
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
La mise en œuvre d'un système anti-Stokes cohérente Raman Scattering (CARS) sur un Ti: saphir et microscope à balayage laser OPO Based Laser Standard
Dans cet article
Résumé
Coherent diffusion anti-Stokes Raman (CARS) microscopie basée sur les vibrations inhérentes de liaisons moléculaires permet chimiquement sélective imagerie des cellules vivantes sans étiquette. Ce travail présente la mise en œuvre d'une technique de microscopie complémentaire sur un microscope à balayage laser multiphotonique standard basé sur un Ti femtoseconde: laser saphir et un laser OPO.
Résumé
microscopes à balayage laser combinant un Ti femtoseconde: laser saphir et un oscillateur paramétrique optique (OPO) pour dupliquer la ligne laser sont devenus disponibles pour les biologistes. Ces systèmes sont conçus principalement pour multivoie microscopie par fluorescence à deux photons. Cependant, sans aucune modification, la microscopie optique non linéaire complémentaire comme deuxième génération de l'harmonique (SHG) ou troisième génération harmonique (THG) peut également être effectuée avec ce set-up, ce qui permet l'imagerie sans étiquette de molécules structurées ou moyen aqueuse les interfaces lipidiques. Ces techniques sont bien adaptés à l' observation in vivo, mais sont limités dans la spécificité chimique. Chimiquement imagerie sélective peut être obtenue à partir de signaux de vibrations inhérentes basées sur la diffusion Raman. Confocale microscopie Raman offre une résolution spatiale en 3D, mais il nécessite une puissance moyenne élevée et de longue durée d'acquisition. Pour surmonter ces difficultés, les progrès récents dans la technologie laser ont permis l'develloppement de la microscopie non linéaire de vibration optique, en particulier anti-Stokes cohérente de diffusion Raman (CARS). Microscopie CARS a donc émergé comme un outil puissant pour l'imagerie cellulaire biologique et en direct, par les lipides de cartographie chimique (par vibration CH stretch), l'eau (via OH vibrations d'étirement), des protéines ou de l'ADN. Dans ce travail, nous décrivons la mise en œuvre de la technique CARS sur un balayage laser multiphotonique microscope OPO couplé standard. Elle est basée sur la synchronisation dans le temps des deux lignes laser en ajustant la longueur de l'un du trajet du faisceau laser. Nous présentons une mise en œuvre étape par étape de cette technique sur un système multiphotonique existant. Une formation de base en optique expérimentale est utile et le système présenté ne nécessite aucun équipement supplémentaire coûteux. Nous illustrons également CARS imagerie obtenus sur les gaines de myéline de nerf sciatique de rongeur, et nous montrons que cette imagerie peut être réalisée simultanément avec d'autres imagerie optique non linéaire, telle que la norme two-photon technique de fluorescence et de génération de seconde harmonique.
Introduction
La microscopie optique est devenue une technique importante pour la visualisation non destructive de processus dynamiques dans des systèmes biologiques vivants avec une résolution subcellulaire. La microscopie à fluorescence est actuellement le contraste d'imagerie le plus populaire utilisé dans les cellules vivantes en raison de sa haute spécificité et la sensibilité 1. Une large palette de sondes fluorescentes a émergé (colorants exogènes, les protéines codées génétiquement, des nanoparticules de semi-conducteurs). Diverses techniques fluorescentes à base illumination de l' échantillon ont fleuri (telles que la microscopie confocale ou à deux photons) pour effectuer l' imagerie 3D et de réduire un inconvénient principal de cette technique qui est photoblanchiment 2. D'autres limitations comprennent l'exigence de l'étiquetage fluorophore parce que la plupart des espèces moléculaires ne sont pas intrinsèquement fluorescent et par conséquent, ces fluorophores doivent être introduits artificiellement dans l'échantillon imagé. Cette manipulation artificielle peut être perturbatrice, en particulier pour les petites molécules ou induit potphoto-toxicité tiel. Ces raisons font microscopie par fluorescence ne conviennent pas pour les observations in vivo. Par conséquent, l'utilisation de techniques d'imagerie optique à haute sensibilité et contrastes moléculaires spécifiques sans l'utilisation de molécules fluorescentes est hautement souhaitable dans la science biomédicale.
Plusieurs techniques d'imagerie optique non linéaire sans étiquetage ou la coloration ont émergé, y compris la deuxième génération de l' harmonique (SHG) 3,4 et troisième génération harmonique (THG) 5. SHG microscopie a été utilisée pour l' image des arrangements structurels au niveau supramoléculaire tels que les microtubules ou le collagène 6. THG est généré à partir hétérogénéités optiques tels que l' interface entre un milieu aqueux et les lipides 7. THG a également été démontrée à l' image de la myéline 8,9. Les deux techniques peuvent être mises en oeuvre sur un microscope à fluorescence à deux photons et ne nécessitent qu'un seul faisceau laser. Cependant, ils nécessitent l'intensité du laser de puissance élevée (typiquement 50mW à 860 nm pour SHG 10, 25 - 50 mW à 1180 nm pour THG 9), qui est délétère dans des échantillons de vie, et ne fournit pas la spécificité chimique qui est nécessaire pour clairement l' image de structures biologiques spécifiques.
Chimiquement imagerie sélective peut être obtenue à partir des signaux de vibration moléculaire inhérentes basées sur la diffusion Raman. Lorsqu'un faisceau de lumière frappe la matière, les photons peuvent être absorbés et dispersés par des atomes ou des molécules. La plupart des photons diffusés auront la même énergie, à savoir la fréquence, que les photons incidents. Ce processus est appelé diffusion Rayleigh. Cependant, un petit nombre de photons seront dispersées à une fréquence optique différente de la fréquence des photons incidents, à savoir, avec un processus de diffusion inélastique appelé diffusion Raman. La différence d'énergie provient d'une excitation des modes de vibration en fonction de la structure moléculaire et l'environnement. Par conséquent, Raman spontanée diffusion provides d'imagerie chimique sélective des molécules différentes ont des fréquences vibratoires spécifiques. Toutefois, il est limité en raison de son signal extrêmement faible. Confocale microscopie Raman a été développé et fournit une résolution spatiale en 3D, mais il nécessite une puissance moyenne élevée et à long temps d' acquisition 11. Pour surmonter ces difficultés, les progrès récents dans la technologie laser ont permis la montée de la microscopie non linéaire de vibration optique, en particulier la diffusion Raman anti-Stokes cohérente (CARS) 11,12,13.
CARS est un processus optique non linéaire du troisième ordre. Trois faisceaux laser, composé d'un faisceau de pompage à la fréquence ω P, un faisceau Stokes à la fréquence ω S et un faisceau de sonde (étant le plus souvent la pompe) sont concentrés dans un échantillon et de générer un faisceau anti-Stokes à la fréquence de AS = ( 2ω P - ω S) 14. Le signal anti-Stokes peut être considérablement améliorée lorsque la différence de fréquenceentre la pompe et les faisceaux de Stokes est accordé à une vibration moléculaire Raman Ω R = (ω P - ω S). le signal CARS est basé sur l'interaction de photons multiples. Il génère donc un ordre de signaux cohérents de grandeur plus forte que la diffusion Raman spontanée.
Microscopie CARS a été démontré expérimentalement par Duncan et al. 15. Zumbusch et al. , Améliore alors la technique, en utilisant deux concentrés dans le proche infrarouge des faisceaux laser femtoseconde avec une lentille d' objectif à grande ouverture numérique, permettant la mise en correspondance l' état de phase de CARS , et en évitant les deux photons non résonante fond 16. Microscopie CARS a donc émergé comme un outil puissant pour l' imagerie cellulaire et les tissus vivants, en détectant chimiquement des molécules telles que les lipides (par vibration CH stretch) 17,18, l' eau (via OH vibrations d'étirement), les protéines, l' ADN dans les cellules vivantes 19,20 mais aussi deutéré composé chimiques pour les produits pharmaceutiques et cosmétiques 21 22.
La principale limitation de la microscopie non linéaire provient de la complexité et le coût des sources optiques. Un système de CARS nécessite deux lasers de longueur d'onde accordables avec de courtes durées d'impulsion et avec des trains d'impulsions temporellement et spatialement synchronisés. Les premiers microscopes CARS reposaient sur deux picoseconde synchronisée Ti: saphir lasers 20. CARS imagerie a également été obtenue à partir d' un seul femtoseconde Ti: laser saphir générer une source de lumière supercontinuum 23. Récemment, des sources laser composées d'un seul femtoseconde Ti: laser saphir pompage d'un oscillateur paramétrique optique accordable (OPO) ont été utilisés pour la microscopie CARS. Cette configuration permet intrinsèquement temporellement synchronisée des faisceaux avec une différence de fréquence entre la pompe et le faisceau Stokes couvrant le spectre de vibration moléculaire complète 24. En outre, les microscopes à balayage laser basé sur un chiffre d'affaireslaser touche fs et une OPO, principalement utilisé pour la fluorescence à deux photons (TPF) sont maintenant disponibles pour les non-physiciens. Le potentiel de ces set-up peut être grandement améliorée sans nécessiter des investissements supplémentaires par l'incorporation d'autres techniques d'imagerie optique non linéaire, puisque chaque non linéaire (NLO) modalité d'imagerie est sensible à des structures ou des molécules spécifiques. Imagerie multimodaux NLO capitalise donc le potentiel de NLO microscopie pour des échantillons biologiques complexes 25. Le couplage de ces techniques a permis à l'enquête sur de nombreuses questions biologiques, en particulier sur le métabolisme des lipides, la peau ou le cancer du développement 26, le développement du muscle squelettique 27, les lésions athéroscléreuses 28. En outre, la mise en œuvre de balayage par faisceau laser avec CARS donne la capacité d'imagerie à haut débit, à savoir, un outil attrayant pour étudier les processus dynamiques in vivo.
Le but de ce travail est de montrer chaque étape pour mettre en œuvre ttechnique il CARS sur un microscope à balayage laser multiphotonique standard. Le microscope est basé sur un alliage Ti fs: laser à saphir et un OPO (pompé par le Ti: saphir laser) commandé par un logiciel pour les biologistes. L'intégration a été réalisée en ajustant la longueur de l'une de la trajectoire du faisceau laser afin de synchroniser dans le temps les deux faisceaux. Nous décrivons la mise en oeuvre étape par étape de cette technique qui ne nécessite qu'une formation de base en optique expérimentale. Nous illustrons également CARS imagerie obtenus sur les gaines de myéline du nerf sciatique des rongeurs, et nous montrons cette imagerie peut être réalisée simultanément avec d'autres imagerie optique non linéaire, comme technique de fluorescence à deux photons standard et la deuxième génération de l'harmonique.
Access restricted. Please log in or start a trial to view this content.
Protocole

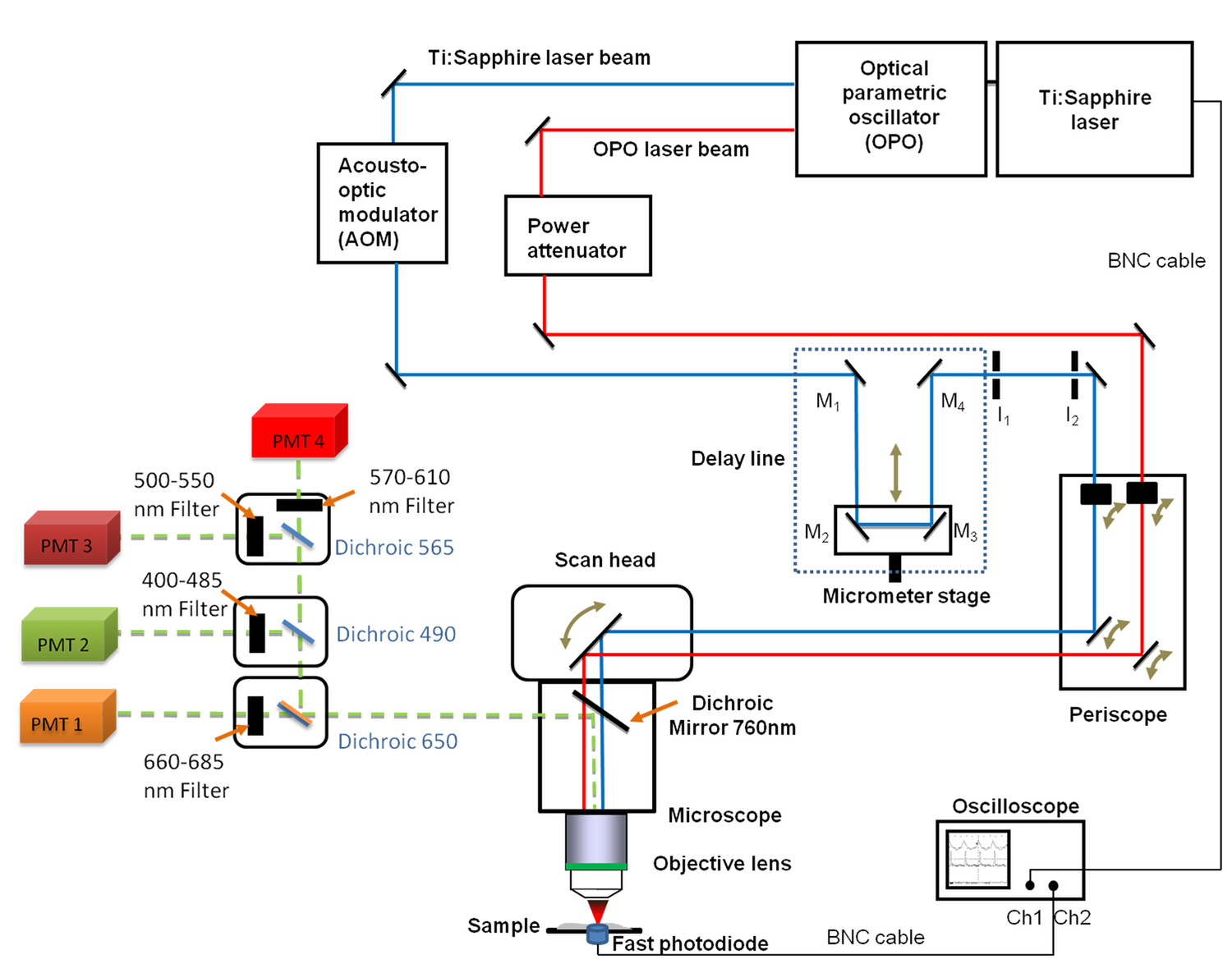
Figure 1. Vue schématique du général set-up Il comprend le Ti:. Saphir (680 - 1,080 nm) et l'OPO (1.050 - 1.300 nm) lasers, la ligne à retard avec les 4 miroirs (M 1 à M 4), l'oscilloscope rapide, la photodiode et deux iris fixes diaphragmes I 1 et I 2. Un miroir M 2 et M 3 sont fixés sur une platine de translation linéaire permettant de modifier la longueur de ligne de retard avec une résolution micrométrique. A 660 -. Bande nm filtre 685 passe a été placé en face du tube photomultiplicateur (PMT) utilisé pour CARS imagerie S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
1. Mise en service du système laser
- Vérifiez que le Ti: saphir longueur d'onde est réglée à 800 nm ou de définircette longueur d'onde sur la Ti: saphir contrôleur d'alimentation. Tournez la clé de veille sur On pour allumer le Ti: saphir laser.
- Allumez le laser OPO à l'arrière du contrôleur OPO et ouvrez le Ti: saphir obturateur sur le Ti: saphir contrôleur d'alimentation.
- Allumez l'ordinateur tablette pour pomper l'OPO. Cliquez sur les icônes OPO connectés et distant connecté sur la tablette. Attendre 30 - 40 min pour se réchauffer.
- Allumez l'ordinateur de microscope et allumer les interrupteurs "Microscope Composants". Démarrez le logiciel en double-cliquant sur l'icône sur le bureau.
- Dans l'onglet Acquisition du logiciel, ouvrez l'outil laser dans le Gestionnaire de configuration pour faire fonctionner les deux lasers à partir du logiciel. Sélectionnez laser Ti: saphir Sur et laser OPO Sur. Vérifiez la valeur de la puissance du laser optique (valeurs typiques de 3.700 mW à 800 nm et 700 mW à 1000 nm).
- Pour configurer la trajectoire du faisceau et les lasers,ouvrir l'outil Light Path dans le groupe d'outils de Gestionnaire de configuration et cochez la case premier tube photomultiplicateur (PMT).
- Pour vérifier le Ti: spot laser saphir à la sortie de l'objectif, ouvrir l'outil de canaux dans le groupe d'outils d' acquisition des paramètres. Sélectionnez le Ti: puissance de saphir à faible valeur (environ 1%), réduire le gain à 0 (aucune image est nécessaire à ce stade) et cliquez sur le bouton Continue pour lancer la procédure de balayage pour lancer le faisceau laser à travers l'objectif du microscope. Vérifier la présence d'une tache rouge par observation directe en positionnant la carte de visualisation laser IR à la sortie de l'objectif du microscope d'air (10X).
- Pour vérifier la tache laser OPO, arrêter le balayage du laser Ti: saphir en cliquant sur le bouton Stop. Sélectionnez la puissance de OPO à faible valeur dans la fenêtre des canaux et cliquez sur le Cbouton ontinuous.
2. Paramètres de microscope
- placer manuellement le miroir dichroïque avec une longueur d'onde de coupure à 760 nm dans le curseur Sideport dans l'espace de l'infini au-dessus du pont objectif de lancer la lumière jusqu'à 760 nm de l'échantillon dans les PMT dans la détection non-descanned Mode (NDD).
- Réglez le filtre étroit passe-bande à 660-685 nm dans le réflecteur cube NDD en face de PMT1 pour enregistrer seulement les CARS signal à 670 nm pour reproduire les résultats présentés dans ce travail.
- Placer un filtre à bande étroite allant de 500 à 550 nm dans le réflecteur cubique NDD devant PMT3 pour l'observation de fluorescence de la myéline. Placer un filtre à bande étroite allant de 565 à 610 nm dans le cube réflecteur devant PMT4 pour l'observation de SHG.
- Pour sélectionner dans le logiciel de l'enregistrement du signal sur le détecteur avec le filtre passe ad hoc du groupe, ouvrez l'outil Light Path dans le menu Setup Manager dans l'onglet Acquisition. Activez le PMT souhaité (case à cocher) et sélectionnez une couleur pour ce canal. Dans ce travail, le vert a été choisi pour CARS, rouge pour fluorescence et magenta pour SHG.
3. Temporal Synchronisation
Remarque: les deux faisceaux laser proviennent du même laser Ti: saphir, mais le faisceau OPO est retardée lorsqu'elle est générée de sorte que les deux faisceaux ne sont pas synchronisés dans le temps lorsqu'ils atteignent le microscope. Le but ici est de retarder l'un des deux faisceaux de re-synchroniser dans le temps avant qu'ils atteignent le microscope.
- Se connecter avec des câbles BNC du canal d'entrée CH1 de l'oscilloscope à la sortie laser BNC électrique (Sync. Out). Connectez le canal d'entrée CH2 de l'oscilloscope à la photodiode et choisissez le canal CH1 comme canal de déclenchement en appuyant sur TRIGGER MENU, puis le menu principal bouton Source, puis sur le bouton menu latéral qui correspond au canal sélectionné CH1.
- Et fixer la position des postes de montage de la photodiode optique dans le plan focal d'un objectif de microscope d'air (10X), ou dans le trajet du faisceau du microscope, après avoir retiré l'objectif. Remarque: Si nécessaire, retirez le condenseur et son support.
- Dans l'outil Chaînes (groupe d'outils d' acquisition des paramètres), définir le Ti: longueur d'onde laser saphir à 830 nm à faible puissance (moins de 1% de la pleine puissance). Dans l'outil Mode d' acquisition, de réduire la zone de numérisation à un seul point pour éclairer la photodiode avec le faisceau minuscule. Allumez le balayage laser en cliquant sur le bouton Continue.
- Appuyez sur AUTOSET sur le panneau avant de l' oscilloscope et manuellement déplacer la position de la photodiode pour obtenir les trains d'impulsions sur l'écran. Appuyez sur le bouton RUN / STOP pour figer l'affichage.
- Pour enregistrer une copie de l'écran de l'oscilloscope, insérez un 3.5pouces disquette dans le lecteur de disquette ou de connecter le port GPIB sur le panneau arrière à un ordinateur. Ensuite , appuyez sur SHIFT HARDCOPY MENU, appuyez sur FORMAT (principal) pour sélectionner le format d'image TIFF et spécifier dans le menu Port du canal de sortie. Appuyez sur la touche HARDCOPY pour enregistrer l'écran de l' oscilloscope des trains d'impulsions du Ti: saphir laser.
- Eteignez le Ti: balayage laser saphir en cliquant sur le bouton Stop. En cliquant sur les canaux outil définir le signal de OPO à 1107 nm puissance et faible. Allumez le balayage laser OPO et enregistrer les trains d'impulsions du laser OPO sur l'oscilloscope. Eteignez le balayage laser OPO.
- Comparez le décalage temporel entre le Ti: saphir et les signaux de OPO.
NOTE: Le décalage temporel t changement donne la longueur de la ligne de retard L DelayLine qui doit être mis en œuvre suivant l'équation: L delayLine = c5; t shift où c est la vitesse de la lumière. - Choisissez l'une des lignes laser.
NOTE: Dans ce travail, le Ti: ligne laser saphir a été choisi parce que l'espace libre est disponible à proximité de cette ligne laser. De plus, ce choix permet d'obtenir le réalignement de la ligne laser avec une lumière laser visible. - Ouvrez la ligne laser en retirant les tubes de protection à l'endroit où la ligne de retard sera mise en œuvre.
Attention! Porter des lunettes de sécurité appropriées et retirer les bracelets de la chaîne ou de regarder de poignets. - Sélectionnez une longueur d' onde dans le domaine visible afin d'être en mesure d'observer facilement le faisceau laser (700 nm par exemple, à une faible puissance dans l'outil Canaux du logiciel). Allumez le balayage laser.
- Placer et fixer avec des postes de montage optiques deux iris diaphragmes le long de la ligne laser ouverte. Placez un iris à la sortie de la ligne à retard et placez les autres iris à l'entrée du périscope.
NOTE: Le pecontrôles riscope par deux miroirs motorisés pilotés par le logiciel de l'angle d'entrée du faisceau laser dans la tête du microscope à balayage laser à balayage. - Diminuer l'ouverture du diaphragme à iris et ajuster la position du diaphragme pour adapter le trajet du faisceau laser. Fixez-les sur la table optique. Ajustez la position verticale d'un troisième diaphragme iris mobile, pour vérifier la hauteur du faisceau laser tout en positionnant successivement les quatre miroirs de la ligne à retard.
NOTE: Ces iris diaphragmes serviront de contrôle de la procédure de réalignement en montrant le chemin à suivre. - Placer le miroir M1 monté sur un miroir cinématique de montage compacte à l'entrée de la ligne à retard (comme représenté sur la figure 1) et d' ajuster sa position et son orientation pour maintenir la hauteur de la poutre avec l'utilisation de l'iris mobiles diaphragme. Lieu miroirs M2 et M3 (également montés sur supports de miroirs compacts cinématiques) à 90 ° sur la scène de traduction qui sera positionné à Midcourse. Placez-les pour correspondre à la longueur de la ligne de retard tel que calculé précédemment.
- Ajuster l'orientation de M2 et M3 de l'utilisation de l'iris mobiles diaphragme. Set M4 (également fixé sur un support compact) à la sortie de la ligne à retard (juste avant l' iris I 1 comme le montre la figure 1) et soigneusement ajuster sa position et l' angle pour adapter la trajectoire du faisceau laser à travers les deux fixes iris diaphragmes.
- Placez la carte de visualisation laser à la sortie de l'objectif du microscope et de vérifier le profil du faisceau laser en cliquant sur Continue pour activer le balayage laser. Observer un disque brillant uniforme. Si nécessaire, ajuster légèrement l'orientation de M4.
- Position de nouveau sur la photodiode rapide sous le faisceau laser dans le plan de focalisation du microscope d'échantillons. Observer le décalage temporel entre le Ti: saphir faisceau laser et le faisceau OPO sur l'oscilloscope.
Remarque: Si nécessaire, changer la longueur de la ligne à retard en déplaçant l'ensemble du système M2, M3 monté sur lestade de la traduction (sans changer le réglage de la phase de traduction) pour synchroniser les deux impulsions. Les changements de quelques centimètres peuvent être nécessaires.
4. Spatial Overlap des faisceaux
Remarque: Pour produire un signal CARS, le chevauchement spatial des deux faisceaux laser est nécessaire. L'éclairage alternatif des deux faisceaux sur les mêmes perles colorées tout au long de deux colorants fluorescents différents peut être utilisé pour indiquer le décalage spatial. Beaux ajustements des positions de miroir peuvent alors minimiser le décalage.
- Utiliser pré-monté microsphères fluorescentes. Ou monter des microsphères en suspension sur des lames de microscope propres comme décrit ci-dessous:
- Avant l'échantillonnage, mélange (sur un mélangeur de cortex ou par sonication) la solution des perles pour être sûr que les perles sont uniformément suspendues.
- Appliquer 5 pi de la suspension de billes à la surface d'une lame et se propager avec la pointe de la pipette. Attendez que la gouttelette à sécher, puis appliquer 5 pi de montageing milieu, tel que le glycerol, l'eau ou l'huile d'immersion au-dessus de l'échantillon sec de billes. Recouvrir l'échantillon avec une lamelle couvre-objet et sceller la lamelle couvre-objet avec de la colle à séchage rapide ou de la paraffine fondue.
- Placez les billes de polystyrène fluorescentes fixées sur une lame de microscope à l'objectif de l'eau 20X. Ajouter quelques gouttes d'eau pour immerger l'objectif.
- Pour réaliser la mise au point sur les billes, ouvrez l'onglet Situer dans le logiciel pour passer du mode de balayage laser à l'observation directe de l'échantillon à l'oeil, en appuyant sur le bouton en ligne. Ouvrez l'outil Ocular pour sélectionner le filtre ad hoc et allumer la lampe halogène en cliquant sur les icônes.
- Supprimez manuellement le miroir dichroïque dans le curseur Sideport dans l'espace infini et utiliser le lecteur de focalisation du microscope pour focaliser le plan de l'échantillon en observant les perles avec les oculaires. Remplacer le miroir dichroïque.
- dans leRepérez onglet, basculer vers le mode de balayage laser en appuyant sur le bouton en mode hors connexion. Allez à l'onglet Acquisition de définir les paramètres pour la numérisation: sélectionnez la taille de l' image à 512 pixels, une vitesse de balayage de 9, une moyenne de 1, une profondeur de bits de 8 bits et d' augmenter la zone de numérisation au maximum.
- Dans l'outil Canaux de l'onglet Acquisition, ajouter une piste (Track 1) si pas déjà créé. Sélectionnez la longueur d' onde à 830 nm et de faible puissance pour le Ti:. Faisceau laser saphir Cochez la couleur verte dans la zone de la voie 1 de la fenêtre des canaux et dans le PMT3 ou la boîte de PMT4 de la fenêtre Light Path.
- Dans l'outil Canaux de l'onglet Acquisition, ajouter une seconde piste (Track 2). Sélectionner la longueur d'onde à 1107 nm et une puissance faible pour que le faisceau laser OPO. Cochez la couleur rouge dans la boîte de la fenêtre des canaux et dans la zone PMT3 de la piste 2 fenêtre Light Path.
- Réglez le gain des deux pistes à 600. Ensuite, appliquer successivement le balayage des deux faisceaux sur l'échantillon en cliquant sur Continue.
- Observez l'image dans la zone d'écran dans la vue 2D. Dans l'affichage Vue bloc de contrôle de l' option, régler l'intensité de l' affichage.
Remarque: Si nécessaire, déplacer légèrement la commande de mise pour trouver le plan de mise au point des perles. Réglez la récolte et agrandir l'image en une seule perle ou dans un groupe de billes adjacentes. - Utilisez le contrôleur de périscope pour chevaucher les faisceaux dans le plan xy. Dans le logiciel, ouvrez l'onglet Gérer. Cliquez sur les options du système et afficher la fenêtre de l' outil motorisé Periscope. Utiliser des ajustements grossiers et fins des miroirs de périscope du Ti: faisceau laser saphir afin de synchroniser dans l'espace les deux images.
- Pour la manipulation du périscope, utilisez les premières barres de réglage pour la verticale et le secoe une pour les mouvements horizontaux du faisceau laser. Déplacer le faisceau par le miroir d'entrée jusqu'à ce que l'image est légèrement visible, puis compenser l'intensité du laser avec le miroir de sortie du périscope en cliquant sur "entrée" et "sortie".
- Afin de se chevaucher verticalement les poutres, dans l'onglet Maintenir, ouvrir l'outil Collimateur et ajuster la valeur de la distance focale de la Ti: faisceau laser saphir.
- Déplacez doucement la position verticale pour objectif de vérifier la différence de concentration sur les deux images. Ou, prendre un z-pile de l'échantillon par l'ouverture dans l'onglet Acquisition l'outil Z-Stack et choisir les différents paramètres (intervalle, le nombre de tranches). Appuyez sur Ortho dans la zone de l'écran d'image pour voir les poutres en coupe axiale. Maximiser le z-recouvrement en faisant la même procédure plusieurs fois.
5. Ajustements finaux et Coherent Anti-Stokes Raman Scattering (CARS) Observation Signal de l'huile d'olive Droplets
- Mettre une goutte d'huile d'olive sur une plaque de verre et le couvrir par une lamelle de verre. Ajouter quelques gouttes d'eau pour immerger un objectif d'immersion 20X d'eau. Concentrez au bord de la lamelle en utilisant les oculaires (comme expliqué précédemment en 4.2).
- Dans l'outil Canaux de l'onglet Acquisition, sélectionnez la piste 1 la longueur d' onde à 830 nm pour le Ti: faisceau laser saphir et à 1107 nm pour l'OPO. Cochez les deux lasers dans la voie 1 pour obtenir un balayage simultané des deux lasers. Définir des pouvoirs à faible valeur pour un début.
- Dans la fenêtre Light Path, sélectionnez PMT1. Allumez les balayages laser en cliquant sur le bouton Continue. Déplacer légèrement la mise au point pour fournir la lumière laser dans la couche mince d'huile.
- Si nécessaire, augmenter la puissance optique des deux lasers. Réglez l'intensité de l'affichage à l'écran Vue bloc de contrôle de l'option. Déplacez lentement la phase de traduction de la ligne de retard jusqu'à ce que le signal devient sigsignificative améliorée.
- Après les beaux alignements sont complets, vérifier si elle est vraiment le signe d'une CARS: Déplacez légèrement l'étape de la traduction; l'intensité du signal doit devenir plus faible. Et / ou couper l'un des faisceaux laser, soit laser Ti: saphir ou OPO. Encore une fois il doit y avoir une forte décroissance de l'intensité par rapport au signal CARS.
- Pour atteindre le signal CARS maximale, sélectionnez l'option sur le logiciel pour fournir une valeur de l'intensité moyenne de l'ensemble de l'image (dans la vue Histo de l'onglet zone de l'écran). Ajuster la longueur d'onde (quelques nm), puis les x, y, z positions du faisceau de mise au point afin de maximiser la valeur moyenne de l'intensité.
6. Enceinte du Chemin Lumière du retard ligne
- Étant donné que le système final est dédié aux non-physiciens, placez le chemin de lumière de la ligne de retard avec des tubes ou une boîte d'enceinte, pour éviter un accès direct à faisceau non visibles nuisibles laser à haute puissance crête. Prenez soin de fournir un accès à l'étape de la traductionbouton.
7. Wavelength Tuning CARS
- Utilisez l'équation
 à régler les longueurs d'onde laser à la vibration Raman souhaitée. Pour reproduire les résultats présentés dans ce travail CARS image signaler de liaisons CH ayant étirement vibration de 3015 cm -1, sélectionnez λ Ti: saphir = 830 nm et λ OPO = 1,095 nm.
à régler les longueurs d'onde laser à la vibration Raman souhaitée. Pour reproduire les résultats présentés dans ce travail CARS image signaler de liaisons CH ayant étirement vibration de 3015 cm -1, sélectionnez λ Ti: saphir = 830 nm et λ OPO = 1,095 nm.
NOTE: fréquences vibratoires caractéristiques Raman observées dans des échantillons biologiques, tels que l' eau, une liaison CH peut être trouvée dans Evans et al 13 ou Ellis et al 29... - Utilisez l'équation
 pour déterminer la longueur d'onde d'émission du signal de CARS. Pour une liaison CH imagerie par CARS, choisissez un filtre à bande étroite à 670 nm depuis CARS X = 670 nm de longueur d' onde laser présenté en 7.1.
pour déterminer la longueur d'onde d'émission du signal de CARS. Pour une liaison CH imagerie par CARS, choisissez un filtre à bande étroite à 670 nm depuis CARS X = 670 nm de longueur d' onde laser présenté en 7.1.
REMARQUE: Une application du téléphone mobile est available pour calculer CARS λ à partir λ P et λ S valeurs (voir référence 30).
8. Observation de CARS Signal et Stained myéline de coupes de nerf sciatique
Remarque: Toutes les expériences sur les animaux ont été menées conformément aux règlements de l'établissement.
- Préparer les coupes du nerf sciatique axiales et longitudinales sur une lame de microscope tel que présenté dans Ozcelik et al. 31.
- Préparer la solution fluoromyelin de coloration rouge en diluant la solution stock de 300 fois dans du PBS. Inonder les coupes nerveuses avec la solution de coloration pendant 20 min à température ambiante. Retirer la solution et laver 3 fois pendant 10 minutes avec du PBS.
- Positionner les coupes dans le cadre de l'objectif d'immersion dans l'eau 20X. Placez une lamelle. Ajouter quelques gouttes de PBS pour immerger l'objectif et la mise au point de l'objectif pour obtenir une image claire des coupes à travers les oculaires (comme détaillé précédemment en 4.2).
- Dans la piste 1, sélectionnez le Ti: saphir et l'OPO lasers et définir leurs longueurs d' onde de 830 nm et 1095 nm, respectivement. Dans la fenêtre Light Path, sélectionnez PMT1 et la couleur verte.
- Dans la piste 2, sélectionnez le laser OPO uniquement (longueur d' onde à 1095 nm). Dans la fenêtre Light Path, sélectionnez PMT4 et la couleur rouge.
- Pour les deux lasers, sélectionnez faible puissance et régler le gain à 600 pour un début. Allumez les balayages laser et d'ajuster les paramètres suivants pour améliorer CARS et le signal de fluorescence contraste: valeurs de puissance, bouton de platine de translation (très légèrement), les longueurs d'onde (quelques nm), l'intensité de l'affichage.
- Pour enregistrer des images finales à haute résolution, sélectionnez dans l'outil Mode d' acquisition des paramètres suivants: taille d'image de 1024 pixels, la vitesse de balayage de 7, la moyenne de 4. Cliquez sur le bouton Magnétisme pour enregistrer une seule image. Enregistrer l'image sous la forme exclusiveà pour enregistrer l'image et les paramètres d'acquisition complète.
9. Observation des CARS et des signaux de SHG de coupes de nerf sciatique
- Préparer le nerf sciatique tel que présenté dans Ozcelik et al. 31.
- Suivez la procédure comme expliqué dans la partie 8 pour obtenir une image à travers les oculaires et pour sélectionner CARS paramètre de signal (voie 1).
- Dans la piste 2, sélectionnez le laser OPO uniquement (longueur d' onde à 1095 nm). Dans la fenêtre Light Path, sélectionnez PMT3 et la couleur magenta.
- Suivez la procédure comme expliqué dans la partie 8 pour passer sur les balayages laser et enregistrer des images haute résolution.
Access restricted. Please log in or start a trial to view this content.
Résultats
La fréquence de la norme Ti de train d'impulsions: laser saphir est généralement autour de 80 MHz. Le BOA a la même fréquence, car il est pompé par le Ti: saphir laser. Un oscilloscope rapide d'au moins 200 MHz est donc nécessaire. Une photodiode rapide dans la gamme de 600 à 1100 nm est également nécessaire. Le décalage temporel maximal se produit lorsque le Ti: saphir et les signaux OPO sont décalés de 1 / (2 × 80 × 10 6) = 6.2 nanosecondes. Il c...
Access restricted. Please log in or start a trial to view this content.
Discussion
La partie la plus difficile du travail est la synchronisation temporelle des faisceaux laser. Elle exige une photodiode rapide combiné avec un oscilloscope rapide, mais seulement un chevauchement rugueux dans le temps peut être réalisée dans un premier temps. Ensuite, un ajustement supplémentaire de quelques cm est nécessaire. Enfin, micromètre se déplace d'une platine de translation linéaire permet d'effectuer le réglage fin final de la longueur de la ligne de retard afin de déclencher le signal CARS...
Access restricted. Please log in or start a trial to view this content.
Déclarations de divulgation
The authors declare that they have no competing financial interests.
Remerciements
The authors want to thank Dr. Philippe Combette (IES, UM, Montpellier, France) for the loan of the fast oscilloscope and acknowledge financial supports from Montpellier RIO Imaging (MRI). HR acknowledges ANR grants France Bio Imaging (ANR-10-INSB-04-01) and France Life Imaging (ANR-11-INSB-0006) infrastructure networks for coherent Raman imaging developments. This work was mainly supported by an European Research Council grant (FP7-IDEAS-ERC 311610) and an INSERM - AVENIR grant to NT.
Access restricted. Please log in or start a trial to view this content.
matériels
| Name | Company | Catalog Number | Comments |
| Oscilloscope | Tektronix | TDS 520D | 500 MHz |
| Photodetector | Thorlabs | DET08C/M, T4290 | 5 GHz InGaAs, 800 - 1,700 nm |
| Ti:Sapphire laser Chameleon Ultra Family II | Coherent | ||
| Optical parametric oscillator OPO Compact Family | APE Berlin | ||
| Axio Examiner microscope LSM 7 MP | Carl Zeiss | ||
| Motorized periscope | Newport | ||
| Objective W Plan-Apochromat 20X/1.0 | Carl Zeiss | ||
| Beam combiner | Carl Zeiss | ||
| Acousto-optic modulator | Carl Zeiss | ||
| OPO power attenuator | Carl Zeiss | ||
| Photomultiplier tube | Carl Zeiss | ||
| ZEN software | Carl Zeiss | ||
| Bandpass filters | Carl Zeiss | LSM BiG 1935-176 | 400 - 480 nm; 500 - 550 nm; 465 - 610 nm |
| Dichroic mirror | Carl Zeiss | Cutoff wavelength 760 nm | |
| Silver mirrors | Newport | 10D20ER.2 | λ/10, 480 - 20,000 nm, Quantity 4 |
| Single-axis translation stage with standard micrometer | Thorlabs | PT1/M | Quantity 1 |
| Aluminium breadboard | Thorlabs | MB1015/M | Quantity 1 |
| Mirror mount | Thorlabs | KMSS/M | Quantity 4 |
| Mirror holder for Ø1" Optics | Thorlabs | MH25 | Quantity 4 |
| Iris diaphragms | Thorlabs | ID8/M | Quantity 3 |
| Protective box | Thorlabs | TB4, XE25L900/M, T205-1.0, RM1S | Quantity 1 |
| Optical posts | Thorlabs | TR40/M, PH50/M, PH75/M, BA2/M | Quantity 8 (lengths depending on the set-up) |
| 661 - 690 nm bandpass filter | Semrock | 676/29 nm BrightLine® single-band bandpass filter | Quantity 1 |
| Fluorescent beads | ThermoFisher | TetraSpeck™ Fluorescent Microspheres Size Kit | |
| Laser viewing card | Thorlabs | IR laser viewing card | |
| Laser safety glass | Newport | LV-F22.P5L07 | |
| FluoroMyelin™ Red Fluorescent Myelin Stain | ThermoFisher | F34652 |
Références
- Valeur, B., Berberan-Santos, M. N. Molecular Fluorescence: Principles and Applications. , 2nd Edition, Wiley-VCH Verlag GmbH. (2012).
- Denk, W., Strickler, J. H., Webb, W. W. Two-photon laser scanning fluorescence microscopy. Science. 248 (4951), 73-76 (1990).
- Moreaux, L., Sandre, O., Mertz, J. Membrane imaging by second-harmonic generation microscopy. JOSA B. 17 (10), 1685-1694 (2000).
- Zoumi, A., Yeh, A., Tromberg, B. J. Imaging cells and extracellular matrix in vivo by using second-harmonic generation and two-photon excited fluorescence. Proc. Natl. Acad. Sci. USA. 99 (17), 11014-11019 (2002).
- Yelin, D., Silberberg, Y. Laser scanning third-harmonic-generation microscopy in biology. Opt. Express. 5 (8), 169-175 (1999).
- Campagnola, P. J., Millard, A. C., Terasaki, M., Hoppe, P. E., Malone, C. J., Mohler, W. A. Three-dimensional high-resolution Second-Harmonic Generation imaging of endogenous structural proteins in biological tissues. Biophys. J. 81 (1), 493-508 (2002).
- Olivier, N., et al. Cell lineage reconstruction of early zebrafish embryos using label-free nonlinear microscopy. Science. 329 (5994), 967-971 (2010).
- Farrar, M. J., Wise, F. W., Fetcho, J. R., Schaffer, C. B. In vivo imaging of myelin in the vertebrate central nervous system using third harmonic generation microscopy. Biophys. J. 100 (5), 1362-1371 (2011).
- Lim, H., Sharoukhov, D., Kassim, L., Zhang, Y., Salzer, J. L., Melendez-Vasquez, C. V. Label-free imaging of Schwann cell myelination by third harmonic generation microscopy. Proc. Natl. Acad. Sci. U.S.A. 111 (50), 18025-18030 (2014).
- Strupler, M., Pena, A. M., Hernest, M., Tharaux, P. L., Martin, J. L., Beaurepaire, E., Schanne-Klein, M. C. Second harmonic imaging and scoring of collagen in fibrotic tissues. Opt. Express. 15 (7), 4054-4065 (2007).
- Cheng, J. X., Xie, X. S. Coherent anti-Stokes Raman scattering microscopy: Instrumentation, theory, and applications. J. Phys. Chem. B. 108 (3), 827-840 (2004).
- Volkmer, A. Vibrational imaging and microspectroscopies based on coherent anti-Stokes scattering microscopy. J. Phys. D: Appl. Phys. 38, R59-R81 (2005).
- Evans, C. L., Xie, X. S. Coherent anti-Stokes Raman scattering microscopy: chemical imaging for biology and medicine. Annu. Rev. Anal. Chem. 1, 883-909 (2008).
- Mukamel, S. Principles of nonlinear optical spectroscopy. , Oxford University Press. New York. (1995).
- Duncan, M. D., Reintjes, J., Manuccia, T. J. Scanning coherent anti-Stokes Raman microscope. Opt. Lett. 7 (8), 350-352 (1982).
- Zumbusch, A., Holtom, G. R., Xie, X. S. Three-dimensional vibrational imaging by coherent anti-Stokes Raman scattering. Phys. Rev. Lett. 82 (20), 4142-4145 (1999).
- Folick, A., Min, W., Wang, M. C. Label-free imaging of lipid dynamics using Coherent Anti-Stokes Raman Scattering (CARS) and Stimulated Raman Scattering (SRS) microscopy. Curr. Opin. Genet. Dev. 21 (5), 585-590 (2011).
- Wang, P., Liu, B., Zhang, D., Belew, M. Y., Tissenbaum, H. A., Cheng, J. X. Imaging lipid metabolism in live Caenorhabditis elegans using fingerprint vibrations. Angew. Chem. Int. Ed. Engl. 53 (44), 11787-11792 (2014).
- Min, W., Freudiger, C. W., Lu, S., Xie, X. S. Coherent nonlinear optical imaging: beyond fluorescence microscopy. Annu. Rev. Phys. Chem. 62, 507-530 (2011).
- Cheng, J. X., Jia, Y. K., Zheng, G., Xie, X. S. Laser-scanning coherent anti-Stokes Raman scattering microscopy and applications to cell biology. Biophys J. 83 (1), 502-509 (2002).
- Chiu, W. S., Belsey, N. A. N., Garrett, L., Moger, J., Delgado-Charro, M. B., Guy, R. H. Molecular diffusion in the human nail measured by stimulated Raman scattering microscopy. Proc Natl. Acad. Sci. U.S.A. 112, 7725-7730 (2015).
- Chen, X., Grégoire, S., Formanek, F., Galey, J. -B., Rigneault, H. Quantitative 3D molecular cutaneous absorption in human skin using label free nonlinear microscopy. J. of Control. Release. 200, 78-86 (2015).
- Kano, H., Hamaguchi, H. In vivo multi-nonlinear optical imaging of a living cell using a supercontinuum light source generated from a photonic crystal fiber. Opt. Express. 14 (7), 2798-2804 (2006).
- Brustlein, S., Ferrand, P., Walther, N., Brasselet, S., Billaudeau, C., Marguet, D., Rigneault, H. Optical parametric oscillator-based light source for coherent Raman scattering microscopy: practical overview. J. Biomed. Opt. 16 (2), 021106(2011).
- Chen, H., et al. A multimodal platform for nonlinear optical microscopy and microspectroscopy. Opt. Express. 17 (3), 1282-1290 (2009).
- Yue, S., Slipchenko, M. N., Cheng, J. X. Multimodal nonlinear optical microscopy. Laser Photonics Rev. 5 (4), 496-512 (2011).
- Sun, Q., Li, Y., He, S., Situ, C., Wu, Z., Qu, J. Y. Label-free multimodal nonlinear optical microscopy reveals fundamental insights of skeletal muscle development. Biomed Opt Express. 5 (1), 158-166 (2013).
- Le, T. T., Langohr, I. M., Locker, M. J., Sturek, M., Cheng, J. X. Label-free molecular imaging of atherosclerotic lesions using multimodal nonlinear optical microscopy. J. Biomed. Opt. 12 (5), 054007(2007).
- Ellis, D. I., Cowcher, D. P., Ashton, L., O'Hagana, S., Goodacre, R. Illuminating disease and enlightening biomedicine: Raman spectroscopy as a diagnostic tool. Analyst. 138, 3871-3884 (2013).
- A•P•E Angewandte Physik & Elektronik GmbH. , Germany. Available from: http://www.ape-berlin.de/en/page/calculator (2015).
- Ozçelik, M., et al. Pals1 is a major regulator of the epithelial-like polarization and the extension of the myelin sheath in peripheral nerves. J Neurosci. 30 (11), 4120-4131 (2010).
- Heinrich, C., Hofer, A., Ritsch, A., Ciardi, C., Bernet, S., Ritsch-Marte, M. Selective imaging of saturated and unsaturated lipids by wide-field CARS-microscopy. Opt. Express. 16 (4), 2699-2708 (2008).
- Kyriakidis, N. B., Skarkalis, P. Fluorescence spectra measurement of olive oil and other vegetable oils. J. AOAC Int. 83 (6), 1435-1439 (2000).
- King, R. Microscopic anatomy: normal structure. Handb. Clin. Neurol. 115, 7-27 (2013).
- Monsma, P. C., Brown, A. FluoroMyelin Red is a bright, photostable and non-toxic fluorescent stain for live imaging of myelin. J. Neurosci. Methods. 209 (2), 344-350 (2012).
- Wang, H., Fu, Y., Zickmund, P., Shi, R., Cheng, J. X. Coherent anti-stokes Raman scattering imaging of axonal myelin in live spinal tissues. Biophys. J. 89 (1), 581-591 (2005).
- Wang, H. W., Fu, Y., Huff, T. B., Le, T. T., Wang, H., Cheng, J. X. Chasing lipids in health and diseases by coherent anti-Stokes Raman scattering microscopy. Vib. Spectrosc. 50 (1), 160-167 (2009).
- Jung, Y., Tam, J., Jalian, H. R., Anderson, R. R., Evans, C. L. Longitudinal, 3D in vivo imaging of sebaceous glands by coherent anti-stokes Raman scattering microscopy: normal function and response to cryotherapy. J. Invest. Dermatol. 135 (1), 39-44 (2015).
Access restricted. Please log in or start a trial to view this content.
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.