
Method Article
Un sang à base d’essai pour la détection des ROS1 et RET transcrits de Fusion de circuler l’acide ribonucléique Using Digital Polymerase Chain Reaction
Dans cet article
Résumé
Détection de l’acide ribonucléique (cRNA) du sang en circulation est un besoin non comblé dans le diagnostic clinique. Nous décrivons ici des méthodes qui caractérisent les cRNA de patients de cancer pulmonaire non à petites cellules en utilisant la réaction en chaîne de polymérase numérique sensible et spécifique. Les tests de satisfaire aux exigences de conception pour détecter les variantes de fusion dans les 72 heures.
Résumé
Nous avons développé de nouvelles méthodes pour l’isolement et la caractérisation des dérivées de tumeurs circulant l’acide ribonucléique (cRNA) pour biopsie liquide à base de sang. Détection robuste de cRNA de sang représente une solution à un besoin non comblé critique dans le diagnostic clinique. Le test commence par la collecte de sang dans les tubes de prélèvement sanguin contenant des conservateurs qui stabilisent les cRNA. Acellulaire, exosomal et l’ARN associés à plaquettes est isolée du plasma dans ce système de test. Le cRNA est inverse, transcrit de l’ADN complémentaire (ADNc) et amplifié à l’aide de la réaction en chaîne de polymérase numérique (dPCR). Échantillons sont analysés pour tant le biomarqueur de cible comme un gène contrôle. Test de validation inclus limite de détection, la précision et des études de robustesse avec échantillons analytiques. La méthode développée à la suite de ces études de façon reproductible détecter plusieurs variantes de fusion pour ROS1 (proto-oncogène C-Ros 1 ; 8 variantes) et retraité (réorganisés au cours du proto-oncogène transfection ; 8 variantes). L’exemple de workflow de traitement a été optimisée afin que les résultats des tests peuvent constamment être générés dans les 72 heures de la réception de l’échantillon.
Introduction
Vers le haut à 25 % d’un cancer du poumon non à petites cellules (NSCLC) patients n’ont pas suffisamment tissus disponibles pour les tests au moment du diagnostic. Même dans les cas où le tissu est disponible, il peut être suffisante quantité ou qualité pour effectuer déconseillé tests moléculaires1,2. Dans les cas où il y a assez tissulaire d’une biopsie pour le profilage moléculaire, les patients peuvent avoir à attendre plusieurs semaines ou plus pour obtenir les résultats, ou de commencer un traitement sans résultats moléculaires3,4. Toutefois, il est crucial que diagnostic moléculaire informative soit disponible, compte tenu de l’avènement de plusieurs options de traitement ciblé pour les patients atteints de CBNPC. Test de l’ADN d’acellulaire circulant (cfDNA) de biopsie liquide est une solution aux défis des tissus traditionnels tests4,5,6. Options de tests actuelles pour une action mutations CPNPC à l’aide de cfDNA et un workflow basé sur dPCR semblable pour la production du résultat rapide, inclure le récepteur du facteur de croissance épidermique (EGFR) sensibiliser les mutations ΔE746-A750 et L858R, mutation de résistance EGFR T790M , Les variantes du proto-oncogène KRAS (SRP) et B-Raf proto-oncogène (BRAF) variante V600E. Bien que ne pas aussi largement adopté par le champ, circulant dérivées de tumeurs l’ARN messager (ARNm) isolés de biopsie liquide peut également fournir des informations cliniques importantes7,8,9. Nous avons déjà développé et a rendu compte des méthodes de détection multiplexée des échinodermes Microtubule Associated Protein comme 4-anaplasique lymphome du récepteur Tyrosine Kinase (EML4-ALK) fusion variantes du plasma sanguin,10. Dans cette étude, nous avons étendu ces méthodes pour inclure ordre supérieur multiplexé RNA cibles pour ROS1 et RET, couvrant huit variantes de fusion au sein de chaque série de tests. L’objectif était de développer une technique rapide, sensible, précise et reproductible pour la détection de ces variantes de la fusion du plasma de patients diagnostiqués auparavant avec NSCLC.
Le processus de test est lancé dans un bureau de médecin à l’aide de RNA stabilisant blood collection tubes11. Ces tubes contiennent une cellule conservateur que les inhibiteurs de la RNase. Les échantillons sont expédiée priorité du jour au lendemain les modifications amélioration laboratoire centralisées accrédités par le College of American Pathologists CAP/cliniques (CLIA)-laboratoire certifié (laboratoire clinique) pour le traitement par personnel compétent. Une fois reçu par le laboratoire clinique, chaque étape du traitement est effectué sous approuvé procédures d’utilisation normalisées (SOP). Sang total est centrifugée pour récupérer le plasma, qui est ensuite utilisée pour isoler l’ARN qui est soit en circulation libre dans le sang ou dans l’encapsulation des moitiés, tels que les exosomes et plaquettes7,8,9. Pour isoler l’ARN de ces compartiments, nous avons choisi le système de récupération de RNA basée sur la comparaison de plusieurs méthodes d’extraction. L’ARN est concentré et inverse, transcrit à l’ARNC. Plusieurs enzymes de la transcriptase inverse et amorces gène-spécifiques ont été évaluées au cours de l’optimisation de la méthode de synthèse de cDNA de maximiser ROS1 et RET cible transcription conversion10. Cela est essentiel pour la faible abondance de transcriptions, comme les variantes dérivées de tumeurs fusion en circulation. Enfin, nous avons optimisé dPCR concentrations primer et sonde pour permettre une détection multiplexée de RET ou ROS1 variantes de fusion et le gène contrôle, glucuronidase-β (GUSB). Puis, nous avons combiné les meilleures conditions de chacune des études d’optimisation de la dans un protocole final verrouillé avant d’effectuer les études de validation analytique décrites dans le présent rapport. Le présent protocole et ces résultats fournissent la base pour un workflow rapid et sensible pour la détection systématique des variantes rares fusion en circulation.
Protocole
Les instructions du fabricant sont suivies pour les réactifs énumérées ci-dessous, sauf indication contraire. Les tests de PCR sont des produits disponibles dans le commerce destinés à détecter les fusions ROS1 et RET.
1. travail avec de l’ARN en vue de la Transcription inverse (RT)-dPCR : bonnes pratiques de laboratoire
- Créer un environnement exempt de RNase lorsque vous travaillez avec de l’ARN.
- Utilisez commercialement disponible en aérosol conçu pour inactiver RNases contaminantes.
- Utilisation certifiée exempte de RNase réactifs, conseils et tubes. Conseils de barrière pour pipettes permet d’empêcher l’introduction des ribonucléases ou contamination croisée des échantillons.
- Toujours porter un manteau de laboratoire afin d’empêcher les particules tombent sur les vêtements de votre échantillon. Désigner un laboratoire manteau spécifique à utiliser avec l’ARN.
- Porter des gants pour éviter la contamination des échantillons de ribonucléases dans la peau. Changer fréquemment.
Remarque : Supposons que des surfaces de laboratoire sont contaminés par la RNase puisqu’elles sont exposées à l’environnement. Gants qui en contact avec la peau, cheveux, poignées de porte, poignées de congélateur, stylos/marqueurs, etc. sont censés pour ne plus être libre de RNase. - Décontaminer les pipettes, benchtops, centrifugeuses et autres surfaces de travail avec une RNase inactivation spray à avant utilisation.
- Si possible, maintenir un ensemble d’équipement pour un usage avec de l’ARN.
- Minimiser les perturbations des flux d’air dans les zones de laboratoire lorsque vous travaillez avec des échantillons d’ARN empêchant les particules de tomber dans des échantillons ou de contaminer la zone de travail.
- Magasin purifiée RNA à-80 ° c.
- Évitez les multiples gel-dégel des échantillons d’ARN, car ceci peut causer la dégradation.
2. génération de matériel d’ARN analytique pour les contrôles positifs
- Conception ADN synthétique à l’aide de séquences d’ADN messagère publiées pour les variantes de la fusion d’intérêts10.
- Pour une variante de fusion donnée, sélectionnez une séquence de fusion d’ARNm qui comprend le site de fusion ainsi qu’une longueur suffisante d’accompagnement de chaque côté pour couvrir l’amplicon PCR.
- Sélectionner des séquences de nucléotides entre 50 et 250 nt pour imiter la taille des RNA capturée à l’aide de plasma enrichi en plaquettes en circulation.
- Ajouter une séquence promotrice de T7 (5'-CAGAGATGCATAATACGACTCACTATAGGGAGA-3') à l’extrémité 5' de la séquence cible.
- Commander des séquences synthétiques que fragments double brin d’acide désoxyribonucléique (ADN).
- Reconstituer l’ADN synthétique dans un tampon Tris-EDTA (TE) à une concentration finale de 10 ng/µL.
- Convertir les ADN synthétique de 60 ng d’ARN utilisant in vitro de transcription.
- Purifier les ARN transcrits à l’aide du réactif à base de phénol/guanidine12.
- Inclure la DNase I, RNase-libre pour supprimer le modèle résiduel ADN.
- Mesurer la concentration de purifiée en vitro ARN utilisant un fluorimètre disponible dans le commerce avec les normes et les colorants propres RNA. S’assurer que l’ARN est dans la plage acceptable pour les normes choisies. Dilution peut être exigée.
- Confirmer la transcription réussie par électrophorèse sur gel à l’aide d’un gel d’agarose 2 % mélangé avec tache de gel RNA et une gamme haute échelle RNA, y compris la gamme de taille 50-250 nt.
- Charge 500 ng de chaque in vitro ARN sur un gel.
- Exécutez le gel à 5 V/cm.
- Visualiser les bandes individuelles à l’aide d’illumination et de documenter les résultats.
- Confirmer la taille de la transcription prévue pour chacune des variantes fusion (basés sur la conception à l’étape 2.1.2).
- Confirmer la détection de chaque in vitro ARN par RT-dPCR utilisant appariés variante spécifique PCR assay (voir les étapes 5 à 8 du présent protocole).
- En option : Préparer un mélange équimolaire de in vitro ARN qui contient chacune des variantes de la fusion et le gène contrôle GUSB.
- Si étape 2.9 est effectuée : confirmer la détection de chacune des variantes fusion inclus dans le mélange de contrôle par dPCR utilisant la PCR variante spécifique (voir les étapes 5 à 8 du présent protocole).
- Déterminer la concentration d’entrée souhaitée pour des contrôles positifs analytiques en testant des concentrations allant de 0,25 à 2,5 fg10. Choisir la concentration basée sur la sortie numéro de copie désirée.
- Après confirmation, préparer 10 µL à usage unique d’extraits de l’ARN analytique à utiliser dans le contrôle positif (étape 4.4) et les conserver à-80 ° c.
3. les donateurs spécimens
- Prélever les échantillons de sang total humain de 10 mL dans les tubes de prélèvement sanguin (BCT) de 10 mL contenant un conservateur de RNA acellulaire.
Remarque : Tous les donneurs humains donneront leur consentement à l’utilisation de la recherche et aucune information d’identification propres donateurs est recueillie ou utilisée au cours des essais. - Traiter des échantillons de sang total dans le délai indiqué par le fabricant de la BCT.
- Pool de plasmas humains normaux peut être acheté d’une source commerciale pour une utilisation dans le contrôle positif analytique. Préparation à usage unique, 1 ml de la pool de plasmas humains normaux et les conserver à-80 ° c pour une utilisation avec le contrôle positif (étape 4.4).
4. récupération des ARN du Plasma en circulation
Remarque : Il est important de travailler rapidement au cours de cette procédure.
- Centrifuger les tubes de sang total à 200 x g pendant 20 min.
- Recueillir jusqu'à 4 mL de plasma du tube de prélèvement de sang centrifugé à l’aide d’une pipette sérologique. Veillez à ne pas déranger ou aspirer la couche leuco.
- Isoler l’ARN circulant à l’aide d’un kit disponible dans le commerce qui peut capturer les RNA acellulaire du plasma, les plaquettes et les exosomes. Isoler l’ARN de l’échantillon de contrôle positif aux côtés de chaque lot.
-
Préparer le contrôle positif pour chaque lot d’échantillons cliniques comme suit :
- Décongeler 1 mL pooled plasma humain normal aliquote (étape 3.3).
- Décongeler la quantité analytique RNA 10 µL (étape 2.12).
- Préparer, contrôle positif en ajoutant 10 µL RNA analytique dans l’échantillon de plasma humain normal une fois que l’éthanol a été ajouté au plasma lysat.
- Éluer les échantillons avec de l’eau exempte de nucléase 100 µL. Procéder immédiatement, nettoyer avec de l’ARN et la concentration.
- Échantillons peuvent être stockés sur la glace humide et à couvert, jusqu'à une heure.
- Concentrer les ARN utilisant la méthode de la colonne en fonction et éluer à 9 µL d’eau exempte de RNase.
- Procéder immédiatement à l’étape 5, ou conserver les échantillons sur la glace humide jusqu'à une heure.
5. reverse Transcription de l’ARN d’ADNc
- Échantillon de RNA concentré circulation convertir cDNA utilisant un kit de réaction de transcription inverse disponibles dans le commerce, y compris amorces aléatoires (voir le tableau 1 pour les composants).
Remarque : Des amorces spécifiques de gènes sont facultatifs et peuvent être conçus pour les variantes de test. Amorces sont conçus selon l’objectif de la séquence d’ARN. Utiliser des séquences variant de fusion du point 2.1.- Inclure aucun échantillon de contrôle de la transcriptase inverse et aucun échantillon de contrôle RNA (voir tableau 1).
- Isoler l’ADNc de la réaction de transcription inverse en utilisant une colonne de spin de concentrateur ADN disponible dans le commerce.
Remarque : Cette étape facilite l’élimination des enzymes, des amorces et gratuit désoxyribonucléosides triphosphates (dNTPs). - Utiliser cDNA immédiatement dans les réactions de PCR ou stocker à-80 ° c.
6. digital PCR
Remarque : Cette PCR est spécifique aux gouttelettes numérique PCR (voir Table des matières).
-
Précautions PCR de mélange.
- Porter un blouson manteau et nitrile de laboratoire jetables.
- Utilisation PCR mélanger les réactifs dans une zone de préparation du réactif dédié. Ne pas manipuler les ADNc dans la zone de préparation de réactif seule.
- Couvrir les sondes alors qu’il travaillait pour les protéger de la lumière. La lumière excessive peut photo le colorant fluorescent relié à la sonde d’eau de Javel.
- Transport mixes, couverts et protégés de la lumière, sur une aire séparée de pré-amplification avant cDNA doit être ajouté.
- Ajouter ADNc à tester au mélange de la PCR dans une hotte de PCR clean situé dans quartier de pré-amplification.
- Préparer les mélanges PCR pour un volume de réaction finale de 20 µL selon le tableau 2.
- Distribuer les mélanges PCR + cDNA pour plaques PCR.
Remarque : Utilisation d’un schéma comme un guide est recommandé. - Couvrir la plaque à l’aide d’un adhésif amovible.
- Centrifuger les plaques brièvement pour prélever des échantillons au fond des puits.
- Mix sur agitateur de plaque sur un réglage bas pendant 10 s.
- Centrifuger les plaques brièvement pour recueillir l’échantillon au fond du puits.
- Retirer l’adhésif. Effectuer la génération de gouttelettes pour PCR-cDNA mélange soit avec un système de génération de gouttelettes manuel ou automatisé.
- Pour la génération manuelle de gouttelettes, virement mélange PCR 20 µL puits d’échantillon sur la cartouche de génération de gouttelettes. Ajouter 70 µL d’huile de génération de gouttelettes. Couvrir avec la cartouche de joint d’étanchéité et de transfert de caoutchouc au générateur de gouttelettes manuel pour lancer la génération de gouttelettes. Après la génération de gouttelettes, transfert gouttelettes sur une plaque PCR fraîche à l’aide de conseils recommandés par le fabricant. Aspirer et distribuer les gouttelettes lentement, en 5 à 6 s chacun, sans toucher l’ouverture de l’embout de la cartouche de gouttelettes ou de la plaque.
- Pour la génération automatisée de gouttelettes, sceller la plaque avec un opercule et le transfert dans le générateur de gouttelettes. S’assurer que tous les conseils, cartouches, et les plaques sont en place avant de commencer la production de gouttelettes.
- Après la génération de gouttelettes et transfert des gouttelettes sur une plaque PCR fraîche, joint avec un adhésif aluminium et plaques de cycle thermique en utilisant les paramètres dans le tableau 3.
- Après le thermocycleur série est terminée, lisez la plaque à l’aide d’un lecteur de gouttelettes. Créer un schéma pour le logiciel de lecteur qui identifie l’emplacement des contrôles, échantillons, etc.et la charge dans le logiciel pour démarrer la lecture.
7. analyse et l’examen et la génération des résultats
- Analyser les résultats à l’aide de logiciels disponibles dans le commerce de lecture de la plaque.
- Accédez au menu analyser pour voir deux dimensions (2D) amplitude de terrains.
- Évaluer la qualité globale des données en examinant les données de gouttelettes.
- Évaluer les données pour les numéros d’événement accepté total en utilisant le menu événements. S’il y a moins de 10 000 événements par puits, évaluer avec soin les données pour des problèmes supplémentaires.
- Vérifier les données pour les amplitudes de fluorescence aberrante. Différences d’amplitude significative et les différences de concentration entre échantillons répétés indiquent la mauvaise manutention ou mélange d’échantillons.
- Prendre des notes, des grappes de gouttelettes avec modèles de pulvérisation sur un axe de 45 degrés, ce qui dénote des gouttelettes de mauvaise qualité ou échantillons problématiques.
- Examiner les données de contrôle positif, N° de la Transcriptase inverse (RT N°) et No RNA contrôle (CNRC) tout d’abord. Sélectionnez tous les échantillons de contrôle et d’analyser des cluster qualité de tracé 2D. Pour le seuil approprié, une séparation nette entre les clusters de gouttelettes devrait être évidente.
- Pour chaque variante de test, définissez le seuil basé sur les puits de contrôle.
- Set de seuils sur les parcelles 2D à l’aide de l’outil de ligne de mire de séparer la population de gouttelettes de double négation de la population de gène témoin (étiqueté avec sonde de 5'-hexachloro-fluorescéine-CE phosphoramidite), axe y et population variant de gène, si présent) marqué avec fluorescéine amidite sonde 6-carboxyfluorescéine OR), axe des abscisses.
- Somme des copies de chaque puits répétées pour un seul échantillon.
- Exprimer les résultats des tests comme le nombre d’exemplaires variant détecté.
Remarque : Pour déterminer la valeur de seuil analytique permettant d’appeler un échantillon positif ou négatif, exécuter une normale échantillon donneur sain mis (au moins 10 échantillons individuels) à travers le processus de finalisation et d’établir le seuil au-dessus de n’importe quel signal détectable de fond pour la mutation de intérêt. En outre, établir le nombre de copies du gène contrôle requis pour appeler un résultat positif ou négatif. Ce seuil de gène de contrôle fonctionne comme un contrôle de qualité interne (QC) pour évaluer la quantité et la qualité de chaque échantillon de RNA qui est traitée.
8. vérification des Conditions de réaction de RT-dPCR utilisant des lignées cellulaires (facultatives)
- Pour vérifier la détection de variants de fusion, utiliser commercialement disponibles lignées cellulaires exprimant la fusion ROS1 ou RET ADN messagère d’intérêt. Procédez comme suit :
- Homogénéiser les cellules congelées flash dans une solution de lyse guanidinium base directement à partir de l’état congelé. Même bref dégel avant homogénéisation peut causer une perte et dégradation de l’ARN.
- Isoler l’ARN à l’aide de colonnes à centrifuger silice-membrane conçues pour l’ARN.
- Mesurer la concentration des échantillons d’ARN à l’aide d’un fluorimètre avec des normes et des réactifs spécifiques de RNA.
- Diluer les ARN dans un fond d’ARN sauvage de plasma ou d’une autre source commerciale.
- Effectuer les étapes de Reverse Transcription de l’ARN à l’ARNC, PCR numérique et l’analyse des données et examen et génération de résultats figurant dans le présent protocole pour confirmer la détection de la variante désirée.
Résultats
Ce protocole décrit un système de test mis au point pour la détection de variants fusion RNA devant servir à la mesure des mutations de pilote dans le plasma des patients CPNPC (Figure 1A). ARNm de fusion ont été produits à partir de l’expression de ces réarrangements RET et ROS1 plus courantes dans la population de NSCLC identifié13,14,15,16,17. Tests de PCR multiplexes ont été ensuite destinés à détecter les huit variantes de transcription plus courantes pour chaque cible dans NSCLC au sein d’une seule réaction. Les translocations plus courantes au locus ROS1 génèrent des associations avec les parties 5' du CD74, SDC4, SLC34A2, EZR ou gènes TPM3 (Figure 1B). Les translocations plus courantes au locus RET entraîner juxtaposition avec KIF5B, pour lesquels le test couvre six jonctions exon. Les partenaires RET supplémentaires qui sont couverts sont ceux avec CCDC6 et TRIM33 (Figure 1C). Au total, les tests couvrent environ 88 % de ROS1 et 99 % des altérations RET connues pour se produire dans la population de patients de NSCLC17.
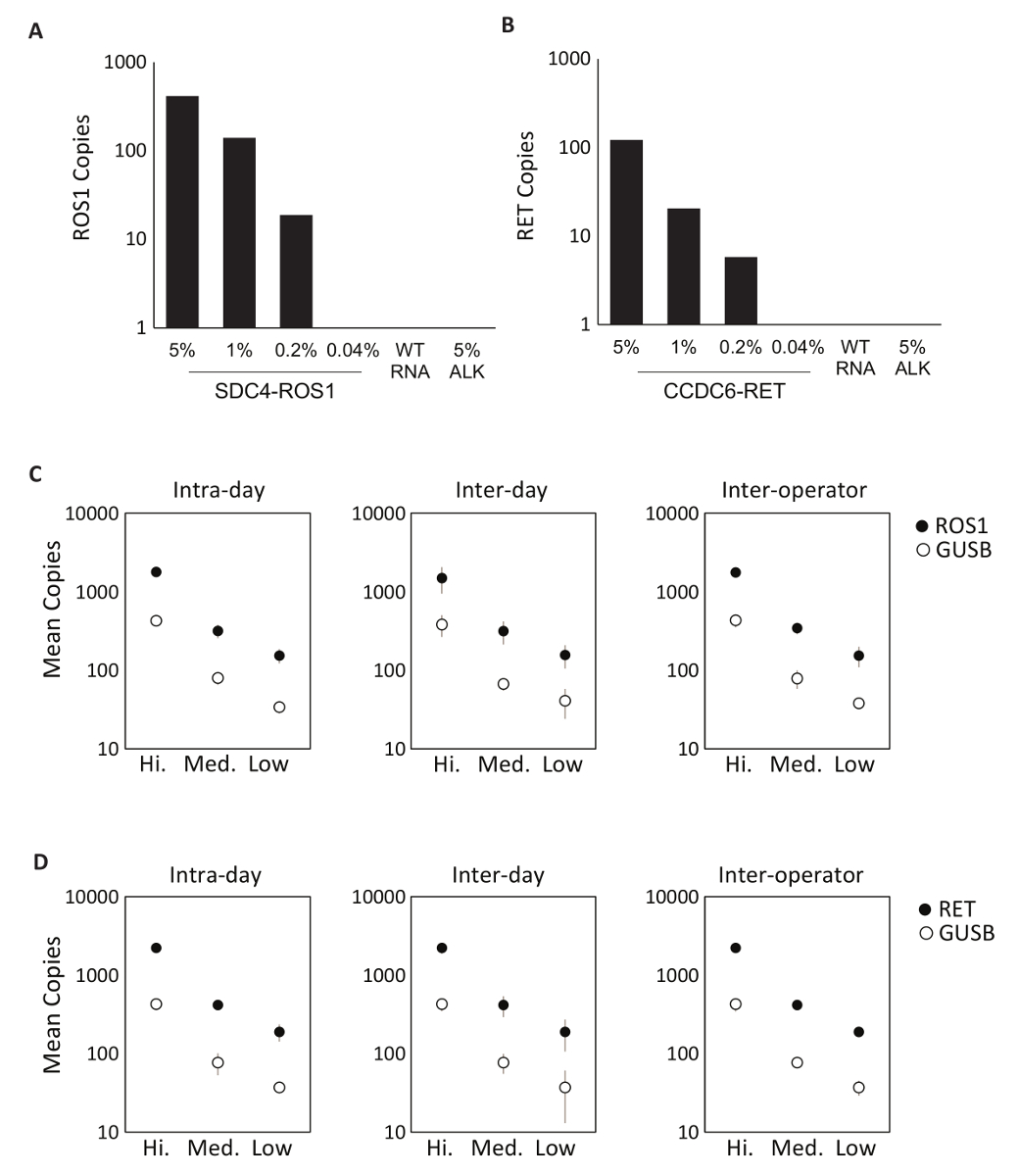
La spécificité des composants dosage a été tout d’abord évaluée en utilisant huit individuels en vitro ARN qui contient la séquence de l’ARNm pour les transcriptions de fusion relevant du ROS1 ou RET multiplexés dosages. Chaque espèces d’ARN a été testée contre chaque dosage variant individuel qui comprend la version multiplexée. Il n’y avait aucune réaction croisée de ces tests, démontrant ainsi 100 % spécificité analytique dans la conception multiplexés dosages (données non présentées). Pour déterminer la limite inférieure de détection du protocole de test, l’ARN total provenant de lignées de cellules exprimant une variante de fusion incluse dans l’essai ont été mélangés dans un fond d’ARN normale à des concentrations de 5 %, 1 %, 0,2 % et 0,04 %. Les multiplex RET et ROS1 variantes analyses par PCR détectés aussi peu comme variante de fusion de 0,2 % (Figure 2A-B). En outre, une préparation de 5 % hors-cible des lignées cellulaires dérivées de RNA (exprimant une transcription de fusion EML4-ALK) n’a pas été détecté avec les dosages ROS1 et RET multiplexés, démontrant ainsi la spécificité (Figure 2A-B).
Tests de précision du processus RT-dPCR a été faite deux ROS1 et matériel de contrôle analytique RET composé d’équimolaire in vitro RNAs a été traitée en trois concentrations (haute, moyenne et faible) par transcription inverse et dPCR sur trois reprises dans la même journée (intra-jour), sur trois jours consécutifs (inter-jour) et avec deux opérateurs (entre opérateurs). Résultats des tests de précision a démontré une détection précise de la transcription de la fusion d’intérêt, tant qu’un gène contrôle, GUSB, qui est inclus dans une métrique QC interne (Figure 2-D).
Outre le contrôle interne GUSB, chaque lot d’échantillons cliniques a été exécuté avec un ensemble de commandes par lot. Un contrôle positif a été développé par un mélange d’analyse in vitro ARN qui représente chacune des variantes fusion testés en RT-dPCR, ainsi qu’analyse in vitro de RNA pour GUSB. Cet ARN a été fortifié dans le plasma humain normal lysat au cours de l’extraction de l’ARN et a été traité avec les échantillons cliniques dans tout le protocole. Sans le contrôle de la transcriptase inverse (RT No) a été un témoin négatif pour confirmer l’absence de matières contaminantes dans le flux de travail RNA extraction et démontrer la spécificité des amorces ARN. Le contrôle N° RT a été généré en utilisant le même matériau que le contrôle positif, mais il n’inclut pas d’enzyme dans la réaction de synthèse de cDNA. Aucun contrôle RNA (CNRC) n’est un témoin négatif pour confirmer l’absence de contamination des transcriptions dans les composants de réaction de transcription inverse. Ce contrôle a été introduit dans le flux de travail à l’étape de la synthèse de cDNA, et l’eau a été ajoutée dans la réaction au lieu d’un descripteur d’ARN. Les contrôles N° RT et le CNRC doivent être négatifs dans les deux voies, si des résultats précis doivent être livrées. Le tableau 1 répertorie les composants de réaction de transcription inverse pour chaque contrôle. Exemples des parcelles 2D pour chacun de ces contrôles sont indiquées pour le ROS1 (Figure 3 A-C) et dosages multiplex RET ( E-Gdela Figure 3 ). Variantes de fusion ont été détectés à l’aide d’une sonde d’amidite (FAM) de fluorescéine et sont représentés le long de l’axe des ordonnées, tandis que le gène contrôle, GUSB, a été détecté à l’aide d’une sonde de 5'-hexachloro-fluorescéine-CE phosphoramidite (HEX) et sont sur l’axe des abscisses. Ces contrôles de lot ont été évaluées au cours des vingt et un jours pour déterminer la robustesse de l’essai. Gouttelettes positives fusion et gouttelettes de gène contrôle GUSB ont été observées pour ROS1 et RET dans toutes les séries de 21 exécutés au cours de l’étude (Figure 3D, H). Tous les contrôles négatifs (N° RT et NRC) a donné des résultats négatifs dans les 21 jours ensemble (données non présentées).
La capacité à résoudre les problèmes est un élément essentiel de n’importe quel protocole de test à exécuter dans le cadre du laboratoire clinique. Ici, nous fournissons des exemples du monde réel des résultats sous-optimaux en utilisant le protocole RT-dPCR. Le premier est un complot 2D exemple démontrant l’importance de l’aucun contrôle de la transcriptase inverse (Figure 4A). Dans cet exemple, mutants gouttelettes positifs étaient présents, même s’il n’y avait aucune conversion de cDNA due à l’absence de l’enzyme. Ce résultat est probablement due à amorces dPCR amplifier l’ADN génomique hors cible. Dans ce cas, conception d’un test s’étendant sur intron empêchera l’amplification de l’ADN génomique. Par ailleurs, une enzyme de la DNase RNase-libre peut être utilisée pour éliminer l’ADN contaminant, mais ce n’est pas recommandé pour la détection des cibles rares, comme une dégradation de l’ARN peut se produire durant l’incubation avec l’enzyme. La prochaine intrigue 2D exemple était un NRC avec gouttelettes positives dans les deux canaux (Figure 4B). Cet état de contamination au cours de l’installation de RT-dPCR. Dans ce cas, la recommandation est de jeter tout potentiellement contaminés réactifs utilisés lors d’essais, soigneusement décontaminer tout le matériel, puis refaire le test avec des composants de réaction fraîches. Le troisième exemple 2D intrigue présentée sous forme de pulvérisation de gouttelettes le long d’une ligne de 45° (Figure 4C). Ceci est souvent causée par la tonte et la coalescence des gouttelettes. Gouttelette attention manipulation avant cyclage thermique est essentiel, comme les gouttelettes ont tendance à endommager. Nous recommandons l’utilisation de génération automatisée de gouttelettes, lorsqu’ils sont disponibles. Si transférer manuellement généré gouttelettes, assurez-vous de choisir les conseils de l’échelle-alésage recommandées et employer la technique de pipetage prudent. Transfert de gouttelettes nécessite lente aspiration et distribution, avec chaque se déroulant sur une période de 5 à 6 secondes, et il est essentiel que l’ouverture de pointe de pipette ne pas toucher la cartouche de gouttelettes ou bien. Lors de la distribution, maintenir l’embout de la pipette au niveau liquid et levez-le lentement comme des gouttelettes sont distribué (voir la vidéo de démonstration). L’exemple de tracé 2D finale montre un manque de séparation entre les populations de gouttelettes positifs et négatifs (Figure 4). Ceci peut avoir plusieurs causes. Inhibiteurs de PCR Strong, tels que les détergents utilisés dans les mémoires tampons de lysis et un excès d’ADN très dégradé, peuvent causer la perte d’espacement. Dans ce cas, envisagez d’ajouter une étape de nettoyage entre la synthèse de cDNA et dPCR (tel que décrit dans l’étape 5 du présent protocole). Enfin, manque de séparation peut également être due à des conditions sous-optimales amplification et optimisation de l’étape de l’ACP devrait également être envisagée.
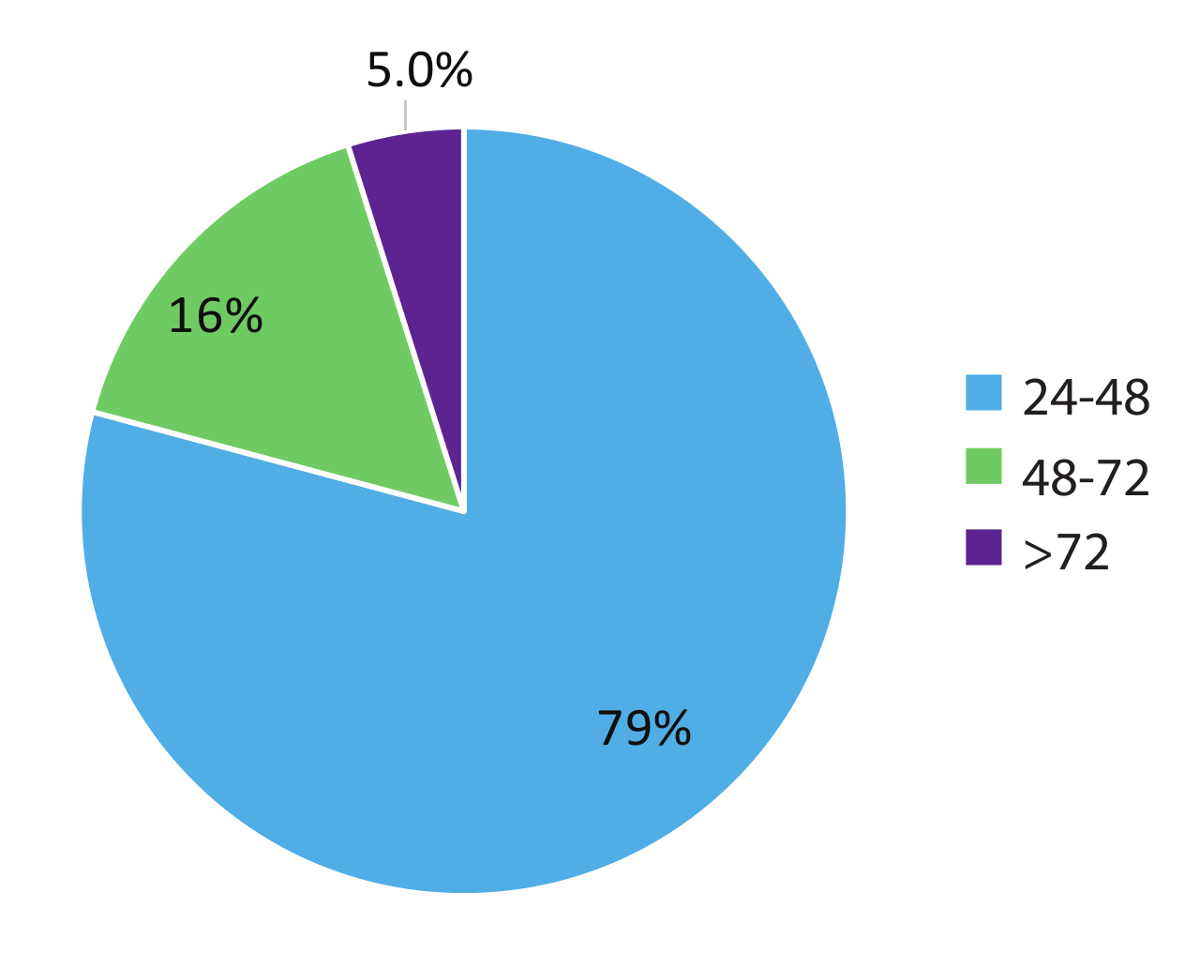
Données dans la Figure 5 représente 984 patient de monde réel retournement fois l’échantillon et montre le caractère rapid de ce flux de travail de test. Résultats ont été signalés au médecin traitant plus tôt que dans les 48 heures (79 % des cas) de la réception de l’échantillon et dans 95 % des cas, dans les 72 heures. En conclusion, l’utilisation de stabilisé circulant des tubes de prélèvement sanguin du RNA, des procédés d’extraction RNA optimisées du sang et RT-dPCR exécuter selon un protocole optimisé avec le cas échéant interne et contrôles de lot, peut fournir un système de test rapide pour la détection précise des variantes de RNA fusion pertinents dans le CBNPC.

Figure 1 : Aperçu des étapes de traitement de l’échantillon de sang pour détection variante de Fusion à l’aide de dosages spécifiques aux plus répandus RET et variantes ROS1 dans CPNPC. (A) essai d’échantillon est initiée lorsque sang entier est dessiné et un BCT est fourni dans la trousse de prélèvement d’échantillon au laboratoire clinique. RNA en circulation est récupéré provenant de plusieurs sources au sein du plasma enrichi en plaquettes, reverse transcrit avec amorçage spécifiques de gènes et purifiées devant servir à dPCR. Les échantillons sont traités en utilisant un système disponible dans le commerce qui se compose de génération de gouttelettes (émulsion), amplification et comptage de gouttelettes. Données sont analysées à l’aide de logiciels disponibles dans le commerce. Les résultats des tests sont ensuite documentés et communiqués au médecin requérant test. Le processus est conçu pour travailler dans un délai de 72 heures suivant la réception de l’échantillon à libération de résultat. Huit variantes pour ROS1 (B) et (C) RET sont couverts dans les dosages multiplexés. Adapté du site Web Biodesix avec permission. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 2 : Validation analytique. Lignées cellulaires exprimant (A) SDC4-ROS1 fusion et (B) CCDC6-RET de fusion ont été dilués dans un contexte de type sauvage humain total (ARN WT). Chaque variante de fusion, la limite de détection a été créée à fréquence variant de 0,2 % à l’aide des critères prédéfinis pour chaque dosage variant. Tous les échantillons au-dessus de ce seuil contenaient également au moins 21 copies du gène de contrôle. Norme EML4-ALK (ALK) 5 % dans un contexte d’ARN sauvage a été testé pour démontrer la spécificité de dosage, ce qui a été confirmée par un résultat négatif. Normes de RNA multiplexés analytiques ont été mesurées à haute, moyenne et faibles concentrations pour ROS1 (C) et de la précision de la retraite (D) a été évaluée au cours de trois passages sur le même jour (Intra-jour), trois passages sur trois jours consécutifs (inter-jour) et avec deux exploitants indépendants (entre opérateurs). Les moyens du nombre de copies et les écarts-types sont indiqués. Adapté du site Web Biodesix avec permission. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 3 : Traitement des exemples de contrôles personnalisés et les données de la robustesse du lot. Tracé 2D de la ROS1 multiplexé test dPCR ne résultats pour contrôle positif (A), (B) aucun contrôle de la transcriptase inverse et (C) aucun contrôle de modèle de RNA. (D) des contrôles ont été effectués sur 21 jours consécutifs (sauf les week-ends et jours fériés). Comme le nombre de copies +/-écart-type pour ROS1 positif de contrôle étaient 439 +/-141. Aucun transcriptase inverse et aucun contrôle de modèle ont été également exécutés chaque jour, et ils étaient tous négatifs (données non présentées). Tracé 2D de la RET multiplexé test dPCR ne résultats pour contrôle positif (E), (F) aucun contrôle de la transcriptase inverse et (G) aucun contrôle de modèle de RNA. (H) des contrôles ont été effectués sur 21 jours consécutifs (sauf les week-ends et jours fériés). Moyenne +/-écart-type des copies pour RET positif de contrôle étaient 586 +/-182. Aucun de la transcriptase inverse et aucun contrôle de modèle qui ont été également exécutés chaque jour et ont été tous négatifs ne sont ne pas présentées. Adapté du site Web Biodesix avec permission. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 4 : Dépannage RT-dPCR. 2D parcelles représentant sous-optimal dPCR résultats obtenus lorsqu’il n’y a (A) la contamination au sein de l’aucun contrôle de la transcriptase inverse, (B) la contamination dans le non contrôle de RNA, (C) cisaillement et coalescence des gouttelettes et (D ) mal optimisé des conditions d’amplification ou l’inhibition de la PCR. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 5 : Délai d’exécution (TAT). TAT (en heures) a été compilée pour tests demandant une variante de RNA (n = 984). Données ne comprennent pas les week-ends, jours fériés et échantillons détenus pour > 24h en raison des informations cliniques incomplètes sur le formules de demande de Test de laboratoire. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Tableau 1 : Préparation des réactifs de la transcriptase inverse pour les contrôles de processus.
| Composant | Volume |
| 2 x dPCR supermix pour sondes (pas 2'-uridine 5'-triphosphate) | 10 ΜL |
| la valeur de x 20 variante cible amorces/sondes (sonde 450 amorces nmol/L, 250 nmol/L FAM) | 1 ΜL |
| 20 x jeu de cible amorces/sonde (sonde 450 amorces nmol/L, 250 nmol/L HEX) | 1 ΜL |
| eau exempte de nucléase | 1 ΜL |
| ADNc | 7 ΜL |
Tableau 2 : Préparation du master mix dPCR.
| Vélo Step | Température | Temps | # Cycles | Vitesse de montée |
| Activation des enzymes | 95 ° C | 10 min | 1 | ~ 2 oC/s |
| Dénaturation | 94 ° C | 30 s | 40 | |
| Recuit/extension | 55 ° C | 1 min | ||
| Désactivation de l’enzyme | 98 ° C | 10 min | 1 | |
| Cale (en option) | 4 ° C | infini | 1 | 1 ~ oC/s |
Tableau 3 : Conditions de cyclisme thermiques.
Discussion
Réarrangements RET et ROS1 forment environ 3 % des mutations au sein de la population de NSCLC18pilote. Bien que rare, la détection de ces altérations génétiques est vitale. Les patients CPNPC avec ces altérations peuvent bénéficier de thérapies ciblées qui inhibent spécifiquement l’activité de la kinase aberrante qui résulte de l' onco-protéines13. Certaines de ces thérapies ont déjà été approuvés par la FDA pour utilisation en ROS1 NSCLC positive, tandis que d’autres sont sont avérées efficaces contre RET dans les essais cliniques,19.
La technologie PCR numérique fournit une sensibilité qui est idéale pour des applications de biopsie liquide20. Il y a eu adoption significative de cette technologie pour une utilisation avec une circulation sans cellule ADN pour la mesure des mutations de la tumeur chez les patients atteints de CBNPC4,6,21,22,23 . En plus de cfDNA, nous avons développé un protocole conçu pour la mesure robuste des variantes fusion plus répandus chez les patients atteints de CBNPC de circuler tumeur RNA (Figure 1A)10.
Notre protocole établi prévoit des limites analytiques de détection à 0,2 % (Figure 2). Alors que la RT-dPCR est exceptionnellement précis et sensibles, les dosages sont limitées au panneau des variantes de fusion connus qui sont choisis et multiplex pour la détection de la PCR. Ainsi, fusions devant figurer dans les essais multiplexes doivent être soigneusement sélectionnées afin d’assurer une couverture adéquate au sein de la population des patients atteints de CBNPC. Nous avons conçu avec succès les essais pour retraité et ROS1 simultanément de détecter huit résultante de variantes de fusion de réarrangements des loci RET ou ROS1 et couvrir 99 % et 88 % de la population positive RET et ROS1, respectivement (Figure 1-B-C )17.
Le flux de travail de test final tel que décrit dans cette étude inclut des contrôles de lot pour assurer la cohérence des résultats. Ces contrôles incluent tous deux un critère analytique positif ainsi que deux contrôles négatifs, qui assurent ensemble il n’y a pas de contamination ou inhibition de PCR survenant dans le lot (Figure 3). Pour assurer la robustesse du dosage, une étude a été réalisée en utilisant les contrôles de lots sur une période de 21 jours (Figure 3D, H). Ces résultats démontrent la cohérence du processus RNA tel qu’établi dans le présent protocole.
Bonnes pratiques de laboratoire et de manipulation de RNA appropriée sont des éléments clés d’assurer des résultats robustes et précis. Espace de laboratoire et équipements dédiés à utiliser avec de l’ARN, nettoyage du matériel après chaque utilisation, l’utilisation des consommables et réactifs RNase-libre et en appliquant un spray d’inactivation de RNase à l’espace de travail tous contribuent à réduire les RNases contaminantes. Gestion consciencieuse des échantillons d’ARN par des techniciens, y compris une blouse de laboratoire dédié, fréquents changements de gant, travailler rapidement par le biais de la procédure d’extraction de RNA, et maintien des échantillons sur la glace sont d’une importance capitale pour préserver l’intégrité de l’échantillon. Une fois que RNA a été inverse, transcrit à l’ARNC, l’échantillon est dans une forme plus stable qui est moins sujette à la dégradation. En complément des méthodes qui prennent en charge l’intégrité RNA, échantillons et composants PCR doivent être maintenus dans des domaines distincts pour prévenir la contamination croisée qui pourrait conduire à des résultats faussement positifs. Le stocks réactifs PCR et la préparation de mélanges maîtres PCR devraient être séparés des modèles PCR et grand soin doit être prélevé pour séparer le modèle amplifié (post-PCR) tous les matériaux pré amplifiés, y compris les réactifs, l’ARN et des échantillons de cDNA. Enfin, la génération correcte et manutention des mélanges PCR émulsionnées avant l’amplification est central à maintenir l’intégrité des gouttelettes et des conditions optimales dPCR. Précautions comme celles-ci sont essentielles lors de l’exécution du présent protocole pour obtenir des résultats cohérents et précis. Toutes les données doivent être examinées par un personnel formé avant la publication des résultats pour être sûr que toute la métrique QC ont été respectée. Dans le cas de résultats sous-optimaux (Figure 4), le lot doit être revu par le personnel technique et le directeur du laboratoire et peut exiger la reprise du traitement.
RT-dPCR résultats peuvent être produites dès 24 heures après réception de l’échantillon et 95 % des résultats de l’échantillon dans le test de la valeur utilisée dans cette étude (n = 984) ont été signalés au médecin commande en moins de 72 heures de la date de réception (Figure 5). Ce temps de rotation fournit aux médecins une grande partie nécessaire des informations moléculaires dans un laps de temps qui permet l’initiation de la thérapie appropriée. Ces résultats sont généralement disponibles plus tôt que ceux obtenus à l’aide d’une biopsie de tissu conventionnel. Biomarqueurs supplémentaires pour NSCLC et d’autres cancers pourraient être développées en utilisant des approches similaires circulants base d’ARN et bénéficieraient du mêmes temps-à-résultats rapides. Par exemple, mesure de la transcription de l’ARNm programmé mort Ligand 1 (PD-L1) à l’aide de RT-dPCR pourrait informer les médecins sur les options de l’immunothérapie. Il y a aussi un intérêt croissant pour l’utilité des liquide biopsie et dPCR dans la surveillance de l’efficacité thérapeutique. Les indications antérieures de résurgence de la tumeur à l’aide de génomique tester des variantes spécifiques pourraient permettent aux médecins d’ajuster les régimes de traitement avant que les patients sont symptomatiques de norme des mesures de soins tels que l’imagerie24. Protocoles comme celle rapportée dans cette étude sont idéales pour la surveillance en raison de leur caractère non invasif, sensibilité, temps de rotation rapide et rapport coût-efficacité. Le test décrit ci-après présente les résultats dans les 72 heures suivant la réception d’échantillon, avec des taux de détection de faux positifs minimale, qui facilite les décisions de traitement rapide et contourner certaines limitations rencontrées avec les tests sur tissus4.
Notre protocole et les données démontrent un système de test robuste pour l’identification des variantes de RNA de faible abondance, mais aussi la possibilité de mutation axée sur le sang, test en pratique clinique. Pour les patients qui n’ont pas un pilote une action mutation identifiée par biopsie liquide ciblée rapide s’approche comme celui-ci, l’ajout de génome plus étendu et proteome essais de tissus et de sang peuvent fournir des informations cliniques encore plus larges pour appuyer la planification du traitement.
Déclarations de divulgation
H.M., L.J., K.A. et G.A.P. sont employés et détiennent des parts dans H.M. Biodesix, Inc., R.D. et G.A.P. sont co-inventeurs sur une demande de brevet déposée par Biodesix, portant sur un système de test de diagnostic pour la détection des variants génétiques dans non à petites cellules en circulation cancer du poumon.
Remerciements
Nous remercions nos collaborateurs, Stephen Jones, Nia Charrington, Dr Dianna Maar et Dr Samantha Cooper depuis le centre de biologie numérique (Bio-Rad Inc. CA) pour leur conception de l’essai en charge ; Nezar Rghei et Dr Moemen Abdalla (Norgen Biotek, Canada) pour obtenir des conseils critiques lors de l’optimisation du protocole d’extraction d’ARN ; et Shannon Campbell, Scott Thurston, Jeff Fensterer, Shannon Martello et Joellyn Enos d’aide pour tester les exigences et le suivi commercial.
matériels
| Name | Company | Catalog Number | Comments |
| Ultrapure Distilled Water (DNAse, RNAse Free) (500 mL) | Life Technologies | 10977-015 | 1604071 |
| Ultrapure Distilled Water (DNAse, RNAse Free) (500 mL) | Life Technologies | 10977-015 | 1809353 |
| Nuclease-free water (molecular grade) | Ambion | AM9938 | 1604071 |
| Nuclease-free water (molecular grade) | Ambion | AM9938 | 1606077 |
| Phosphate Buffered Saline 1X, Sterile | Amresco | K812-500mL | 1446C189 |
| Phosphate Buffered Saline 1X, Sterile – 500 mL | Invitrogen | 10010023 | 1916C092 |
| RNase Zap (Life Tech) (250 mL) | Ambion | AM9780 | 353952 |
| Beta-Mercaptoethanol (BME) (250 mL) | CalbioChem | 6050 | W105B |
| OmniPur Ethyl Alcohol | CalbioChem | 4455-4L | 56054611 |
| OmniPur Ethyl Alcohol | CalbioChem | 4455-4L | 56238638 |
| Isopropyl Alcohol | VWR | 0918-4L | 2116C416 |
| TranscriptAid T7 High Yield Transcription Kit | Thermo Scientific | K0441 | 403648 |
| TranscriptAid T7 High Yield Transcription Kit | Thermo Scientific | K0441 | 288461 |
| DNase I | Thermo | K0441 | 371299 |
| QIAzol Lysis Reagent | Qiagen | 79306 | 54809699 |
| 20x TE buffer pH 8.0 | Alfa Aesar | J62388 | R13C548 |
| UltraPure Agarose | Invitrogen | 16500-100 | 552730 |
| 10x TBE buffer | Invitrogen | AM9863 | 353065 |
| Cell-Free RNA BCT | Streck | 218976 | 60110327 |
| Cell-Free RNA BCT | Streck | 218976 | 61900327 |
| Cell-Free RNA BCT | Streck | 218976 | 61480327 |
| Cell-Free RNA BCT | Streck | 218976 | 62320327 |
| Plasma/Serum Circulating and Exosomal RNA Purification Kit (Slurry Format) 50 preps | Norgen | 42800 | 585849 |
| Plasma/Serum Circulating and Exosomal RNA Purification Kit (Slurry Format) 50 preps | Norgen | 42800 | 588308 |
| Lysis Buffer | Norgen | 21205 | A5F61E |
| RNA Cleanup and Concentration Micro-Elute Kit (Norgen) 50 preps | Norgen | 61000 | 585848 |
| RNA Cleanup and Concentration Micro-Elute Kit (Norgen) 50 preps | Norgen | 61000 | 588309 |
| DNA Clean and ConcentratorTM- 5 200 preps (samples) | Zymo | D4014 | ZRC186976 |
| DNA Clean and ConcentratorTM- 5 200 preps (samples) | Zymo | D4014 | ZRC188077 |
| DNA Clean and ConcentratorTM- 5 200 preps (samples) | Zymo | D4014 | ZRC188413 |
| Collection Tubes 500 pack | Zymo | C1001-500 | N/A |
| SuperScript IV First Strand Synthesis System 200 rxn (samples) | Life Technologies | 18091200 | 391657 |
| SuperScript IV First Strand Synthesis System 200 rxn (samples) | Life Technologies | 18091200 | 392504 |
| SuperScript IV First Strand Synthesis System 200 rxn (samples) | Life Technologies | 18091200 | 448001 |
| SuperScript IV Reverse Transcriptase | Life Technologies | 18090200 | 451702 |
| Qubit HS RNA Assay Kit (500) | Life Technologies | Q32854 | 1745264 |
| Qubit assay tubes (500) | Life Technologies | Q32856 | 13416Q311 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64031651 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64063941 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64065740 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64065741 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64079083 |
| ddPCR Buffer Control for Probes | Bio-Rad | 1863052 | 64025320 |
| ddPCR Buffer Control for Probes | Bio-Rad | 1863052 | 64052358 |
| gBlock KIF5B-RET K15:R12 | IDT | 151004172 | 4-Oct-16 |
| gBlock KIF5B-RET K16:R12 | IDT | 151004173 | 4-Oct-16 |
| gBlock KIF5B-RET K22:R12 | IDT | 151004174 | 4-Oct-16 |
| gBlock KIF5B-RET K23:R12 | IDT | 151004175 | 4-Oct-16 |
| gBlock KIF5B-RET K24:R11 | IDT | 151004176 | 4-Oct-16 |
| gBlock KIF5B-RET K24:R8 | IDT | 151004177 | 4-Oct-16 |
| gBlock CCDC6-RET C1:R12 | IDT | 151004178 | 4-Oct-16 |
| gBlock TRIM33-RET T14:R12 | IDT | 151004179 | 4-Oct-16 |
| RET exon 8 RT Gene Specific Primer | IDT | 150554385 | 28-Sep-16 |
| 5’-CTCCACTCACACCTG-3’ | IDT | 150554385 | 28-Sep-16 |
| RET exon 11 RT Gene Specific Primer | IDT | 150554384 | 28-Sep-16 |
| 5’-GCAAACTTGTGGTAGCAG-3’ | IDT | 150554384 | 28-Sep-16 |
| RET exon 12 RT Gene Specific Primer | IDT | 150554383 | 28-Sep-16 |
| 5’-CTGCCTTTCAGATGGAAG-3’ | IDT | 150554383 | 28-Sep-16 |
| gBlock CD74-ROS1 C6:R34 | IDT | 152324366 | 15-Nov-16 |
| gBlock CD74-ROS1 C6:R32 | IDT | 152324367 | 15-Nov-16 |
| gBlock SDC4-ROS1 S2:R32 | IDT | 152324368 | 15-Nov-16 |
| gBlock SDC4-ROS1 S2:R34 | IDT | 152324369 | 15-Nov-16 |
| gBlock S13del2046-ROS1 S13del2046:R32 | IDT | 152324370 | 15-Nov-16 |
| gBlock S13del2046-ROS1 S13del2046:R34 | IDT | 152324371 | 15-Nov-16 |
| gBlock EZR-ROS1 E10:R34 | IDT | 152324372 | 15-Nov-16 |
| gBlock TPM3-ROS1 T8:R35 | IDT | 152324373 | 15-Nov-16 |
| ROS1 exon 34 RT Gene Specific Primer | IDT | 152704983 | 21-Nov-16 |
| 5’-CCTTCCTTGGCACTTT-3’ | IDT | 152704983 | 21-Nov-16 |
| ROS1 exon 35 RT Gene Specific Primer | IDT | 152704985 | 21-Nov-16 |
| 5’-CTCTTGGGTTGGAAGAGTATG-3’ | IDT | 152704985 | 21-Nov-16 |
| ALK Gene Specific Primer | IDT | 140035422 | 26-Aug-16 |
| 5’-CAGTAGTTGGGGTTGTAGTCG-3’ | IDT | 140035422 | 26-Aug-16 |
| EML4-ALK Cell line pellet | Horizon Discovery | N/A | 11-Jun-15 |
| SLC34A2-ROS1 Cell line pellet | Horizon Discovery | N/A | 11-Jun-15 |
| CCDC6-RET Cell line pellet | Horizon Discovery | N/A | 11-Jun-15 |
| Human Brain Total RNA | Ambion | AM7962 | 1703548 |
| PrimePCR ddPCR Expert Design Assay: K15:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K16:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K22:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K23:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K24:R11 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K24:R8 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: C1:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: T14:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: C6:R34 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: C6:R32 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: S2:R32 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: S2:R34 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: S13del2046:R32 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: S13del2046:R34 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: E10:R34 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: T8:R35 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| (version 2) | Bio-Rad | 12003909 | 213939881 |
| PrimePCR ddPCR Expert Design Assay: ROS1 Multiplex (version 3.2) | Bio-Rad | N/A | 13-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: ROS1 Multiplex (version 3.2) | Bio-Rad | N/A | 20170112v3.2 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 212851151 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 207383915 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 195995635 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 212851152 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 213949301 |
| PrimePCR ddPCR Expert Design Assay: EML4-ALK | Bio-Rad | 12003909 | 20160914 |
| PrimePCR ddPCR Expert Design Assay: EML4-ALK | Bio-Rad | 12003909 | 211383227 |
| Droplet Generation Oil for Probes | Bio-Rad | 186-3005 | 1065C220 |
| Droplet Generation Oil for Probes | Bio-Rad | 186-3005 | 64052953 |
| Droplet Generation Oil for Probes | Bio-Rad | 186-3005 | 64052358 |
| Automated Droplet Generation Oil for Probes (20x96) | Bio-Rad | 186-4110 | 1065C320 |
| Automated Droplet Generation Oil for Probes (20x96) | Bio-Rad | 186-4110 | 64052952 |
| Automated Droplet Generation Oil for Probes (20x96) | Bio-Rad | 186-4110 | 64064127 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000065883 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000084276 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000079928 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000084395 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000084634 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20160627 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20161107 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20161206 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20161216 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20170125 |
| Pipet Tips for Automated Droplet Generator | Bio-Rad | 1864120 | PR125340 |
| DG32 Cartridge for Automated Droplet Generator (10-96 well plates) | Bio-Rad | 186-4108 | 206894 |
| DG32 Cartridge for Automated Droplet Generator (10-96 well plates) | Bio-Rad | 186-4108 | 206893 |
| Pierceable Foil Heat Seal | Bio-Rad | 1814040 | 1409850 |
| Pierceable Foil Heat Seal | Bio-Rad | 1814040 | 100402 |
| Pierceable Foil Heat Seal | Bio-Rad | 1814040 | 145851 |
| Microseal 'B' seals | Bio-Rad | MSB1001 | BR00428490 |
| ddPCR Droplet Reader Oil | Bio-Rad | 186-3004 | 64039089 |
| ddPCR Droplet Reader Oil | Bio-Rad | 186-3004 | 64049253 |
| ddPCR Droplet Reader Oil | Bio-Rad | 186-3004 | 64049255 |
| ddPCR Droplet Reader Oil | Bio-Rad | 186-3004 | 64081870 |
| DNA Lo Bind Tube 0.5 mL | Eppendorf | 22431005 | E1629620 |
| DNA Lo Bind Tube 1.5 mL | Eppendorf | 22431021 | F16698K |
| DNA Lo Bind Tube 2 mL | Eppendorf | 22431048 | E160610I |
| 50 mL Conicals, Polypropylene (25) | Thermo | 339652 | G5ZF5W8118 |
| TempAssure PCR 8-Strips, Optical Caps, Natural, polypropylene (120) | USA Scientific | 1402-4700 | 16202 |
| For Rainin LTS Pipettors 0.5-20 µL tips | Pipette.com | LF-20 | 40155-642C4-642C |
| For Rainin LTS Pipettors 5-200 µL tips | Pipette.com | LF-250 | 40154-642C4-642B |
| Tips LTS 200 ul Filter 960/10 RT-L200F (10 boxes) | Rainin | 17002927 | 1635 |
| Pipet Tips, 10 ul TipOne RPT ultra low retention filter tip refill cassette, sterile | USA Scientific | 1181-3710 | F1175551-1108 |
| Pipet Tips, 10 ul TipOne RPT ultra low retention filter tip refill cassette, sterile | USA Scientific | 1181-3710 | F118054L-1720 |
| Pipet Tips, 100 ul TipOne RPT ultra low retention filter tip refill cassette, sterile (10x96) | USA Scientific | 1180-1740 | 0014961Q-2501 |
| Pipet Tips, 200 ul TipOne RPT ultra low retention filter tip refill cassette, sterile | USA Scientific | 1180-8710 | E116684P-1540 |
| Pipet Tips, 1000 ul XL TipOne RPT ultra low retention filter tip refill cassette, sterile (10x96) | USA Scientific | 1182-1730 | F118815P |
| 5 mL Standard Racked Gilson-fit Reference Tips | Scientific Specialties | 4411-00 | 14312 |
| Combitips advanced, 0.1 mL Biopur | Eppendorf | 003 008 9618 | F165414H |
| Combitips advanced, 0.2 mL Biopur | Eppendorf | 0030 089.626 | F166689J |
| Combitips advanced, 5 mL Biopur | Eppendorf | 0030.089 669 | F166054J |
| Combitips advanced, 50 mL Biopur | Eppendorf | 003.008.9693 | F166055I |
| Reagent Reservoir | VWR | 89094-680 | 141500 |
| Twin tec PCR Plate 96, semi-skirted, Clear | Eppendorf | 951020303 | E163697P |
| Twin tec PCR Plate 96, semi-skirted, Clear | Eppendorf | 951020303 | F165029I |
| Twin tec PCR Plate 96, semi-skirted, Clear | Eppendorf | 951020303 | F165028G |
| Twin tec PCR Plate 96, semi-skirted, Clear | Eppendorf | 951020303 | E163697P |
| Twin tec PCR Plate 96, semi-skirted, Green | Eppendorf | 951020346 | F166183K |
| Equipment Type | Equipment ID | ||
| Analytical Balance | EQP0125 | ||
| Cryogenic Freezer 1, -80oC | EQP0095 | ||
| Refrigerator 6.1 cu ft GP06W1AREF | EQP0139 | ||
| -20oC Freezer | EQP0140 | ||
| Beckman Coulter Microfuge 22R | EQP0025 | ||
| Beckman Coulter Microfuge 22R | EQP0124 | ||
| Thermo Scientific Hereaus Megafuge 8 | EQP0104 | ||
| Mini Centrifuge | EQP0131 | ||
| Mini Centrifuge | EQP0136 | ||
| Mini Centrifuge | EQP0134 | ||
| Mini Centrifuge | EQP0235 | ||
| Mini Centrifuge | EQP0216 | ||
| Thermo Scientific HeraTherm Incubator | EQP0105 | ||
| Pipette 0.1 - 2.5 μL | EQP0182 | ||
| Pipette 0.1 - 2.5 μL | EQP0072 | ||
| Pipette 0.1 - 2.5 μL | EQP0070 | ||
| Pipette 0.5-10 μL | EQP0218 | ||
| Pipette 0.5-10 μL | EQP0075 | ||
| Pipette 0.5-10 μL | EQP0169 | ||
| Pipette 0.5-10 μL | EQP0074 | ||
| Pipette 0.5-10 μL | EQP0147 | ||
| Pipette 2 - 20 μL | EQP0128 | ||
| Pipette 2 - 20 μL | EQP0160 | ||
| Pipette 2 - 20 μL | EQP0018 | ||
| Pipette 2 - 20 μL | EQP0146 | ||
| Pipette 10 - 100 μL | EQP0079 | ||
| Pipette 10 - 100 μL | EQP0181 | ||
| Pipette 10 - 100 μL | EQP0085 | ||
| Pipette 10 - 100 μL | EQP0077 | ||
| Pipette 20 - 200 μL | EQP0088 | ||
| Pipette 20 - 200 μL | EQP0087 | ||
| Pipette 20 - 200 μL | EQP0231 | ||
| Pipette 100 - 1000 μL | EQP0050 | ||
| Pipette 100 - 1000 μL | EQP0158 | ||
| Pipette 100 - 1000 μL | EQP0217 | ||
| Pipette 100 - 1000 μL | EQP0082 | ||
| Pipette 100 - 1000 μL | EQP0183 | ||
| Pipette 100 - 1000 μL | EQP0083 | ||
| Pipette 5 mL | EQP0153 | ||
| Timer | S/N 140623950 | ||
| Hamilton SafeAire VAV Fume Hood | EQP0206 | ||
| Biosafety Cabinet | EQP0205 | ||
| Biosafety Cabinet | EQP0204 | ||
| Qubit 3.0 | EQP0102 | ||
| Benchmark Digital Heat Block | EQP0108 | ||
| Benchmark Digital Heat Block | EQP0231 | ||
| Polaroid Z2300 Instant Print Digital Gel Camera with WiFi and 16GB SDHC memory card | EQP0111 | ||
| Electrophoresis Power Unit | EQP0113 | ||
| Electrophoresis Small Gel Box | EQP0116 | ||
| Maestro Transilluminator | EQP0118 | ||
| Microwave | EQP0215 | ||
| Multichannel 8-well Pipette 2 - 20 μL | EQP0207 | ||
| Multichannel 8-well Pipette 10 - 100 μL | EQP0090 | ||
| Rainin Multichannel 8-well Pipette 50 μL | EQP0094 | ||
| Rainin Multichannel 8-well Pipette 50 μL | EQP0161 | ||
| Rainin Multichannel 8-well Pipette 50 μL | EQP0162 | ||
| Rainin Multichannel 8-well Pipette 50 μL | EQP0163 | ||
| Vortex Genie 2 | EQP0052 | ||
| Vortex Genie 2 | EQP0007 | ||
| Vortex Genie 2 | EQP0132 | ||
| Vortex Genie 2 | EQP0137 | ||
| Vortex Genie 2 | EQP0135 | ||
| Air Clean PCR Workstation | EQP0203 | ||
| Air Clean PCR Workstation | EQP0096 | ||
| Air Clean PCR Workstation | EQP0148 | ||
| Air Clean PCR Workstation | EQP0097 | ||
| QX200 Droplet Generator | EQP0202 | ||
| QX200 Droplet Generator | EQP0121 | ||
| Automated Droplet Generator | EQP0179 | ||
| PX1 PCR Plate Sealer | EQP0123 | ||
| PX1 PCR Plate Sealer | EQP0186 | ||
| C1000 Touch Cycler w/96W FS RM | EQP0120 | ||
| S1000 Cycler w/96W FS RM | EQP0174 | ||
| S1000 Cycler w/96W FS RM | EQP0173 | ||
| T100 Thermal Cycler | EQP0180 | ||
| T100 Thermal Cycler | EQP0175 | ||
| QX200 Droplet Reader | EQP0194 | ||
| QX200 Droplet Reader | EQP0122 |
Références
- Ignatiadis, M., Lee, M., Jeffrey, S. S. Circulating Tumor Cells and Circulating Tumor DNA: Challenges and Opportunities on the Path to Clinical Utility. Clin Cancer Res. 21 (21), 4786-4800 (2015).
- Alix-Panabieres, C., Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. , (2016).
- Paxton, A. Is Molecular AP testing in sync with guidelines. CAP Today. , (2014).
- Sacher, A. G., et al. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. , (2016).
- Sozzi, G., et al. Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol. 21 (21), 3902-3908 (2003).
- Oxnard, G. R., et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 20 (6), 1698-1705 (2014).
- Best, M. G., et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell. 28 (5), 666-676 (2015).
- Rodriguez, M., et al. Different exosome cargo from plasma/bronchoalveolar lavage in non-small-cell lung cancer. Genes Chromosomes Cancer. 53 (9), 713-724 (2014).
- Kalluri, R. The biology and function of exosomes in cancer. J Clin Invest. 126 (4), 1208-1215 (2016).
- Mellert, H., et al. Development and Clinical Utility of a Blood-Based Test Service for the Rapid Identification of Actionable Mutations in Non-Small Cell Lung Carcinoma. J Mol Diagn. 19 (3), 404-416 (2017).
- Qin, J., Williams, T. L., Fernando, M. R. A novel blood collection device stabilizes cell-free RNA in blood during sample shipping and storage. BMC Res Notes. 6, 380 (2013).
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques. 15 (3), 532-537 (1993).
- Kohno, T., et al. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl Lung Cancer Res. 4 (2), 156-164 (2015).
- Takeuchi, K., et al. ROS1 and ALK fusions in lung cancer. Nat Med. 18 (3), 378-381 (2012).
- Rimkunas, V. M., et al. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clin Cancer Res. 18 (16), 4449-4457 (2012).
- Tsuta, K., et al. RET-rearranged non-small-cell lung carcinoma: a clinicopathological and molecular analysis. Br J Cancer. 110 (6), 1571-1578 (2014).
- Forbes, S. A., et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45 (1), 777-783 (2017).
- Salgia, R. Diagnostic challenges in non-small-cell lung cancer: an integrated medicine approach. Future Oncol. 11 (3), 489-500 (2015).
- Cagle, P. T., Raparia, K., Portier, B. P. Emerging Biomarkers in Personalized Therapy of Lung Cancer. Adv Exp Med Biol. 890, 25-36 (2016).
- Vogelstein, B., Kinzler, K. W. Digital PCR. Proc Natl Acad Sci U S A. 96 (16), 9236-9241 (1999).
- Oxnard, G. R., et al. Association Between Plasma Genotyping and Outcomes of Treatment With Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 34 (28), 3375-3382 (2016).
- Reckamp, K. L., et al. A Highly Sensitive and Quantitative Test Platform for Detection of NSCLC EGFR Mutations in Urine and Plasma. J Thorac Oncol. 11 (10), 1690-1700 (2016).
- Yanagita, M., et al. A prospective evaluation of circulating tumor cells and cell-free DNA in EGFR mutant non-small cell lung cancer patients treated with erlotinib on a phase II trial. Clin Cancer Res. , (2016).
- Abbosh, C., et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 545 (7655), 446-451 (2017).
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. BioTechniques. 15 (3), 532-534 (1993).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.