
Method Article
Análisis qPCRTag - Un alto rendimiento, Tiempo real ensayo PCR para Sc2.0 Genotipado
En este artículo
Resumen
Designer chromosomes of the Synthetic Yeast Genome project, Sc2.0, can be distinguished from their native counterparts using a PCR-based genotyping assay called PCRTagging, which has a presence/absence endpoint. Here we describe a high-throughput real time PCR detection method for PCRTag genotyping.
Resumen
El Proyecto Genoma levadura sintética (Sc2.0) tiene como objetivo construir 16 cromosomas de la levadura de diseño y combinarlos en una sola célula de levadura. Hasta la fecha, un cromosoma sintético, synIII 1, y un brazo cromosoma sintético, synIXR 2, se han construido y su función in vivo validado en ausencia de los correspondientes cromosomas de tipo salvaje. Una característica importante del diseño de los cromosomas Sc2.0 es la introducción de PCRTags, que son secuencias cortas, re-codificado dentro de los marcos de lectura abierta (ORFs) que permiten la diferenciación de los cromosomas sintéticos a partir de sus homólogos de tipo salvaje. PCRTag cebadores hibridan selectivamente a cualquiera de cromosomas sintéticos de tipo o salvajes y la presencia / ausencia de cada tipo de ADN se puede probar con un simple ensayo de PCR. La lectura estándar del ensayo PCRTag es evaluar la presencia / ausencia de amplicones por electroforesis en gel de agarosa. Sin embargo, con una densidad media amplicón PCRTag de uno por 1,5 kb y agenome tamaño de ~ 12 Mb, el genoma Sc2.0 completado codificará aproximadamente 8.000 PCRTags. Para mejorar el rendimiento, hemos desarrollado un ensayo de detección basado en la PCR en tiempo real para PCRTag genotipado que llamamos análisis qPCRTag. El flujo de trabajo especifica 500 reacciones nl en una placa de múltiples pocillos 1536, lo que nos permite probar hasta 768 PCRTags con ambos pares de cebadores de tipo sintético y salvajes en un solo experimento.
Introducción
Sc2.0, o el Proyecto Genoma levadura sintética ( www.syntheticyeast.org ), ha fijado el objetivo de diseñar y construir un genoma eucariota totalmente sintético. El uso de la secuencia del genoma altamente comisariada de Saccharomyces cerevisiae 3 como punto de partida, cada uno de los cromosomas lineales dieciséis ha sido rediseñado para satisfacer un conjunto de principios de diseño que especifican mantener la condición física de células, mejorando la estabilidad del genoma, y el aumento de la flexibilidad genética. Por ejemplo, elementos desestabilizadores tales como repeticiones se eliminan de cromosomas Sc2.0. Todas las instancias de los codones de parada TAG son re-codificados para TAA para 'liberar' un codón en la cepa final para la introducción de un aminoácido codificado no genéticamente. Además un sistema de evolución inducible, Scramble, habilitado por el sistema Cre-lox, permite la capacidad sin precedentes para generar genomas derivados con nuevas estructuras 4.
Otro elemento importante del diseño en el genoma Sc2.0 es la introducción de PCRTags, que sirven como marcas de agua de ADN para permitir el seguimiento del ADN de tipo sintético y salvaje. PCRTags son segmentos cortos, re-codificado en ORFs en los cromosomas sintéticos; mientras que las secuencias PCRTag difieren en el nivel de ADN entre los cromosomas tipo sintético y salvajes, las proteínas codificadas son idénticos en secuencia de aminoácidos y, por tanto, presumiblemente, la función. Secuencias PCRTag están diseñados específicamente como sitios de unión del cebador para facilitar la amplificación selectiva (Figura 1A). Diseño PCRTag se lleva a cabo utilizando el algoritmo 'más diferente' en GeneDesign 5,6, obteniéndose recodificada secuencias sintéticas que son típicamente ~ 60% diferente de las secuencias nativas (mínimo 33%) con temperaturas de fusión entre 58 ° C y 60 ° C y amplicón longitudes de entre 200-500 pares de bases 2. Recodificación no está permitido dentro de la primera 100 pb de cada ORF, ya que estas regiones son conocidos portienen preferencias especiales en términos de uso de codones 7. En conjunto, estas reglas de diseño a favor de un alto rendimiento de casi todos PCRTags bajo un único conjunto de condiciones de la PCR mediante el cual pares sintéticos y salvajes imprimación tipo PCRTag unen exclusiva y amplifican el ADN sintético y nativo, respectivamente (Figura 1B).

Figura 1:. PCRTag esquemática PCRTags (A) son secuencias dentro de los marcos de lectura abierta (ORF) de los genes en los cromosomas Sc2.0 re-codificadas. (B) sintético (SYN) y de tipo salvaje (WT) primers PCRTag unen exclusivamente y amplifican el ADN genómico de tipo sintético y salvaje (gDNA), respectivamente. Aquí se muestra un análisis de un segmento kb ~ 30 del brazo izquierdo del cromosoma seis, probando trece pares de cebadores PCRTag utilizando ya sea WT o semi-synVIL2 gDNA como plantilla. En muchos casos un solo ORF codifica más de un PCRTag. Presencia / ausencia de amplicones PCRTag se evaluó a través de electroforesis en gel de agarosa. Amplicones PCRTag varían en tamaño desde 200 pb a 500 pb. Las especies que migran más rápidamente en la parte inferior de los paneles son dímeros de cebadores.
Análisis PCRTag ha demostrado ser una herramienta importante en el conjunto de cromosomas Sc2.0. En un experimento típico de 30-50 kb de ADN sintético, que codifica 20-30 PCRTags, se transforma en células de levadura para reemplazar el correspondiente ADN de tipo salvaje 1,2,8. Análisis PCRTag se utiliza para identificar los transformantes que codifican PCRTags tipo sintéticos pero no salvaje que abarcan ese segmento de ADN, o los llamados "ganadores". Por lo general es necesario probar varios transformantes para identificar "ganadores", por lo que el rendimiento y costo del análisis PCRTag son consideraciones importantes. Actualmente dos cromosomas Sc2.0 se han completado (synIII 1 y synIXR 2), que representa menos del 10% del genoma Sc2.0, although más de la mitad de los cromosomas restantes están actualmente en proceso de síntesis y ensamblaje. La escala de análisis PCRTag requerida para este proyecto está superando rápidamente la capacidad de ejecutar geles y manualmente anotar la presencia de ADN sintético y ausencia de ADN de tipo salvaje.
Para mejorar el rendimiento del ensayo PCRTag hemos desarrollado un flujo de trabajo utilizando PCR en tiempo real para eludir el uso de electroforesis en gel de agarosa. El flujo de trabajo hace uso de un dispensador de líquido a granel para distribuir qPCR mezcla maestra en cada pocillo de una placa de múltiples pocillos 1536, un dispensador de líquido acústica nanoescala para transferir ADN molde y los cebadores, y un termociclador 1536 qPCR, lo que nos permite miniaturizar reacciones a 500 nl y maximizar el rendimiento. Además análisis puede automatizarse. Este tipo de protocolo de genotipado de alto rendimiento debe ser generalizables a cualquier proyecto que requiera análisis de muchos clones en múltiples loci.
Protocolo
1. Preparar el ADN genómico de la levadura (gDNA)
- Preparar un stock de levadura Lysis Buffer 9 que contiene 50 mM Tris pH 8,0, NaCl 100 mM, SDS al 1% (w / v), 2% de Triton X-100 (v / v), 1 mM EDTA pH 8,0.
- Como a partir del uso de material ~ 2 x 10 6 células, que normalmente corresponde a ~ 100 l de la cultura saturada de cepas de laboratorio del BY4741 10 de fondo. Las células cultivadas se recogen por centrifugación a 1000 xg durante 3 min a temperatura ambiente en un tubo de microcentrífuga de 1,5 ml. Aspirar el sobrenadante.
- Añadir 200 l de tampón de lisis de levadura y volver a suspender el sedimento celular con la pipeta.
- Añadir ~ 300 l de ácido lavó perlas de vidrio de tal manera que el nivel de perlas es de aproximadamente 1 mm por debajo del nivel de la suspensión celular.
- En una campana de humos añadir 200 l de fenol / cloroformo / alcohol isoamílico (25: 24: 1). Tape el tubo de microcentrífuga y asegurarse de que no hay cuentas están atrapados entre la tapa y el tubo ya que esto dará lugar a fugas durante la agitación(Paso 1.6). Invierta 3 veces para mezclar y comprobar la tapa está cerrada.
- Agitar el tubo durante 10 min a temperatura ambiente usando un mezclador de sobremesa tal como un Eppendorf 5432.
- Centrifugar a 20800 xg durante 10 min a 4 ° C.
- Transferencia de 75 l de la fase acuosa a un nuevo tubo de microcentrífuga que contiene 1 ml de etanol al 100%. Invierta 10 veces para mezclar.
- Sedimentar las ADN por centrifugación a 20.800 xg durante 20 min a 4 ° C.
- Aspirar el sobrenadante sin desalojar el sedimento de ADN. Lavar el sedimento con 500 l de etanol al 70%.
- Centrifugar a 20.800 xg durante 5 min a 4 ° C.
- Aspirar el sobrenadante sin desalojar el sedimento de ADN y aire secar el pellet con la tapa abierta hasta microcentrífuga exceso de etanol se ha evaporado, que es generalmente alrededor de 10 min.
- Resuspender el sedimento de ADN en 75 l de 10 mM Tris pH 7,4 y agitar para mezclar. Conservar la muestra a -20 ° C indefinidamente. Medir la concentración de ADN en la muestra utilizando unabenchtop fluorímetro como el qubit, que distingue el ADN a partir de ARN. Asegúrese de que la concentración final de ADNg es ~ 1.2 ng / l.
- Preparar una dilución 1:10 de ADN genómico utilizando agua y agitar para mezclar.
- Alícuota de 30 l de gDNA diluida en el pocillo apropiado de una placa de fuente que es compatible con un dispensador de líquido acústica nanoescala (paso 3). Si se utiliza un Labcyte Echo 550, utilice una placa de polipropileno de 384 pocillos (placa 384PP).
NOTA: Cuando sellada apropiadamente este plato se puede almacenar durante al menos un mes a -20 ° C.
2. Preparar y dispensación qPCR Master Mix en una placa de múltiples pocillos 1536
- Preparar 850 l de mezcla maestra qPCR, como LightCycler 1536 ADN Verde Maestro, en un tubo de microcentrífuga de 1,5 ml y agitar para mezclar. Centrifugar la mezcla maestra qPCR durante 2 min a 20.800 xg a temperatura ambiente para eliminar cualquier burbuja. Los componentes de la mezcla maestra qPCR requerido para una sola placa de múltiples pocillos 1536 son los siguientess:
- Combine 170 l de mezcla maestra enzima (Maestro Verde Tubo 1, 5x concentrado), 42,5 l de SYBR (Tubo Maestro Verde 4, 20x concentrado), y 637,5 l de agua.
- Dispensar 500 nl / pocillo en una placa de múltiples pocillos 1536 usando un dispensador de líquido a granel tal como el Art Robbins Cobra. (Tenga en cuenta que los pasos 2.2.1-2.2.4 se refieren específicamente a la Cobra.)
- Crear un programa de dispensación con los siguientes valores: clase de líquido, el agua; volumen aspirado, 800 l; dispensar el volumen, 500 nl; tiempo de lavado, 20 seg. En la configuración de dispensación, compruebe la velocidad de dispensación se ajusta a 49,6 mm / seg, el desplazamiento de dispensación es 0,25 mm, y el exceso de líquido aspirado se dispensará de nuevo en la fuente bien.
- Coloque el tubo de microcentrífuga que contiene la mezcla maestra qPCR en pocillo A1 del soporte del tubo de microcentrífuga, que se encuentra en la posición de la cubierta 1. Cortar la tapa del tubo de microcentrífuga o simplemente dejarlo abierto. Ajuste el 'Aspirar bien "a A1 editando el Aspiratconfiguración e.
- Realice una etapa de lavado antes de la dispensación mezcla maestra qPCR por los primeros pasos de-seleccionar el aspirado y prescindir del programa creado en el paso 2.2.2 y luego golpear 'run'. Una vez completado, vuelva a seleccionar el aspirado y dispensar pasos antes de ejecutar el programa completo para dispensar mezcla maestra qPCR. Si la Cobra se ha sentado inactivo durante menos de 10 minutos de la etapa inicial de lavado, que sirve para cebar el sistema, es innecesario.
- Hit 'run' para dispensar mezcla maestra qPCR a cada pocillo.
- Recoger la mezcla maestra qPCR en la parte inferior de cada pocillo mediante una breve centrifugación de la placa de múltiples pocillos 1536 (30 seg usando un de baja velocidad PCR placa spinner).
3. ADN y cebadores en el 1536 multipocillos placa de la plantilla de dispensación
- Prescindir de 5 nl diluida gDNA (paso 1.3) en los pocillos deseados de la placa de múltiples pocillos 1536 utilizando un sistema de transferencia de líquido acústico, como el Echo 550.
- Para el Echo 550, utilice el software de formatear la Plata o bien proporcionar una hoja de cálculo Excel personalizada para programar la transferencia. Asegúrese de que la fuente seleccionada también para el gDNA en el programa coincida con la fuente y en la que ha dispensado físicamente el gDNA (paso 1.3). Seleccione el tipo de placa de fuente '384PP_AQ_BP2', lo que indica que el líquido en la placa 384PP es un tampón sin surfactantes.
- Dispense 10 nl de cebadores, pre-mezclado adelante y atrás a una concentración final de 50 mM cada uno, en los pocillos deseados de la placa de múltiples pocillos 1536 utilizando un sistema de transferencia de líquido acústico, como el Echo 550.
- Para el sistema de transferencia de líquido, utilizar el software de reformateo Plate o bien proporcionar una hoja de cálculo Excel personalizada para programar la transferencia.
NOTA: Aquí es posible prescindir de cebadores de tal manera que el diseño de la placa de múltiples pocillos 1536 coincide con el orden cromosómico de los cebadores por lo que es fácil de inspeccionar visualmente los resultados de la PCR en tiempo realdespués (paso 6). - Piensos compuestos los cebadores en una placa que es compatible con el dispensador de líquido acústico. Para el Echo 550, utilice una placa Labcyte 384LDV (bajo volumen muerto) y elegir el tipo de placa fuente '384LDV_AQ_B' al programar el Echo para indicar que el líquido es un buffer simple sin proteínas.
- Para el sistema de transferencia de líquido, utilizar el software de reformateo Plate o bien proporcionar una hoja de cálculo Excel personalizada para programar la transferencia.
4. Sello y Centrifugar el 1536 multipocillos Plate
- Utilizando un sistema sellador de microplacas tal como el PlateLoc de microplacas térmica sellador, sellar inmediatamente la placa de múltiples pocillos 1536 usando sello ópticamente claro que es compatible con el instrumento sellador térmico. No toque la superficie de la junta para evitar marcas de borrones.
- Si se utiliza el PlateLoc térmica Sellador de microplacas, utilice los siguientes ajustes: sellar tiempo 2,0 seg, la temperatura de sellado 162 ° C, presión de aire 82 psi. Este instrumento toma ~ 5 minutos para calentar y así debe ser activado por adelantado.
- Si se utiliza el PlateLoc microplacas térmica sellador, una ADAPTEr debe ser utilizado para elevar la altura de la placa de múltiples pocillos 1536 en el escenario del instrumento para asegurar un buen sellado.
NOTA: Si bien es preferible comprar una placa de la etapa para la PlateLoc microplacas térmica sellador, una solución alternativa es insertar cuatro ~ 2 mm de espesor arandelas estándar disponibles en cualquier ferretería en las esquinas para elevar la altura de la placa de múltiples pocillos 1536.
- Inmediatamente centrifugar la placa de 1536 pocillos múltiples sellado a 2000 xg durante 3 min a temperatura ambiente para recoger todos los reactivos en la parte inferior de cada pocillo.
5. PCR en tiempo real
- Programa de un instrumento 1536 qPCR, tales como el LightCycler 1536, con un protocolo de amplificación de dos etapas seguido de un análisis de la curva de fusión. Usando el software LightCycler 1536 estableció un programa de la siguiente manera:
- Pre-incubación: 95 ° C, 1 min de espera, la tasa de rampa de 4,8 ° C / seg, no adquisición.
- Dos etapa de amplificación: 30 ciclos de [95 ° C, no espera,rampa tasa de 4,8 ° C / seg, ninguna adquisición; 64 ° C, espera 30 seg, velocidad de rampa de 2.5 ° C / seg, sola adquisición].
- Derretir curva: 95 ° C, 10 seg, la tasa de rampa de 4,8 ° C / seg, no adquisición; 60 ° C, 1 min, la tasa de rampa de 2,5 ° C / seg, no adquisición; 97 ° C, la tasa de rampa de 0,1 ° C / seg, 5 adquisiciones / ° C (continua).
- Cool: 40 ° C, 30 seg, la tasa de rampa de 2,5 ° C / seg, no adquisición.
- Ajuste el formato de detección de 'Green intercalando Tinte' en la pestaña Run Definición.
- Ajuste el control de pipeteo de 'Master Control' en la pestaña Run Definición. Esta característica sirve como un control interno y detecta la presencia de mezcla maestra qPCR en cada pocillo de la placa de múltiples pocillos 1536, que es útil para identificar falsos negativos putativos.
- Inserte la placa de múltiples pocillos 1536 preparado en los pasos 2-4 en el instrumento 1536 qPCR y ejecutar el programa.
Análisis 6. Datos
- Perform inspección visual de los datos dentro del software qPCR.
NOTA: El software de 1536 permite la visualización tanto de amplificación y derretir curvas, así como un mapa de calor que muestra la llamada (positivo, negativo, no válida, N / A, Figura 2), el valor del punto de cruce (deslizamiento escala de colores para el positivo o gris para el negativo; Figura 3), o un punto final de fluorescencia (EPF) Valor (escala móvil de color). - Exportar datos del 1536 del software en forma de un archivo .txt (tabla de resultados) o un archivo .xml, ya sea con los datos brutos o datos calculados para el procesamiento en línea del instrumento.
NOTA: El punto de cruce automatizado específico (Cp) algoritmo llamando integrado en el software de 1536 tiene en cuenta la forma y la pendiente de la curva de amplificación, mientras que la determinación de las llamadas de valores positivos / negativos y Cp con alta confianza, en volúmenes de reacción microlitro bajo sub con relativamente baja los valores de fluorescencia. - Si el verde Maestro ADN LightCycler era usod para qPCR, compruebe que todos los pozos pasaron el "control maestro". Un Despiste indica el bien no recibió mezcla maestra qPCR y el punto de datos faltantes se debe considerar una posible falso negativo. Verifique que todos los pozos pasaron el "control maestro". Un Despiste indica el bien no recibió mastermix ADN y considerar el punto de datos que falta un potencial de falsos negativos.
Resultados
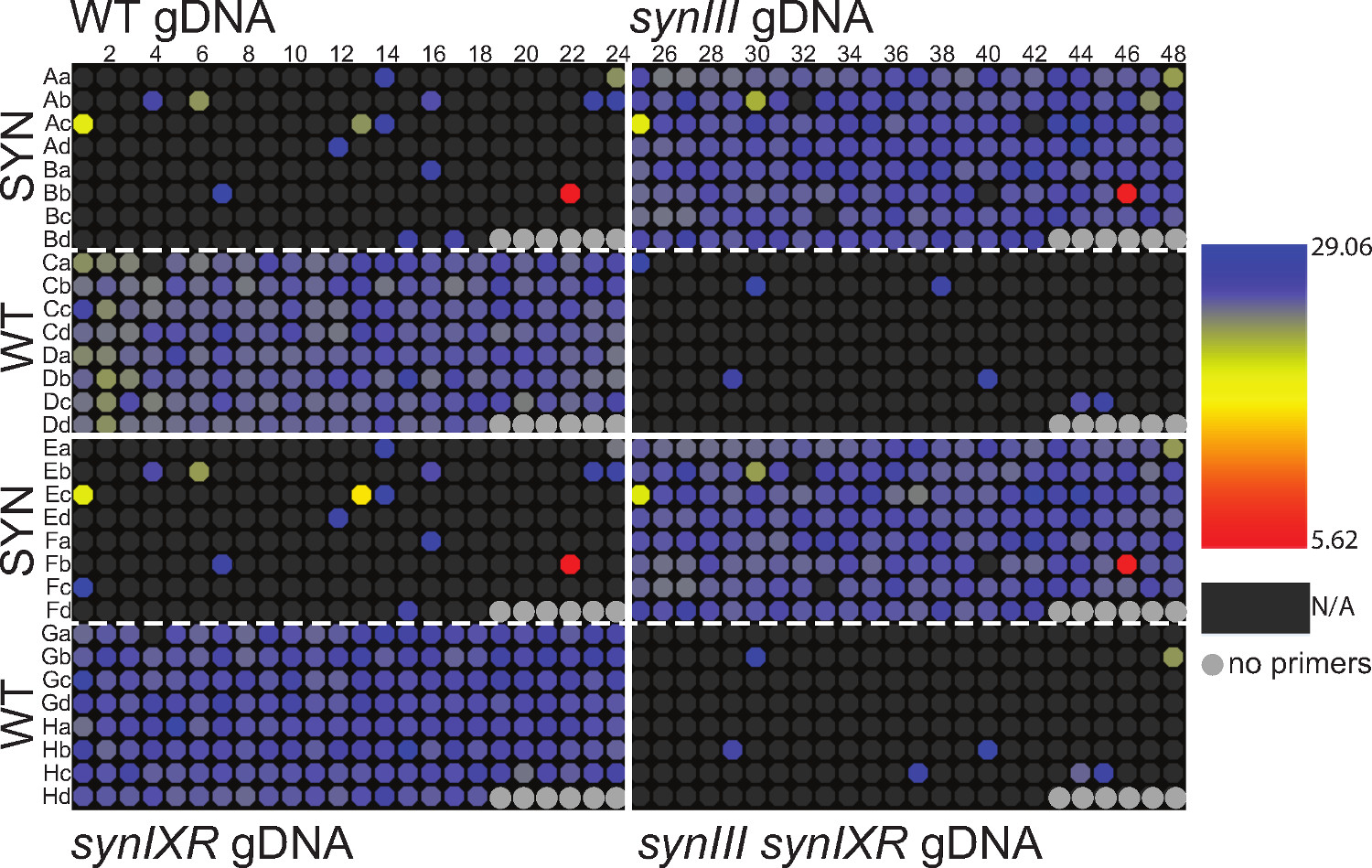
Probamos sintético y de tipo salvaje cromosoma 3 pares de cebadores PCRTag 1 con la levadura de ADN genómico (ADNg) extraído de cuatro cepas diferentes. Cromosoma 3 tiene 186 pares PCRTag de cebadores que abarcan la longitud del cromosoma (synIII es ~ 270 kb y de tipo salvaje del cromosoma 3 es ~ 315 kb). Para probar cada una de las cuatro cepas con ambos conjuntos de cebadores, dividimos la placa de múltiples pocillos en cuatro cuadrantes, uno para cada tipo de gDNA, asignando cebadores PCRTag sintéticos para la mitad superior de cada cuadrante y de tipo salvaje de la mitad inferior. El ADN genómico se extrajo de las cepas de levadura, dos de los cuales codifican tipo salvaje cromosoma 3 (tipo salvaje, synIXR 2), mientras que los dos codificar cromosoma sintético 3 restante (synIII 1, synIII synIXR). mezcla maestra qPCR se dispensó en cada pocillo de una placa de múltiples pocillos 1536 usando un dispensador de líquidos a granel, seguido de gDNA y PCRTag cebadores utilizando los Echo 550. Los cebadores fueron dispuestos de manera idéntica encada cuadrante de la placa de múltiples pocillos para la comparación visual fácil. La placa de múltiples pocillos fue luego térmicamente sellada con sello ópticamente claro y se sometió a análisis de PCR en tiempo real.
En este experimento se observó qPCRTag, en su mayor parte, la amplificación como se esperaba, por lo que amplifican exclusivamente cebadores sintéticos de ADN sintético y viceversa (Figuras 2 y 3). Sin embargo, también se observó varias desviaciones del patrón esperado, lo que sugiere falsos negativos y falsos positivos en el conjunto de datos. En este experimento, se detectó el control maestro en 100% de los pozos, lo que indica el dispensador de líquido a granel dispensado con éxito Mastermix en cada pocillo de la placa (datos no mostrados). Esto descarta una fuente potencial de falsos negativos. Además, algunos de los cromosomas 3 PCRTags se conocen a fallar (mostrado en las Figuras S6 y S7 de Annaluru et al. 1), incluyendo al menos 2 SYN y 1 WTprimer pares; por lo tanto estos pozos pueden ser ignorados en cada cuadrante. Los verdaderos falsos negativos podrían surgir de la falta de transferencia de gDNA plantilla o cebadores, sin embargo, en nuestra experiencia, dada la correcta calibración del Echo 550, así como la preparación de gDNA y cebadores como se describe, esto no ha sido una importante fuente de error. En general, en este experimento, la tasa de falsos negativos fue extremadamente baja para cebadores WT con plantilla WT (~ 2%), aunque algo mayor para cebadores con SYN SYN ADN (~ 8%).
Los falsos positivos, la detección de la señal en los pocillos en los cebadores SYN se mezclan con WT gDNA (y viceversa), pueden surgir de la amplificación cruz o dímeros de cebadores. De hecho, los dímeros de cebadores son a menudo visible por electroforesis en gel (Figura 1B) y representan una fuente de error razonable. Para la aplicación de qPCR, el examen de las curvas de fusión puede ser útil para determinar si diferentes especies, tales como dímeros de cebadores, pueden estar contribuyendo a una señal. Además, performing un experimento de control mediante el cual cebadores se dispensan en ausencia de plantilla de ADN puede ayudar a identificar cebadores con una propensión a dimerizar. Cross amplificación pueden ser observados por electroforesis en gel, en particular si demasiados ciclos de PCR se llevan a cabo o si la temperatura de recocido es demasiado bajo. Hemos tratado de minimizar el número de falsos positivos debido a la amplificación cruzada mediante la optimización de estos dos parámetros para el protocolo qPCRTag. Por último, el examen de los valores de punto de cruce (Cp) para cada pozo puede ayudar a identificar cebadores que no son adecuados para la detección basada en tiempo real (Figura 3, por ejemplo, BB22, FB22, BB46, Fb46). En general, en este experimento, la tasa de falsos positivos fue baja para cebadores SYN con plantilla WT (~ 5%) y más alto para los cebadores WT con plantilla SYN (~ 10%).

Figura 2: Placa mapa de calor que muestra la presencia / abllamada sence para un experimento qPCRTag. Cuatro tipos diferentes de ADN genómico (cuadrantes separados por líneas blancas sólidas) se sometieron a análisis PCRTag usando sintético (SYN) y de tipo salvaje (WT) cromosoma 3 cebadores PCRTag (líneas blancas discontinuas para separar SYN (arriba ) y WT (abajo) en cada cuadrante). WT y synIXR gDNA codifican tipo salvaje cromosoma 3, produciendo la amplificación con cebadores WT PCRTag. SynIII y synIII synIXR gDNA codificar cromosoma sintético 3, produciendo la amplificación con cebadores SYN PCRTag. Los cebadores están dispuestos de acuerdo con su posicionamiento cromosómica de izquierda a derecha y colocados de forma idéntica en los cuatro cuadrantes para la comparación. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: calor de placasmapa que muestra punto de cruce (Cp) valor para un experimento de qPCRTag. Este es el mismo conjunto de datos como en la Figura 2 y la disposición de la placa es por lo tanto idéntica. N / A se refiere a 'sin amplificación ". Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
La incorporación de la detección por PCR en tiempo real en el ensayo de genotipado PCRTag es un avance importante para el proyecto Sc2.0 ya que permite un mayor rendimiento significativamente. El flujo de trabajo anterior especifica 2,5 reacciones mu l en placas de 384 pocillos de PCR, 1,5 horas de tiempo térmica de ciclo de ejecución, electroforesis en gel de agarosa, y anotación manual de la gel.
El flujo de trabajo que aquí se presenta, llamado análisis qPCRTag, supera varios cuellos de botella importantes. En primer lugar, una carrera qPCRTag condensa 4 x 384 y placas en un único experimento que puede ser procesada, de principio a fin (placa de configurar más el tiempo de ejecución), en aproximadamente una hora. Es importante señalar que el costo de reactivo por pocillo para el análisis qPCRTag está a la par con el enfoque basado en gel de agarosa rendimiento inferior. Sin embargo, eludiendo electroforesis en gel y anotación manual, el flujo de trabajo qPCRTag proporciona ahorros sustanciales en el tiempo y mano de obra, que son las principales ventajas. Es importante destacar que el flujo de trabajo es en qPCRTagenteramente compatible con la automatización.
Una preocupación importante con el uso de la detección en tiempo real como la salida del ensayo PCRTag es la tasa de falsos positivos y falsos negativos. Dado que los cebadores PCRTag no fueron diseñados originalmente para su uso en PCR en tiempo real, se espera que no todos los pares de cebadores serán apropiados para su uso con esta salida. Para ello es importante para validar la función y la especificidad de todos los pares de cebadores PCRTag excluir el subconjunto que no funcionan en el ensayo basado en tiempo real en la delantera. Por ejemplo, el cromosoma 3 PCRTag falsos positivos y negativos están haciendo ya gran reproducible en el tiempo real de datos de PCR (Figuras 2 y 3) y estos pueden ser excluidos de los nuevos análisis. Además, los pares de cebadores conocidos a fallar (evaluados por electroforesis en gel y se muestran en las figuras S6 y S7 de Annaluru et al. 1) También se puede excluir. Esto se logra fácilmente, simplemente excluding los pares de cebadores defectuosas al configurar el protocolo de transferencia acústica.
Al igual que la mayoría de los ensayos de alto rendimiento, tenemos la intención de utilizar la detección PCRTag tiempo real como pantalla principal para identificar transformantes que merecen una mayor validación aguas abajo. Posteriormente, el estándar de oro para la investigación secundaria permanecerá análisis PCRTag con electroforesis en gel como la lectura. Más allá de la aplicación del análisis PCRTag para Sc2.0, combinando los sistemas de manejo de líquidos a nanoescala con tecnología de última generación con un alto rendimiento en tiempo real la tecnología PCR permite un análisis rápido y automatizado y tiene potencial de impactar muchos campos. Por ejemplo, este flujo de trabajo se podría aplicar a cribado de alto rendimiento de la biblioteca, el diagnóstico de enfermedades infecciosas, el análisis microbioma, y edición genoma de vanguardia enfoques intentar modificar múltiples loci simultáneamente.
Divulgaciones
LAM, NAP, JAM y JDB tienen nada que revelar. RC y AL son empleados de Roche Ciencias de la Vida, de Estados Unidos y trabajó en el desarrollo de la aplicación con LAM como parte de una prueba de principio experimento.
Agradecimientos
Esta obra fue financiada en parte por la Fundación Nacional de Ciencias de subvención MCB-0718846 y Defensa Agencia de Proyectos de Investigación Avanzada Contrato N66001-12-C-4020 (a JDB). LAM fue financiado por una beca postdoctoral de Ciencias Naturales e Ingeniería de Investigación de Canadá. La publicación de este artículo es patrocinado por Roche.
Materiales
| Name | Company | Catalog Number | Comments |
| Yeast gDNA prep | |||
| yeast gDNA | custom | custom | template for real time PCR |
| Acid-washed glass beads (0.5 mm) | Sigma | G8772 | yeast cell lysis |
| Ultrapure Phenol:Chloroform:Isoamyl Alcohol (25:24:1, v/v) | Invitrogen | 15593-031 | yeast cell lysis |
| 5432 Mixer | Eppendorf | 5432 | yeast cell lysis |
| Microcentrifuge 5417R | Eppendorf | 22621807 | yeast cell lysis |
| Qubit 3.0 Fluorometer | Life Technologies | Q33216 | gDNA quantification |
| Qubit dsDNA BR Assay Kit | Life Technologies | Q32850 | gDNA quantification |
| Labcyte 384PP plate | Labcyte | P-05525 | gDNA source plate |
| DNA Green Mastermix prep and dispensing | |||
| LightCycler 1536 DNA Green | Roche | 5573092001 | real time PCR mastermix |
| 1.5 ml Microfuge Tube Holder | ARI | EST BD060314-1 | microfuge tube holder for deck of Cobra |
| LightCycler 1536 Multiwell Plate | Roche | 5358639001 | |
| Cobra liquid handling system | Art Robbins Instruments | 630-1000-10 | dispense qPCR master mix into 1536 plate |
| PCR Plate Spinner | VWR | 89184-608 | cenrifugation of 1536 plate |
| Template and primer dispensing | |||
| PCRTag primers | IDT | custom | premixed forward and reverse, 50 μM each |
| TempPlate pierceable sealing foil, sterile | USA Scientific | 2923-0110 | temporary seal for PCRTag primer plates |
| Labcyte LDV 384 well plate | Labcyte | LP-0200 | pre-mixed primer source plate |
| Echo 550 Liquid Handler | Labcyte | transfer 2.5 nl drops of primer and template DNA into 1536 plate | |
| Plateloc Thermal Microplate Sealer | Agilent | G5402-90001 | heat seal for 1536 plate prior to LC1536 run |
| Clear Permanent Seal | Agilent | 24212-001 | optically clear heat seal for LC1536 multiwell plate |
| PlateLoc Roche/LightCycler 1536 Plate Stage | Agilent | G5402-20008 | can substitute ~2 mm thick washers |
| Sorvall Legend XTR | Thermo Scientfic | 75-004-521 | centrifuge heat sealed 1536 plate |
| Industrial Air Compressor | Jun Air | 1795011 | to run the Echo and Heat Sealer |
| Real Time PCR | |||
| LightCycler 1536 | Roche | requires 220 V outlet | |
Referencias
- Annaluru, N., et al. Total synthesis of a functional designer eukaryotic chromosome. Science. 344 (6179), 55-58 (2014).
- Dymond, J. S., et al. Synthetic chromosome arms function in yeast and generate phenotypic diversity by design. Nature. 477 (7365), 471-476 (2011).
- Goffeau, A., et al. Life with 6000 genes. Science. 274 (5287), 546-547 (1996).
- Dymond, J., Boeke, J. The Saccharomyces cerevisiae SCRaMbLE system and genome minimization. Bioeng Bugs. 3 (3), 168-171 (2012).
- Richardson, S. M., Nunley, P. W., Yarrington, R. M., Boeke, J. D., Bader, J. S. GeneDesign 3.0 is an updated synthetic biology toolkit. Nucleic Acids Res. 38 (8), 2603-2606 (2010).
- Richardson, S. M., Liu, S., Boeke, J. D., Bader, J. S. Design-A-Gene with GeneDesign. Methods Mol Biol. 852, 235-247 (2012).
- Lajoie, M. J., et al. Probing the limits of genetic recoding in essential genes. Science. 342 (6156), 361-363 (2013).
- Jovicevic, D., Blount, B. A., Ellis, T. Total synthesis of a eukaryotic chromosome: Redesigning and SCRaMbLE-ing yeast. Bioessays. 36 (9), 855-860 (2014).
- Dymond, J. S. Preparation of genomic DNA from Saccharomyces cerevisiae. Methods Enzymol. 529, 153-160 (2013).
- Brachmann, C. B., et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 14 (2), 115-132 (1998).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados