
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Computergestützte Vorhersage der Aminosäurepräferenzen potenziell multispezifischer Peptidbindungsdomänen, die an Protein-Protein-Wechselwirkungen beteiligt sind
In diesem Artikel
Zusammenfassung
Wir beschreiben eine auf Sequenzdiversifikation basierende Methodik zur Abschätzung der Aminosäurepräferenzen multispezifischer Bindungsstellen in Protein-Protein-Wechselwirkungen (PPIs). Bei dieser Strategie werden Tausende von potentiellen Peptidliganden erzeugt und in silico gescreent, wodurch einige Einschränkungen der verfügbaren experimentellen Methoden überwunden werden.
Zusammenfassung
Viele Protein-Protein-Wechselwirkungen beinhalten die Bindung kurzer Proteinsegmente an Peptidbindungsdomänen. In der Regel erfordern solche Wechselwirkungen das Erkennen linearer Motive mit variabler Konservierung. Die Kombination von hochkonservierten und variableren Regionen in denselben Liganden trägt oft zur Multispezifität der Bindung bei, eine gemeinsame Eigenschaft von Enzymen und zellulären Signalproteinen. Die Charakterisierung der Aminosäurepräferenzen von Peptidbindungsdomänen ist wichtig für das Design von Mediatoren von Protein-Protein-Wechselwirkungen (PPIs). Computergestützte Methoden sind eine effiziente Alternative zu den oft kostspieligen und umständlichen experimentellen Techniken und ermöglichen das Design potenzieller Mediatoren, die später in nachgelagerten Experimenten validiert werden können. In dieser Arbeit haben wir eine Methodik beschrieben, die die Pepspec-Anwendung des molekularen Modellierungspakets von Rosetta verwendet, um die Aminosäurepräferenzen von Peptidbindungsdomänen vorherzusagen. Diese Methode ist nützlich, wenn sowohl die Struktur des Rezeptorproteins als auch die Art des Peptidliganden bekannt sind oder abgeleitet werden können. Die Methodik beginnt mit einem gut charakterisierten Anker aus dem Liganden, der durch zufällige Zugabe von Aminosäureresten erweitert wird. Die Bindungsaffinität der auf diese Weise erzeugten Peptide wird dann durch flexibles Rückgrat-Peptid-Docking bewertet, um die Peptide mit den besten vorhergesagten Bindungswerten auszuwählen. Diese Peptide werden dann zur Berechnung von Aminosäurepräferenzen und optional zur Berechnung einer Positionsgewichtsmatrix (PWM) verwendet, die in weiteren Studien verwendet werden kann. Um die Anwendung dieser Methodik zu veranschaulichen, verwendeten wir die Interaktion zwischen Untereinheiten des humanen Interferon-Regulationsfaktors 5 (IRF5), von dem bisher bekannt war, dass er multispezifisch ist, aber global von einem kurzen konservierten Motiv namens pLxIS geleitet wird. Die geschätzten Aminosäurepräferenzen stimmten mit den bisherigen Erkenntnissen über die IRF5-Bindungsoberfläche überein. Positionen, die von phosphorylierbaren Serinresten eingenommen wurden, wiesen eine hohe Häufigkeit von Aspartat und Glutamat auf, wahrscheinlich weil ihre negativ geladenen Seitenketten denen von Phosphoserin ähneln.
Einleitung
Die Interaktion zwischen zwei Proteinen beinhaltet oft die Bindung kurzer Segmente von Aminosäuren an Peptid-Bindungsdomänen, die Protein-Peptid-Grenzflächen ähneln. Rezeptorproteine, die an solchen Protein-Protein-Wechselwirkungen (PPI) beteiligt sind, haben oft die Fähigkeit, einen bestimmten Satz überlappender, aber divergierender Ligandensequenzen zu erkennen, eine Eigenschaft, die als Multispezifität bekannt ist 1,2. Die multispezifische Erkennung ist ein Merkmal vieler zellulärer Proteine, aber besonders bemerkenswert ist sie bei Enzymen und zellulären Signalproteinen3. Proteine, die mit multispezifischen Bindungsstellen interagieren, haben oft eine Kombination aus mehr und weniger konservierten Regionen in ihrer Sequenz 4,5,6. In diesem Szenario sind die stärker konservierten Sequenzmotive an stringenten molekularen Wechselwirkungen beteiligt. Umgekehrt interagieren die variableren Sequenzen mit irgendwie permissiven Oberflächen in der Rezeptorbindungsstelle. In der Regel handelt es sich bei diesen weniger konservierten, aber immer noch funktionell relevanten Segmenten um Schleifen ohne definierte Sekundärstrukturmuster oder um noch dynamischere Konformationen, wie sie für intrinsisch ungeordnete Proteine typisch sind7.
Die Identifizierung potenzieller Peptidliganden von Bindungsstellen ist in der Regel der erste Schritt bei der Entwicklung von Mediatoren, die in der Lage sind, die entsprechenden PPIs zu interferieren8. Es ist jedoch oft unwahrscheinlich, einen einzigen der häufigsten Aminosäurereste an den meisten Sequenzpositionen in Liganden multispezifischer Bindungsstellen zu finden. Stattdessen können diese Stellen besondere Präferenzen für eine bestimmte Klasse von Aminosäuren entsprechend ihren chemischen Eigenschaften haben, z. B. saure und negativ geladene Aminosäuren wie Aspartat oder Glutamat, sperrige aromatische Aminosäuren wie Phenylalanin oder mehr hydrophobe Reste wie aliphatische Aminosäuren Alanin, Valin, Leucin oder Isoleucin3. Verschiedene experimentelle Methoden können Einblicke in die Aminosäurepräferenzen von Proteinbindungsstellen liefern, darunter die gerichtete Evolution9, die Multicodon-Scanning-Mutagenese10 und die tiefeMutations-Scanning 11. Alle diese Methoden folgen dem Ansatz der Sequenzdiversifizierung, der darauf basiert, Mutationen in ursprüngliche Liganden einzuführen und deren Wirkung auf die Funktion des Rezeptorproteins weiter zu analysieren (siehe Bratulic und Badran12 für eine umfassende Übersicht). Diese Methoden erfordern jedoch oft die Durchsicht großer Sequenzbibliotheken, was sie umständlicher, kostspieliger und zeitaufwändiger macht.
Computergestützte Methoden zur Ableitung der Aminosäurepräferenzen multispezifischer Bindungsstellen haben das Potenzial, die Einschränkungen von Nasslabormethoden zu umgehen. Unter diesen bewertet der In-silico-Sequenzdiversifikationsansatz den energetischen Einfluss einer Vielzahl von Aminosäureersatzstoffen in der Ligandensequenz, um die strukturelle Plastizität des PPI13 zu charakterisieren. Diese Methode beginnt mit der Struktur oder dem Modell des Peptidliganden, der an die Rezeptorbindungsstelle gebunden ist, und führt anschließend Mutationen in die Ligandensequenz ein. Statistische und Energie-Scoring-Funktionen werden dann verwendet, um den Einfluss dieser Mutationen auf die Stabilität und Bindungsaffinität zu bewerten. Der Satz der Ligandensequenzen mit der besten Punktzahl, der sich aus der Evaluierungsphase ergibt, kann dann zur Berechnung der Aminosäurepräferenzen verwendet werden. Diese Strategie hat das Potenzial, eine sehr große Anzahl von Ligandensequenzen effizient zu verarbeiten. Daher kann es eine vollständigere und konsistentere Rückschluss auf die Aminosäurepräferenzen liefern, verglichen mit denen, die aus der begrenzteren Anzahl von Sequenzen berechnet werden, die normalerweise in Nasslaboransätzen verarbeitet werden können.
Die Pepspec-Anwendung der Rosetta Molecular Modeling Suite14 ist ein Werkzeug, das die Sequenzdiversifizierung als wichtigen Schritt seines Peptiddesignmodus durchführt. Diese Anwendung erfordert eine Struktur oder ein Modell des Rezeptorproteins mit einem gebundenen Peptid bis zu einem einzigen Aminosäurerest in der Länge, das als Anker für die nächsten Schritte verwendet wird. Die Sequenz des gebundenen Peptids wird dann (falls erforderlich) verlängert und diversifiziert, um eine große Anzahl von mutmaßlichen Peptidliganden zu erzeugen. Die Bindungsaffinität dieser Peptide wird dann durch Peptid-Docking mit flexiblem Rückgrat bewertet, um diejenigen mit den besten vorhergesagten Bindungswerten auszuwählen. Obwohl das Hauptergebnis dieser Anwendung die besten Peptidkandidaten sind, die am Ende der Designphase ausgewählt werden, kann die viel größere Menge von Peptiden, die in dieser Phase akzeptiert werden, auch zur Berechnung der Aminosäurepräferenzen der Zielbindungsstelle verwendet werden. Aminosäurepräferenzen werden als Häufigkeit jedes Aminosäurerests pro Position der Ligandensequenz berechnet, die entweder als Positionsgewichtsmatrix (PWM) oder als visuelleres Sequenzlogo dargestellt wird.
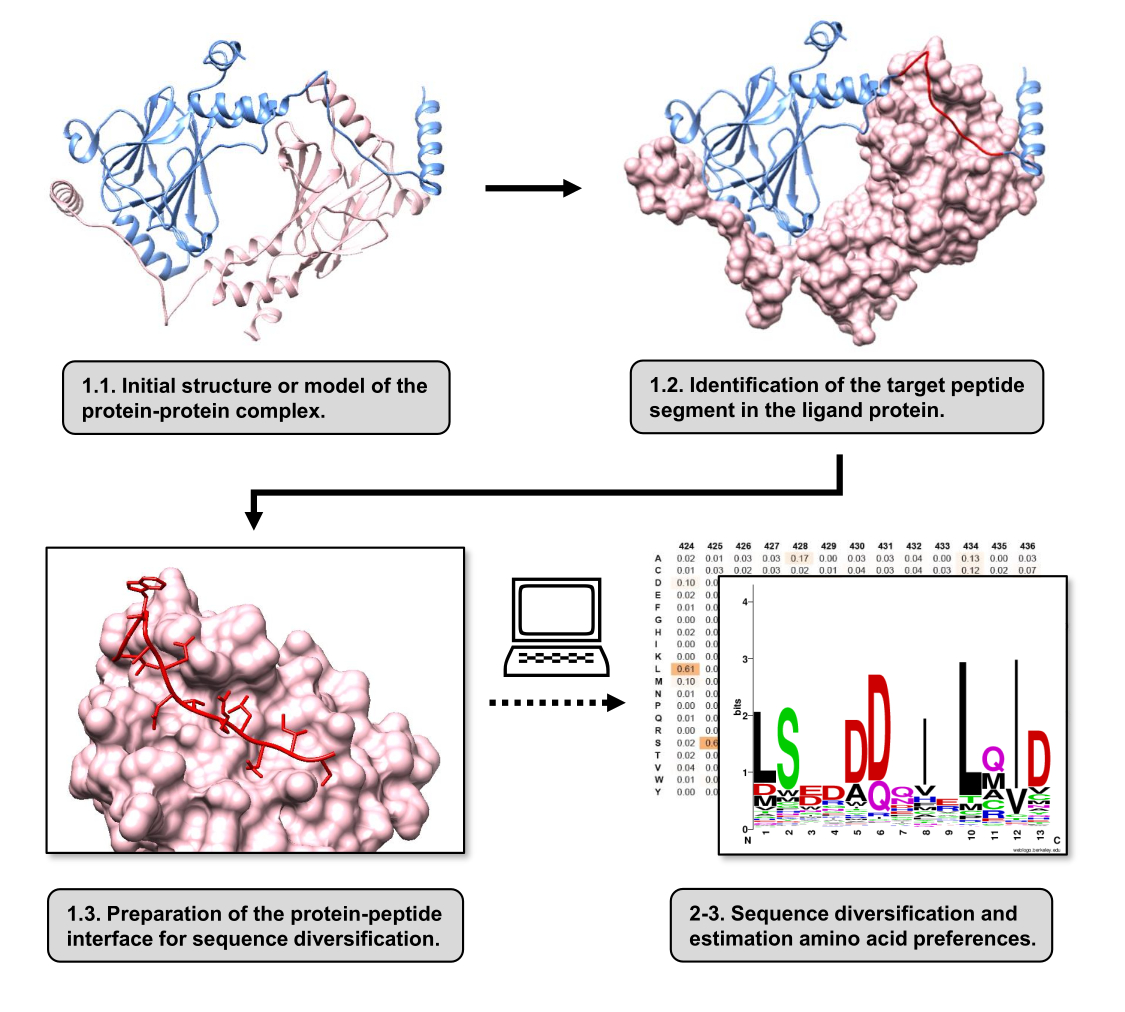
In diesem Artikel beschreiben wir ein Protokoll zur Abschätzung der Aminosäurepräferenzen der Bindungsoberfläche eines Rezeptorproteins, das an einem PPI beteiligt ist. Das Protokoll konzentriert sich auf PPIs, bei denen bekannt ist, dass ein lineares Segment des Protein-Liganden an das Rezeptorprotein bindet, so dass das Szenario als Protein-Peptid-Schnittstelle modelliert werden kann. In diesem Szenario interagieren konservierte Motive aus dem Liganden typischerweise mit definierten Taschen in der Rezeptorbindungsstelle, obwohl das gesamte Ligandensegment, das an der PPI beteiligt ist, weniger konservierte Regionen enthalten kann. Ein Flussdiagramm, das die wichtigsten Schritte des Protokolls zusammenfasst, ist in Abbildung 1 dargestellt. Das Protokoll beginnt mit der 3D-Struktur des Protein-Protein-Komplexes und reduziert das Ligandenprotein weiter auf das potenziell am besten interagierende Segment, wobei das Rezeptorprotein intakt bleibt. Das am besten interagierende Segment wird unter Verwendung des BUDE Alanin-Scan-Servers15 abgeleitet, der eine rechnergestützte Alanin-Scanning-Mutagenese durchführt, um Hot-Spot-Reste zwischen den beiden interagierenden Proteinen zu identifizieren. Bei diesem Ansatz werden Reste aus dem Liganden einzeln durch Alanin ersetzt, und die geschätzte Änderung der freien Energie oder Stabilität des Komplexes (ΔΔG) wird dann verwendet, um auf die Relevanz des entsprechenden Rests für den Ziel-PPI zu schließen. Sobald das am besten interagierende Segment abgeleitet ist, wird sein Komplex mit dem Rezeptorprotein als Basisstruktur verwendet, die an Pepspec übermittelt wird, um eine Sequenzdiversifizierung durchzuführen.

Abbildung 1: Überblick über die wichtigsten Schritte des Protokolls, das in dieser Arbeit vorgeschlagen wird. Die Nummern stimmen mit den Schrittnummern im Protokollabschnitt überein. Die Abbildungen wurden mit dem Protein-Protein-Komplex erstellt, der als Beispiel für das im Text beschriebene Beispiel verwendet wird. In diesem Komplex ist die Proteinkette, die als Rezeptor betrachtet wird, rosa dargestellt, während die Kette, die als Ligand betrachtet wird, in hellblau dargestellt ist, wobei ihr vorhergesagtes Segment mit der besten Wechselwirkung rot hervorgehoben ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Eine der Einschränkungen des vorgeschlagenen Protokolls ist die Anforderung einer aufgelösten Struktur der Protein-Peptid-Grenzfläche. Das Protokoll kann alternativ mit einem Modell der Zielprotein-Peptid-Grenzfläche beginnen, obwohl die spezifischen Modellierungsschritte hier nicht beschrieben werden. Obwohl das Protokoll auf einem PC mit einem beliebigen Betriebssystem ausgeführt werden kann, ist für die Schritte mit den Rosetta-Anwendungen eine Linux-Umgebung erforderlich. Ein Computercluster wird auch für den Schritt der Sequenzdiversifizierung dringend empfohlen, da Pepspec typischerweise eine große Anzahl von Iterationen durchführt.
Die Anwendung des vorgeschlagenen Protokolls wird durch die Schätzung der Aminosäurepräferenzen der Gebotsfläche von IRF5, einem Mitglied der Familie der humanen Interferon-Regulationsfaktoren (IRF), veranschaulicht. Wir haben dieses Protein als Beispiel gewählt, weil sich während seiner Aktivierung zwei Untereinheiten zu einem Dimer verbinden, dessen Struktur gut charakterisiert ist16. In IRF-Dimeren kann die Bindung als Protein-Peptid-Grenzfläche modelliert werden, bei der eine Untereinheit die Bindungsoberfläche bildet und die andere durch eine Region interagiert, die ein kurzes konserviertes Motiv namens pLxIS17,18 enthält. Darüber hinaus ist die Bindung an IRF-Untereinheiten multispezifisch; Daher können sie Homodimere, Heterodimere und Komplexe mit anderen zellulären Proteinen bilden, die als Koaktivatoren bekannt sind18.
Protokoll
1. Erste Vorbereitung der Protein-Peptid-Grenzfläche
- Herunterladen der Struktur des Protein-Protein-Komplexes
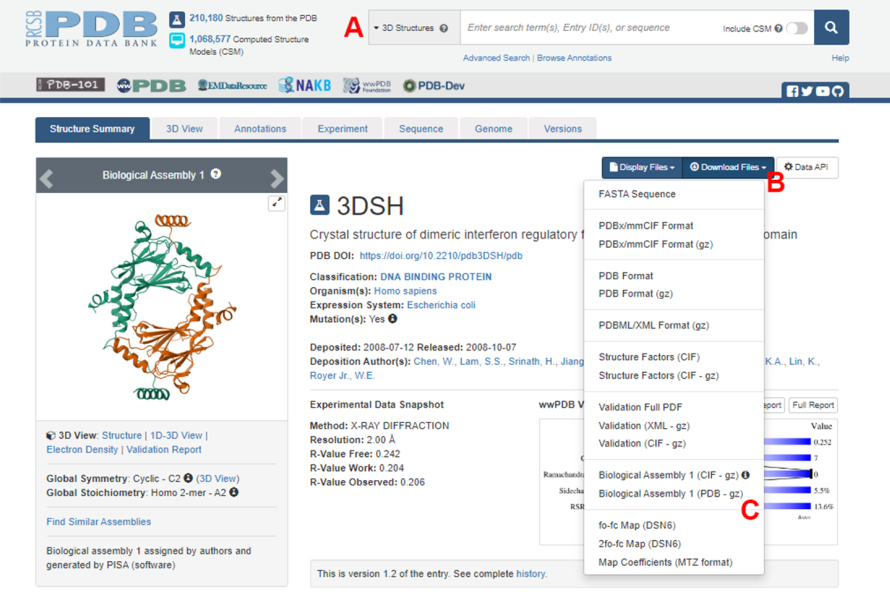
- Navigieren Sie zur Homepage der Protein Data Bank (PDB) (https://www.rcsb.org/) und geben Sie die PDB-ID für die Struktur des Protein-Protein-Komplexes in das Hauptsuchfeld ein (Abbildung 2A). Die PDB-ID für die Struktur des IRF5-Dimers, die in dieser Arbeit als Beispiel verwendet wird, ist 3DSH19.
- Klicken Sie auf der Hauptseite für die gewünschte Struktur auf Download Files (Abbildung 2B) und dann auf Biologische Anordnung 1 (PDB - gz) (Abbildung 2C).
HINWEIS: In der PDB-Datenbank werden Strukturen vieler Proteinkomplexe, die aus identischen Monomeren bestehen, als biologische Anordnungen dargestellt, bei denen nur die Struktur eines Monomers (asymmetrische Einheit) in der PDB-Datei gespeichert ist. Die Struktur des Multimers, in diesem Fall das IRF5-Dimer, muss als biologische Anordnung heruntergeladen werden, die zwei Instanzen der asymmetrischen Einheit enthält. Um die nächsten Schritte dieses Protokolls zu erleichtern, werden zunächst die beiden Monomere getrennt und ihnen unterschiedliche Ketten-IDs zugewiesen. - Öffnen Sie die heruntergeladene Struktur in UCSF Chimera20 und klicken Sie auf Tools > Strukturbearbeitung > Ketten-IDs ändern. In diesem Beispiel erhalten beide Ketten in der biologischen Baugruppe den Namen A. Benennen Sie die zweite Kette (mit der Bezeichnung #0.2) in B um, und klicken Sie auf OK.
- Klicken Sie auf Favoriten > Modellgruppe, und wählen Sie dann das Modell aus, das die beiden Ketten enthält. Klicken Sie auf die Schaltfläche Gruppieren/Gruppierung aufheben , um jede Kette in ein anderes Modell zu unterteilen. Wählen Sie dann die beiden Modelle aus und klicken Sie auf die Schaltfläche Kopieren/Kombinieren . Geben Sie einen neuen Namen für das kombinierte Modell ein, aktivieren Sie die Option Quellmodelle schließen, und klicken Sie auf OK.
- Klicken Sie auf > Kette auswählen und bestätigen Sie, dass jede Kette im Dimer nun durch einen anderen Buchstaben gekennzeichnet ist, nämlich A und B.
- Verwenden Sie Datei > PDB speichern , um die bearbeitete Struktur in einer anderen PDB-Datei zu speichern, die in den nächsten Schritten des Protokolls verwendet wird (hier wurde der Name IRF5_dimer.pdb verwendet).

Abbildung 2: Die Seite der Protein Data Bank (PDB) für die Struktur, die in dieser Arbeit als repräsentatives Beispiel verwendet wird. (A) Suchfeld zur Eingabe des PDB-Zugangscodes der Zielstruktur. (B) Menü zum Herunterladen der Struktur in verschiedenen Formaten. (C) Optionen zum Herunterladen biologischer Anordnungen, wenn die Struktur als asymmetrische Einheit gespeichert wurde (siehe Schritt 1.1.2 für weitere Details). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Identifizierung des Zielsegments im Ligandenprotein
- Navigieren Sie zum BUDE Alanine Scan Server (https://pragmaticproteindesign.bio.ed.ac.uk/balas/). Klicken Sie unter Struktur-Upload auf die Schaltfläche Datei auswählen und wählen Sie die in Schritt 1.1.6 gespeicherte PDB-Datei aus.
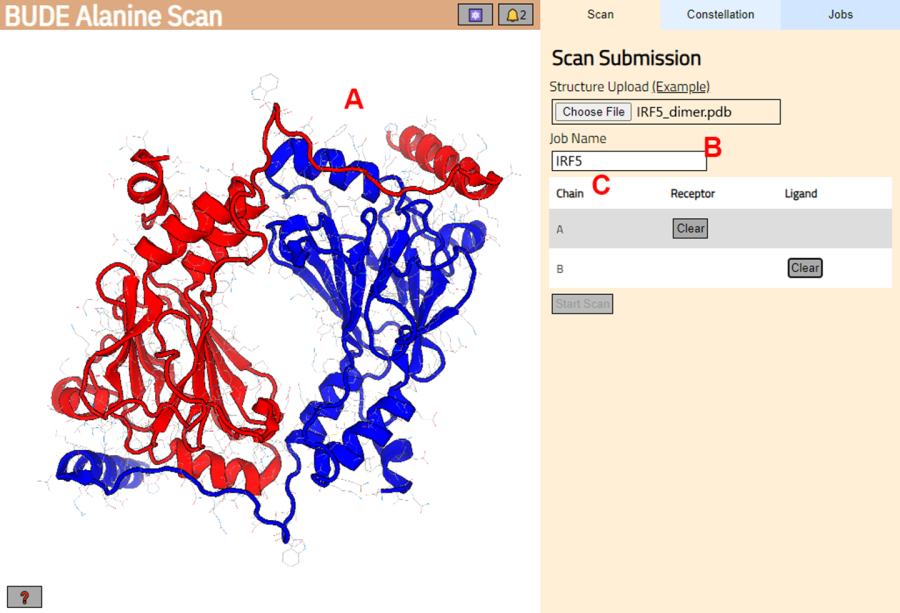
- Überprüfen Sie auf der nächsten Seite, ob die Struktur korrekt geladen wurde (Abbildung 3A), und geben Sie einen Namen für den Job auf dem Server ein (Abbildung 3B).
- Legen Sie die Ketten aus dem PDB fest, die als Rezeptor (A) und Ligand (B) behandelt werden (Abbildung 3C). Klicken Sie dann auf die Schaltfläche Scan starten , um den Auftrag zu senden.
- Sobald der Auftrag abgeschlossen ist, klicken Sie auf Ergebnisse anzeigen , um die Ergebnisseite zu öffnen (Abbildung 4).
HINWEIS: Auf der Ergebnisseite sind die Reste der Ligandenstruktur entsprechend ihrer geschätzten Änderung der freien Energie (ΔΔG) eingefärbt, und die Reste mit höheren Werten sind rot eingefärbt. - Wählen Sie aus der Rückstandsliste den Abschnitt der Rückstände aus, der voraussichtlich besser mit der Zielbindungsoberfläche interagiert. Stellen Sie sicher, dass diese Reste die höheren Werte für die Differenz der freien Energie (ΔΔG) gruppieren. In diesem Beispiel wurde das Segment zwischen den Resten Leu424 und Ser436 ausgewählt (hervorgehoben durch ein rotes Feld im rechten Bereich von Abbildung 4).
- Vorbereitung der Protein-Peptid-Grenzfläche für die Sequenzdiversifizierung
- Öffnen Sie die in Schritt 1.1.6 gespeicherte PDB-Datei in Chimera und überprüfen Sie, ob keine Atome oder Bindungen in der Struktur der Zieluntereinheiten fehlen.
- Löschen Sie alle kleinen Moleküle, Ionen und Lösungsmittel, die mit der ursprünglichen Struktur cokristallisiert wurden. Klicken Sie dazu auf Select > Residues und wählen Sie dann alle Moleküle außer Standardaminosäuren aus. Klicken Sie dann auf Aktionen > Atome/Bindungen und Löschen.
- Schneiden Sie die Ligandenkette auf das am besten interagierende Segment zu, das in Schritt 1.2.5 ausgewählt wurde. Klicken Sie dazu auf Favoriten und Sequenz und dann auf die Kette, die als Ligand gilt (B). Ziehen Sie im Sequenzfenster die Maus, um alle Rückstände mit Ausnahme der Rückstände zwischen den Positionen 424 und 436 auszuwählen. Um diese Rückstände zu löschen, klicken Sie auf Aktionen > Atome/Bindungen und Löschen.
- Verwenden Sie Datei > PDB speichern , um die bearbeitete Struktur in einer anderen PDB-Datei zu speichern, die in den nächsten Schritten des Protokolls verwendet wird (hier wurde der Name IRF5_interface.pdb verwendet).

Abbildung 3: Selektion von Rezeptor und Ligand im BUDE Alanine Scan Server. (A) Grafische Darstellung des Protein-Protein-Komplexes. (B) Textfeld zur Eingabe des Namens des Jobs auf dem Server. (C) Panel zur interaktiven Auswahl der Ketten, die als Rezeptor und Ligand betrachtet werden (siehe Schritt 1.2 für weitere Details). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Ergebnisseite des BUDE Alanine Scan Servers. Das potenziell am besten interagierende Segment in der Ligandensequenz ist mit einem roten Kästchen gekennzeichnet. Im linken Feld ist der Rückstand mit dem höheren prognostizierten Energiebeitrag (Leu433) grün hervorgehoben. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
2. Diversifizierung der Sequenz
HINWEIS: In den folgenden Schritten bezieht sich rosetta_main auf das Hauptinstallationsverzeichnis von Rosetta, das sich in der Regel unter /opt/rosetta_src__bundle/main/ befindet, wobei die installierte Rosetta-Version angibt. Außerdem wird davon ausgegangen, dass Rosetta-Anwendungen systemweit zugänglich sind. Ist dies nicht der Fall, muss der vollständige Pfad zu den ausführbaren Dateien angegeben werden. Wenn diese ausführbaren Dateien aus dem Quellcode kompiliert werden, befinden sie sich im Verzeichnis /rosetta_main/source/bin/.
- Erste Optimierung von Aminosäure-Seitenketten
- Kopieren Sie die in Schritt 1.3.4 gespeicherte bearbeitete Struktur an einen Linux-Speicherort, auf den die Rosetta-Anwendungen zugreifen können.
- Verwenden Sie die FixBB-Anwendung von Rosetta, um ein Repack aller Aminosäureseitenketten der Basenstruktur vor der Sequenzdiversifizierung durchzuführen. Bei diesem Vorgang wird die Ausrichtung aller Aminosäureseitenketten optimiert, um den Energieaufwand zu minimieren und die Stabilität des Komplexes zu verbessern. Führen Sie dazu den folgenden Befehl aus:

HINWEIS: Dieser Befehl gibt eine PDB-Datei aus, die nach der ursprünglichen Struktur benannt ist und ein zusätzliches numerisches Suffix enthält (in diesem Beispiel IRF5_interface_0001.pdb ). - Um den nächsten Schritt des Protokolls zu erleichtern, benennen Sie die neu gepackte PDB-Datei mit dem Suffix _repack mit dem folgenden Befehl um:
MV IRF5_interface_0001.PDB IRF5_repack.PDB
- Diversifizierung der Sequenz
- Führen Sie Pepspec im Designmodus aus, um den eigentlichen Schritt zur Sequenzdiversifizierung mit dem folgenden Befehl auszuführen:

Im Folgenden finden Sie allgemeine Optionen:- -s gibt die Eingabedatei an (die neu gepackte PDB-Datei, die in Schritt 2.1.3 generiert wurde).
- -o gibt das Präfix an, um Ausgabedateien zu benennen.
- - database gibt den Pfad zur Hauptdatenbank von Rosetta 3 an.
- -ex1, -ex2 und extrachi_cutoff sind Rotamer-Bibliotheksoptionen (weitere Informationen finden Sie in der Pepspec-Dokumentation).
- -overwrite weist die Anwendung an, mögliche bereits vorhandene Ausgaben zu überschreiben, die von früheren Iterationen generiert wurden.
- -pepspec:pep_chain gibt die PDB-Ketten an, die als Ligand betrachtet werden (in diesem Beispiel 'b').
- -pepspec:native_pep_anchor gibt den Aminosäurerest an, der als Anker verwendet wird (in diesem Beispiel der Leu-Rest an Position 10 des Ligandenpeptids).
- -pepspec:n_peptides gibt die Anzahl der auszugebenden Peptidstrukturen an.
- -pepspec:no_prepack_prot weist die Anwendung an, das erneute Packen in der Eingabebasisstruktur zu überspringen (da dies zuvor in Schritt 2.1 durchgeführt wurde).
HINWEIS: Die Hauptausgabe von Pepspec ist ein Verzeichnis, das die PDB-Dateien für Peptide enthält, die aus der Designphase resultieren und mit dem Ausgabepräfix mit dem Suffix .pdbs benannt sind (IRF5.pdbs im Beispiel). Darüber hinaus gibt Pepspec alle akzeptierten Peptidsequenzen, die im Rahmen des Sequenzdiversifizierungsschritts getestet wurden, und die entsprechenden Rosetta-Energiewerte in einer tabulatorgetrennten Textdatei aus, die nach dem Ausgabepräfix benannt ist, wobei die . spec-Suffix (IRF5.spec im Beispiel). Da das in dieser Arbeit beschriebene Protokoll eher darauf abzielt, Aminosäurepräferenzen als das tatsächliche Peptiddesign zu schätzen, wird in den nächsten Schritten IRF5.spec anstelle der PDB-Strukturen im .pdbs-Verzeichnis verwendet.
- Führen Sie Pepspec im Designmodus aus, um den eigentlichen Schritt zur Sequenzdiversifizierung mit dem folgenden Befehl auszuführen:
3. Schätzung der Aminosäurepräferenzen
- Berechnen einer PWM
- Um eine PWM zu generieren, verwenden Sie das gen_pepspec_pwm.py-Skript , das in der Rosetta-Suite enthalten ist. Verwenden Sie den folgenden Befehl, um dieses Skript auszuführen:

wo:- IRF5.spec ist die Pepspec-Ausgabedatei, die in Schritt 2.2 generiert wurde.
- -1 gibt an, dass es keine zusätzlichen N-terminalen Reste in der Sequenz gibt und daher Positionen in der PWM 1-basiert sind.
- 0,2 weist das Skript an, nur die Peptide mit den besten 20 % aus der Pepspec-Ausgabe zu berücksichtigen (der Standardwert ist 0,1, entsprechend 10 %)
- interface_score weist das Skript an, Peptide basierend auf dem Interface-Score zu ordnen, der einer der verschiedenen Rosetta-Scores ist, die in der Pepspec-Ausgabedatei enthalten sind.
HINWEIS: Dieses Skript generiert zwei Ausgabedateien, eine für die berechnete PWM (mit dem Suffix .pwm ) und die andere für die Sequenzen der Teilmenge von Peptiden, die zur Berechnung der PWM verwendet werden (mit dem Suffix .seq ). Die Namen dieser Dateien enthalten auch den Score und den Anteil der Peptide, die für die Rangfolge verwendet werden. In diesem Beispiel haben diese Dateien die Namen IRF5_interface_score_0.2.pwm bzw. IRF5_interface_score_0.2.seq.
- Um eine PWM zu generieren, verwenden Sie das gen_pepspec_pwm.py-Skript , das in der Rosetta-Suite enthalten ist. Verwenden Sie den folgenden Befehl, um dieses Skript auszuführen:
- Generieren eines Sequenzlogos
- Navigieren Sie zum WebLogo-Server (https://weblogo.berkeley.edu/logo.cgi)21 und klicken Sie auf die Schaltfläche Datei auswählen neben Sequenzdaten hochladen. Laden Sie die Datei mit den Peptidsequenzen hoch, die in Schritt 3.1.1 generiert wurden (in diesem Beispiel IRF5_interface_score_0.2.seq ).
- Wählen Sie das gewünschte Format und die Größe des Logos entsprechend der Eingabelänge. Im Beispiel wird das PDF-Format und eine Größe von 15 cm x 12 cm verwendet. Klicken Sie auf Logo erstellen.
Ergebnisse
In diesem Artikel haben wir ein Protokoll zur Vorhersage der Aminosäurepräferenzen der Bindungsoberfläche von IRF5 beschrieben, einem Mitglied einer Familie von Transkriptionsfaktoren, die als humane Interferon-Regulationsfaktoren bekannt sind. Diese Proteine sind Regulatoren der angeborenen und adaptiven Immunantwort und beteiligen sich an der Differenzierung und Aktivierung mehrerer Immunzellen. IRF-Untereinheiten weisen hochplastische und multispezifische Bindungsoberflächen auf u...
Diskussion
Der vorliegende Artikel beschreibt ein Protokoll zur Abschätzung der Aminosäurepräferenzen potentiell multispezifischer Bindungsstellen auf der Grundlage einer in silico Sequenzdiversifikation. Es wurden nur wenige computergestützte Werkzeuge entwickelt, um die Aminosäurepräferenzen von Protein-Peptid-Grenzflächen abzuschätzen 14,25,26. Diese Tools haben einen prädiktiven Charakter, unt...
Offenlegungen
Autoren haben nichts offenzulegen.
Danksagungen
Die finanzielle Unterstützung durch das Sistema Nacional de Investigación (SNI) (Förderkennzeichen SNI-043-2023 und SNI-170-2021), die Secretaría Nacional de Ciencia, die Tecnología e Innovación (SENACYT) von Panama und das Instituto para la Formación y Aprovechamiento de Recursos Humanos (IFARHU) werden dankbar gewürdigt. Die Autoren danken Dr. Miguel Rodríguez für die sorgfältige Durchsicht des Manuskripts.
Materialien
| Name | Company | Catalog Number | Comments |
| BUDE Alanine Scan Server | University of Edinburgh | https://pragmaticproteindesign.bio.ed.ac.uk/balas/ | doi: 10.1021/acschembio.9b00560 |
| Rosetta Modeling Software | Rosetta Commons | https://www.rosettacommons.org/software | doi: 10.1002/prot.22851 |
| UCSF Chimera | University of California San Francisco | https://www.cgl.ucsf.edu/chimera/ | doi: 10.1002/jcc.20084 |
Referenzen
- Kim, P. M., Lu, L. J., Xia, Y., Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 314 (5807), 1938-1941 (2006).
- Schreiber, G., Keating, A. E. Protein binding specificity versus promiscuity. Current Opinion in Structural Biology. 21 (1), 50-61 (2011).
- Erijman, A., Aizner, Y., Shifman, J. M. Multispecific recognition: Mechanism, evolution, and design. Biochemistry. 50 (5), 602-611 (2011).
- Fromer, M., Shifman, J. M. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Computational Biology. 5 (12), e1000627 (2009).
- Xie, T., Zmyslowski, A. M., Zhang, Y., Radhakrishnan, I. Structural basis for multispecificity of MRG domains. Structure. 23 (6), 1049-1057 (2015).
- Hendler, A., et al. Human SIRT1 multispecificity is modulated by active-site vicinity substitutions during natural evolution. Molecular Biology and Evolution. 38 (2), 545-556 (2021).
- Teilum, K., Olsen, J. G., Kragelund, B. B. On the specificity of protein-protein interactions in the context of disorder. The Biochemical Journal. 478 (11), 2035-2050 (2021).
- Pelay-Gimeno, M., Glas, A., Koch, O., Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angewandte Chemie (International ed. in English). 54 (31), 8896-8927 (2015).
- Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., Zhao, H. Directed evolution: Methodologies and applications. Chemical Reviews. 121 (20), 12384-12444 (2021).
- Liu, J., Cropp, T. A. Rational protein sequence diversification by multi-codon scanning mutagenesis. Methods in Molecular Biology. 978, 217-228 (2013).
- Wei, H., Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Frontiers in Genetics. 14, 1087267 (2023).
- Bratulic, S., Badran, A. H. Modern methods for laboratory diversification of biomolecules. Current Opinion in Chemical Biology. 41, 50-60 (2017).
- Humphris, E. L., Kortemme, T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 16 (12), 1777-1788 (2008).
- King, C. A., Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 78 (16), 3437-3449 (2010).
- Ibarra, A. A., et al. Predicting and experimentally validating hot-spot residues at protein-protein interfaces. ACS Chemical Biology. 14 (10), 2252-2263 (2019).
- Chen, W., Srinath, H., Lam, S. S., Schiffer, C. A., Royer, W. E., Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. Journal of Molecular Biology. 379 (2), 251-260 (2008).
- Mancino, A., Natoli, G. Specificity and function of IRF family transcription factors: Insights from genomics. Journal of Interferon & Cytokine Research. 36 (7), 462-469 (2016).
- Schwanke, H., Stempel, M., Brinkmann, M. M. Of keeping and tipping the balance: Host regulation and viral modulation of IRF3-dependent IFNB1 expression. Viruses. 12 (7), 33 (2020).
- Chen, W., et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural & Molecular Biology. 15 (11), 1213-1220 (2008).
- Pettersen, E. F., et al. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Crooks, G. E., Hon, G., Chandonia, J. -. M., Brenner, S. E. WebLogo: a sequence logo generator. Genome Research. 14 (6), 1188-1190 (2004).
- Panne, D., McWhirter, S. M., Maniatis, T., Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of Biological Chemistry. 282 (31), 22816-22822 (2007).
- Weihrauch, D., et al. An IRF5 decoy peptide reduces myocardial inflammation and fibrosis and improves endothelial cell function in tight-skin mice. PloS One. 11 (4), e0151999 (2016).
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of Biological Chemistry. 279 (11), 9698-9702 (2004).
- Smith, C. A., Kortemme, T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PloS One. 6 (7), e20451 (2011).
- Rubenstein, A. B., Pethe, M. A., Khare, S. D. MFPred: Rapid and accurate prediction of protein-peptide recognition multispecificity using self-consistent mean field theory. PLoS Computational Biology. 13 (6), e1005614 (2017).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten