
Method Article
Méthodes de transfert localisé précisément de cellules ou de l'ADN dans des embryons de souris après l'implantation précoce
Dans cet article
Résumé
We demonstrate a method for grafting cultured cells into defined sites of early mouse embryos to determine their in vivo potential. We also introduce an optimized electroporation method that uses glass capillaries of known diameter, allowing the precise delivery of exogenous DNA into a few cells in the embryos.
Résumé
La manipulation et la culture d'embryons de souris début est encore un puissant largement sous-utilisé la technologie améliorant la valeur de ce système modèle. En revanche, la culture cellulaire a été largement utilisé dans les études de biologie du développement. Cependant, il est important de déterminer si les cellules cultivées in vitro représentent réellement dans des types cellulaires in vivo. Le greffage des cellules dans des embryons, suivi par une évaluation de leur contribution au cours du développement est un procédé utile pour déterminer le potentiel des cellules en culture in vitro. Dans cette étude, nous décrivons un procédé de greffage de cellules dans un site défini d'embryons de souris post-implantation précoce de, suivie de la culture ex vivo. Nous présentons également un procédé d'électroporation optimisé qui utilise des capillaires en verre de diamètre connu, ce qui permet une localisation précise et l'ajustement du nombre de cellules recevant de l'ADN exogène à la fois avec l'efficacité de transfection élevée et faible la mort cellulaire. Ces techniques, qui ne nécessitent pas de spéquipements ecialized, rendre des manipulations expérimentales de la gastrulation et de l'embryon possible de la souris au début de l'organogenèse-scène, permettant l'analyse de l'engagement dans les sous-populations de cellules en culture et l'effet des manipulations génétiques in situ sur la différenciation cellulaire.
Introduction
La culture cellulaire a été largement utilisé dans les études de biologie du développement. Souris cellules souches embryonnaires (CSE) et les cellules souches épiblastique (EpiSCs) peuvent se différencier en tous les trois feuillets embryonnaires in vitro et sont un modèle utile pour la différenciation cellulaire au début de l'embryogenèse chez les mammifères. La dérivation de ces lignées cellulaires a ouvert une opportunité pour la manipulation in vitro et étude détaillée des événements de signalisation localisée et des réseaux de transcription d'exploitation au début de structuration embryonnaire. Toutefois, il demeure important de déterminer la pertinence in vivo de toutes manipulations effectuées dans la culture. Le potentiel in vivo des CES de souris provenant de l'embryon préimplantatoire a été évaluée en les introduisant de nouveau dans embryons préimplantatoires (de morulae ou blastocystes) 1. Cependant, EpiSCs qui représentent les cellules épiblastiques dans des embryons de post-implantation ne peuvent pas intégrer efficacement dans des embryons préimplantatoires 2,3. Notre previonous avons montré que les résultats EpiSCs peut efficacement générer des chimères et de contribuer à toutes les couches germinales, lorsque greffé dans des embryons post-implantation 4. Ainsi, la meilleure façon d'évaluer les cellules cultivées in vitro est de les initier à leur environnement correspondant in vivo.
L'électroporation est un procédé largement utilisé pour délivrer des molécules exogènes dans des cellules ciblées, tant in vivo et des expériences in vitro. L'énergie électrique peut générer un grand nombre de pores dans la membrane cellulaire, ce qui permet à l'acide exogène désoxyribonucléique (ADN) ou de l'acide ribonucléique (ARN) pour entrer dans les cellules. Un des plus grands défis pour cette technique consiste à combiner la viabilité cellulaire optimale avec une grande efficacité électrotransfection 5,6. Pour électroporation d'acides nucléiques dans les tissus embryonnaires, plaqué or électrodes ont le plus souvent été utilisé, ce qui permet le ciblage des cellules dans un large éventail spatiale 7-9. Pour parvenir à une plus locale transfert de gènes lisée, une électrode en forme d'aiguille a été utilisée pour réaliser un champ électrique focal 10,11. En utilisant cette méthode, les auteurs ont montré que, après l'électroporation, les cellules autour de 30 à 60 avaient pris la 11 construction d'ADN. Néanmoins, il semble que le réglage avec précision le nombre de cellules électroporées reste difficile avec une électrode de largeur fixe. La technique d'électroporation capillaire a été utilisée pour fournir des plasmides de cellules individuelles 12-14. Cependant, cette technique n'a pas été appliquée pour l'électroporation plasmides aux embryons ex vivo. Plus récemment, un microdispositif a été rapporté que l'électroporation localement un petit nombre de cellules de l'endoderme viscéral distales (moins de 4 cellules) en post-implantation précoce des embryons de souris 15. Cependant, il est encore impossible de savoir si ce dispositif peut cibler efficacement ectoderme et mésoderme ex vivo.
Dans cette étude, nous décrivons deux nouvelles méthodes pour évaluer la fonction cellulaire et génique au début de poste-implantation embryons. Nous démontrons d'abord comment greffer des cellules cultivées in vitro dans des sites définis dans des embryons de souris tôt pour évaluer leur potentiel in vivo. L'intégration des cellules greffées et de leurs descendants, tous étiquetés par une étiquette génétique (par exemple, une protéine fluorescente verte (GFP), peut encore être examiné par immunocoloration des protéines spécifiques de tissu 4. Deuxièmement, nous décrivons une méthode améliorée pour offrir précisément l'ADN pour des sites localisés dans l'embryon par électroporation. Plutôt que d'utiliser une électrode en forme d'aiguille, nous avons inséré un mince fil à l'intérieur d'un capillaire à pointe fine de verre, et de démontrer que cette modification peut fournir l'ADN à un petit nombre de cellules à haut rendement et limité la mort cellulaire. En outre, nous montrons que l'utilisation de capillaires en verre avec différentes tailles d'ouverture, nous pouvons contrôler le nombre de cellules par électroporation. Par conséquent, nous croyons que cette méthode peut être d'une grande utilité pour étudier la structuration embryonnaire précoce impliquant de petits nombres ocellules f.
Protocole
Toutes les expériences sur les animaux ont été effectuées en conformité avec UK Accueil Règlement sur le bureau comme spécifié dans les Animaux (Scientific Procedures) Act (1986) sous le numéro de licence Project 60/4435. Pour recueillir des embryons à des stades de développement spécifiques, accouplements chronométrés ont été mis en place O / N. Midi le jour de la recherche d'un bouchon vaginal a été désigné jour embryonnaire (E) de 0,5.
1. Dissection de la E7.5 ou E8.5 après l'implantation d'embryons pour la Culture ex vivo
- Sacrifiez les souris femelles enceintes par dislocation cervicale.
- Isoler l'utérus à l'aide de ciseaux, le tenant avec une pince, et le placer dans un plat de 30 mm rempli de milieu M2.
- Détachez avec soin en dehors du myomètre en utilisant deux paires de pinces fines.
- Décollez la caduque, en faisant attention à ne pas percer les cavités extra-embryonnaires.
- Retirer la membrane de Reichert en la pinçant avec une pince et lentement le séparant de l'embryon.
- Vérifiez les embryons sous une dissectionstéréomicroscope pour garantir que le sac vitellin, amnios et cône ectoplacentaire sont intacts.
- Transférer les embryons à un plat propre de M2 à l'aide d'une pipette et le placer sur un couvercle de boîte de Pétri de 30 mm en plastique sur la glace (la «plate-forme de glace») à des embryons partiellement refroidissement.
- Si nécessaire, la conservation des embryons en M2 sur une plate-forme de glace pour un maximum de 1,5 heures, par exemple., Lors de la préparation des médias ou de la manipulation de petits lots de ~ 3-4 embryons à TA.
Remarque: La récupération et la dissection des embryons de rongeurs a été décrite en détail précédemment 7,8,16.
2. Préparer la culture d'embryons moyen
- Fraîchement dégeler soit du sérum de rat disponible dans le commerce (voir Glanville-Jones et al. 13 pour les spécifications), ou le sérum de rat préparé en interne selon Copp et Cockroft 16 qui a été inactivé par la chaleur pendant 30 min à 56 ° C et congelés dans 1 ml des aliquotes à -80 ° C
Remarque: le sérum de rat disponibles dans le commerce est acceptable pour les périodes de culture de 24-36 h, même si le sérum préparés à l'interne est, dans notre expérience, de qualité supérieure pour les périodes de culture jusqu'à 48 h. - Fraîchement préparer un excès (par exemple., 10 ml) de suppléments définis consistant en un milieu essentiel minimal de Glasgow (GMEM), 1% acides aminés non essentiels (NEAA), 2 mM de L-glutamine et pyruvate de sodium 1 mM.
- Calculez le volume de milieu de culture qui est nécessaire en fonction du nombre d'embryons de chaque étape qui vont être cultivées (voir section 3). Mélanger le sérum de rat avec les suppléments définis pour constituer un milieu de culture de 50% (1: 1 (v / v) de sérum de rat: Les suppléments définies) et / ou du milieu de culture de 75% (3: 1 (v / v) de sérum de rat: défini suppléments).
- Faire passer le milieu de culture d'embryons à travers un filtre de 0,45 pm et ajouter 10 000 UI / ml de pénicilline et 10 pg / ml de streptomycine.
Remarque: solution de L-glutamine et pyruvate de sodium fraîchement décongelé est cruciale pour le développement de l'embryon au cours de culture ex vivo.
- Pour les embryons de E7.5: culture statique en utilisant des plaques 4 puits dans un incubateur fourni avec 5% de CO 2 dans l'air à 37 ° C pendant 24 heures. Culture jusqu'à deux embryons par puits dans du milieu de culture 1 ml de 50%.
- Pour E8.5 embryons: utiliser un appareil de culture de galet 8 tournant à 35 tours / min incorporant gazage continu avec 5% de CO 2 dans l'air à 37 ° C pendant 24 heures (1 ml de 50% de milieu de culture par embryon).
- Pour les embryons de E9.5: utiliser un appareil de culture de rouleaux tournant à 35 tours / min incorporant gaz fournis avec 5% de CO 2, 40% O 2 55% de N 2 à 37 ° C pendant 24 heures (1 ml de 75% de milieu de culture par embryon).
Remarque: les embryons Culture à moins de 3 heures après l'euthanasie des souris comme des périodes prolongées dans M2 nuire au développement. Voir Copp Cockroft et 16 pour le procédé de culture ex vivo de l'embryon.
4. Grafting cellules cultivées dans E7.5 ou E8.5 embryons de souris
- Racler physiquement EpiSCs, qui expriment la GFP ubiquitaire, d'une plaque de culture de 6 puits à l'aide d'une pipette 20-200 pi et placez-les dans un plat de 30 mm contenant les embryons
Remarque: Pour insérer amas de cellules dans des embryons, les cellules doivent être physiquement au rebut plutôt que trypsinisées. - Fixez une greffe capillaire tiré à la main sur le tube de l'aspirateur pour faire une pipette de bouche.
- Aspirer délicatement la pipette de la bouche de tirer un ou plusieurs amas de cellules de taille> 20 cellules dans le capillaire de greffage.
- Soufflez doucement sur les cellules, pour disperser partiellement grosses touffes.
- Sélectionnez un bouquet de cellules contenant ~ 10-20 cellules et sucer dans le capillaire greffer à nouveau, en le gardant à proximité de l'ouverture du capillaire. Veillez à ne pas déplacer le bouquet de cellules dans et hors du capillaire à plusieurs reprises, afin d'éviter la débâcle en petits morceaux.
- Maintenez l'embryon lâche en place avec une paire de pinces et insérez le greffagecapillaire dans la région d'intérêt pour créer une ouverture.
- Expulser doucement le massif du capillaire greffage, laissant la courte chaîne de 10-20 cellules déposées dans l'embryon.
- Répétez la procédure de greffe pour le nombre désiré d'embryons. Utilisez les tailles de 3-4 embryons de lots pour plus de commodité.
- Laissant embryons dans la même boîte de milieu M2, l'image des embryons greffés en utilisant un composé de fluorescence microscope de dissection avec la caméra, garder le temps d'imagerie à un minimum pour éviter l'exposition des embryons à la lumière et de la chaleur excessive.
Remarque: déterminer les temps de formation d'image de façon empirique que ceux-ci dépendent des caractéristiques de la caméra et microscope, ainsi que la nature et l'intensité de fluorescence du fluorophore. - Transférer l'embryon dans un pastette avec un volume minimal de milieu M2 à pré-équilibrée milieu de culture (voir section 3) immédiatement après l'imagerie.
Remarque: Examiner attentivement la morphologie des embryons après la greffe. Seule la culture de la INTACt embryons.
5. main électroporation Matériaux et configuration d'appareil (Préparer la suite de l'avance des expériences d'électroporation):
- Pour pipettes d'injection ADN: tirez pipettes d'injection d'ADN à l'aide d'un extracteur micropipette horizontale. Pipettes d'injection doivent avoir une pointe fine avec une ouverture inférieure à 10 um pour éviter des dommages aux tissus lors de l'injection d'ADN dans les cavités embryonnaires.
- Pour le verre capillaire électroporation: utiliser un microforge à découper l'ouverture de pipettes d'injection de l'ADN à un diamètre intérieur de 20 ou 30 um. Pour éviter tout dommage cellulaire lorsque le capillaire est en contact avec l'embryon pour l'électroporation, la pointe du capillaire en verre doit être coupé proprement et ne pas contenir bords tranchants, cassées.
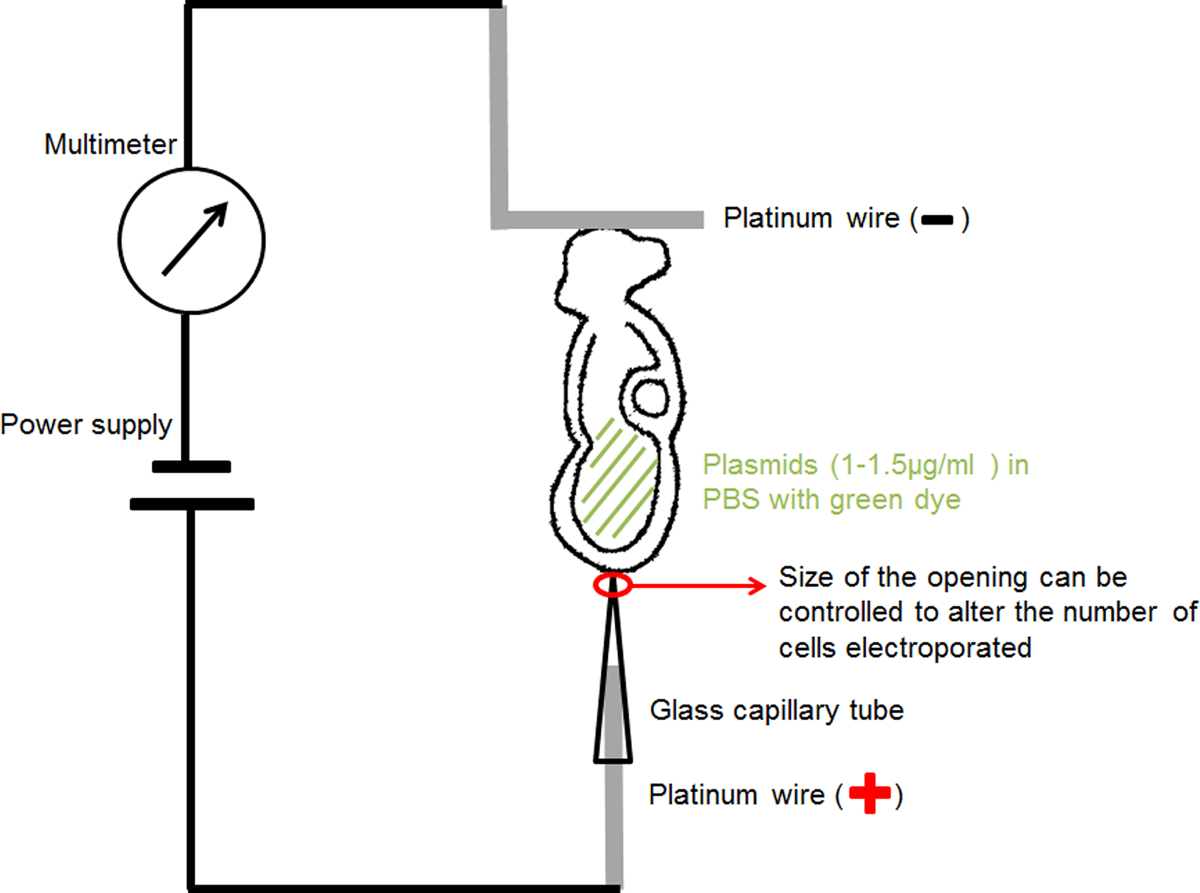
- Pour l'électrode capillaire à la main (anode) (figure 1): insérer un fil de platine de diamètre 0,2 mm dans un capillaire de verre d'électroporation avec une ouverture fixe de 20 ou 30 um de diamètre se concentrer les élusric courant et délivrer l'ADN plasmidique à une petite région d'intérêt dans l'embryon.
- Pour la main en forme de L-électrode (cathode) (Figure 1): plier un diamètre de fil de platine de 0,2 mm créant une forme de "L", avec la partie horizontale du "L" autour de 1 mm de longueur.
- Fixez chaque électrode de platine à un fil isolé mince et l'insérer dans un porte-aiguille de microinjection couverts par du ruban isolant.
- Montez les supports d'aiguille sur les détenteurs d'instruments de micromanipulation standard.
- Connecter le circuit comme représenté sur la Figure 1: connecter l'électrode capillaire à l'anode de la source d'alimentation; connecter l'électrode en forme de L à l'anode du multimètre; connecter la cathode de la mulimeter à la cathode de l'alimentation électrique.
6. L'électroporation E7.5 ou E8.5 embryons de souris
- Remplir le capillaire en verre d'électroporation avec du PBS à l'intérieur de 1-2 mm de la partie supérieure et insérer le strailutte électrode de platine (anode) dans le capillaire de verre jusqu'à ce qu'elle atteigne le fond du capillaire.
- Ancrer l'électrode en forme de L (cathode) sur la surface d'une boîte de Pétri de 30 mm rempli avec du PBS.
- Transférer l'embryon à partir du milieu M2 dans le plat d'électroporation PBS-rempli.
- Insérer l'aiguille d'injection de épiblaste latérale dans la cavité amniotique de l'embryon. En utilisant une pompe de pico pneumatique, injecter la solution d'ADN (pCAG-Cre: GFP ou pCAG-GFP 1-1,5 pg / ml avec 0,01% nourriture verte coloration colorant) dans la cavité jusqu'à ce qu'il soit complètement plein. Pour les embryons E7.5-E8.5, solution d'ADN à moins de 5 ul est requis pour une embryon. Utilisation du colorant vert comme un indicateur, veillez à ne pas éclater l'embryon.
- Positionner soigneusement l'embryon entre les électrodes et déplacer l'électrode capillaire à l'endroit précis où l'ADN doit être livré.
Remarque: L'orientation de l'embryon dépend de la région où l'ADN est à une électroporation. - Electropoévaluer l'embryon en utilisant 200 volts (V) dans 6 impulsions, chaque 50 ms de durée avec un intervalle d'une seconde entre chaque impulsion.
- Transférer l'embryon pré-équilibrée milieu de culture immédiatement après l'électroporation. Si vous le souhaitez, répétez le processus pour la prochaine embryon.
Note: Ajouter le milieu de culture dans un récipient stérile et le mettre dans l'incubateur de culture de pré-équilibrer le moyen. - Pour détecter les cellules électroporées 2 heures après la culture, de transférer les embryons à un 30 mm propre boîte de Pétri de milieu M2 en utilisant un pastette. Image que les embryons dans l'étape 5.9 en utilisant un composé de fluorescence microscope de dissection.
- Transférer les embryons de retour à la culture en utilisant un pastette immédiatement après imagerie.
- Pour détecter les cellules mortes causées par électroporation, colorer les embryons avec une cellule colorant rouge lointain de la membrane imperméable nucléaire (1: 200 dans du milieu de culture d'embryons) à 37 ° C pendant 10 min 2 heures après électroporation (facultatif)
- Pour compter les cellules électroporées, fixer le EmbryOS dans 4% de paraformaldehyde (PFA) pour 2-4 heures à 4 ° C, le noyau tache avec un ultraviolets ou rouge lointain de contraste nucléaire fluorescent et l'image les embryons en utilisant un microscope confocal (Facultatif).
Remarque: la croissance embryonnaire est affectée si elle reste trop longtemps dans PBS. Par conséquent, assurez-vous que le temps pris pour l'électroporation chaque embryon est minimisé (<5 min par embryon).
Résultats
Greffage
EpiSCs qui expriment de manière ubiquitaire EGFP (r04-GFP, dérivé de la E6.5 épiblaste, et C2, dérivé in vitro à partir de mESCs) 4 ont été grattées manuellement à partir de la boîte de culture et greffées dans les différents sites d'embryons de E7.5 (figure 2A). Les embryons ont été cultivées ex vivo et analysées après 24 h. La distribution des cellules du donneur a été évaluée par microscopie à fluorescence. Si les cellules du donneur incorporées, elles ont proliféré et leurs dérivés dispersées à l'intérieur des embryons hôtes (figure 2B). Il a été observé que les greffes de cellules contenant 10-16 incorporés efficacement dans des embryons hôtes (figure 2A, et 2B), cependant, plusieurs cellules greffage ne conduit pas à une meilleure chimérisme. Au lieu de cela, les cellules greffées produites touffes individuelles (figure 2C et 2D).
Électroporation
Pour uné valuer l'efficacité de notre système d'électroporation, nous avons livré des plasmides exprimant la GFP (pCAG-GFP et pCAG-Cre: GFP) à des sites spécifiques dans l'embryon. Conformément à une étude précédente 11, les cellules GFP + ont été détectés chez les embryons 1-2 heures après l'électroporation (Figure 2E et 2G). Lorsque les cellules épiblastiques distales à la fin de l'embryon primitif de stade de la ligne ont été électroporation, les cellules marquées ont contribué à l'ectoderme neural après 24 h en culture (figure 2E et 2F). Ce résultat correspond bien à cartes de sort connu de cellules épiblastiques dans des embryons au stade de gastrulation 17. De même, lorsque le plasmide d'expression de la GFP a été électroporé dans la ligne primitive à E8.5 (2-5 somites), cellules GFP + ont contribué à la mésoderme paraxial (Figure 2G et 2H), compatible avec les cartes de sort connus de la fin du sillon primitif 18 . De plus, nous avons observé contribution à chacune des trois couches germinales de cellules électroporées (Figure 2I-K),ce qui suggère que la procédure d'électroporation ne compromet pas le comportement des cellules in vivo. Cependant, nous avons également remarqué que si épiblaste (E7.5) ou des cellules de ligne primitive (de E8.5) ont été ciblés, certaines cellules de l'endoderme ont également été électroporation (figure 3C et le tableau 1).
L'un des principaux avantages de l'utilisation d'une électrode capillaire est que le nombre de cellules électroporées peut être commandé, simplement en changeant le diamètre de son ouverture. Pour déterminer le nombre de cellules par électroporation, les embryons ont été fixés 2 heures après l'électroporation et imagés dans wholemount sur un microscope confocal. Le nombre de cellules GFP + a été d'compté manuellement dans les z piles confocales. Le tableau 1 montre que, pour un étage donné, l'augmentation de la taille de l'ouverture du capillaire en verre de 20 à 30 um conduit à l'absorption d'ADN par plusieurs cellules. Lorsque la taille de l'ouverture d'un seul a été comparée entre les étapes (E7.5 contre E8.5), plusieurs cellules se sont révélées être une électroporation à la dernière étape. Cet effet peut être dû à une plus forte concentration d'ADN présent dans la cavité amniotique à E8.5. Parce que la solution d'ADN a été mélangé avec le colorant alimentaire vert, nous pouvons utiliser la couleur verte pour évaluer la concentration de l'ADN dans la cavité amniotique. Dans le microscope, il est clair que, par rapport aux embryons de E8.5, la couleur verte après l'injection d'ADN est beaucoup plus léger dans la cavité d'embryons E7.5. Bien que la même concentration de solution d'ADN a été injecté dans des embryons E7.5 et E8.5, plus solution d'ADN a été dans la cavité amniotique d'embryons E8.5 afin de remplir complètement, car ils sont plus grandes dans la taille. Après avoir retiré l'aiguille d'injection, il y a toujours un certain degré de fuite d'une solution d'ADN à partir de la cavité amniotique, et depuis le trou de perforation est plus grande par rapport à la taille de la cavité amniotique dans les embryons antérieures, il est probable qu'il y avait proportionnellement plus de fuite de E7.5 que les embryons E8.5, conduisant à une concentration en ADN inférieure. Le nombre différent de transfectéescellules pourraient aussi être due aux différents diamètres ou tension transmembranaire induite (ITV) seuils de cellules à différents stades.
Un inconvénient de l'électroporation est la mort cellulaire associée. Semblable à la plaqué or traditionnel ou des électrodes en forme d'aiguilles, une électroporation en utilisant une électrode capillaire provoque également la mort cellulaire. Après électroporation la région ciblée est apparu de couleur plus foncée par rapport aux régions voisines (Figure 3a et 3b), indiquant qu'un certain degré de mort cellulaire doit avoir eu lieu dans ce domaine. Pour déterminer en outre le nombre de cellules mortes causées par la procédure d'électroporation, les embryons ont été colorées avec un colorant nucléaire cellule à membrane imperméable fluorescent. Les noyaux des cellules mortes ont été marquées avec un colorant rouge lointain de fluorescence membrane imperméable. La coloration a confirmé que cette technique d'électroporation capillaire résulte que dans un petit nombre de cellules mortes à proximité du site d'électroporation (Figure 3D et Table 1).
Nous avons remarqué que, bien que les cellules mortes apparaissent sur le site d'électroporation, cellules GFP + et les cellules mortes sont aussi les plus exclusifs les uns des autres (figure 3E et 3F). En outre, lorsque l'épiblaste caudale latérale à E8.5 a été électroporé avec pCAG-GFP et une ouverture capillaire en verre de 20 um, un grand nombre de cellules GFP + a été détecté après 48 heures de culture (figure 3G et 3H). Pris ensemble, ces résultats suggèrent la plupart des cellules GFP + détectées 2 h après l'électroporation sont encore viables au cours de la culture de plus.
Nous avons marqué le nombre de cellules GFP + après 24 h la culture ex vivo. Six embryons ont été électroporées avec pCAG-GFP à E7.5, en utilisant une ouverture capillaire d'un diamètre de 20 um. 107 ± 31 (moyenne ± écart type) cellules GFP + / embryons ont été détectés. Depuis au début de la culture (2 h), 9 cellules ont été électroporés en moyenne par embryon ( trong> Tableau 1), ce qui suggère que les cellules électroporées ont subi 3-4 divisions au sein de 2 h. Le temps de doublement cellulaire moyen de E7.5 à E8.5 embryons est d'environ 6-7 h dans toutes les cellules en dehors de ceux dans le noeud ventral 19,20. Ceci suggère que la procédure d'électroporation ne gêne pas la croissance cellulaire normale.

Figure 1. Schéma montrant la configuration de l'électroporation. La solution d'ADN contenant de l'embryon dans sa cavité amniotique a été positionné entre les deux électrodes. Courant à des paramètres choisis a été fourni par un générateur d'impulsions d'onde carrée (d'alimentation). Un multimètre a été connectée en série pour détecter le courant électrique passant à l'embryon. S'il vous plaît cliquez ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 2. La distribution des cellules greffées ou électroporation d'embryons d'accueil. (AH) superpositions de fluorescence de la GFP (vert) sur des images en fond clair d'embryons wholemount (niveaux de gris) (A) 10-16 GFP + EpiSCs ont été greffées dans la région distale d'un retard embryon -streak de scène. (C) Une plus grande de touffe de GFP + EpiSCs a été greffée dans la région distale d'un embryon au stade de mi-série. (B et D) La répartition des EpiSCs cellules dérivées (vert) dans les embryons hôtes (représenté en A et C) après 24 h dans la culture. (B) cellules GFP + dispersées dans l'embryon d'accueil, ce qui suggère l'intégration correcte des cellules du donneur. (D) greffage grands amas cellulaires ont abouti à la formation d'amas non constituée en société dans l'embryon hôte. (EK) pCAG-Cre: GFP plasmide ele ctroporated dans des domaines spécifiques d'embryons de type sauvage. Électroporation la région distale d'un embryon précoce des boutons de l'étape (E) ou la ligne primitive d'un stade embryonnaire 2-5 somites (G) a entraîné cellules GFP + dans ces régions 2 heures après la procédure. (F et H) La distribution des cellules GFP + dans les embryons hôtes après 24 h dans la culture, montrant que les cellules électroporées contribuent à la neuroectoderme (flèche noire) (F) et le mésoderme paraxial (flèche blanche) (H). (IK) DAB immunocoloration des cellules GFP + montrant que les cellules électroporées peuvent donner lieu à la neuro-ectoderme (I), mésoderme (J) et l'endoderme (J et K) après 24 heures en culture. La barre d'échelle (AH) = 250 um; barre d'échelle (IK) = 100 um. REMARQUE: la figure 1A et 1B sont reproduites à partir de notre publication précédente 4.href = "https://www.jove.com/files/ftp_upload/53295/53295fig2large.jpg" target = "_ blank"> S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.

Figure 3. Distribution des cellules GFP + et des cellules mortes dans les embryons après électroporation (AC) pCAG-Cre. Plasmide GFP dans les cellules par électroporation épiblastiques latérales caudale d'une E8.5 (05/02 stade de somite) embryon (la taille de l'ouverture capillaire :. 20 um) (A) deux heures après l'intervention, la région ciblée a montré une couleur foncée (flèche blanche) par rapport à d'autres parties de l'embryon. Encart montre un agrandissement de la région électroporation. (B) l'image en fond clair (niveaux de gris) couvrit avec le canal fluorescente verte montrant les cellules électroporées (vert). (C) A confocale z-tranche montrant quedeux cellules de l'endoderme (en vert) ont également pris le plasmide lorsque les cellules latérales caudales épiblastiques ont été ciblés. Les noyaux cellulaires sont présentés dans le rouge (DF) pCAG-Cre. Plasmide GFP a été électroporé dans la partie caudale du noeud d'un embryon de E8.5 (capillaire de la taille de l'ouverture: 30 um). L'embryon a été cultivé pendant 2 heures. Cellules électroporées sont présentés dans les cellules vertes et morts en rouge. (D) La zone électroporation contient à la fois des cellules GFP + ainsi que les cellules mortes. La zone dans la case blanche a également été analysée dans un microscope confocal. Comptage manuel de la z-stack a montré qu'il y avait 33 cellules GFP + 23 et les cellules mortes dans ce domaine. Seules deux cellules étaient à la fois positive pour les deux fluorophores. (E et F) XYZ vue d'un z-tranche confocale de la région en boîte blanche dans D montrant des cellules GFP + sont séparés des cellules mortes. Les noyaux sont représentées en bleu (G et H) pCAG-Cre:. GFP plasmide a été introduit par électroporation dans quelques-uns cells dans l'épiblaste latérale caudale d'un embryon de E8.5 (capillaire de taille d'ouverture: 20 um) et imagée après deux (G) et 48 (H) heures ex vivo culture Note: (H) L'embryon ont été sectionnée en deux après culture. Les régions de la tête et le coeur ont été enlevés. La barre d'échelle (A, B, D, G et H) = 250 um; barre d'échelle (C, E et F) = 100 um. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
| Diamètre de l'ouverture du tube capillaire | Stade embryonnaire | L'efficacité d'électroporation: non. embryons contenant cellules GFP + après 2h / pas totale. embryons de l'électroporation (pas. GFP + embryons qui se sont développés normalement après 24 ou la culture de 48h) | Nombre moyen de cellules GFP + par embryon ± sd (n = non. D'embryons examinés) | Nombre deGFP + endodermiques cellules par chaque embryon ± sd (n = non. D'embryons examinés) |
| 20 pm | E7.5 (LS-LB) | 9.7 (7) | 9 ± 3 (n = 4) | 4 ± 2 (n = 4) |
| 30 pm | E7.5 (LS-LB) | 13/15 (12) | 17 ± 2 (n = 4) | 6 ± 1 (n = 4) |
| 20 pm | E8.5 (2-5 somites) | 12/13 (10) | 21 ± 4 (n = 4) | 11 ± 4 (n = 4) |
| 30 pm | E8.5 (2-5 somites) | 2/2 (2) | 33 et 26 (n = 2) | 14 et 16 (n = 2) |
Tableau 1. L'électroporation efficacité de pCAG-Cre: plasmide GFP dans les embryons de souris.
Abréviation: LS, stade de la ligne primitive fin; LB: stade tardif des bourgeons. Les embryons sont mis en scène selonDowns et Davies 12
Discussion
Greffage
L'étape critique pour les expériences de greffe de cellules est l'insertion d'une chaîne cohérente de cellules idéalement dans une seule action, pour éviter la rupture de la touffe. Cette technique nécessite un peu de pratique dans le contrôle de la bouche pipette. Si les cellules du donneur intègrent bien dans l'hôte, leurs dérivés se disperseront dans l'embryon. Pour déterminer en outre si les cellules provenant de donneurs dispersée se différencient de manière appropriée dans l'hôte, une immunocoloration peut être effectuée sur les sections de l'embryon. Si les cellules du donneur ne sont pas compatibles avec l'environnement hôte, soit ils ne peuvent pas être détectés (comme ils sont expulsés de l'embryon) ou former des amas non constituées en société dans les embryons après la culture. Si les deux cellules dispersées et amas de cellules ont été observées, cela peut indiquer que trop de cellules ont été greffées et les cellules excessives des bailleurs de fonds qui ne peuvent pas interagir avec les cellules hôtes environnantes abouti à la formation bouquet. Dans ce cas, les greffes supplémentaires contenantun plus petit nombre de cellules peut être effectuée.
La principale limite de la technique de greffage de la cellule est qu'il est impossible de déterminer le potentiel complet in vivo de cellules de souris depuis culture ex vivo sur des périodes plus longues que 48 heures n'a pas été atteint. Cependant, si elle est combinée avec l'injection de cellules guidée par échographie, il peut être possible de transférer des cellules en culture à des embryons dans l'utérus. Pour résumer, les expériences de greffe de cellules ont été largement utilisés dans notre groupe et nous ont donné de précieux indices sur le potentiel in vivo de divers types de cellules 4,21,22. Il est une technique d'utilité générale pour évaluer le potentiel in vivo de cellules en culture in vitro dans des embryons précoces de post-implantation.
Électroporation
Bien que dans cette étude, nous avons seulement montré qu'il est efficace d'utiliser la technique d'électroporation capillaire pour cibler l'épiblaste, it est également possible de cibler intentionnellement autres couches germinales telles que des cellules de l'endoderme. L'étape critique pour la technique d'électroporation capillaire est de minimiser le temps nécessaire pour l'électroporation chaque embryon (<5 min par embryon) depuis PBS est sous-optimal pour les embryons de souris au début. Nos données ci-dessus a montré que, dans la plupart des zones dans les embryons, l'électroporation ne modifie pas la croissance de l'embryon. Cependant, l'électroporation dans le noeud causé un développement anormal et a conduit à la mort prématurée de l'embryon. Cela est probablement dû à des lésions ou la mort des cellules qui forment les centres de signalisation importants 23. Par conséquent, cette région devrait être évité avec cette technique. Un autre inconvénient est que, comme mentionné dans la section des résultats, tandis que épiblaste ou des cellules de ligne primitive ont été ciblés, certaines cellules de l'endoderme ont également été électroporation. Cela peut être parce que l'ADN atteint à l'endoderme travers les espaces sous l'épithélium épiblastique. Endoderme est composé de cellules épithéliales et dans notre expérience èmeESE cellules ont une plus forte propension à absorber l'ADN. Par conséquent, lors de l'application de cette technique pour la cartographie du destin, il est important de déterminer quelles cellules prendre d'abord l'ADN.
Il convient également de noter que, bien que pCAG-GFP et pCAG-Cre: plasmides GFP peuvent être efficacement administrés en utilisant les paramètres d'électroporation montrés dans cette étude, l'efficacité d'autres constructions d'ADN peut varier et optimisation besoin individuel. Les modifications de la concentration d'ADN, l'électroporation ou la tension de la nombre d'impulsions peuvent être faites si les plasmides sont avérés difficiles à transfecter.
Pour résumer, notre système d'électroporation capillaire optimisé peut efficacement et de façon reproductible livrer GFP ou Cre: plasmides GFP en très peu de cellules de l'embryon à la mort cellulaire limitée. Depuis cette méthode ne nécessite pas de matériel coûteux ou très spécialisé, il peut être d'une grande utilité pour les études de suivi de la cellule ou à tester l'effet de l'expression ectopique ou conditionnelle suppression of gènes dans les embryons précoces, si électroporation est effectuée dans des embryons porteurs d'allèles mutants conditionnels floxés. Par conséquent, cette technique d'électroporation fournit un outil utile pour la compréhension fonctionnel sur une base cellule par cellule les rôles des facteurs de cellules intrinsèque dans le contexte d'un environnement de type sauvage embryonnaires localisée.
Déclarations de divulgation
The authors have no conflicts of financial or other interest to declare.
Remerciements
We thank Filip Wymeersch and Anestis Tsakiridis for comments on the manuscript, staff in the SCRM animal unit for help with animal maintenance and Prof. Stuart Forbes for immunohistochemistry reagents. This work was supported by MRC grant Mr/K011200/1 and the China Scholarship Council
matériels
| Name | Company | Catalog Number | Comments |
| Forceps | Dumostar | T5390 | |
| Dissecting stereomicroscope | Zeiss | Stemi 2000-C | |
| Stereomicroscope system with fluorescence | Nikon | AZ100 | |
| Inverted microscope with a digital camera | Olympus | Olympus BX61 | |
| Inverted confocal microscope | Leica Microsystems | Leica TCS SP8 | |
| Low melting point agarose | Life Technologies | 16520-050 | |
| Pasteur pipettes | Fisher Scientific | 11397863 | |
| 30mm Petri dishes | Fisher Scientific | 121V | |
| 4-well plates | Thermo scientific | 179820 | |
| M2 medium | Sigma-Aldrich | M7167 | |
| Phosphate Buffered Saline (PBS) | Life Technologies | 10010015 | |
| Paraformaldehyde (PFA) | Sigma-Aldrich | P6148 | |
| Pipettes | NICHIRYO | Nichipet | |
| tips | Greiner Bio One | 685280 | |
| Cell culture incubator | SANYO | MCO-17AIC | |
| Roller culture apparatus | BTC Engineering | ||
| Syringe filters 0.45µm, sterile | Sigma-Aldrich | 10462100 | |
| Glasgow Minimum Essential Medium (GMEM) | Sigma-Aldrich | G5154 | |
| non-essential amino acids (NEAA) | Life Technologies | 11140050 | |
| L-glutamine | Fisher Scientific | SH30549.01 | |
| Sodium pyruvate solution | Fisher Scientific | SH30239.01 | |
| Penicillin and Streptomycin 10.000UI/ml | Lonza | DE17-602E | |
| Gas Cartridge for Portable Meker Burner | COLEMAN | COLEMAN 250 | |
| Thin Wall Borosilicate Capillary Glass with Fillament, OD 1.0 mm, ID 0.78 mm | Harvard Apparatus | 640798 | |
| Aspirator tube assemblies for calibrated microcapillary pipettes | Sigma-Aldrich | A5177-5EA | |
| Flaming/Brown micropipette puller | Sutter Instrument Company | P-97 | |
| Microforge | De Fonbrune | BS030301 | |
| Pneumatic pico pump | World Precision Instruments | PV830 | |
| Microloader tips | Eppendorf | 5242956.003 | |
| ECM 830 square wave pulse generator | BTX | 45-0002 | |
| Green food coloring dye | Sigma-Aldrich | C.I. 42053 | |
| A far-red cell membrane-impermeable nuclear dye | Biotium | 40060-T | |
| pCAG-Cre:GFP | Addgene | #13776 | |
| pCAG-GFP | Addgene | #16664 | |
| Multimeter | Excel | XL830L | |
| Micromanipulators | Leitz | ||
| 0.2mm diameter platinum wire | Agar Scientific | E404-2 | |
| Anti-GFP antibody | Abcam | ab13970 | |
| Goat anti-Chicken IgY, HRP | Santa Cruz | sc-2428 | |
| Liquid DAB+ Substrate Chromogen System | Dako | K3467 | |
| 4',6-diamidino-2-phenylindole (DAPI) | Life Technologies | D21490 | |
| A far-red fluorescence nuclear counterstain | Life Technologies | T3605 |
Références
- O'Hagan, A. R., Morton, R., Eid, N. Loss of asthma control in pediatric patients after discontinuation of long-acting Beta-agonists. Pulmonary med. , 894063 (2012).
- Brons, I. G., et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 448, 191-195 (2007).
- Tesar, P. J., et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 448, 196-199 (2007).
- Huang, Y., Osorno, R., Tsakiridis, A., Wilson, V. In Vivo differentiation potential of epiblast stem cells revealed by chimeric embryo formation. Cell rep. 2, 1571-1578 (2012).
- Sadik, M. M., et al. Scaling relationship and optimization of double-pulse electroporation. Biophys. J. 106, 801-812 (2014).
- Kaestner, L., Scholz, A., Lipp, P. Conceptual and technical aspects of transfection and gene delivery. Bioorg. Med. Chem. Lett. , (2015).
- Soares, M. L., Torres-Padilla, M. E., Zernicka-Goetz, M. Bone morphogenetic protein 4 signaling regulates development of the anterior visceral endoderm in the mouse embryo. Dev. Growth Differ. 50, 615-621 (2008).
- Pierreux, C. E., Poll, A. V., Jacquemin, P., Lemaigre, F. P., Rousseau, G. G. Gene transfer into mouse prepancreatic endoderm by whole embryo electroporation. JOP. 6, 128-135 (2005).
- Falk, J., et al. Electroporation of cDNA/Morpholinos to targeted areas of embryonic CNS in Xenopus. BMC Dev. Biol. 7, 107 (2007).
- Davidson, B. P., Tsang, T. E., Khoo, P. L., Gad, J. M., Tam, P. P. Introduction of cell markers into germ layer tissues of the mouse gastrula by whole embryo electroporation. Genesis. 35, 57-62 (2003).
- Khoo, P. L., Franklin, V. J., Tam, P. P. Fate-Mapping Technique: Targeted Whole-Embryo Electroporation of DNA Constructs into the Germ Layers of Mouse Embryos 7-7.5 Days Post-coitum. CSH protocols. 2007. , pdb.prot4893 (2007).
- Tawk, M., Bianco, I. H., Clarke, J. D. Focal electroporation in zebrafish embryos and larvae. Methods Mol Biol. 546, 145-151 (2009).
- Haas, K., Jensen, K., Sin, W. C., Foa, L., Cline, H. T. Targeted electroporation in Xenopus tadpoles in vivo--from single cells to the entire brain. Differentiation. 70, 148-154 (2002).
- Nolkrantz, K., et al. Electroporation of single cells and tissues with an electrolyte-filled capillary. Anal. Chem. 73, 4469-4477 (2001).
- Mazari, E., et al. A microdevice to locally electroporate embryos with high efficiency and reduced cell damage. Development. 141, 2349-2359 (2014).
- Copp, A. J., Cockroft, D. L. . Postimplantation mammalian embryos : a practical approach. , (1990).
- Tam, P. P., Behringer, R. R. Mouse gastrulation: the formation of a mammalian body plan. Mech. Dev. 68, 3-25 (1997).
- Wilson, V., Beddington, R. S. Cell fate and morphogenetic movement in the late mouse primitive streak. Mech. Dev. 55, 79-89 (1996).
- Tzouanacou, E., Wegener, A., Wymeersch, F. J., Wilson, V., Nicolas, J. F. Redefining the progression of lineage segregations during mammalian embryogenesis by clonal analysis. Dev Cell. 17, 365-376 (2009).
- Bellomo, D., Lander, A., Harragan, I., Brown, N. A. Cell proliferation in mammalian gastrulation: the ventral node and notochord are relatively quiescent. Dev. Dynam. 205, 471-485 (1996).
- Tsakiridis, A., et al. Distinct Wnt-driven primitive streak-like populations reflect in vivo lineage precursors. Development. 141, 1209-1221 (2014).
- Gouti, M., et al. In vitro generation of neuromesodermal progenitors reveals distinct roles for wnt signalling in the specification of spinal cord and paraxial mesoderm identity. PLoS biology. 12, e1001937 (2014).
- Beddington, R. S. Induction of a second neural axis by the mouse node. Development. 120, 613-620 (1994).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.