
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Experimentelle Quantifizierung von Wechselwirkungen zwischen Wirkstoffabgabesystemen und Zellen in vitro: Ein Leitfaden für die präklinische Bewertung der Nanomedizin
In diesem Artikel
Zusammenfassung
Es wird ein Workflow für die absolute Quantifizierung von Wirkstoffträger-Zell-Interaktionen mittels Durchflusszytometrie demonstriert, um eine bessere rationale Bewertung neuartiger Wirkstoffabgabesysteme zu ermöglichen. Dieser Workflow gilt für Arzneimittelträger aller Art.
Zusammenfassung
Eine wichtige Komponente bei der Entwicklung von Medikamentenabgabesystemen betrifft die Verstärkung oder Abschwächung von Interaktionen mit bestimmten Zelltypen. Zum Beispiel könnte ein Chemotherapeutikum mit einem Antikörper funktionalisiert werden, um die Bindung an Krebszellen zu verbessern ("Targeting") oder mit Polyethylenglykol funktionalisiert werden, um die Immunzellerkennung zu umgehen ("Stealth"). Selbst auf zellulärer Ebene ist die Optimierung der Bindung und Aufnahme eines Wirkstoffträgers ein komplexes biologisches Designproblem. Daher ist es wertvoll zu trennen, wie stark ein neuer Träger mit einer Zelle interagiert, von der funktionellen Wirksamkeit der Fracht eines Trägers, sobald er an diese Zelle geliefert wurde.
Um das chemotherapeutische Beispiel fortzusetzen, "wie gut es an eine Krebszelle bindet" ist ein separates Problem von "wie gut es eine Krebszelle tötet". Quantitative In-vitro-Assays für letztere sind gut etabliert und beruhen in der Regel auf der Messung der Lebensfähigkeit. Die meisten veröffentlichten Forschungsergebnisse zu Zellträgerinteraktionen sind jedoch qualitativ oder semiquantitativ. Im Allgemeinen beruhen diese Messungen auf fluoreszierender Markierung des Trägers und berichten folglich über Wechselwirkungen mit Zellen in relativen oder beliebigen Einheiten. Diese Arbeit kann jedoch standardisiert und mit einer kleinen Anzahl von Charakterisierungsexperimenten absolut quantitativ gemacht werden. Eine solche absolute Quantifizierung ist wertvoll, da sie rationale, klassen- und klasseninterne Vergleiche verschiedener Wirkstoffabgabesysteme ermöglicht - Nanopartikel, Mikropartikel, Viren, Antikörper-Wirkstoff-Konjugate, technisch hergestellte therapeutische Zellen oder extrazelluläre Vesikel.
Darüber hinaus ist die Quantifizierung Voraussetzung für nachfolgende Metaanalysen oder In-silico-Modellierungsansätze . In diesem Artikel werden Videoanleitungen sowie ein Entscheidungsbaum für das Erreichen einer In-vitro-Quantifizierung für Trägerarzneimittelabgabesysteme vorgestellt, die Unterschiede in der Trägergröße und der Kennzeichnungsmodalität berücksichtigen. Darüber hinaus werden weitere Überlegungen zur quantitativen Bewertung fortgeschrittener Wirkstoffabgabesysteme diskutiert. Dies soll als wertvolle Ressource dienen, um die rationale Bewertung und das Design für die nächste Generation von Medikamenten zu verbessern.
Einleitung
Das Design von Medikamentenabgabekonstrukten, die je nach Zelltyp ein spezifisches, entworfenes Verhalten aufweisen, hat erhebliches Forschungsinteresse geweckt. Potenzielle Wirkstoffabgabekonstrukte oder "Träger" umfassen Lipidformulierungen, nanogewachsene anorganische Substanzen, polymere Anordnungen, extrazelluläre Vesikel, funktionalisierte Bakterienzellen oder modifizierte Viren. Alle diese können Organ-, Gewebe- oder Zellspezifität aufgrund physikalischer Eigenschaften, Oberflächeneigenschaften oder technischer chemischer Funktionalisierungen wie Antikörperbindung 1,2 aufweisen.
Ein fast allgegenwärtiger Schritt in der In-vitro-Trägerbewertung besteht darin, Zellen mit einer Suspension zu inkubieren, die den medikamentenbeladenen Träger enthält. Nach der Inkubation wird die Trägerleistung über eine funktionale Anzeige der Leistung der Arzneimittelladung gemessen, z. B. Transfektionseffizienz oder Toxizität. Funktionale Anzeigen sind nützlich, da sie ein nachgelagertes Maß für die Wirksamkeit des Trägers sind. Für komplexere Wirkstoffabgabekonstrukte wird es jedoch immer wichtiger, über funktionale Messwerte hinauszugehen und den Grad der Trägerinteraktion mit der interessierenden Zelle separat zu quantifizieren. Dafür gibt es mehrere Gründe.
Erstens besteht ein zunehmendes Interesse an der Entdeckung (und iterativen Verbesserung) von "Plattform"-Trägertechnologien, die eine Vielzahl von Fracht transportieren können. Zum Beispiel können Lipid-Nanopartikel (LNPs), die RNA verkapseln sollen, eine RNA-Sequenz mit wenigen Einschränkungen gegen eine andere austauschen3. Um die Trägertechnologie iterativ zu verbessern, ist es daher wichtig, ihre Leistung unabhängig von der Frachtfunktionalität zu quantifizieren. Zweitens sind funktionale Anzeigen für die Ladung von Interesse möglicherweise nicht einfach und beeinträchtigen die Fähigkeit, Trägerformulierungen schnell zu iterieren und zu bewerten. Während man eine In-vitro-Optimierung mit einer Modellladung mit einer einfachen funktionellen Anzeige (z. B. Fluoreszenz) durchführen könnte, kann das Ändern der Ladung die biologische Reaktion auf einen Träger4 verändern und daher möglicherweise keine repräsentativen Ergebnisse liefern. Drittens sind viele Träger so konzipiert, dass sie mit einem bestimmten Zelltyp interagieren und von ihm aufgenommen werden. Eine solche Zielerfassung eines Trägers kann und sollte von der Leistung seines therapeutischen Frachtpostenziels unterschieden werden. Um das LNP-Beispiel fortzusetzen, könnte eine RNA-Fracht extrem potent sein, aber wenn das LNP nicht in der Lage ist, an die Zelle zu binden, internalisiert zu werden und die RNA freizusetzen, wird kein nachgeschalteter funktioneller Effekt beobachtet. Dies kann insbesondere für Träger, die schwer zu transfizierende Zelltypen wie T-Zellen5 anvisieren sollen, ein Problem darstellen. Umgekehrt könnte ein LNP extrem effektiv angreifen, aber die RNA-Fracht funktioniert möglicherweise nicht. Ein nachgeschalteter Assay, der nur die Frachtfunktionalität misst, wird nicht in der Lage sein, zwischen diesen beiden Situationen zu unterscheiden, was die Entwicklung und Optimierung von Carrier-Drug-Delivery-Systemen erschwert.
In dieser Arbeit wird diskutiert, wie die Trägerassoziation absolut quantifiziert werden kann. Assoziation ist ein Begriff, der sich auf den experimentell gemessenen Grad der Interaktion zwischen einem Träger und einer Zelle bezieht. Die Assoziation unterscheidet nicht zwischen Membranbindung und Internalisierung - ein Träger kann assoziiert sein, weil er an die Zelloberfläche gebunden ist oder weil die Zelle ihn verinnerlicht hat. Die Assoziation wird häufig im Rahmen von Zellträger-Inkubationsexperimenten gemessen. In der Vergangenheit wurde die Assoziation entweder in beliebigen fluoreszierenden Einheiten (typischerweise "mediane Fluoreszenzintensität" oder MFI) oder als "prozentuale Assoziation" berichtet, Metriken, deren Einschränkungen zuvor diskutiert wurden6. Kurz gesagt, diese Messungen sind aufgrund von Unterschieden in den experimentellen Protokollen, Durchflusszytometereinstellungen und den Markierungsintensitäten verschiedener Träger nicht zwischen Experimenten, Laboratorien und Wirkstoffträgern vergleichbar. Ersteres wurde durch Kalibrierung des Zytometers überwunden, wodurch das relative Maß des MFI in ein absolut quantitatives Maß für Fluoreszenz umgewandeltwurde 7. Diese Methode berücksichtigt jedoch nicht die Variabilität in der Markierungsintensität verschiedener Träger und erlaubt daher keinen rationalen Vergleich verschiedener Trägerleistungen in einer Zielzelle der Wahl8.
Hier wird gezeigt, wie man praktisch von relativen, beliebigen fluoreszierenden Einheiten in die absolute quantitative Metrik der "Anzahl der Träger pro Zelle" umrechnen kann, indem eine kleine Anzahl zusätzlicher Charakterisierungsexperimente durchgeführt wird. Wenn eine andere Metrik der Trägerkonzentration gewünscht wird (z. B. Trägermasse pro Zelle oder Trägervolumen pro Zelle), ist es einfach, von Trägern pro Zelle umzurechnen, sofern eine Trägercharakterisierung durchgeführt wurde. Aus Gründen der Kürze und um Jargon zu vermeiden, wird das Wort "Träger" in dieser Arbeit verwendet, um sich auf die große Auswahl an Medikamentenabgabekonstrukten zu beziehen. Diese Quantifizierungstechniken sind gleichermaßen anwendbar, unabhängig davon, ob sie auf ein nanotechnologisch hergestelltes Goldpartikel oder ein biotechnologisch hergestelltes Bakterium angewendet werden.
Einige wenige Fakten ermöglichen die Umwandlung von beliebigen fluoreszierenden Einheiten in Träger pro Zelle. Erstens ist die gemessene Fluoreszenzintensität proportional zur Konzentration eines Fluorophors9 (oder eines fluoreszenzmarkierten Trägers), vorausgesetzt, die Fluoreszenz liegt innerhalb der Nachweisgrenzen des Instruments und die Instrumentierungseinstellungen sind gleich. Wenn also die Fluoreszenz eines Trägers und die Fluoreszenz einer Probe bekannt sind, kann man bestimmen, wie viele Träger in dieser Probe vorhanden sind, wenn alle Messungen unter den gleichen Einstellungen und Bedingungen durchgeführt wurden. Insbesondere bei kleineren Trägern ist es jedoch möglicherweise nicht möglich, Trägerfluoreszenz, Zellautofluoreszenz und zellassoziierte Trägerfluoreszenz auf demselben Instrument mit den gleichen Einstellungen zu messen. In diesem Fall gibt es eine zweite Anforderung, um die Umrechnung zwischen gemessener Fluoreszenz auf einem Instrument und gemessener Fluoreszenz auf einem anderen Instrument zu ermöglichen. Zu diesem Zweck kann eine Standardkurve der Fluorophorkonzentration erstellt werden, um die Fluoreszenzintensität auf beiden Instrumenten zu messen, wobei der MESF-Standard9 (Molecules of Equivalent Soluble Fluorochrome) genutzt wird. Dies ermöglicht dann die Messung der Trägerfluoreszenz in großen Mengen auf einem Nicht-Zytometer, eine Messung, die an Trägern beliebiger Größe oder Eigenschaft durchgeführt werden kann. Wenn eine solche Massenquantifizierung an einer Trägersuspension bekannter Konzentration durchgeführt wird, kann die Anzahl der Träger pro Zelle einer Probe erneut berechnet werden.
Während diese Arbeit den Prozess zur Messung der Trägerassoziation (bestimmt durch die gemessene Fluoreszenzintensität) demonstriert, könnte ein analoges Protokoll für andere Messungen der Zell-Träger-Interaktion durchgeführt werden (z. B. ein experimentelles Protokoll, das internalisierte und membrangebundene Träger unterscheidet). Darüber hinaus wäre dieses Protokoll weitgehend gleich, wenn die Assoziation durch einen nicht-fluoreszierenden Assay (z. B. durch Massenzytometrie) gemessen würde.
Protokoll
1. Auswahl des passenden Streams
- Befolgen Sie die in Abbildung 1 dargestellte Entscheidungsstruktur, um den besten Workflow (Stream) (Abbildung 2) für den verwendeten Versuchsaufbau zu ermitteln. Weitere Kommentare zu dieser Stream-Auswahl finden Sie in der Diskussion.
- Wenn Sie dem Zytometer-Stream folgen, fahren Sie mit den Schritten 2.1.1-2.2.7 fort. Wenn Sie dem Massenstream folgen, fahren Sie mit den Schritten 3.1.1.1-3.1.5.7 fort.

Abbildung 1: Entscheidungsbaum des Workstreams. Die Entscheidung, welcher Stream verwendet werden soll, hängt in erster Linie von der Art des interessierenden Trägers ab. Größere Träger und Träger mit hohen Streueigenschaften können auf Zytometern leichter einzeln detektiert werden und eignen sich somit für die Quantifizierung mit dem Cytometer Stream. Der Bulk Stream ist für alle anderen Trägertypen geeignet. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 2: Übersicht der Workstreams. Dieses Protokoll ist in zwei verschiedene Streams aufgeteilt. Der Cytometer Stream verwendet ein empfindliches Zytometer, um die Träger in Suspension zu zählen, ihre individuelle Fluoreszenz zu messen und dann die Fluoreszenz von Zellen zu bestimmen, die mit Trägern inkubiert wurden. Der Bulk Stream verwendet nicht-zytometrische Techniken wie die Nanopartikel-Tracking-Analyse, um die Träger in Suspension zu zählen. Die individuelle Trägerfluoreszenz wird dann mit einem Mikrotiterplatten-Reader oder Spektrofluorometer quantifiziert. Die Verwendung des Durchflusszytometers beschränkt sich daher auf die Messung der endgültigen Fluoreszenz von Zellen, die mit Trägern inkubiert wurden, eine Messung, die auf einem breiteren Bereich von Zytometern durchgeführt werden kann und unabhängig vom verwendeten Trägertyp ist. Abkürzungen: MESF = Molecules of Equivalent Soluble Fluorochrome; MFI = mediane Fluoreszenzintensität. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
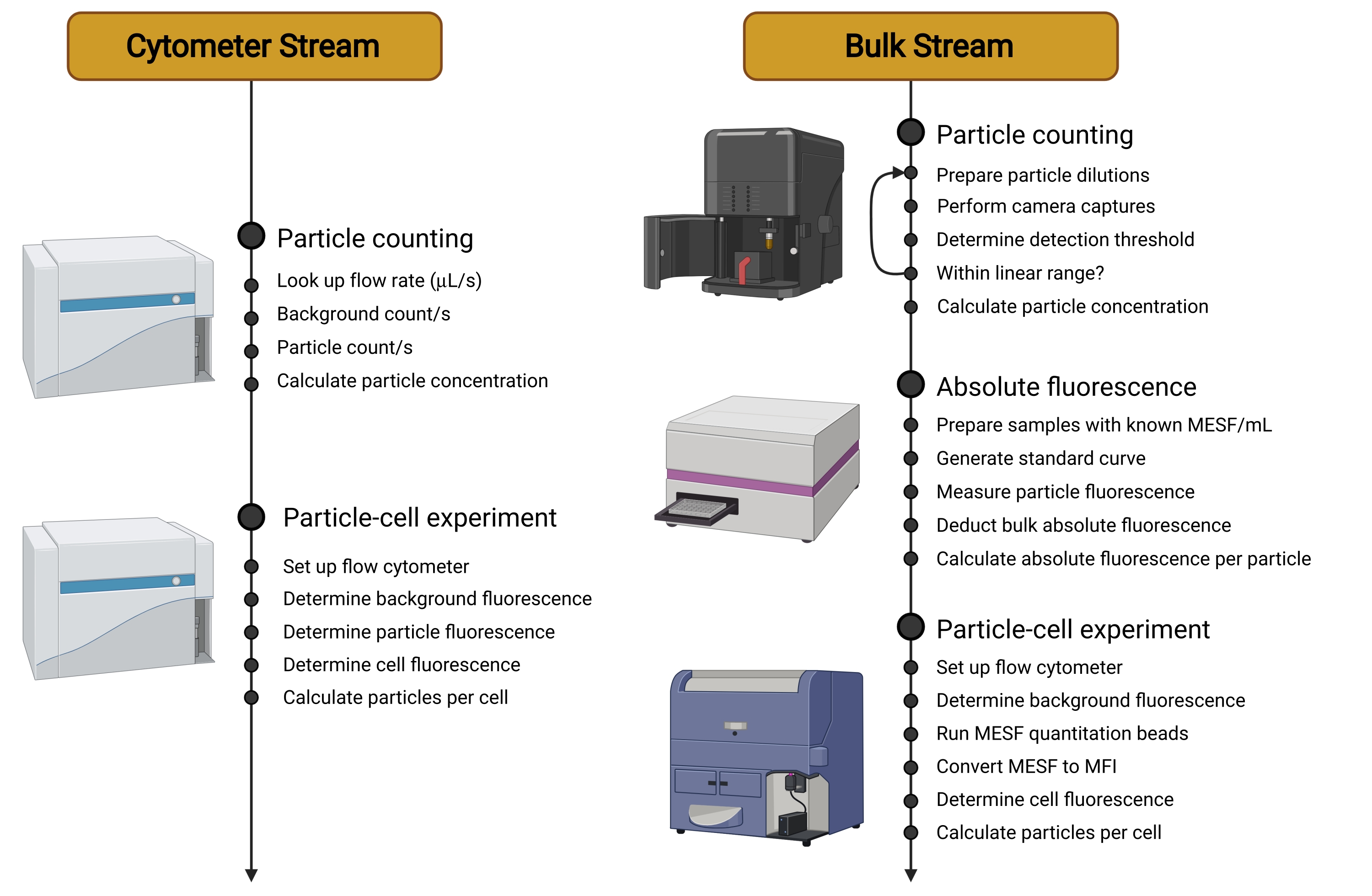{kind=link}
2. Der Zytometerstrom
- Carrier-Zählung
HINWEIS: Jedes Durchflusszytometer kann für diese Messung verwendet werden, sofern die Durchflussrate (μL/s) bekannt ist. Wenn die Durchflussrate unbekannt ist und nicht bestimmt werden kann, fahren Sie mit diesem Schritt nicht fort. Fahren Sie stattdessen mit Schritt 3.1 fort. Die Zählung der Träger in Suspension ermöglicht eine genaue und reproduzierbare Bestimmung der Anzahl der in jedem Zellexperiment inkubierten Träger.- Richten Sie das Zytometer so ein, dass Träger sowohl durch einen optischen Streukanal (typischerweise Seitenstreuung [SSC]) als auch durch Fluoreszenz erkannt werden. Stellen Sie sicher, dass Sie den Schwellenwert anpassen, um die Erkennung der Träger zu ermöglichen.
HINWEIS: Iterationen durch verschiedene optische Streukanäle können erforderlich sein, wenn SSC kein klares Signal liefert (z. B. Vorwärtsstreuung [FSC]). - Führen Sie eine reine Verdünnungsprobe aus, um die Anzahl der Hintergrundereignisse sowohl im SSC- als auch im Fluoreszenzkanal zu quantifizieren.
HINWEIS: Die ideale Anzahl von Hintergrundereignissen beträgt <100 Ereignisse/s. - Bereiten Sie die Träger für die Durchflusszytometrie vor.
- Stellen Sie sicher, dass die Träger je nach Trägersystem durch Wirbeln oder Beschallung gut aufgehängt sind.
- Wenn möglich, stellen Sie sicher, dass die Trägerkonzentration zwischen 1.000 Trägern/μL und 10.000 Trägern/μL liegt. Eine Ereignisanzahl von ein bis zwei Größenordnungen höher als der Hintergrund ist ein guter Anfang. Wenn die Größenordnung der Trägerkonzentration unbekannt ist, ist es ein guter Anfang, eine Verdünnung von 1:1.000 aus dem Bestand herzustellen. Verwenden Sie die ersten Ergebnisse als Feedback, um zukünftige Probenverdünnungen zu informieren.
HINWEIS: Eine trübe Suspension ist im Allgemeinen zu konzentriert.
- Laden Sie die erste Trägerprobe auf das Zytometer und starten Sie die Aufnahme.
- Vergleichen Sie die Anzahl der Ereignisse, die sich sowohl aus dem SSC- als auch aus dem Fluoreszenzkanal ergeben. Diese sollten ungefähr gleich sein (<10% Unterschied). Wenn nicht, überprüfen Sie die Zytometereinstellungen, z. B. die Einstellungen der Photomultiplierröhre (PMT) und die Laserintensität für den Fluoreszenzkanal. Alternativ können Sie andere Methoden wie konfokale Mikroskopie verwenden, um zu validieren, dass die fluoreszierende Markierung der Träger vorhanden und einheitlich ist.
- Wiederholen Sie die Schritte 2.1.3-2.1.5 zwei oder mehr weitere Male mit unterschiedlichen Verdünnungen aus der Aktie. Stellen Sie sicher, dass die Ereignisanzahl in jeder Stichprobe mindestens eine Größenordnung höher ist als die Anzahl der Hintergrundereignisse.
- Stellen Sie sicher, dass drei oder mehr Proben einen linearen Trend zeigen, d. h., eine zweifache Probenverdünnung sollte zu einer entsprechenden zweifachen Verringerung der gemessenen Trägerkonzentration führen.
- Verwenden Sie die Proben innerhalb des linearen Bereichs, die entsprechenden Verdünnungsfaktoren und die bekannte Zytometer-Flussrate, um die Stoffträgerkonzentration gemäß Gleichung (1) zu berechnen:
 (1)
(1)
wobei C die Trägerkonzentration in Trägern/ml ist. Es wird empfohlen, die Ereignisanzahl zu verwenden, die von der optischen Streudetektion abgeleitet wird, anstelle von Fluoreszenz.
- Richten Sie das Zytometer so ein, dass Träger sowohl durch einen optischen Streukanal (typischerweise Seitenstreuung [SSC]) als auch durch Fluoreszenz erkannt werden. Stellen Sie sicher, dass Sie den Schwellenwert anpassen, um die Erkennung der Träger zu ermöglichen.
- Durchflusszytometrie-Auslesung des Trägerzellexperiments, einschließlich Bestimmung der Fluoreszenzintensität pro Träger
HINWEIS: Idealerweise wird die Fluoreszenzintensität pro Träger so nah wie möglich am Trägerzellexperiment bestimmt. Damit soll sichergestellt werden, dass die für einzelne Träger erhaltenen MFIs direkt mit den MFIs der mit den Trägern assoziierten Zellen verglichen werden können. In der Praxis liefert ein Zytometer in der Regel ähnliche Ergebnisse, wenn es an aufeinanderfolgenden Tagen mit den gleichen PMT-Spannungen verwendet wird, dies kann jedoch nicht garantiert werden.- Entwerfen Sie das Trägerzellexperiment. Verwenden Sie die in Schritt 2.1 bestimmte Trägerkonzentration, um die gewünschte Dosis von Trägern zu verabreichen.
- Richten Sie das Durchflusszytometer für das abschließende Trägerzellexperiment ein, indem Sie die optimalen PMT-Spannungseinstellungen in den relevanten Kanälen bestimmen. Legen Sie die Schwellenwerte fest, um die Netzbetreibererkennung zuzulassen.
- Führen Sie die Träger in Suspension aus, um die Fluoreszenzintensität pro Träger unter den aktuellen PMT-Einstellungen zu bestimmen.
- Ändern Sie bei Bedarf die Zytometerschwellen, um die Zellen und nicht die Träger zu erkennen.
- Führen Sie eine Negativkontrollprobe durch - Zellen, die nicht mit Trägern inkubiert wurden -, um die Hintergrundfluoreszenz (Autofluoreszenz) der Zellen zu bestimmen.
- Führen Sie die Trägerzellenproben durch, um die Fluoreszenzintensität pro Zelle zu bestimmen. Diese Fluoreszenz ist eine lineare Kombination aus zellulärer Autofluoreszenz und dem Vorhandensein von fluoreszierenden Trägern.
- Berechnen Sie die Anzahl der Träger pro Zelle mit der folgenden Gleichung (2):
 (2)
(2)
wobei P-assoc die Anzahl der pro Zelle assoziierten Träger ist, FI-Zelle das MFI von Zellen, die mit Trägern inkubiert wurden, FI-Hintergrund das MFI von Zellen, die nicht mit Trägern inkubiert wurden, und FI-Träger das MFI von Trägern in Suspension ist.
3. Der Massenstrom
- Trägerzählung: Nanopartikel-Tracking-Analyse
HINWEIS: Im Bulk Stream ist die Ladungsträgerzählung ein notwendiger Schritt, um die absolute Fluoreszenzintensität pro Träger zu quantifizieren (siehe Schritt 3.1.4). Darüber hinaus ermöglicht die Zählung der Träger in Suspension die genaue und reproduzierbare Bestimmung der Anzahl der in jedem Zellexperiment inkubierten Träger.- Präparat
- Montieren Sie die Durchflusszelle auf dem Lasermodul und verriegeln Sie das gesamte Lasermodul im Instrument.
- Spülen Sie die Durchflusszelle langsam (nicht schneller als 0,1 ml / s) mit ~ 1 ml destilliertem Wasser. Wenn sich Blasen in der Durchflusszelle bilden, ziehen Sie die Suspension teilweise zurück, um die Blase mit der Luft-Flüssigkeits-Grenzfläche zu verschmelzen, bevor Sie fortfahren.
- Starten Sie die Kamera ungefähr auf halbem Weg durch die Spülung; Stellen Sie sicher, dass Trägerablagerungen ausgewaschen sind. Wählen Sie Aufnahme , um die Registerkarte Aufnahmeeinstellungen zu öffnen, und klicken Sie auf Kamera starten.
- Trocknen Sie das System mit 1 mL Luft. Wenn statische Träger auf dem Bildschirm sichtbar sind, reinigen Sie die Durchflusszelle gemäß den Anweisungen des Herstellers.
- Bereiten Sie die Träger für die Nanopartikel-Tracking-Analyse vor, indem Sie sicherstellen, dass die Träger je nach Trägersystem durch Wirbeln oder Beschallung gut suspendiert sind. Wenn die Größenordnung der Stammkonzentration unbekannt ist, bereiten Sie eine Verdünnung von 1:100 aus der Brühe vor und verwenden Sie die ersten Ergebnisse als Rückmeldung, um zukünftige Probenverdünnungen zu informieren. Die Träger werden in Wasser und nicht in phosphatgepufferter Kochsalzlösung (PBS) verdünnt, um mindestens ~0,6-1 ml jeder Probe mit einer Trägerkonzentration zwischen 1 × 107 Trägern /ml und 1 × 109 Trägern/ml herzustellen.
HINWEIS: Eine trübe Suspension ist im Allgemeinen zu konzentriert. Puffer und Salze können hohe Hintergrundgeräusche erzeugen.
- Messung
- Nehmen Sie das Lasermodul heraus und stellen Sie es aufrecht.
- Ziehen Sie die erste Trägerprobe in eine 1-ml-Spritze. Befestigen Sie die Spritze am Schlaucheinlass und laden Sie die Probe vorsichtig in die Durchflusszelle. Wenn sich Blasen in der Durchflusszelle bilden, ziehen Sie die Suspension teilweise zurück, um die Blase mit der Luft-Flüssigkeits-Grenzfläche zu verschmelzen, bevor Sie fortfahren. Stellen Sie sicher, dass die gesamte Durchflusszelle mit Flüssigkeit gefüllt ist, und halten Sie dann an.
- Passen Sie den Kamerafokus bei Bedarf an, um einzelne Träger zu visualisieren. Nehmen Sie grobe Fokuseinstellungen mit dem Drehknopf auf der rechten Seite des Instruments vor. Nehmen Sie feinere Anpassungen vor, indem Sie die Registerkarte Hardware | Pumpen/Stufe. Ändern Sie den Fokus, indem Sie den Fokusschieberegler anpassen.
- Stellen Sie die Kamerastufe ein, um sicherzustellen, dass es keine Übersättigung gibt. Wählen Sie auf der Registerkarte Aufnahme die optimale Kamerastufe aus, indem Sie den Schieberegler anpassen.
- Wenn das Gerät mit diesem Zubehör ausgestattet ist, legen Sie die Spritze mit der Trägerprobe in die Spritzenpumpe , um einen kontinuierlichen Probenfluss während der Messungen sicherzustellen.
- Wählen Sie auf der Registerkarte SOP die Option Standardmessung aus, um fünf Aufnahmen von jeweils 30 s zu machen. Geben Sie den Basisdateinamen ein und fügen Sie bei Bedarf zusätzliche Beispielinformationen hinzu, indem Sie auf die Schaltfläche Erweitert klicken (wodurch ein modaler Dialog mit einer Vielzahl von Auswahlmöglichkeiten geöffnet wird).
HINWEIS: Wenn ein Verdünnungsfaktor eingegeben wird, wird die endgültige Messung der Trägerkonzentration automatisch von der Software angepasst. Von der Eingabe dieses Faktors wird abgeraten. Führen Sie stattdessen die Einstellung manuell durch, was die Analyse erleichtert und die Beurteilung ermöglicht, ob jede Verdünnung in den dynamischen Bereich des Geräts fällt (Schritt 3.1.3.4). - Klicken Sie auf Create (Skript erstellen ) und Run Script (Skript ausführen ) und warten Sie, bis ein Popup-Fenster mit der Aufforderung Please advance sample (Bitte erweiterte Beispiel) angezeigt wird.
- Wenn Sie die Spritzenpumpe verwenden, wählen Sie die Registerkarte Hardware | Registerkarte " Spritzenpumpe" | Stellen Sie die Infusionsrate auf 30-80 ein und drücken Sie Infusion. Wenn Sie die Spritzenpumpe nicht verwenden, ziehen Sie die Probe manuell vor.
- Wählen Sie im Popup-Fenster OK aus, um die Aufnahme zu starten. Überprüfen Sie nach jeder der fünf Aufnahmen, wenn das Popup-Fenster Please advance sample (Bitte erweiterte Probe) erneut angezeigt wird, entweder manuell oder über die Spritzenpumpe, ob sich die Probe noch durch die Durchflusszelle bewegt. Wählen Sie dann OK , um mit der nächsten Aufnahme fortzufahren.
HINWEIS: Nach fünf Aufnahmen öffnet die Software automatisch die Registerkarte Prozess und öffnet ein Popup-Fenster, in dem Sie aufgefordert werden, die Prozesseinstellungen anzupassen.
- Analyse
- Passen Sie auf der Registerkarte Prozess den Schieberegler Erkennungsschwellenwert (zwischen 4 und 8) an, um verschiedene auf dem Bildschirm sichtbare Träger korrekt zu identifizieren. Passen Sie auch die Bildschirmverstärkung an, um die Visualisierung zu unterstützen. Es hat keinen Einfluss auf die nachgelagerte Analyse. Verwenden Sie den Schieberegler unter dem Aufnahmebildschirm, um durch mehrere Frames des Videos zu scrollen, um die Einstellung des Erkennungsschwellenwerts zu unterstützen.
HINWEIS: Die Nachweisschwelle sollte einmal festgelegt und anschließend nicht zwischen Messungen oder Proben geändert werden. - Drücken Sie im Popup-Fenster (Hinweis nach Schritt 3.1.2.9) OK , um die Tracking-Analyse zu starten. Überwachen Sie den Fortschritt der Analyse, indem Sie auf die Registerkarte Analyse | Einzelne Analyseregisterkarte .
- Sobald die Analyse abgeschlossen ist, suchen Sie nach einer Eingabeaufforderung für Exporteinstellungen, in der standardmäßig PDF einschließen und Experimentzusammenfassung einschließen ausgewählt sein sollte. Wählen Sie beliebige andere Exportformate aus.
- Um sicherzustellen, dass die gemessene Konzentration zuverlässig ist, überprüfen Sie im Abschnitt Ergebnisse des PDF-Datenexports, ob die gemessene Trägerkonzentration zwischen 1 × 107 Trägern/ml und 1 × 109 Trägern/ml liegt - dem Dynamikbereich des Geräts - und überprüfen Sie, ob unter dem Konzentrationsmessergebnis Fehlermeldungen oder Warnmeldungen angezeigt werden.
- Wiederholen Sie die Schritte 3.1.2.1-3.1.3.4 zwei oder mehr Male mit unterschiedlichen Verdünnungen aus der Aktie. Stellen Sie sicher, dass die Konzentration jeder Probe innerhalb des linearen Bereichs des Geräts liegt.
- Wählen Sie drei oder mehr Proben aus, die einen linearen Trend zeigen, d.h. eine zweifache Probenverdünnung sollte zu einer entsprechenden zweifachen Verringerung der gemessenen Trägerkonzentration führen. Verwenden Sie die ausgewählten Proben und die entsprechenden Verdünnungsfaktoren, um die Trägerkonzentration zu berechnen.
- Passen Sie auf der Registerkarte Prozess den Schieberegler Erkennungsschwellenwert (zwischen 4 und 8) an, um verschiedene auf dem Bildschirm sichtbare Träger korrekt zu identifizieren. Passen Sie auch die Bildschirmverstärkung an, um die Visualisierung zu unterstützen. Es hat keinen Einfluss auf die nachgelagerte Analyse. Verwenden Sie den Schieberegler unter dem Aufnahmebildschirm, um durch mehrere Frames des Videos zu scrollen, um die Einstellung des Erkennungsschwellenwerts zu unterstützen.
- Bestimmung der absoluten Fluoreszenzintensität pro Träger
HINWEIS: Da die Fluoreszenz einzelner Träger in diesem Strom nicht direkt charakterisiert werden kann, wird die Fluoreszenzintensität in großen Mengen quantifiziert. Diese Methode beruht auf der Tatsache, dass die Fluoreszenzintensität linear mit der Fluorochromkonzentration nach dem Lambert-Beer-Gesetz zusammenhängt. Wenn eine solche Massenquantifizierung von Trägern in Suspension an einer Suspension bekannter Trägerkonzentration durchgeführt wird (siehe Schritt 3.1), kann die Fluoreszenz pro Träger abgeleitet werden. Dieser Schritt kann entweder auf einem Fluoreszenzplattenleser oder einem Spektrofluorometer durchgeführt werden. Die Fluoreszenzintensität wird mit einer Standardkurve von Proben mit bekannter absoluter Fluoreszenz verglichen, die in der Anzahl der MESFs angegeben ist.- Verwenden Sie eine Lösung des freien Fluorochroms, um den Träger zu markieren: Resuspendieren Sie den Farbstoff im entsprechenden Puffer (z. B. DMSO) und führen Sie weitere Verdünnungen im selben Puffer wie das Trägerverdünnungsmittel durch. Alternativ kann eine Lösung eines Antikörpers verwendet werden, der mit dem Fluorochrom konjugiert ist. Berechnen Sie die Konzentration der Stammlösung (MESF/ml) aus der Konzentration in mg/ml, dem Molekulargewicht in mg/Mol und der Avogadro-Zahl unter Verwendung von Gleichung (3). Führen Sie eine serielle Verdünnung im Trägerverdünnungsmittel durch, um Standardkurvenproben zu erzeugen.
 (3)
(3)
HINWEIS: Verwenden Sie einen fluorochromkonjugierten Antikörper nur, wenn der Grad der Markierung, d. h. das molare Verhältnis zwischen Fluorochrom und Antikörper in der Lösung, bekannt ist. Erzeugen Sie zunächst eine Standardkurve mit einem weiten Bereich, da die Fluoreszenzintensität der Trägerprobe noch unbekannt ist. Von hier aus schränken Sie es ein, um den erforderlichen Bereich einzubeziehen. - Bereiten Sie die Trägerproben vor.
HINWEIS: Es empfiehlt sich, zwei oder mehr Trägerverdünnungen zu testen, um zu validieren, dass die Messungen linear sind und in den Bereich der Standardkurve fallen. - Messen Sie die Fluoreszenz gleicher Volumina jeder Probe, d. H. Sowohl Träger- als auch Standardkurven.
- Generieren Sie eine Standardkurve und ziehen Sie die absolute Fluoreszenzintensität in MESF/ml für die gemessenen Trägerproben ab.
- Berechnen Sie die absolute Fluoreszenzintensität pro Träger (MESF/Träger), indem Sie die Massenfluoreszenz (MESF/ml) durch die Trägerkonzentration (Träger/ml) dividieren, wie in Gleichung (4):
 (4)
(4)
- Verwenden Sie eine Lösung des freien Fluorochroms, um den Träger zu markieren: Resuspendieren Sie den Farbstoff im entsprechenden Puffer (z. B. DMSO) und führen Sie weitere Verdünnungen im selben Puffer wie das Trägerverdünnungsmittel durch. Alternativ kann eine Lösung eines Antikörpers verwendet werden, der mit dem Fluorochrom konjugiert ist. Berechnen Sie die Konzentration der Stammlösung (MESF/ml) aus der Konzentration in mg/ml, dem Molekulargewicht in mg/Mol und der Avogadro-Zahl unter Verwendung von Gleichung (3). Führen Sie eine serielle Verdünnung im Trägerverdünnungsmittel durch, um Standardkurvenproben zu erzeugen.
- Zellexperiment (einschließlich Bestimmung der äquivalenten Fluoreszenzintensität pro Träger)
HINWEIS: In diesem Schritt werden Durchflusszytometrie-Quantifizierungsperlen verwendet, um eine Standardkurve der Beziehung zwischen MESF und MFI zu generieren. Diese Quantifizierungskügelchen bestehen aus mehreren Perlenpopulationen mit einer bekannten Anzahl von MESF pro Perle, und diese einzelnen Perlen können von jedem Zytometer detektiert werden. Idealerweise wird die MESF-Standardkurve gleichzeitig mit dem Auslesen der Trägerzellexperimente bestimmt. Dadurch soll sichergestellt werden, dass die für einzelne Träger berechneten MFI-Werte direkt mit dem MFI von Zellen verglichen werden können, die den Trägern zugeordnet sind. In der Praxis liefert ein Zytometer in der Regel ähnliche Ergebnisse, wenn es an aufeinanderfolgenden Tagen mit den gleichen PMT-Spannungen verwendet wird, dies kann jedoch nicht garantiert werden.- Entwerfen Sie das Trägerzellexperiment. Verwenden Sie die in Abschnitt 3.1.3 bestimmte Trägerkonzentration, um die gewünschte Dosis von Trägerstoffen zu verabreichen.
- Richten Sie das Durchflusszytometer für das abschließende Trägerzellexperiment ein, indem Sie die optimalen PMT-Spannungseinstellungen in den relevanten Kanälen bestimmen.
- Führen Sie eine negative Kontrollprobe durch, dh Zellen, die nicht mit Trägern inkubiert wurden, um die Hintergrundfluoreszenz zu bestimmen.
- Bereiten Sie die Durchflusszytometrie-Quantifizierungsperlen vor und resuspendieren Sie sie. Verwenden Sie den gleichen Puffer wie für die Zellproben (z. B. PBS). Wenn die Perlenpopulationen separat bereitgestellt werden, bündeln Sie sie zusammen.
- Führen Sie die Durchflusszytometrie-Quantifizierungsperlenprobe durch.
- Führen Sie die Trägerzellenproben durch, um die Fluoreszenzintensität pro Zelle zu bestimmen.
- Verwenden Sie die Quantifizierungsperlenprobe, um eine Standardkurve zu erstellen, die die absolute Fluoreszenzintensität (MESF) in MFI umwandelt. Verwenden Sie diese Standardkurve und die Ergebnisse aus Schritt 3.1.4, um den theoretischen MFI der Träger zu berechnen. Berechnen Sie die Anzahl der Träger pro Zelle mit Gleichung (5):
 (5)
(5)
Dabei ist P-Assoc die Anzahl der pro Zelle assoziierten Träger, FI-Zelle ist das MFI von Zellen, die mit Trägern inkubiert wurden, FI-Hintergrund ist das MFI von Zellen, die nicht mit Trägern inkubiert wurden, und FI-Träger ist der berechnete MFI von Trägern in Suspension (Schritt 3.1.4).
- Präparat
Ergebnisse
Wie bereits erwähnt, erfordern verschiedene Wirkstoffträgertypen die Verwendung unterschiedlicher Techniken zur absoluten Quantifizierung der Zellträgerassoziation. Zum Beispiel sind 633 nm disulfidstabilisierte Poly(methacrylsäure) (PMA SH) Kernschalenpartikel groß und dicht genug für die Detektion mit einem empfindlichen Durchflusszytometer. Als solche wurden diese Partikel fluoreszierend markiert, dann mit Seitenwinkellichtstreuung (SALS, analog zu SSC) sowie dem entsprechenden Fluoreszenzkanal angegu...
Diskussion
Die Charakterisierung der Wechselwirkungen zwischen Wirkstoffträgern und Zellen gewinnt bei der Entwicklung neuartiger Wirkstoffabgabesysteme zunehmend an Bedeutung. Insbesondere um die rationale Bewertung und den Vergleich verschiedener Trägerkonstrukte zu ermöglichen, ist die absolute Quantifizierung der Leistung des Trägers bei der Interaktion mit Ziel- und Off-Target-Zellen von entscheidender Bedeutung. Dieses Protokoll beschreibt eine Zwei-Stream-Methodik, die es jedem Forscher, der mit einem Wirkstoffträger ar...
Offenlegungen
Die Autoren haben keine Interessenkonflikte offenzulegen.
Danksagungen
Diese Arbeit wurde vom Australian National Health and Medical Research Council (NHMRC; Program Grant No. GNT1149990), dem Australian Centre for HIV and Hepatitis Virology Research (ACH2), sowie eine Schenkung aus dem Nachlass von Réjane Louise Langlois. F.C. erkennt die Vergabe eines National Health and Medical Research Council (NHMRC) Senior Principal Research Fellowship (GNT1135806) an. Abbildung 1 und Abbildung 2 wurden mit BioRender.com erstellt.
Materialien
| Name | Company | Catalog Number | Comments |
| Alexa Fluor 647 C2 Maleimide | Invitrogen | A20347 | pH-stable dye used to label 150 nm, 235 nm, or 633 nm PMASH carriers; example of good dye to use in cell-carrier association studies |
| Apogee A50 Microflow | Apogee | Sensitive flow cytometer capable of detecting small carriers for counting | |
| CytoFLEX S Flow Cytometer | Beckman Coulter | Sensitive flow cytometer capable of detecting small carriers for counting and read out for final cell-barrier experiments | |
| FCS Express | De Novo Software | Software used to analyze flow cytometry data, i.e., perform gating and derive median fluorescence intensity values of populations of choice. Alternatives include FlowJo, OMIQ, Python | |
| Infinite 200 PRO | Tecan Lifesciences | Standard microplate reader instrument used for bulk fluorescence measurements of carriers in solution | |
| LSRFortessa Cell Analyzer | BD Biosciences | Less sensitive flow cytometer, but one more generally available to researchers. Can be used to read out final cell-carrier experiment | |
| NanoSight NS300 | Malvern Panalytical | Instrument used for Nanoparticle Tracking Analysis | |
| Prism 8 | GraphPad | Software used to graph and calculate standard curves. Alternatives include Microsoft Excel, Origin, Minitab, Python amongst many others | |
| Quantum MESF kits Alexa Fluor 647 | Bangs Laboratories | 647 | Absolute quantitation beads for flow cytometery. Used to convert fluorescence intensities measured in bulk on a microplate reader to fluorescence intensities measured on a flow cytometer using the MESF standard |
Referenzen
- Conde, J., et al. Revisiting 30 years of biofunctionalization and surface chemistry of inorganic nanoparticles for nanomedicine. Frontiers in Chemistry. 2, 48 (2014).
- Cheng, Q., et al. Selective ORgan Targeting (SORT) nanoparticles for tissue specific mRNA delivery and CRISPR/Cas gene editing. Nature Nanotechnology. 15 (4), 313-320 (2020).
- Jackson, N. A. C., Kester, K. E., Casimiro, D., Gurunathan, S., DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective. npj Vaccines. 5 (1), 1-6 (2020).
- Press, A. T., et al. Cargo-carrier interactions significantly contribute to micellar conformation and biodistribution. NPG Asia Materials. 9 (10), 444 (2017).
- Cevaal, P. M., et al. In vivo T cell-targeting nanoparticle drug delivery systems: Considerations for rational design. ACS Nano. 15 (3), 3736-3753 (2021).
- Faria, M., Johnston, S. T., Mitchell, A. J., Crampin, E., Caruso, F. Bio-nano science: Better metrics would accelerate progress. Chemistry of Materials. 33 (19), 7613-7619 (2021).
- Shin, H., Kwak, M., Geol Lee, T., Youn Lee, J. Quantifying the level of nanoparticle uptake in mammalian cells using flow cytometry. Nanoscale. 12 (29), 15743-15751 (2020).
- Lozano-Andrés, E., et al. Considerations for MESF-bead based assignment of absolute fluorescence values to nanoparticles and extracellular vesicles by flow cytometry. bioRxiv. , (2021).
- Schwartz, A., et al. Formalization of the MESF unit of fluorescence intensity. Cytometry. Part B, Clinical Cytometry. 57 (1), 1-6 (2004).
- Faria, M., et al. Revisiting cell-particle association in vitro: A quantitative method to compare particle performance. Journal of Controlled Release. 307, 355-367 (2019).
- Chen, A. K., Cheng, Z., Behlke, M. A., Tsourkas, A. Assessing the sensitivity of commercially available fluorophores to the intracellular environment. Analytical Chemistry. 80 (19), 7437-7444 (2008).
- Comfort, N., et al. Nanoparticle tracking analysis for the quantification and size determination of extracellular vesicles. Journal of Visualized Experiments. (169), e62447 (2021).
- Cui, J., et al. Immobilized particle imaging for quantification of nano- and microparticles. Langmuir. 32 (14), 3532-3540 (2016).
- Shang, J., Gao, X. Nanoparticle counting: Towards accurate determination of the molar concentration. Chemical Society Reviews. 43 (21), 7267-7278 (2014).
- Thomas, D. G., et al. ISD3: A particokinetic model for predicting the combined effects of particle sedimentation, diffusion and dissolution on cellular dosimetry for in vitro systems. Particle and Fibre Toxicology. 15 (1), 6 (2018).
- Johnston, S. T., Faria, M., Crampin, E. J. Isolating the sources of heterogeneity in nano-engineered particle-cell interactions. The Journal of the Royal Society Interface. 17 (166), 20200221 (2020).
- Ahmed-Cox, A., et al. Spatio-temporal analysis of nanoparticles in live tumor spheroids impacted by cell origin and density. Journal of Controlled Release. 341, 661-675 (2022).
- Faria, M., et al. Minimum information reporting in bio-nano experimental literature. Nature Nanotechnology. 13 (9), 777-785 (2018).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten