
Method Article
Un ensayo de HS-MRM para la cuantificación de las proteínas de la célula huésped en productos biofarmacéuticos de proteínas por cromatografía líquida QTOF masa espectrometría por movilidad iónica
* Estos autores han contribuido por igual
En este artículo
Resumen
Aquí, describimos un análisis cromatográfico juntado con la separación de movilidad de iones precursores de péptidos seguida por la alta resolución (~ 30.000) MS-detección de fragmentos de péptidos para la cuantificación de normas péptido con púas en un anticuerpo monoclonal Digest.
Resumen
El análisis de impurezas de proteínas de bajo nivel (1-100 ppm) (e.g., proteínas de la célula huésped (EP)) en proteína biotherapeutics es un desafiante ensayo que requieren alta sensibilidad y un amplio rango dinámico. Ensayos de cuantificación basada en espectrometría de masa de proteínas implican típicamente la digestión de proteínas, seguida por la reacción de reacción selectiva múltiple monitoreo monitoreo de cuantificación (SRM/MRM) de péptidos con un tándem (Rs ~ 1.000) baja resolución Espectrómetro de masas cuadrupolar. Una de las limitaciones de este enfoque es el fenómeno de interferencia observado cuando el péptido de interés tiene el "mismo" precursor y fragmento de masa (en términos de valores de m/z) como otros péptidos Co liberador presentes en la muestra (dentro de una ventana de 1-Da). Para evitar este fenómeno, se propone un enfoque alternativo de la espectrometría de masa, un ensayo MRM de alta selectividad (HS) que combina la separación de movilidad de iones precursores de péptidos con alta resolución (Rs ~ 30.000) MS la detección de fragmentos de péptido. Exploramos las capacidades de este enfoque para cuantificar estándares de baja abundancia péptido tacon en una síntesis de anticuerpo monoclonal (mAb) y demostró que tiene la sensibilidad y rango dinámico (por lo menos 3 órdenes de magnitud) alcanzado típicamente en HCP Análisis. Todos los seis estándares de péptido se detectaron en concentraciones tan bajas como 0.1 nM (1 femtomole cargado en una columna cromatográfica de 2,1 mm ID) en presencia de un fondo de alta abundancia péptido (2 μg de una mAb digest cargado en columna). Al considerar el MW de la fosforilasa de conejo (97.2 kDa), de la cual se derivaron los péptidos con picos, LOQ de este ensayo es inferior a 50 ppm. Desviación estándar relativa (RSD) de zonas de pico (n = 4 repeticiones) eran menos del 15% en toda la gama de concentración todo investigada (0.1-100 nanómetro o 1-1.000 ppm) en este estudio.
Introducción
Cuantificación de grandes biomoléculas (proteínas) en ajustes industriales se basa actualmente en inmunoensayos (p. ej., ELISA), principalmente debido a varias ventajas: sensibilidad, alto rendimiento, facilidad de uso y bajo costo por muestra. Cuando se aplica para analizar las impurezas de la proteína de baja abundancia (ppm de 1-100 de las proteínas de la célula huésped (EP)) presentes en proteínas terapéuticas, estos ensayos biológicos proporcionan típicamente la concentración total de HCP (generalmente expresada en ppm o ng mAb HCP/mg), pero no se puede identificar y medir los contaminantes HCP. Recientemente se han desarrollado varios ensayos de MS para complementar a ELISAs o proporcionar información que Elisa no ofrecen1,2,3,4,5,6 , 7 , 8 , 9. debido a la complejidad de la muestra y el requisito para detectar péptidos HCP a través de un amplio rango dinámico en la concentración (al menos 3 órdenes de magnitud), tienen métodos cromatográficos multidimensionales licitación fraccionamiento muestra amplia tradicionalmente utilizado para ayudar a identificar la abundancia baja EP1,2,3,4,5,6,7.
Un natural paso siguiente HCP identificación y validación es PCH seguimiento (monitoreo) a través de múltiples lotes de productos biofarmacéuticos. En esta situación, la sola dimensión LC/MS métodos se han propuesto para mejorar muestra rendimiento8,9. Sin embargo, la precisión y las mediciones de rango dinámico de HCP pueden verse afectadas en un análisis de LC/MS 1D por la abrumadora presencia de péptidos de la biofarmacéutica. En comparación con una separación multidimensional, el potencial de señal interferencia19,20,21,22 se incrementa en una sola dimensión separación cromatográfica porque la probabilidad de más precursores de péptidos que co liberador se incrementa. La incorporación de medios orthogonal para separar precursores de péptidos sin extender el tiempo de separación cromatográfica claramente sería ventajosa. Viajes onda ion movilidad (TWIM)10 tiene la capacidad para resolver los espectros de MS congestionados en milisegundos. Aproximadamente 500 separaciones de movilidad pueden realizarse durante la elución de un único péptido, suponiendo una anchura del pico cromatográfico completo de 10 s y teniendo en cuenta que el tiempo de ejecución de una separación de IM en el instrumento de movilidad de iones es ms 20.
Análisis de espectrometría de masa para la cuantificación de proteínas se han desarrollado con éxito en los últimos decenio mediante la bien aceptada seleccionado (múltiples) reacción control de enfoque (método de SRM/MRM) implementado en el tándem espectrómetros de masas11, 12,13,14,15,16,17,18,19,20,21 ,22,23. Una de las limitaciones de este análisis de espectrometría de masa baja es la interferencia fenómeno19,20,21,22 cuando el péptido de interés tiene el "mismo" precursor y fragmento de masa como otros péptidos Co liberador presentan en la muestra (dentro de una ventana de 1-Da). Hay dos formas para mejorar la precisión de los métodos SRM/MRM: una opción implica un paso adicional de la separación a nivel precursor para eliminar iones interferentes del precursor, mientras que la otra opción es aumentar la resolución de la MS de la detección de precursores/fragmento a evitar la superposición de las señales de MS. El modo de adquisición de alta selectividad (HS) MRM descrito aquí aprovecha de ambos de estos enfoques por la separación de movilidad de iones precursores de péptidos de acoplamiento con las alta resolución (Rs ~ 30.000) MS la detección de fragmentos de péptido. El ensayo descrito aquí cubiertas por lo menos tres órdenes de magnitud, que es el rango dinámico normalmente observadas en SRM/MRM proteómica experimentos17,18,24.
La utilidad de la prueba de HS-MRM para la cuantificación de la HCP se demostró mediante el control de la linealidad de la señal producida por las seis normas de péptido tacon a diferentes concentraciones (rango de 0.1 a 100 nM) en una síntesis de anticuerpo monoclonal.
Protocolo
1. preparación de la recopilación de infliximab (procedimiento de ~ 24 h)
- Preparar soluciones frescas de 50 mM de bicarbonato de amonio, 1% de surfactante aniónico en 50 mM NH4HCO3, 500 mM Ditiotreitol (DTT) en 50 mM NH4HCO3y 500 mM Yodoacetamida (IAM) en 50 mM NH4HCO3.
- A 750 μl de 50 mM NH4HCO3, añadir 200 μL de mAb de infliximab (10 mg/mL) y 50 μl de solución al 1% surfactante aniónico y desnaturalizar la proteína durante 15 min a 60 ° C en presencia de surfactante aniónico de 0.05%.
- Agregar 40 μl de 500 mM DTT y reducir la muestra por 60 min a 60 ° C en presencia de 20 mM TDT.
- Añadir 20 μl de 500 mM IAM y alkylate la muestra durante 30 min a temperatura ambiente en la oscuridad en presencia de ~ 10 m m IAM.
- Añadir el contenido del tubo de microcentrífuga a un frasco de vidrio que contiene 20 μg de MS grado secuencia Lys C/tripsina y digerir la muestra 3 h a 37 ° C.
- Añadir una segunda enzima alícuota (20 μg) mediante la transferencia de la muestra digerida a otro frasco de cristal que contiene 20 μg de MS grado secuencia Lys C/tripsina y digerir la muestra durante la noche (12-15 h) a 37 ° C.
- Después de la proteólisis durante la noche, añadir 5 μl del 100% ácido fórmico (FA) e incubar la digestión durante 30 min a 37 ° C para descomponer el ácido lábil tensioactivador aniónico.
- Desactivación de la recopilación durante 15 min a 4.000 x g en una centrífuga para precipitar el componente insoluble del Tensioactivo aniónico.
- Recuperar ~ 1.000 μl de la recopilación de infliximab y colocar en un frasco de muestreador automático de LC; la concentración de recopilación debe ser ~ 2 mg/mL. Guardar el resumen en un congelador a-20 ° C hasta que el sistema LC/MS está listo para el análisis.
2. clavar de normas del péptido (~ 30 min)
- Descongelar el Resumen de infliximab colocando la muestra congelada en el frasco de cristal en un banco a temperatura ambiente.
- Añadir 1 mL de 0.1% FA en frasco de digest de H2O a un conejo fosforilasa b (PHO) para preparar 1 μm soluciones para todos los seis estándares péptido de la PHO contenida en el vial (frasco contiene 1 nmol de cada péptido).
- Preparar diluciones de 4 x 1 mL de la solución madre de PHO para obtener soluciones que contienen 0.1, 1, 10 y 100 péptidos nM, utilizando 0.1% FA como el solvente de dilución. Preparar todas las diluciones en viales de vidrio (frascos del muestreador automático de LC) y añadir 100 μl de infliximab digest como la matriz de fondo a todos los frascos. Carga 2 μg de digest fondo en columna con cada inyección de 10 μl. Preparar una muestra en blanco que contiene el Resumen de infliximab diluido (el mismo 1:10 dilución), con no péptidos con picos.
3. configuración del método de adquisición de datos LC/HDMS E
Nota: El flujo de trabajo que resume los pasos necesarios para configurar una adquisición de HS-MRM es representado en la figura 1 y se describe en detalle en las secciones 3-6. La adquisición de un conjunto de datos HDMSE independiente de los datos es necesaria para establecer el tiempo de retención de cada péptido monitoreado, el padre m/z del ion del péptido más abundante después de la ionización por electrospray, y el correspondiente CCS (choque Cruz sección) deriva de la separación de movilidad de iones. Además, el conjunto de datos independiente de los datos proporciona información sobre el m/z de los tres iones del fragmento más abundantes de cada precursor del péptido. En el segundo paso del flujo de trabajo (optimización de la CE), se incrementa la sensibilidad del ensayo de sintonización de la energía de la celda de colisión para obtener la intensidad más alta del ion para cada ión fragmento. Finalmente, en el paso final, todos los parámetros descritos anteriormente se introducen en el editor de método de HS-MRM para cada péptido monitoreado.
- Realizar experimentos de LC/MS en un cuadrupolo tiempo de vuelo (QTOF) Espectrómetro de masas acoplado a un sistema de cromatografía líquida ultra utilizando un método de adquisición de datos-independiente (HDMSE).
Nota: Información más detallada sobre la configuración del equipo está incluido en la sección Material. Este instrumento utiliza un dispositivo de movilidad de iones onda que viaja para la separación de péptidos precursores10. - Preparar dos fases móviles que contiene 0,1% FA en agua (solvente A) y 0.1% FA en acetonitrilo (solvente B).
Nota: Utilizar una columna de C18 híbrido superficie cargada (CSH) (2.1 x 150 mm, lleno de partículas de 1.7 μm) para la separación de los compendios mAb claveteados. - En el software de adquisición de datos, haga clic en "Crear/análisis de método" y elegir "Generar una adquisición y método de proceso". Escriba el nombre de método, el método carpeta (directorio) y haga clic en "Siguiente".
- Para "Tipo de análisis," elegir "péptido mapa (IMS)" y en la pestaña de "Sistema de instrumento", seleccione el instrumento de "Gestor de solvente cuaternario". Edite la configuración del degradado para lograr la separación del péptido con un caudal de 200 μL/min uso una elución gradiente de 1 a 40% de disolvente B en 30min seguido de un lavado de columna de 2 min y un equilibrado de 9 min, con un tiempo de ejecución total de 50 minutos introducir estos experimental parámetros en el editor de método de LC.
- En la pestaña de "Sistema de instrumento", seleccione el instrumento de "Gestor de muestra". Ajustar la temperatura de la muestra a 10 ° C y la temperatura de la columna a 60 ° C.
- Operar el espectrómetro de masas QTOF en ion positivo modo de sensibilidad de ESI, con un poder de resolución típico de 30.000 FWHM.
Nota: LC/HDMSE es un modo de adquisición de datos-independiente (DIA) que funciona recogiendo rápidamente exploraciones con energías de colisión (en la celda de colisión) se alterna entre una energía baja (6 V para los análisis de canal 1 de MS) y una energía elevada (15 - 40 V rampa a producen los espectros de fragmentación sin selección de precursor en el canal 2 de la MS).- Ingrese los parámetros de MS relacionada con la fuente por defecto en el software de adquisición de datos: "Mi obra/instrumento sistema," seleccione la pestaña "Vion IMS Qtof" y en el menú "Herramientas", introduzca estos parámetros: 3.0 kV tensión capilar, temperatura de 100 ° C fuente, fuente de V 100 offset, 50 L/h cono de flujo de gas y 40 V tensión de cono. Ajustar la temperatura del desolvation a 250 ° C, el caudal de gas desolvation a 500 L/h y la tensión capilar de referencia 3.0 kV.
- En el software de adquisición de datos, haga clic en "Crear/análisis de método" y elegir "Generar una adquisición y método de proceso". Escriba el nombre del método y el método de carpeta (directorio) y haga clic en "Siguiente".
- Para "Tipo de análisis," elegir "péptido mapa (IMS)" y en la pestaña de "Sistema de instrumento", seleccione el instrumento "Vion IMS QTof". Haga clic en "Finalizar".
Nota: El método LC/HDMSE recién creado se abre automáticamente en la pantalla "Configuración de método de instrumento". - En la pestaña "Settings", entrar en estos parámetros: 3,0 kV tensión capilar, temperatura de fuente de 100 ° C, 250 ° C desolvation temperatura, flujo de gas de 50 L/h cono y flujo de gas de 500 L/h desolvation.
- En la configuración de la pestaña de "Experimento", elige "MSEde alta definición" y escoge "Tiempo del método de análisis." En la ficha "Configuración de exploración", introduzca estos parámetros: baja masa, 100; la Misa, 2.000; tiempo, 400 ms. en la pestaña de "CE" de exploración, tipo "6 V" para la configuración de baja energía y elegir una rampa de gran energía de 15 a 40 V. Asegúrese que "Reducción de datos desactivar" comprobada.
- Infundir una solución de encefalina leucina ng/mL 50 (LE) en 50% acetonitrilo con 0,1% FA a un flujo de 10 μl/min para la calibración de la masa de bloqueo durante la adquisición de datos.
Nota: Los datos de bloqueo masa adquiere cada 5 min utilizando la misma tasa de adquisición sobre el mismo rango de masa.
- Analizar la muestra de 10 nM, tacon de PHO usando el análisis de LC/HDMSE descrito por inyección 10 μl de muestra (el importe cargado en la columna es 100 fmol para cada péptido estándar).
4. configuración del método Tof-MRM para optimización de energía (CE) de colisión
- Del conjunto de datos LC/HDMSE , obtener el tiempo de retención, el precursor y el fragmento iónico m/z para cada péptido PHO. Conservar la m/z y el estado de carga de los más abundantes iones del fragmento precursor ion y las correspondientes tres más abundantes.
Nota: Un ejemplo de los espectros de baja energía y alta energía típicamente grabado por el análisis de LC/HDMSE se presenta en la figura 2. - Uso de optimización energética (CE) de colisión en un experimento SRM/MRM para obtener la mejor señal para cada péptido23.
- En el software de adquisición de datos, haga clic en "Crear/análisis de método" y elegir "Generar una adquisición y método de proceso". Escriba el nombre de método, el método carpeta (directorio) y haga clic en "Siguiente".
- "Tipo de análisis," seleccione "Cuantificar". Elegir "Cuantificar ensayo cromatográfico de Tof 2D" y haga clic en "Siguiente". En la pestaña de "Sistema de instrumento", seleccione "Vion IMS QTof" y haga clic en "Finalizar".
Nota: El método recién creado de Tof-MRM se abre automáticamente en la pantalla "Configuración de método de instrumento". - En la pestaña "Settings", entrar en estos parámetros: 3,0 kV tensión capilar, temperatura de fuente de 100 ° C, 250 ° C desolvation temperatura, flujo de gas de 50 L/h cono y flujo de gas de 500 L/h desolvation.
- En la configuración de la pestaña de "Experimento", seleccione "Tabla de funciones" y elija "uno o varios MS, MSM o MRM funciones." Usando las flechas hacia abajo, de configurar una función de "Tof-MRM" para cada péptido introduciendo tres "transiciones" (combinaciones del precursor más abundante y tres de sus más abundantes iones fragmento).
Nota: En modo Tof-MRM, la mayor sensibilidad se logra utilizando un enfoque MRM programado. - En el editor de función "Tof-MRM", elegir una ventana de tiempo de retención para cada péptido (al menos 1 minuto de largo), dispuestas según el orden de elución de péptido. En el editor de función "Tof-MRM", inserte el péptido precursor m/z y el producto de m/z; Seleccione la ventana de aislamiento quad como "Low" (ventana 4-Da) para el precursor del péptido; Seleccione un tiempo de exploración fija de 100 ms; y elegir doce valores de la energía de colisión diferentes en el rango de 14-36 V, como sigue: 14, 16, 18, 20, 22, 24, 26, 28, 30, 32, 34 y 36, como se muestra en el ejemplo de la figura 3.
Nota: Con el método de Tof-MRM descrito anteriormente, por lo menos 10 puntos por pico cromatográfico se recogen para cada transición en cada CE especificado. Para reducir el tamaño del archivo de datos, seleccione la opción "Ancha" (Da 6 ventana) para la señal del ión fragmento. En la tabla 2 se muestran ejemplos de los valores de CE-optimizado. - Analizar la muestra de 10 nM, tacon de PHO usando el ensayo de Tof-MRM descrito por vía parenteral 10 μl de muestra (el importe cargado en la columna es 100 fmol para cada péptido estándar).
5. configuración del método de adquisición de MRM capítulo final para la cuantificación del péptido mediante separación de movilidad de iones precursores de péptidos
- En el menú de "Investigar" el software de adquisición de datos, Mostrar todas las trazas cromatográficas 11 generadas para cada CE para cada péptido e integrar los picos utilizando el botón de "Integración" de la barra de herramientas "opciones de procesamiento y operaciones"; el área de pico más alto indicará el mejor CE para cada transición.
- Comparar las mejores zonas de pico para los 3 fragmentos de cada péptido visualmente y conservar sólo los mejores "transición" (el fragmento de iones que produce la señal más intensa); La tabla 2 muestra el mejor "transiciones" de cuatro péptidos PHO.
- Configuración de un método de HS-MRM que contiene sólo un ión fragmento por péptido. Utilice los valores del CCS de cada precursor del péptido del LC/HDMSE conjunto de datos.
- Modificar el método de Tof-MRM creado en la sección anterior (sección 4) seleccionando la función "HS-MRM" en lugar de la función de "Tof-MRM".
- En el editor de función "HS-MRM", especifique los siguientes parámetros relacionados con el precursor: m/z, resolución de cuadrupolo: bajo (Da 4), cargar estado, precursor CCS valor y resolución de CCS precursor: bajo. Para el ion del producto, ingrese su m/z, la energía de colisión óptimo, elegir un tiempo de exploración de 0,4 s y seleccione una configuración de "Amplio" espectro (6 Da) para incluir todos sus isótopos. Un ejemplo del método de adquisición capítulo MRM final para todos cuatro péptidos se presenta en la figura 4.
- Analizar todas las muestras utilizando el método de HS-MRM, a partir de la recopilación de mAb en blanco (muestra sin tacon), seguidas de 4 inyecciones replicadas (10 μl) de las siguientes concentraciones: 0.1, 1, 10 y 100 nM PHO péptidos.
6. creación de un método de procesamiento para el análisis del conjunto de datos capítulo MRM
- Crear un método de procesamiento para el análisis del conjunto de datos capítulo-MRM.
- En el método de análisis para el método de adquisición de datos "HS-MRM", haga clic en la pestaña de "Propósito" y elija "Administrar componentes."
- En el menú desplegable debajo de "Tipo de experimento", seleccione la opción "capítulo MS/MS/HS-MRM".
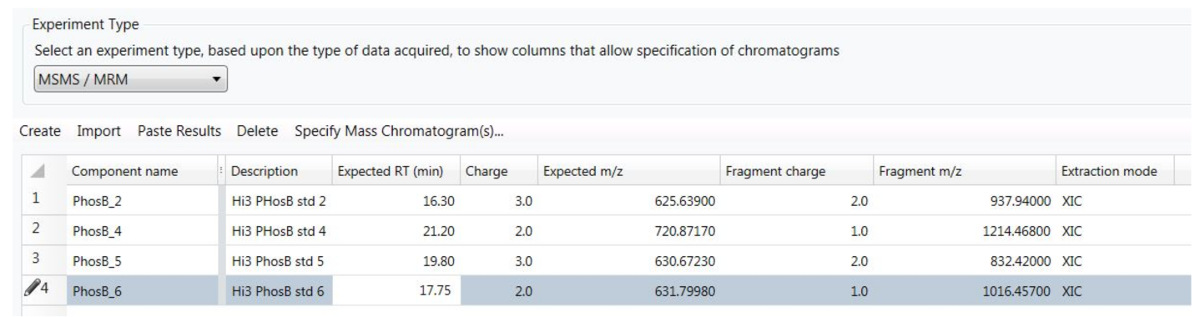
- Haga clic en "Crear nueva entrada" e introducir los siguientes parámetros en el editor de métodos de procesamiento: tiempo de retención del péptido (RT), estado de carga de precursor, precursor m/z y precursor deriva tiempo (DT). Para el ión fragmento, entrar en su estado m/z y carga y especificar el rastro "XIC" como el modo de extracción.
- En la pestaña de "Propósito" del mismo método de adquisición "HS-MRM", haga clic en"predeterminado" y entrar en las siguientes concentraciones: 0.1, 1, 10 y 100 nM PHO péptidos.
- Especifique los siguientes parámetros en los "ajustes de procesamiento y extracción": tolerancia total predeterminado, 10 ppm (para el m/z de todos los iones del fragmento) y por defecto deriva del tiempo, 5%; en la figura 5 se muestra un ejemplo del método de procesamiento del péptido PHO.
- Para el tipo de calibración, seleccione el ajuste de curvas lineales con 1 /2 carga.
- Procesar los datos de HS-MRM para integrar todos los cromatogramas y construir las curvas de calibración de cada péptido PHO.
Resultados
Las secuencias individuales de seis fosforilasa b péptido las normas contenidas en la mezcla de péptidos PHO se muestran en la tabla 1, junto con sus tiempos de retención y sus precursores más abundantes en el HDMSE experimentan. El primer paso en el desarrollo de un ensayo MRM (HS) de alta selectividad es la adquisición de un conjunto de datos HDMSE establecer los tiempos de elución de cada péptido PHO, junto con su correspondiente mayoría precursor abundante y los tres fragmentos más abundantes. La figura 2 muestra los espectros HDMSE adquiridos para uno de los péptidos PHO (Pep 6) dispararon en la recopilación de infliximab. Después de establecer el 3 mejor "transiciones" (combinaciones de masas precursor y fragmento) para cada péptido, se realiza un experimento de Tof-MRM para encontrar la energía de colisión óptima para aprovechar al máximo las señales generadas para cada péptido. Los resultados del experimento de optimización de CE se resumen en la tabla 2. El ensayo final de capítulo-MRM (ver figura 4) conserva sólo los mejores «transición» para cada péptido y se utiliza para el análisis de las muestras inoculadas. Ejemplos de cromatogramas de HS-MRM generados para péptidos PHO 4 a través de todas las concentraciones investigadas se presentan en la figura 7. Cuatro curvas de calibración obtengan para cada siguiente péptido la integración de los picos de HS-MRM resaltado en la figura 7 se muestran en la figura 8. Además, la desviación de estándar relativa de pico zona, calculada en base a 4 replicar las inyecciones, se resume en 4 mesas que se muestra en la tabla 3.
| Péptido | Péptido | Retención de | Estados de carga | ||||
| ID | Secuencia de | tiempo (min) | + 1 | + 2 | + 3 | + 4 | |
| Pep 1 | VLYPNDNFFEGK | 19.4 | 1442.6951 | 721.8512 | 481.5699 | 361.4292 | |
| Pep 2 | TCAYTNHTVLPEALER | 16.0 | 1874.9065 | 937.9569 | 625.6404 | 469.4821 | |
| Pep 3 | IGEEYISDLDQLRK | 18.9 | 1678.8646 | 839.9360 | 560.2931 | 420.4716 | |
| Pep 4 | LLSYVDDEAFIR | 21.1 | 1440.7369 | 720.8721 | 480.9172 | 360.9397 | |
| Pep 5 | LITAIGDVVNHDPVVGDR | 19.7 | 1890.0080 | 945.5076 | 630.6742 | 473.2574 | |
| Pep 6 | VFADYEEYVK | 17.7 | 1262.5939 | 631.8006 | 421.5362 | 316.4039 | |
Tabla 1. Normas PHO péptido contenidas en la mezcla de masa PREP tacon en la recopilación de infliximab. Tiempos de retención de péptido y sus precursores más abundantes (resaltados en negrita) se muestran en formato tabular.
| Péptido | Péptido | Retención de | Precursor de péptidos | Fragmento más abundantes iones/carga | Óptimo | ||||
| ID | Secuencia de | tiempo (min) | carga y m/z | Tiempo de deriva (ms) | Me | II | III | CE (V) | |
| Pep 2 | TCAYTNHTVLPEALER | 16.0 | 625.6404 (+ 3) | 6.2 | 714.3781 (+ 1) | 807.4177 (+ 2) | 827.4621 (+ 1) | 24 | |
| Pep 4 | LLSYVDDEAFIR | 21.1 | 720.8721 (+ 2) | 7.5 | 865.4050 (+ 1) | 964.4734 (+ 1) | 1214.5688 (+ 1) | 22 | |
| Pep 5 | LITAIGDVVNHDPVVGDR | 19.7 | 630.6742 (+ 3) | 6.3 | 642.3570 (+ 1) | 689.8391 (+ 2) | 832.4236 (+ 2) | 20 | |
| Pep 6 | VFADYEEYVK | 17.7 | 631.8006 (+ 2) | 7.0 | 830.3931 (+ 1) | 945.4200 (+ 2) | 1016.4571 (+ 1) | 24 | |
Tabla 2. Resultados del experimento de optimización de Tof-MRM: se indican los tres más abundantes fragmentos de cada péptido PHO cuantificada en este estudio, junto con la correspondiente energía de colisión optimizado.
| Conc | Cantidad | Pep 2 zonas de pico (tabla 3A) | |||||
| (nM) | en la columna (fmoles) | Rep01 | Rep02 | Rep03 | Rep04 | Significa | RSD (%) |
| 0.1 | 1 | 490 | 439 | 462 | 431 | 456 | 5.8 |
| 1 | 10 | 5121 | 4842 | 5198 | 4842 | 5001 | 3.7 |
| 10 | 100 | 63853 | 64279 | 66111 | 62509 | 64188 | 2.3 |
| 100 | 1000 | 612392 | 605553 | 613229 | 611004 | 610545 | 0.6 |
| Conc | Cantidad | Pep 4 áreas de pico (tabla 3B) | |||||
| (nM) | en la columna (fmoles) | Rep01 | Rep02 | Rep03 | Rep04 | Significa | RSD (%) |
| 0.1 | 1 | 275 | 359 | 325 | 288 | 312 | 12.2 |
| 1 | 10 | 3559 | 3694 | 3287 | 3754 | 3574 | 5.8 |
| 10 | 100 | 45259 | 45775 | 42976 | 45548 | 44890 | 2.9 |
| 100 | 1000 | 459374 | 467927 | 436272 | 458994 | 455642 | 3.0 |
| Conc | Cantidad | Pep a 5 áreas de pico (tabla 3) | |||||
| (nM) | en la columna (fmoles) | Rep01 | Rep02 | Rep03 | Rep04 | Significa | RSD (%) |
| 0.1 | 1 | 3194 | 3243 | 3202 | 3257 | 3224 | 1.0 |
| 1 | 10 | 31464 | 31150 | 31464 | 31433 | 31378 | 0.5 |
| 10 | 100 | 313638 | 320712 | 311943 | 311943 | 314559 | 1.3 |
| 100 | 1000 | 2845736 | 2840031 | 2882006 | 2864052 | 2857956 | 0,7 |
| Conc | Cantidad | Pep 6 áreas de pico (cuadro 3D) | |||||
| (nM) | en la columna (fmoles) | Rep01 | Rep02 | Rep03 | Rep04 | Significa | RSD (%) |
| 0.1 | 1 | 490 | 583 | 440 | 440 | 488 | 13.8 |
| 1 | 10 | 6429 | 6429 | 6848 | 6623 | 6582 | 3.0 |
| 10 | 100 | 71295 | 70400 | 71563 | 70400 | 70915 | 0,9 |
| 100 | 1000 | 707640 | 707640 | 694461 | 729490 | 709808 | 2.0 |
Tabla 3. Tabla que contiene las áreas pico de cromatogramas de MRM capítulo grabado para 4 péptidos PHO (Pep 2, 4, 5 y 6) para cada inyección de LC/MS (16 carreras de LC/MS y 4 concentraciones probadas).
La desviación estándar relativa era mejor que 15% de los péptidos en el rango de concentración todo investigado.

Figura 1. Diagrama de flujo de trabajo que resume los tres pasos necesarios para configurar un método de adquisición capítulo MRM. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2. Ejemplo de datos HDMSE:
(A) de baja energía espectro mostrando el ion del precursor Pep 6. (B) espectro de fragmentación energía del mismo péptido, mostrando los 3 primeros más abundante fragmento iones (en círculo) seleccionados para la optimización de energía de colisión de Tof-MRM. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3. Experimentan de parámetros que se utilizan para configurar una optimización de Tof-MRM.
Para cada transición se probaron once energías de colisión (en el rango de 16 a 36 V). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4. Ejemplo del método final de capítulo-MRM.
Varios parámetros son necesarios para cada «transición», incluyendo el péptido precursor m/z, su estado de carga y tiempo de deriva de movilidad de iones, la m/z del ion más abundante del fragmento, la energía de colisión óptima y el tiempo de la adquisición de MS. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5. Configuración utilizada por el método de procesamiento para el análisis del conjunto de datos de HS-MRM.
Cada péptido es controlada por un solo «transición» descrito por el péptido precursor m/z, cargar estado y esperar el tiempo de retención, junto con la m/z del fragmento más abundante y su estado de carga. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

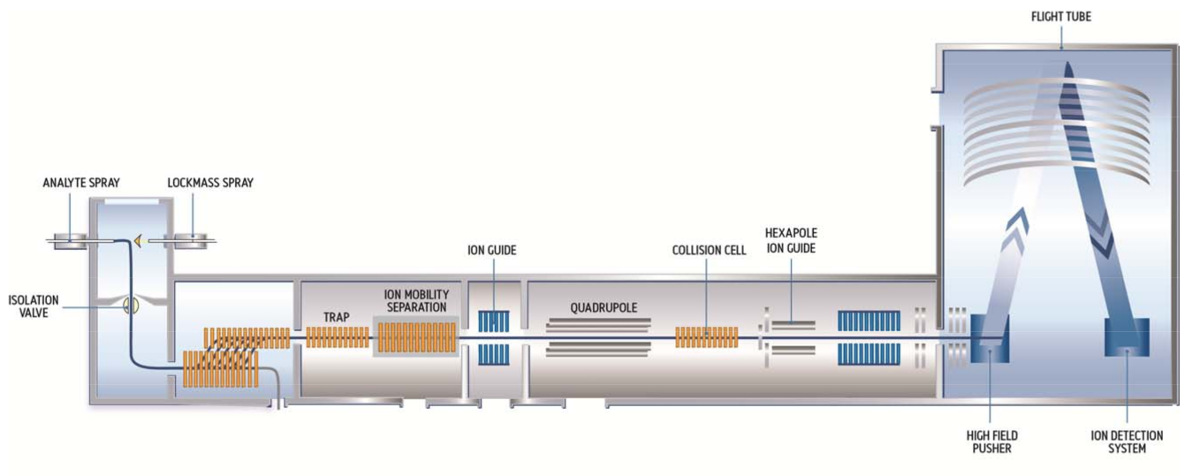
Figura 6. Diagrama del espectrómetro de masas de movilidad de iones.
En el modo de adquisición capítulo-MRM, los precursores del péptido que está siendo cuantificado se separan de otros precursores de péptidos (interferencia) Co liberador en la célula de movilidad de iones, aislados por el cuadrupolo y fragmentado con una energía de colisión fija en el celda de colisión. La señal producida por los iones fragmento del péptido se realza mediante el ajuste de la frecuencia del empujador, y cuantificación del péptido se realiza mediante la resolución de alta MS (> 30.000) señales producidas por el ion del fragmento más intenso de cada péptido. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 7. Cromatogramas de MRM capítulo grabado para 4 péptidos PHO en 4 diferentes concentraciones que abarca 3 órdenes de magnitud (0.1, 1, 10 y 100 nM).
(A) pep 2 cromatogramas, cromatogramas (B) Pep 4, cromatogramas de Pep (C) 5 y 6 (D) Pep cromatogramas. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 8. Curvas de calibración para 4 péptidos PHO en 4 diferentes concentraciones (0.1, 1, 10 y 100 nM).
Las tablas debajo de cada curva muestran las áreas de cada pico (valores) registradas para cada inyección, mientras que la segunda columna de cada tabla muestra la desviación porcentual de la respuesta lineal esperada. (A) pep 2 calibración, calibración de 4 (B) Pep, Pep (C) 5 calibración y calibración (D) Pep 6. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
Alta resolución (Rs > 20.000) espectrometría de masas se utiliza rutinariamente para la caracterización estructural de proteínas terapéuticas en una variedad de plataformas de instrumento. En cambio, cuantificación de proteínas basada en MS se realiza típicamente por SRM/MRM en baja resolución (Rs ~ 1.000) tándem cuadrupolo espectrómetros de masas utilizando péptidos de firma generados por el clivaje enzimático de proteínas11,12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23. separaciones cromatográficas unidimensional no pueden resolver completamente mezclas de complejos péptido producidos por la digestión enzimática, elución Co péptido es una ocurrencia común, incluso en el caso de un resumen de solo proteína. Por resúmenes muy complejas de proteínas (por ejemplo, para la cuantificación de PMP de 1-100 de EP en presencia de un fondo rico en péptidos producidos por la proteína terapéutica), la sensibilidad, exactitud y linealidad del SRM/MRM ensayo puede ser afectado por interferencia.
Los ensayos SRM/MRM tienen selectividad unidimensional, confiando sólo en una combinación "única" de las masas precursor/fragmento. Por este motivo, estos análisis fallan en situaciones cuando el fondo de péptido cambia inesperadamente (por ejemplo, para las muestras de la biofarmacéutica Obtenido de procesos de purificación diferentes). Para superar estas limitaciones, proponemos aquí un ensayo de alta selectividad (HS) MRM aplicado sobre un ion permitió movilidad alta resolución tiempo de vuelo (QTOF) híbrido espectrómetro de masas tetrapolar (para el esquema del instrumento, ver figura 6).
El instrumento se separa los precursores de los péptidos de interés a partir de otros precursores de péptidos (interferencia) Co liberador en la celda de movilidad de iones, aísla la envolvente completo isotópica del precursor en el cuadrupolo y fragmentos con un CE fija en el celda de colisión. La señal producida por su fragmento péptido más abundante es mejorarse ajustando la frecuencia del empujador (mejora de destino), que empuja selectivamente masivas regiones de interés en el tubo de vuelo, en lugar de todos los iones, como con un análisis completo. Cuantificación del péptido se realiza utilizando las señales de alta resolución MS (Rs ~ 30.000) producidas por este ión fragmento. En comparación con los ensayos SRM/MRM, el análisis de capítulo-MRM ofrece dos niveles adicionales de selectividad: uno proviene de la separación de la movilidad del ion precursor-nivel, mientras que el segundo se ofrece la mayor resolución masiva del analizador TOF. Los resultados de estas mejoras de selectividad son visibles en los cromatogramas de HS-MRM mostrados en la figura 7, que está libres de interferencias a través de tres órdenes de magnitud.
A diferencia de los ensayos SRM/MRM, hay varios parámetros que pueden ajustarse para optimizar el análisis de capítulo-MRM: la ventana de RT en el precursor del péptido (típicamente establecida en 0,2 min), la ventana de aislamiento de cuadrupolo (4 Da), la ventana de tiempo de la deriva que rodea el precursor (± FWHM de la cima del precursor de la mobilogram ion correspondiente) y la resolución MS del ion del fragmento (20.000-40.000). Los ensayos de HS-MRM son muy sensibles: el importe más bajo detectado para cada péptidos PHO es 1 femtomole en la columna (o 0,1 nM en términos de concentración de péptido). Cuando se considera péptido MW (ver tabla 1 para MWs exacta), la cantidad detectada en columna es del orden de 1-2 pg, mientras que la columna está cargada con una cantidad significativamente mayor (2 μg) de péptidos de fondo de la mAb digest.
Teniendo en cuenta el peso molecular de la proteína integral de PHO (97.2 kDa) de la cual se derivaron los péptidos con picos, el ensayo es capaz de detectar 50 ppm de una impureza de la proteína en presencia de iones de alta abundancia de fondo. Límites mínimos de detección (5-10 ppm) son alcanzables para un peso molecular más bajo EP (10-20 kDa). El ensayo abarca tres órdenes de magnitud (como se muestra en las curvas de calibración de la figura 8), lo que significa que puede medir la EP en la gama 1-1.000 ppm. Además, la reproducibilidad de los ensayos de HS-MRM, ilustrado en la tabla 3, coincide muy bien con la reproducibilidad de los ensayos SRM/MRM de moléculas pequeñas, con área de pico RSD mejor del 15%.
Exploraron las posibilidades de un nuevo análisis para la cuantificación de normas péptido con púas en un anticuerpo monoclonal (mAb) Resumen y demostraron su sensibilidad y su utilidad para cubrir el rango dinámico amplio (por lo menos tres órdenes de magnitud) típicamente encontrado en el análisis HCP. Todos los seis estándares de péptido se detectaron en concentraciones tan bajas como 0.1 nM (1 femtomole cargado en una columna cromatográfica de 2,1 mm ID) en presencia de un fondo de alta abundancia péptido (2 μg de una mAb digest cargado en columna). Al incorporar dirigida HRMS y separación de precursor de movilidad de iones, el análisis de capítulo-MRM tiene gran potencial para convertirse en un ensayo de seguimiento rápido y alto rendimiento para varios profesionales a través de múltiples lotes de productos biofarmacéuticos.
Divulgaciones
Todos los autores son empleados de la Corporación de aguas, que es el productor de varios reactivos y los instrumentos utilizados en este artículo.
Agradecimientos
Los autores desean agradecer a Lesley Malouin y Tony Catlin de aguas Corporation para la preparación de la figura 6 del manuscrito.
Materiales
| Name | Company | Catalog Number | Comments |
| Vion IMS Qtof mass spectrometer | Waters | 186009214 | |
| Acquity H-Class Quaternary solvent manager (QSM) | Waters | 186015041 | |
| Acquity H-Class FTN Sample Manager (SM) | Waters | 186015040 | |
| Acquity H-Class Column Manager (CM) | Waters | 186015043 | |
| Acetonitrile (ACN) | Fisher Chemical | A996-4 | |
| Ammonium bicarbonate | Sigma Aldrich | 40867-50G-F | |
| 2.1 x 150 mm CSH C18 UPLC column, 1.8 µm particles | Waters | 186005298 | |
| Dithiothreitol (DTT) | Sigma Aldrich | D5545-5G | |
| Formic acid, eluent additive for LC/MS | Sigma Aldrich | 56302-10X1ML-F | |
| Hi3 rabbit phosphorylase (PHO) MassPREP standard | Waters | 186006011 | |
| Iodoacetamide (IAM) | Sigma Aldrich | I-1149-5G | |
| LC vials (12x32 mm glass vials, screw neck) | Waters | 186000327c | |
| Leucine Enkephalin acetate salt hydrate | Sigma Aldrich | L9133-10MG | |
| Protein LoBind 2.0 mL tubes (2x50) | Eppendorf | 22431102 | |
| RapiGest SF | Waters | 186001861 | |
| Trypsin/Lys-C enzyme mix, mass spec grade (5 x 20 µg) | Promega | V5073 | |
| Water, LC/MS grade | Sigma Aldrich | 39253-1L-R |
Referencias
- Doneanu, C. E., et al. Analysis of host-cell proteins in biotherapeutic proteins by comprehensive online two-dimensional liquid chromatography/mass spectrometry. MAbs. 4, 24-44 (2012).
- Schenauer, M. R., Flynn, G. C., Goetze, A. M. Identification of host-cell protein impurities in biotherapeutics using mass spectrometry. Anal Biochem. 428, 150-157 (2012).
- Zhang, Q., et al. Comprehensive tracking of host-cell proteins during monoclonal antibody purifications using mass spectrometry. MAbs. 6, 659-670 (2014).
- Farrell, A., et al. Quantitative host-cell protein analysis using two-dimensional data independent LC-MS(E). Anal Chem. 87, 9186-9193 (2015).
- Doneanu, C. E., et al. Enhanced detection of low-abundance host cell protein impurities in high-purity monoclonal antibodies down to 1 ppm using ion mobility mass spectrometry coupled with multidimensional liquid chromatography. Anal Chem. 87, 10283-10291 (2015).
- Doneanu, C. E., et al. Improved identification and quantification of host cell proteins (HCPs) in biotherapeutics using liquid chromatography mass spectrometry. Technologies for therapeutic monoclonal antibody characterization, vol. 3: Defining the next generation of analytical and biophysical techniques. , 357-393 (2015).
- Zhang, Q., et al. Characterization of the co-elution of host-cell proteins with monoclonal antibodies during protein A purification. Biotechnol Prog. 32, 708-717 (2016).
- Reisinger, V., Toll, H., Mayer, R. E., Visser, J., Wolschin, F. A mass spectrometry based approach to host cell protein identification and its application in a comparatibility exercise. Anal Biochem. , 1-6 (2014).
- Madsen, J. A., et al. Toward the complete characterization of host-cell proteins in biotherapeutics via affinity depletions, LC/MS/MS and multivariate analysis. MAbs. 7, 1128-1137 (2015).
- Giles, K., et al. Applications of a trevelling wave-based radio-frequency-only stacked ring ion guide. Rapid Commun Mass Spectrom. 18, 2401-2414 (2004).
- Anderson, V., Hunter, C. L. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 5, 573-588 (2006).
- Lange, V., Picotti, P., Domon, B., Aebersold, R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol. 4, 1-14 (2008).
- Picotti, P., et al. High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat Methods. 7, 43-46 (2010).
- Zhao, L., et al. Quantification of proteins using peptide immunoaffinity enrichment coupled with mass spectrometry. J Vis Exp. (53), (2011).
- Liebler, D. C., Zimmerman, L. J. Targeted quantitation of proteins by mass spectrometry. Biochemistry. 52, 3797-3806 (2013).
- Manes, N. P., Mann, J. M., Nita-Lazar, A. Selected reaction monitoring mass spectrometry for absolute protein quantification. J Vis Exp. (102), (2015).
- Ebhardt, H. A., Sabido, E., Huttenhain, R., Collins, B., Aebersold, R. Range of protein detection by selected/multiple reaction monitoring mass spectrometry in an unfractioned human cell culture lysate. Proteomics. 12, 1185-1193 (2012).
- Picotti, P., Bodenmiller, B., Mueller, L. N., Domon, B., Aebersold, R. Full dynamic range proteome analysis of S. cerevisiae by targetted proteomics. Cell. 138, 795-806 (2009).
- Sherman, J., McKay, M. J., Ashman, K., Molloy, M. P. How specific is my SRM: the issue of precursor and product ion reductancy. Proteomics. 9, 1120-1123 (2009).
- Abbatiello, S. E., Mani, D. R., Keshishian, H., Carr, S. A. Automated detection of innacurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin Chem. 56, 291-305 (2010).
- Rost, H., Malmstrom, L., Aebersold, R. A computational tool to detect and avoid redundancy in selected reaction monitoring. Mol Cell Proteomics. 11, 540-549 (2012).
- Bao, Y., et al. Detection and correction of interference in SRM analysis. Methods. 61, 299-303 (2013).
- Maclean, B., et al. Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Anal Chem. 82, 10116-10124 (2010).
- Mbasu, R. J., et al. Advances in quadrupole and time-of-flight mass spectrometry for peptide MRM based translational reserch analysis. Proteomics. 16, 2206-2220 (2016).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados