B. MSPE) (siehe Materialtabelle) mit Qualitätssicherung/Qualitätskontrolle (QA/QC) und HeLa verwenden.
- Laden Sie Autosampler-Fläschchen an den entsprechenden Positionen im LC-Autosampler. Analysieren Sie einzelne Fraktionen mit einer geeigneten Gradienten-/MS-Methode. Streuen Sie einen "Waschgang" mit Peptidstandard (z. B. Peptidretentionszeitkalibrierung [PRTC]) ein, um die chromatographische und massenspektrale Leistung zu bewerten. Führen Sie nach der Analyse jeder TMT-Multiplex-Probenfraktionsserie QA/QC TMT-Benchmark-Standards aus, um die Systemleistung zu bewerten.
- Führen Sie Massenspektrometer-Auswertungsroutinen nach QA/QC TMT-Benchmark-Standards durch, um die Leistung nach der Probe zu bewerten, und kalibrieren Sie dann das System wie in Schritt 10.1 für den nächsten Probensatz.
11. Bioinformatische Datenanalyse
HINWEIS: Ungefähre Zeit: Experimentelles Design abhängig.
- Übertragen Sie alle Beispieldaten (z. B. .raw Dateien) auf einen geeigneten Netzwerkspeicher/ein geeignetes Computerlaufwerk.
- Suchen Sie alle Fraktionen zusammen mit der gewünschten Datenanalyseanwendung (z. B. Proteome Discover, Mascot) mit geeigneten Parametern9 gegen eine speziesspezifische Proteinreferenzdatenbank, um Peptidspektralübereinstimmungen (PSMs) zu erzeugen und TMT-Reporter-Ionensignalintensitäten zu extrahieren. Filtern Sie PSMs basierend auf geeigneten Qualitätskontrollmetriken und aggregieren Sie normalisierte, mittlere log2-transformierte TMT-Reporter-Ionenverhältnishäufigkeiten in globale Proteinfüllmengen, wie zuvor beschrieben 3,9.
- Vergleichen Sie Proteinveränderungen unter interessanten Bedingungen mit der gewünschten Differentialanalysesoftware.
Ergebnisse
Frisch gefrorene Gewebedünnschnitte von zwei HGSOC- und zwei OCCC-Patienten wurden mit diesem integrierten KI-gesteuerten Workflow zur Identifizierung des ROI, Segmentierung, LMD und quantitativen Proteomikanalyse von Gewebe analysiert (Abbildung 1). Repräsentative H & E-gefärbte Gewebeabschnitte für jeden Tumor wurden von einem Board-zertifizierten Pathologen überprüft; Die Zellularität des Tumors reichte von 70% bis 99%. Die Gewebe wurden dünn geschnitten auf PEN-Membranobjektträger (Supplemental File 2) und mit Kalibrator-Fiducials (Supplemental File 1) vorgeschnitten, was die Integration von Positionsorientierungsdaten aus Anmerkungen, die in der Bildanalysesoftware generiert wurden (siehe Materialverzeichnis), mit kartesischer Koordinatenorientierung in der LMD-Software ermöglichte. Nach der H&E-Färbung wurden hochauflösende Bilder (20x) der PEN-Objektträger aufgenommen, die das Gewebe sowie Kalibratoren enthielten.
Tumor- und Stromazellpopulationen in den Mikroaufnahmen wurden mit Hilfe von Bildanalysesoftware (siehe Materialtabelle) für die selektive Ernte durch LMD segmentiert, zusammen mit Ernten, die den gesamten Gewebedünnschnitt (z. B. ganzes Tumorgewebe) darstellten (Abbildung 1). Nicht-diskriminierende Annotationen für ganze Tumorgewebesammlungen wurden erzeugt, indem der gesamte Gewebeabschnitt mit Kacheln von 500 μm2 geteilt wurde, wodurch eine Lücke von 40 μm zwischen den Kacheln verblieb, um die Integrität der PEN-Membran aufrechtzuerhalten und zu verhindern, dass sich die Membran während der LMD kräuselt. Auf Objektträgern für die histologieaufgelöste LMD-Anreicherung wurde der KI-Klassifikator in der Bildanalysesoftware (siehe Materialverzeichnis) darauf trainiert, zwischen Tumor- und Stromazellen zu unterscheiden, zusammen mit dem leeren Glasobjektträgerhintergrund. Repräsentative Tumor-, Stroma- und Blankglasbereiche wurden manuell hervorgehoben, und das Klassifikatorwerkzeug wurde verwendet, um diese ROIs über den gesamten Gewebeabschnitt zu segmentieren. Die segmentierten Schichten, die das ganze Gewebe, das Tumorepithel und das Stroma darstellen, wurden separat als einzelne .annotation-Dateien (Supplemental File 3 und Supplemental File 6) gespeichert. In einer separaten Kopie der Image-Datei (ohne die partitionierten ROI-Anmerkungen) wurde eine kurze Zeile von der mittleren Spitze jedes der drei treuhänderischen Kalibratoren kommentiert und als .annotation-Datei mit demselben Dateinamen wie jede der LMD-Annotationsschichtdateien gespeichert, jedoch mit dem Suffix "_calib" (Supplemental File 4) angehängt. Diese Linien wurden verwendet, um die Position der PEN-Membrankalibratoren mit den in der Bildanalysesoftware gezeichneten Anmerkungsformlistendaten zu registrieren.
Die vorliegende Studie stellt zwei Algorithmen zur Verfügung, "Malleator" und "Dapọ" in Python, um diesen KI-gesteuerten LMD-Workflow zu unterstützen, die bei https://github.com/GYNCOE/Mitchell.et.al.2022 verfügbar sind. Der Malleator-Algorithmus extrahiert die spezifischen kartesischen Koordinaten für alle einzelnen Annotationen (Gewebe-ROI und Kalibratoren) aus den gepaarten .annotation-Dateien und führt diese in einer einzigen XML-Importdatei (Extensible Markup Language) zusammen (Supplemental File 5). Insbesondere verwendet der Malleator-Algorithmus den Verzeichnisnamen aus einem übergeordneten Ordner als Eingabe, um alle Unterverzeichnisordner zu durchsuchen, und generiert .xml Dateien für alle Unterordner, die noch keine .xml zusammengeführte Datei haben. Der Malleator-Algorithmus führt alle Annotationsschichten in der Bildanalysesoftware (siehe Materialtabelle) zu einer einzigen Ebene zusammen und konvertiert die KI-generierten Formlistendaten, die als proprietärer .annotation-Dateityp gespeichert werden, in .xml Format, das mit der LMD-Software kompatibel ist. Nach dem Zusammenführen der Annotations- und Kalibratordateien wird die algorithmusgenerierte .xml Datei gespeichert und in die LMD-Software importiert. Leichte Anpassungen sind notwendig, um die Ausrichtung von Anmerkungen manuell anzupassen, was auch dazu dient, die vertikale (Z-Ebene) Position des Objektträgertisches auf dem Lasermikroskop zu registrieren. Der Dapọ-Algorithmus wird speziell für LMD-angereicherte Sammlungen verwendet. Partitionierte Kacheln werden von der Bildanalysesoftware automatisch einzelnen Annotationsebenen zugewiesen. Der Dapọ-Algorithmus führt vor der Verwendung des Classifier-Tools alle partitionierten Kacheln zu einer einzigen Anmerkungsebene zusammen und reduziert so die Laufzeit der Classifier-Analyse für LMD-angereicherte Sammlungen.
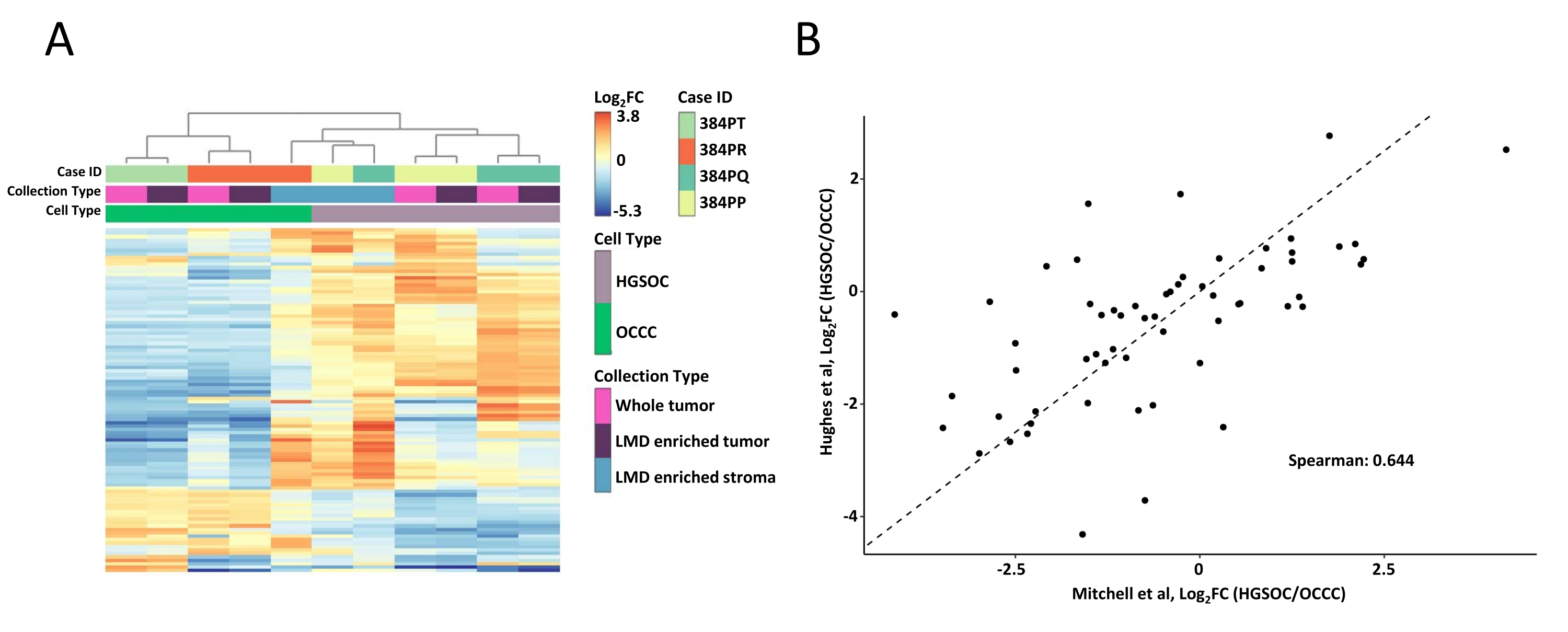
Der gesamte Tumor und die mit LMD angereicherten Gewebeproben wurden verdaut, mit TMT-Reagenzien markiert, gemultiplext, offline fraktioniert und mittels quantitativer MS-basierter Proteomik analysiert, wie zuvor beschrieben9. Die mittlere Peptidausbeute (43-60 μg) und die Ausbeute (0,46-0,59 μg/mm2) für Proben, die mit diesem KI-gesteuerten Workflow entnommen wurden, waren vergleichbar mit früheren Berichten 9,10. Insgesamt wurden 5.971 Proteine über alle Proben koquantifiziert (Supplemental Table S1). Unüberwachtes hierarchisches Clustering unter Verwendung der 100 variabelsten Proteine führte zu einer Segregation der HGSOC- und OCCC-Histotypen aus den LMD-angereicherten und ganzen Tumorproben (Abbildung 2A), ähnlich der zuvor beschriebenen11. Im Gegensatz dazu gruppierten sich die LMD-angereicherten Stromaproben von HGSOC und OCCC zusammen und unabhängig von den LMD-angereicherten Tumor- und ganzen Tumorproben. Unter den 5.971 quantifizierten Proteinen waren 215 signifikant verändert (LIMMA adj. p < 0,05) zwischen ganzen Tumorsammlungen aus HGSOC- und OCCC-Proben (Supplemental Table S2). Diese veränderten Proteine wurden mit denen verglichen, die von Hughes et al.11 zur Differenzierung von HGSOC- und OCCC-Tumorgewebe identifiziert wurden. Von den 76 Signaturproteinen, die von Hughes et al. quantifiziert wurden, wurden 57 in diesem Datensatz koquantifiziert und waren stark korreliert (Spearman Rho = 0,644, p < 0,001) (Abbildung 2B).

Abbildung 1: Zusammenfassung des integrierten Workflows für die automatisierte Auswahl von Geweberegionen von Interesse für die Lasermikrodissektion für die nachgelagerte quantitative Proteomik. Kalibrierfizierer werden auf PEN-Membranobjektträger geschnitten, um Positionsorientierungsdaten von KI-abgeleiteten Segmenten des Gewebe-ROI in der Bildanalysesoftware HALO mit horizontaler Positionierung auf dem LMD-Mikroskop zu registrieren. Der Malleator-Algorithmus wird verwendet, um die kommentierten Segmentierungsdaten über alle Anmerkungsebenen für eine Folie mit der _calib Referenzdatei zusammenzuführen und in eine .xml Datei zu konvertieren, die mit der LMD-Software kompatibel ist. LMD-geerntetes Gewebe für die Proteomikanalyse wird wie zuvor beschriebendurch quantitative Proteomik mit hohem Durchsatz verdaut und analysiert. Abkürzungen: LMD = Laser-Mikrodissektion; ROI = Region des Interesses; TMT = Tandem-Massen-Tag; Quant. = Quantifizierung; Identifizieren. = Identifikation; LC-MS/MS = Flüssigkeitschromatographie-Tandem-Massenspektrometrie. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
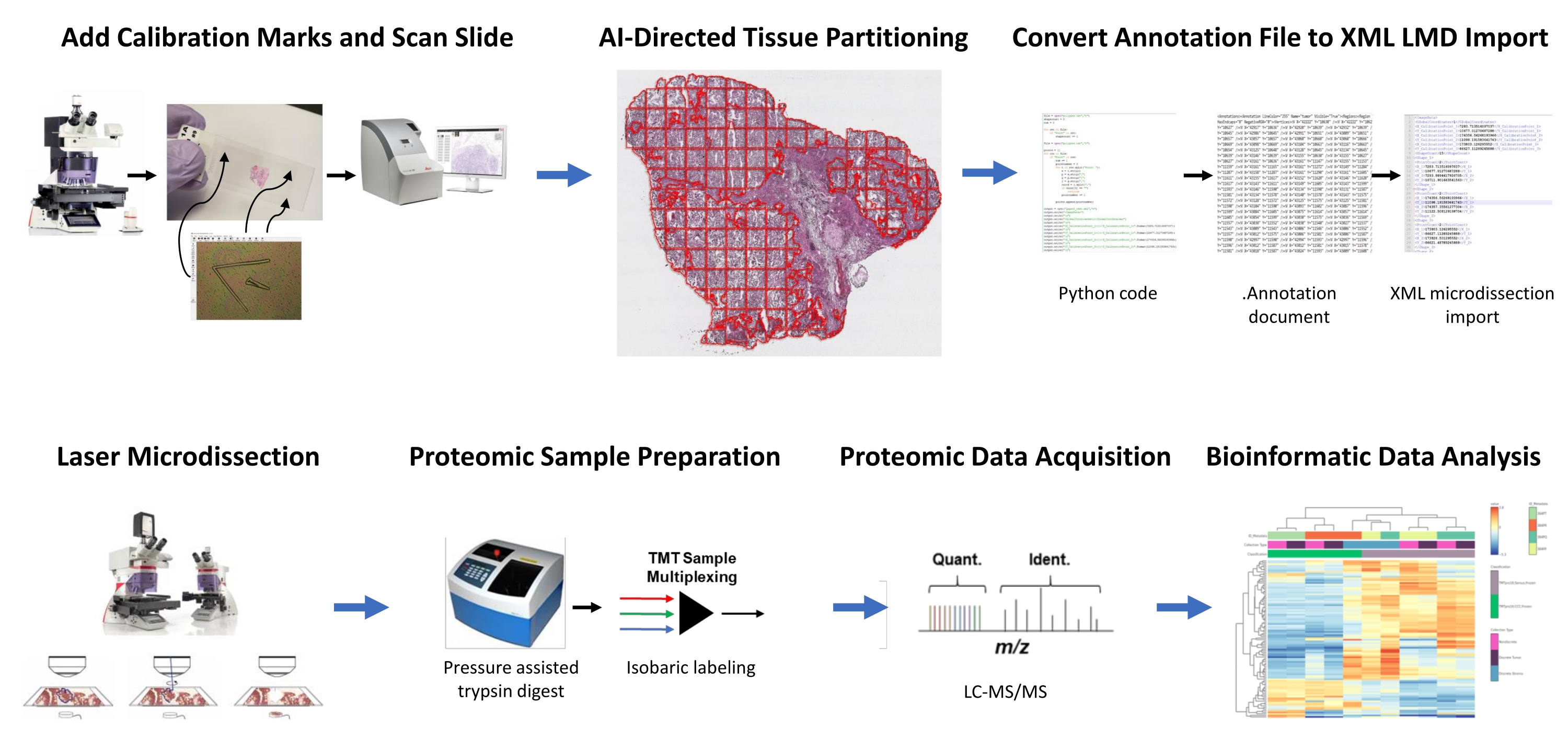{kind=link}

Abbildung 2: Analyse der Proteine in LMD-angereicherten und ganzen Tumorproben. (A) Unüberwachte hierarchische Clusteranalyse der 100 variabelsten Proteine in HGSOC- und OCCC-LMD-angereicherten und ganzen Tumorproben. (B) Korrelation of log2 fold-change protein abundances between HGSOC and OCCC whole tumor harvests in the present study (Mitchell et al., x-axis) and a similar study by Hughes et al. (y-axis)11. Abkürzungen: LMD = Laser-Mikrodissektion; HGSOC = hochgradiger seröser Eierstockkrebs; OCCC = Ovarialkarzinom der klaren Zelle; log 2 FC = log2-transformierteproteomische Häufigkeit. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Ergänzungstabelle S1: Häufigkeiten von 5.971 Proteinen, koquantifiziert über alle LMD-angereicherten und ganzen Tumorproben von HGSOC- und OCCC-Gewebeproben. Abkürzungen: LMD = Laser-Mikrodissektion; HGSOC = hochgradiger seröser Eierstockkrebs; OCCC = Ovarialkarzinom der klaren Zelle. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Supplemental Table S2: Differentially expression proteins (215) in whole tumor collections from HGSOC vs OCCC (LIMMA adj. p < 0.05). Abkürzungen: HGSOC = hochgradiger seröser Eierstockkrebs; OCCC = Ovarialkarzinom der klaren Zelle. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Ergänzende Datei 1: Repräsentative Formlistendatendatei (.sld) mit Standardkalibrator-Fiducials für vier Objektträgerpositionen. Die Datei kann in die LMD-Software importiert werden. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 2: Repräsentative .svs-Bilddatei eines H & E-gefärbten hochauflösenden (20x) Gewebeabschnitts. Die Datei kann mit einer Bildanalysesoftware oder LMD-Software geöffnet und angezeigt werden. Abkürzung: H&E = Hämatoxylin und Eosin; LMD = Laser-Mikrodissektion. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 3: Repräsentative .annotation-Datei von partitionierten ganzen Tumorsegmenten. Die Datei kann in eine Bildanalysesoftware importiert werden. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 4: Repräsentative _calib.annotation-Datei der treuhänderischen Kalibratorsegmente. Koordinateninformationen stellen die orientalische Positionierung der kurzen Kalibratorlinien dar, die von jedem Pfeilspitzen-Fiducial gezeichnet werden. Die Datei kann in eine Bildanalysesoftware importiert werden. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 5: Repräsentative .xml-Datei (Extensible Markup Language), die vom Malleator-Algorithmus generiert wird. Die Datei kann in die Laser-Mikrodissektionssoftware importiert werden. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 6: Repräsentative .annotation-Datei mit partitionierten KI-klassifizierten Segmenten für LMD-angereicherte Sammlungen. Die Datei kann in eine Bildanalysesoftware importiert werden. Abkürzungen: AI = künstliche Intelligenz; LMD = Laser-Mikrodissektion. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
Während es mehrere Präzedenzfälle für Studien gab, die darauf abzielten, Arbeitsabläufe für die Anreicherung von zellulären Zielsubpopulationen aus FFPE und / oder frisch gefrorenem Gewebe und Methoden zur Aufrechterhaltung der Probenqualität während der Verarbeitung zu entwickeln 9,12,13,14,15, besteht ein erheblicher Bedarf, automatisierte Strategien für die Vorbereitung klinischer Gewebeproben für molekulare Analysenzu entwickeln, um die Variabilität zu verringern und erhöhen die Reproduzierbarkeit. Dieser Workflow beschreibt ein standardisiertes, halbautomatisiertes Protokoll, das bestehende Bildanalyse-Softwaretools (siehe Materialtabelle) für die histologieaufgelöste Ernte diskreter Zellpopulationen durch LMD aus klinischen Gewebeproben integriert.
Die räumlich aufgelöste LMD-Anreicherung von ROIs zur Erfassung diskreter Zellpopulationen stellt einen Gewebeverarbeitungsschritt der nächsten Generation vor multiomischen Analysen dar, um die molekulare Charakterisierung und Identifizierung zu verbessern und die Entdeckung zellselektiver Biomarker zu erleichtern. Dieses Protokoll verbessert bestehende Methoden, indem es die oft lange Exposition von Gewebeabschnitten gegenüber der Umgebungsumgebung reduziert, die mit einer manuellen Segmentierung des ROI durch einen Histologen verbunden ist (was >1-2 Stunden vor der LMD-Sammlung dauern kann). Dieser Workflow ermöglicht es stattdessen, den ROI durch KI-gesteuerte Klassifizierung und Segmentierung vorab zu identifizieren. Die Begrenzung der Gewebeverweilzeit verringert falsche Variationen bei der Beurteilung von hochlabilen molekularen Zielen wie Phosphopeptiden und mRNA oder für Antikörper-basierte Analysetechniken, die darauf angewiesen sind, dass sich ein Zielprotein in seiner nativen Konformation befindet, um nachgewiesen zu werden.
Eine Einschränkung der aktuellen Version des Malleator-Algorithmus besteht darin, dass sie nicht mit den vordefinierten Anmerkungsformwerkzeugen kompatibel ist, die von der Bildanalysesoftware bereitgestellt werden (siehe Materialverzeichnis), obwohl zukünftige Updates/Versionen des Algorithmus darauf abzielen, diese Kompatibilität zu verbessern. Die ANMERKUNGSDATEI für Formen, die mit diesen Werkzeugen gezeichnet wurden, enthält nur zwei Sätze von gepaarten x- und y-Koordinaten für jede Anmerkung, ohne die vollständige räumliche Ausrichtung um diese Punkte. Die aktuelle Verwendung dieser Werkzeuge führt dazu, dass die Anmerkungen während des Importvorgangs in gerade Linien umgewandelt werden, die nur durch zwei Punkte definiert sind. Die manuelle Definition von Tissue-ROI-Segmenten ist für eine erfolgreiche Konvertierung in das XML-Format und den LMD-Import erforderlich. Dies kann entweder durch manuelle Definition jedes ROI mit individuellen freihändigen polygonalen Annotationen, die für den Zielbereich spezifisch sind, oder durch Anwendung einer angenäherten kreisförmigen oder rechteckigen Annotation auf alle ROI-Segmente des Gewebes, falls gewünscht, erfolgen und ist mit diesem Workflow kompatibel.
Während der hier vorgestellte Workflow für die proteomische Analyse von frisch gefrorenen menschlichen Krebsgewebeproben demonstriert wurde, kann dieser KI-gesteuerte LMD-Workflow äquivalent mit FFPE-Geweben, nicht-krebsartigen Gewebetypen und solchen aus nicht-menschlichen Quellen verwendet werden. Es kann auch andere nachgelagerte molekulare Profiling-Workflows unterstützen, einschließlich transkriptomischer, genomischer oder phosphoprogeomischer Analysen. Dieser Workflow kann auch andere Anwendungen der Bildanalysesoftware nutzen (siehe Materialtabelle), einschließlich der Funktionen im Zusammenhang mit der Zellzählung oder anderen Analysemodulen, einschließlich des Moduls "Multiplex IHC" oder des "Tissue Microarray (TMA) Add-on". Zukünftige Anwendungen dieses Workflows können auch davon profitieren, die Anzahl der Zellen pro ROI-Segment vorab zu definieren und dadurch äquivalente zelluläre Inputs über mehrere Sammlungen hinweg sicherzustellen, oder indem alternative Methoden zur Definition von zellulären ROIs von Interesse verwendet werden, z. B. durch Immunhistochemie oder Zellsoziologie.
Offenlegungen
T.P.C. ist Mitglied der ThermoFisher Scientific, Inc SAB und erhält Forschungsgelder von AbbVie.
Danksagungen
Die Finanzierung dieses Projekts erfolgte zum Teil durch das Defense Health Program (HU0001-16-2-0006 und HU0001-16-2-00014) an die Uniformed Services University for the Gynecologic Cancer Center of Excellence. Die Sponsoren spielten keine Rolle bei der Gestaltung, Durchführung, Interpretation oder dem Schreiben der Studie. Verzichtserklärung: Die hierin geäußerten Ansichten sind die der Autoren und spiegeln nicht die offizielle Politik des Department of Army / Navy / Air Force, des Department of Defense oder der US-Regierung wider.
Materialien
| Name | Company | Catalog Number | Comments |
| 1260 Infinity II System | Agilent Technologies Inc | Offline LC system | |
| 96 MicroCaps (150uL) in bulk | Pressure Biosciences Inc | MC150-96 | |
| 96 MicroPestles in bulk | Pressure Biosciences Inc | MP-96 | |
| 96 MicroTubes in bulk (no caps) | Pressure Biosciences Inc | MT-96 | |
| 9mm MS Certified Clear Screw Thread Kits | Fisher Scientific | 03-060-058 | Sample vial for offline LC frationation and mass spectrometry |
| Acetonitrile, Optima LC/MS Grade | Fisher Chemical | A995-4 | Mobile phase solvent |
| Aperio AT2 | Leica Microsystems | 23AT2100 | Slide scanner |
| Axygen PCR Tubes with 0.5 mL Flat Cap | Fisher Scientific | 14-222-292 | Sample tubes; size fits PCT tubes and thermocycler |
| Barocycler 2320EXT | Pressure Biosciences Inc | 2320-EXT | Barocycler |
| BCA Protein Assay Kit | Fisher Scientific | P123225 | |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 11836170001 | |
| Easy-nLC 1200 | Thermo Fisher Scientific | Liquid Chromatography | |
| EasyPep Maxi Sample Prep Kit | Thermo Fisher Scientific | NCI5734 | Post-label sample clean up column |
| EASY-SPRAY C18 2UM 50CM X 75 | Fisher Scientific | ES903 | Analytical column |
| Eosin Y Solution Aqueous | Sigma Aldrich | HT110216 | |
| Formic Acid, 99+ % | Thermo Fisher Scientific | 28905 | Mobile phase additive |
| ggplot2 version 3.3.5 | CRAN | https://cran.r-project.org/web/packages/ggplot2/ | |
| HALO | Indica Labs | Image analysis software | |
| IDLE (Integrated Development and Learning Environment) | Python Software Foundation | ||
| iheatmapr version 0.5.1 | CRAN | https://cran.r-project.org/web/packages/iheatmapr/ | |
| iRT Kit | Biognosys | Ki-3002-1 | LC-MS QAQC Standard |
| limma version 3.42.2 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/limma.html | |
| LMD Scanning stage Ultra LMT350 | Leica Microsystems | 11888453 | LMD stage model outfitted with PCT tube holder |
| LMD7 (software version 8.2.3.7603) | Leica Microsystems | LMD apparatus (microscope, laser, camera, PC, tablet) | |
| Mascot Server | Matrix Science | Data analysis software | |
| Mass Spec-Compatible Human Protein Extract, Digest | Promega | V6951 | LC-MS QAQC Standard |
| Mayer’s Hematoxylin Solution | Sigma Aldrich | MHS32 | |
| PEN Membrane Glass Slides | Leica Microsystems | 11532918 | |
| Peptide Retention Time Calibration Mixture | Thermo Fisher Scientific | 88321 | LC-MS QAQC Standard |
| Phosphatase Inhibitor Cocktail 2 | Sigma Aldrich | P5726 | |
| Phosphatase Inhibitor Cocktail 3 | Sigma Aldrich | P0044 | |
| Pierce LTQ Velos ESI Positive Ion Calibration Solution | Thermo Fisher Scientific | 88323 | Instrument calibration solution |
| PM100 C18 3UM 75UMX20MM NV 2PK | Fisher Scientific | 164535 | Pre-column |
| Proteome Discoverer | Thermo Fisher Scientific | OPTON-31040 | Data analysis software |
| Python | Python Software Foundation | ||
| Q Exactive HF-X | Thermo Fisher Scientific | Mass spectrometer | |
| R version 3.6.0 | CRAN | https://cran-archive.r-project.org/bin/windows/base/old/2.6.2/ | |
| RColorBrewer version 1.1-2 | CRAN | https://cran.r-project.org/web/packages/RColorBrewer/ | |
| Soluble Smart Digest Kit | Thermo Fisher Scientific | 3251711 | Digestion reagent |
| TMTpro 16plex Label Reagent Set | Thermo Fisher Scientific | A44520 | isobaric TMT labeling reagents |
| Veriti 60 well thermal cycler | Applied Biosystems | 4384638 | Thermocycler |
| Water, Optima LC/MS Grade | Fisher Chemical | W6-4 | Mobile phase solvent |
| ZORBAX Extend 300 C18, 2.1 x 12.5 mm, 5 µm, guard cartridge (ZGC) | Agilent Technologies Inc | 821125-932 | Offline LC trap column |
| ZORBAX Extend 300 C18, 2.1 x 150 mm, 3.5 µm | Agilent Technologies Inc | 763750-902 | Offline LC analytical column |
Referenzen
- Motohara, T., et al. An evolving story of the metastatic voyage of ovarian cancer cells: cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene. 38 (16), 2885-2898 (2019).
- Aran, D., Sirota, M., Butte, A. J. Systematic pan-cancer analysis of tumour purity. Nature Communications. 6, 8971 (2015).
- Hunt, A. L., et al. Extensive three-dimensional intratumor proteomic heterogeneity revealed by multiregion sampling in high-grade serous ovarian tumor specimens. iScience. 24 (7), 102757 (2021).
- Dou, Y., et al. Proteogenomic characterization of endometrial carcinoma. Cell. 180 (4), 729-748 (2020).
- Zhang, H., et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 166 (3), 755-765 (2016).
- Gillette, M. A., et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell. 182 (1), 200-225 (2020).
- Silvestri, A., et al. Protein pathway biomarker analysis of human cancer reveals requirement for upfront cellular-enrichment processing. Laboratory Investigation. 90 (5), 787-796 (2010).
- Echle, A., et al. Deep learning in cancer pathology: a new generation of clinical biomarkers. British Journal of Cancer. 124 (4), 686-696 (2021).
- Lee, S., et al. Molecular analysis of clinically defined subsets of high-grade serous ovarian cancer. Cell Reports. 31 (2), 107502 (2020).
- Xuan, Y., et al. Standardization and harmonization of distributed multi-center proteotype analysis supporting precision medicine studies. Nature Communications. 11 (1), 5248 (2020).
- Hughes, C. S., et al. Quantitative profiling of single formalin fixed tumour sections: proteomics for translational research. Scientific Reports. 6 (1), 34949 (2016).
- Espina, V., et al. A portrait of tissue phosphoprotein stability in the clinical tissue procurement process. Molecular & Cellular Proteomics. 7 (10), 1998-2018 (2008).
- Espina, V., Heiby, M., Pierobon, M., Liotta, L. A. Laser capture microdissection technology. Expert Review of Molecular Diagnostics. 7 (5), 647-657 (2007).
- Havnar, C. A., et al. Automated dissection protocol for tumor enrichment in low tumor content tissues. Journal of Visualized Experiments. (169), e62394 (2021).
- Mueller, C., et al. One-step preservation of phosphoproteins and tissue morphology at room temperature for diagnostic and research specimens. PLoS One. 6 (8), (2011).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten