
Method Article
Une plate-forme de Microfluidic ultra haute capacité pour le séquençage du génome de cellules individuelles
Dans cet article
Résumé
Unicellulaire séquençage révèle une hétérogénéité génotypique dans les systèmes biologiques, mais les technologies actuelles n’ont pas le débit nécessaire pour la détermination des caractéristiques profondes de la fonction et la composition de la communauté. Nous décrivons ici un "workflow" microfluidique pour séquençage > 50 000 unicellulaires génomes de diverses populations de cellules.
Résumé
Technologies de séquençage ont subi un changement de paradigme de vrac à cellule unique résolution faisant suite à une compréhension en évolution du rôle de l’hétérogénéité cellulaire dans les systèmes biologiques. Cependant, monocellulaires séquençage de vastes populations a été entravé par limites dans le traitement des génomes de séquençage. Dans cet article, nous décrivons une méthode de séquençage des génomes unicellulaires (SiC-seq) qui utilise des gouttelettes microfluidique à isoler, amplifier et code à barres des génomes de cellules individuelles. Encapsulation de cellules en microgels permet la purification compartimentée et tagmentation de l’ADN, alors qu’une fusion de microfluidique paires efficacement chaque génome avec un code à barres unique oligonucléotide unicellulaires, permettant à > 50 000 cellules individuelles à séquencer par course. Les données de séquençage sont démultiplexage par code à barres, générer des groupes de lectures provenant de cellules individuelles. Une méthode d’unicellulaires séquençage haut-débit et trop faible, SiC-seq afin de permettre à un plus large éventail d’études génomiques destiné aux populations de diverses cellules.
Introduction
Le génome sert comme un modèle d’identité cellulaire et fonction, contenant l’intégralité d’un organisme de codification potentiel. Une compréhension de la biologie cellulaire au niveau du génome peut expliquer la diversité phénotypique observée au sein de populations de cellules hétérogènes. Cette hétérogénéité est apparente dans les systèmes biologiques et a de grandes implications pour la santé humaine et la maladie. Par exemple, gène copie numéros écarts entre les cellules tumorales sont liés à l’évolution et la propagation du cancer1,2. Dans les infections bactériennes, îles de pathogénicité présents dans une petite fraction des génomes horizontalement, peuvent être transférés et conduisent à la prolifération des bactéries résistantes aux antibiotiques3,4. Un défi principal dans l’étude des génomes au niveau unicellulaire est les faibles quantités d’ADN disponible, ainsi que la nécessité d’analyser des milliers de cellules à déguster toute la gamme des génotypes. Pour ces raisons, limitations de débit expérimental ont entravé l’efficacité des études unicellulaires, polarisation des résultats vers les cellules plus abondants. Techniques d’isolement de la cellule unique comme flux tri5,6, pinces optiques7, un enfoncement en vrac gels8et9 de la microfluidique sont capables de traiter des centaines de cellules pour le séquençage ; Toutefois, cela représente seulement une petite fraction de la plupart des échantillons. Une méthode pour le séquençage du génome de la cellule unique avec un débit nettement plus élevé permettrait plus profonde et plus complète de profilage de populations cellulaires, ainsi élucider le rôle de la diversité génotypique au sein de ces communautés.
Gouttelette microfluidique permet la manipulation de haut débit de cellules et de réactifs biologiques dans des millions de réacteurs picolitre. À ce jour, les microgouttelettes technologies ont été utilisées pour étudier l’expression différentielle entre les cellules de tissus hétérogènes10,11,12, profondément des séquences longues molécules13,14 ,15et conduite chromatine immunoprécipitation séquençage (ChiP-seq) analyses monocellules16. En effet, les gouttelettes sont capables d’opérations de haut-débit, compartimentées, ce qui les rend propices aux applications en génomique des cellules individuelles. Le développement de cette technologie présente ses propres défis technologiques uniques, cependant. Cellules doivent être lysées, purifiées et amplifiés avec polarisation minimale, à uniformément les populations de cellules échantillon. En outre, contrairement à polyadénylé transcriptions d’ARNm dans les cellules de mammifères, il n’y a aucun motif moléculaire comparable dans le génome pour faciliter la capture de l’acide nucléique cible. Pour ces raisons, le séquençage du génome de cellules individuelles a été difficile à mettre en œuvre dans les plates-formes de microgouttelettes.
Dans ce travail, nous fournissons un protocole détaillé de notre approche rapportées antérieurement unicellulaires microfluidique capable de séquencer les génomes de dizaines de milliers de cellules dans une seule expérience17. Avec cette technologie, appelée CTI-seq, cellules bactériennes sont encapsulés dans échelle micron hydrogels et individuellement lysées, tagmented et a fusionné avec un microdroplet contenant un code-barres unique oligonucléotide, qui est raccordé sur l’ADN génomique de la cellule via une chevauchement unique extension chaîne par polymérase (PCR). Les hydrogels servent de conteneurs isolés où l’ADN génomique de high-molecular-weight est stériquement enfermé, permettant à des molécules plus petites comme les détergents et enzymes lytiques d’accéder et de purifier l’ADN avant barcoding18. Ce protocole traite > 50 000 cellules uniques en quelques heures, ce qui entraîne une bibliothèque avec code à barres prête pour le séquençage. Après le séquençage, les lectures sont demultiplexed selon leur séquence de barcode unicellulaires, résultant en un ensemble de données composé de millions de lectures, chacun avec un indice de cellulaire.
Protocole
1. Fabrication de dispositifs microfluidiques
- Préparer la microfluidique dessins de masque en utilisant le logiciel de conception assistée par ordinateur (CAO) (fourni dans le. DWG ; Voir Des fichiers complémentaires). A ces conceptions imprimées par le vendeur avec une résolution de 10 µm sur un film de platine.
NOTE : Pour les dispositifs microfluidiques multi-couches, les masques correspondants contiennent des repères. -
Pour chaque périphérique, fabriquer le moule principal SU-8 (Figure 1 a) comme suit.
- Préparer une plaquette de silicium de diamètre de 3 po en versant environ 1 mL de résine photosensible 3025 SU-8 vers le centre de la plaquette. Fixez la plaquette sur la coucheuse spin mandrin en appliquant d’aspiration.
- Voir le tableau 1 pour une liste des épaisseurs de couche et faites tourner les vitesses pour chaque périphérique. Pour tous les périphériques, commencer l’enduction centrifuge avec 30 s à 500 tr/min, suivie de 30 s à la vitesse indiquée.
- Retirer la plaquette de SU-8-silicium de la coucheuse spin et douce cuisson au four sur une plaque chauffante sur 135 ° C pendant 30 min. permettre la gaufrette refroidir à température ambiante après la cuisson.
- Exposer la plaquette de SU-8-silicium avec le masque de la microfluidique approprié sous un collimaté 190 mW, 365 nanomètre UV LED pendant 3 min.
- Après l’exposition, dur de faire cuire la galette sur une plaque chauffante réglée à 135 ° C pendant 1 min. Après cette étape de cuisson, laisser la gaufrette refroidir à température ambiante.
- Pour un dispositif microfluidique seule couche, passez à l’étape 1.2.7. Pour un dispositif microfluidique multi-couches, répétez les étapes 1.2.1 - 1.2.5 pour la deuxième couche de la résine photosensible (Figure 1 b).
- Après la première cuisson dure pour un dispositif simple couche (ou la deuxième cuisson dure pour un dispositif multicouche), développer la plaquette en l’immergeant dans un bain d’acétate d’éther monométhylique propylèneglycol (PGMEA) pendant 30 min.
- Après le développement de la gaufrette, utilisez un vaporisateur contenant le PGMEA pour rincer la plaquette. Rincer ensuite la plaquette avec un vaporisateur contenant isopropanol avant de le placer sur une plaque chauffante de 135 ° C pendant 1 min sécher.
- Placer la plaquette (ci-après dénommée "le maître") dans une boîte de Pétri pour la coulée avec polydiméthylsiloxane (PDMS).
-
Avec le maître préparé à l’étape 1.2, faire effectuer la fabrication de l’appareil avec un casting de PDMS.
- Préparer le PDMS en combinant une base de silicone avec un adjuvant de salaison dans un rapport de 11:1 en masse. Mélanger à la main la base de silicone et de salaison avec un bâton de remuer.
- Dégazer le PDMS en plaçant dans une chambre de dégazage et en appliquant un vide. Permettre le PDMS à dégazer jusqu'à ce que les bulles d’air ne sont plus visibles (en général 30 min).
- Verser délicatement le PDMS dégazé sur le maître, pour une épaisseur finale de PDMS-couche d’environ 5 mm. Degas le PDMS à nouveau pour assurer l’élimination des bulles d’air.
- Après le dégazage, faites cuire le PDMS et le maître à 80 ° C pendant 80 minutes.
- L’accise soigneusement la dalle PDMS guérie du maître au four à l’aide d’une lame de rasoir. S’assurer que toutes les découpes sont sur le dessus de la plaquette de silicium.
Remarque : Toutes les découpes faites au large de la plaquette de silicium peuvent entraîner une lèvre empêchant un collage uniforme. - Perforez les entrées et sorties à l’aide d’un poinçon de biopsie de 0,75 mm. Enlever toute poussière et PDMS errants à l’aide d’un ruban d’emballage sur le côté de la fonctionnalité de l’appareil.
- Avant plasma traitant de l’appareil, nettoyer une lame de verre de 50 mm x 75 mm en rinçant avec de l’isopropanol et asséchant.
- Pour le traitement du plasma, mettre la lame de verre et la dalle PDMS dans le bonder de plasma avec les fonctionnalités vers le haut. Exécuter le traitement par plasma à l’aide de 1 mbar O2 plasma pour 1 min. Bond l’appareil sur le verre glisser en apportant les exposés ou relever, côtés ensemble.
- Suite au traitement du plasma, cuire l’appareil à 80 ° C pendant 40 min.
- Enfin, injecter liquide de traitement de surface de verre dans l’une prises pour rendre les canaux microfluidiques hydrophobes de. S’assurer que tous les canaux sont complètement inondés de la solution et renouveler l’injection pour chaque dropmaker. Faire cuire l’appareil traitée à 80 ° C pendant 10 min évaporer l’excès de solvant.
2. encapsulation des cellules en gel d’Agarose Microgels
Remarque : Voir la Figure 2 a.
- Préparer 1 mL de 3 % p/v bas point de fusion température d’agarose dans 1 x tampon Tris-EDTA (TE). Conserver la solution d’agarose sur un bloc de chaleur de 90 ° C jusqu'à ce que juste avant la seringue de chargement.
-
Préparer la suspension cellulaire.
Remarque : Le présent protocole et ses dispositifs microfluidiques associées ont été validés pour travailler avec des cellules bactériennes, soit d’un stock congelée ou fraîche préparation. Les cellules de mammifères, selon le type de cellule, peuvent exiger un ajustement des dimensions canal microfluidique pour accueillir les grandes tailles de cellule.- Remettre en suspension les cellules dans 1 mL de solution saline tamponnée au phosphate (PBS).
- Compter les cellules dans un hémocytomètre ou en triant les flux. Pour 25 µm microgels avec un taux d’encapsulation de cellule cible de 1 à 10, préparer 1 mL de suspension de cellules à une concentration finale de 2. 4 x 107 cellules/mL.
- Faites tourner les cellules vers le bas à 3 000 x g pendant 3 min. aspirer le surnageant et Resuspendre le culot dans 1 mL de milieu dégradé de 17 % v/v densité (voir Table des matières) dans du PBS. Gardez-le sur la glace jusqu'à ce que le chargement de la seringue.
- Charger une seringue de 3 mL avec de l’huile fluorés (HFE) contenant un agent de surface 2 % w/w perfluoropolyéthers-polyéthylène glycol (PEG-PFPE), fixez-le à l’aide d’une aiguille de 27 G et placez-le dans un pousse-seringue.
Nota : Placer toutes les seringues avec une aiguille de 27 G pour la microfluidique étapes dans le présent protocole. Pour réduire le risque de piqûre accidentelle, garder casquettes sur toutes les aiguilles jusqu'à ce que le fonctionnement de la pompe commence. - Charger la suspension cellulaire et l’agarose fondu en seringues de 1 mL, s’adapter à tous les deux avec des aiguilles de calibre 27 et placer ces derniers dans les pompes à seringue.
- Réchauffer la seringue de gel d’agarose et la pompe avec un petit radiateur pour empêcher la gélification dans la seringue et le tuyau d’arrivée de l’agarose. L’appareil de chauffage trop élevé et positionnez-le de sorte que la surface de chauffe est d’environ 10 cm de la seringue. S’assurer que la température mesurée à la seringue est environ 80 ° C.
Remarque : Les utilisateurs sont conseillés pour maintenir l’appareil à la distance recommandée de l’appareil de pompage pour réduire le risque de dommages matériels, y compris la fusion de la tubulure. -
Générer 25 µm microgel gouttes à l’aide de l’appareil de dropmaking de débit Co.
Remarque : Voir la Figure 3 a pour un dispositif schématique indiquant l’emplacement des entrées de réactif et prise de courant.- Connecter les aiguilles de seringue pour les piquages des dispositifs microfluidiques à l’aide de morceaux de tuyaux en polyéthylène (PE). Avant d’insérer les tubes dans le dispositif, amorcer la pompe pour enlever l’air de la conduite.
- Raccorder un bout de tube de sortie d’et placer l’extrémité libre dans un tube de prélèvement de 15 mL.
- Utiliser les taux de débit (recommandé) suivant pour dropmaking : 800 µL/h pour le HFE 2 % w/w PFPE-PEG ; 200 µL/h pour la suspension de cellules dans du PBS ; et 200 µL/h pour le 3 % p/v d’agarose.
- Après le dropmaking, placer le tube de prélèvement à 4 ° C pendant 30 min assurer la complète gélification de l’agarose.
3. rupture et laver l’Agarose Microgels
- Retirer le tube de prélèvement à l’aide d’une seringue de 3 mL munie d’une aiguille de 20 G, en prenant soin de ne pas pour perturber la couche supérieure des gouttelettes d’agarose de la couche inférieure d’huile.
-
Briser les émulsions avec perfluorooctanol (PFO).
- Ajouter 1 mL de 10 % v/v PFO dans HFE dans les gouttes de gel d’agarose. Pipette de cette solution de haut en bas pendant 1 min pour bien enrober les émulsions.
Remarque : Pour tous le microgel laver étapes, pipette les solutions avec une pointe de 1 000 µL. La suspension microgel devrait apparaître homogène et exempt de touffes après pipetage il. - Tourner le tube conique à 2 000 x g pendant 1 min recueillir les microgels d’agarose. Retirez le surnageant PFO/HFE par aspiration ; les microgels sont désormais libres de leur couche de surfactant et apparaîtra claires.
- Ajouter 1 mL de 10 % v/v PFO dans HFE dans les gouttes de gel d’agarose. Pipette de cette solution de haut en bas pendant 1 min pour bien enrober les émulsions.
-
Laver le microgels avec un tensioactif dans l’hexane.
ATTENTION : Hexane est un solvant organique volatil, et le lavage à l’étape 3.3 devrait être mené sous une hotte.- Ajouter 2 mL de sorbitane monooléate non ioniques tensioactif de 1 % v/v (voir Table des matières) dans l’hexane de la microgels d’agarose. Pipette de haut en bas de 10 x à mélanger, assurant la séparation complète de la microgel-pellet.
- Tourner le tube à 1 000 x g pendant 1 min recueillir les microgels. Aspirer le surnageant pour enlever la solution de tensioactif/hexane.
- Répéter le lavage de surfactant/hexane.
-
Laver le microgels dans un tampon aqueux pour enlever n’importe quel solvant organique résiduelle.
- Ajouter 5 mL de tampon de TET [0,1 % v/v octylphénol éthoxylé un détergent non ionique (voir Table des matières) dans 1 x TE] au tube conique. Pipette de haut en bas de 10 x à mélanger.
- Tourner le tube conique à 2 000 x g pendant 2 min recueillir les microgels. Aspirer le surnageant pour retirer le tampon TET.
- Répétez la TET laver 2 x.
- Ajouter 5 mL de tampon de TE 1 x dans le tube conique. Pipette de haut en bas de 10 x à mélanger.
- Tourner le tube conique à 2 000 x g pendant 2 min recueillir les microgels. Aspirer le surnageant pour retirer le tampon TE.
- Répéter le lavage TE.
- Vérifier l’encapsulation de cellules dans la microgels sous un microscope à un grossissement de X 400 par coloration une quantité de 10 µL de gels avec 1 x nucleic acid stain (voir Table des matières).
Remarque : Microgels apparaîtra clairement dans le canal transparent tout en cellule QU'ADN sera une fluorescence sous la moulure de la GFP (longueurs d’onde 497/520 excitation/émission nm). Une superposition de couches transparentes et fluorescents montrera les cellules dans le microgels comme illustré dans la Figure 4 a.
4. la lyse des cellules en gel d’Agarose par l’intermédiaire des Enzymes lytiques
- Préparer 1 mL d’une enzyme lytique cocktail à l’aide de 800 µL de tampon TE (1 x), 2 µL d’enzyme lytique de la levure (5 U/µL cartonné, 10 U/mL final) 30 µL de dithiothréitol (stock 1 M, 30 mM de finale), 60 mg de lysozyme (poudre lyophilisée), 15 µL de l’EDTA (0,5 M stock final 7,5 mM), 2 µL de mutanolysine (stock de 100 U/µL, 200 U/mL final), 2 µL de lysostaphine (stock de 10 U/µL, 20 U/mL final) et 30 µL de NaCl (stock 1 M, 30 mM final).
- Ajouter TE supplémentaire pour porter le volume à 1 mL. Mélanger la solution au vortex.
- Ajouter l’ensemble 1 mL de la solution d’enzyme lytique à pas plus de 1 mL de la microgels lavé. La composition de pipetage x 10. Incuber le mélange à 37 ° C pendant > 2 h dans un shaker (le maximum est l’incubation durant la nuit).
5. base de détergent Microgel traitement
-
Suite à la lyse par l’intermédiaire d’enzymes lytiques (étape 4), laver le microgels.
- Tournez en bas la microgels à 2 000 x g pendant 2 min et aspirer le surnageant. Les gouttelettes apparaîtra blanc opaque à cause de débris de cellules dispersées dans le culot.
- Resuspendre le microgels dans 5 mL de tampon Tris-HCl 10 mM et pipette de haut en bas de 10 x à mélanger.
- Faites tourner le mélange vers le bas à 2 000 x g pendant 2 min, puis retirez le surnageant par aspiration.
-
Effectuer un traitement de détergent sur le microgels.
- Les gels de remettre en suspension dans un tampon de lyse au lithium dodécyl sulfate (couvercles) (0,5 % p/v couvercles en 20 mM Tris-HCl) et 60 µL d’EDTA 0,5 M pour un volume final de 3 mL.
- Ajouter 5 µL de l’enzyme protéinase K (800 U/mL de bouillon). Pipette de haut en bas de 10 x à mélanger, puis incuber le mélange à 42 ° C sur un bloc chauffant pendant 1 h à solubiliser les membranes cellulaires et à digérer les protéines.
-
Suite au traitement de détergent, laver la microgels.
- Tourner le tube à fond conique avec le microgels vers le bas à 2 000 g pendant 2 min. Retirez le surnageant par aspiration.
- Laver le microgels avec 10 mL de polysorbate de 2 % v/v 20 dans l’eau. Pipette de haut en bas de 10 x à mélanger.
- Faites tourner le tube à fond conique vers le bas à 2 000 x g pendant 2 min, puis retirez le surnageant par aspiration.
- Laver le microgels avec 10 mL d’éthanol à 100 % à inactiver tout enzymatique résiduelle. Pipette de haut en bas de 10 x à mélanger.
- Faites tourner le tube à fond conique vers le bas à 2 000 x g pendant 2 min, puis retirez le surnageant par aspiration.
- Laver le microgels avec 10 mL de polysorbate de 0,02 % v/v 20 dans l’eau. Pipette de haut en bas de 10 x à mélanger.
- Faites tourner le tube à fond conique vers le bas à 2 000 x g pendant 2 min, puis retirez le surnageant par aspiration.
- Répéter le polysorbate 20 laver 3 x. Passer la solution à travers un tamis de cellule de 100 µm avant le dernier lavage pour enlever les grosses touffes.
- Resuspendre le microgels dans 5 mL de tampon Tris-HCl 10 mM à prévenir la dégradation de l’ADN. Les microgels peuvent être conservés à 4 ° C pendant 1 semaine avant la tagmentation (étape 7).
6. générer des gouttelettes de codes à barres par PCR numérique
- Préparation d’un stock de 500-pM de BAR apprêt (tableau 2) 1 x tampon TE dans un tube de faible-lier. Avant chaque utilisation, diluer l’amorce d’un stock de travail de 13:00 et chauffer à 70 ° C pendant 1 min sur un bloc chauffant.
- Préparer un mélange 150 µL de la réaction PCR à l’aide de 75 µL de démarrage à chaud haute fidélité master mix (voir Table des matières) (x 2), 42 µL d’eau PCR-grade, 3 µL d’apprêt DNA_BAR (10 µM stock, finale de 0,2 µM), 3 µL de P7_BAR primer (stock µM 10 Final de 0,2 µM), 6 µL de la dilution de codes à barres (stock de 13:00, final 40 fM), 6 µL de polysorbate 20 (50 % v/v stock, 2 % final) et 15 µL de PEG 6 k (50 % p/v stock, 5 % final). La composition de pipetage et descendre de 10 x.
- Préparer une seringue HFE-soutenu par le dessin 200 µL de HFE huile dans une seringue et fixez-le à l’aide d’une aiguille. Attacher une section de tube PE à l’aiguille et amorcer la ligne à la main. Insérez l’extrémité du tube PE dans la solution cible et tracer soigneusement tous les 150 µL du mélange PCR dans les tubes PE et la seringue. Charger la seringue dans un pousse-seringue.
- Charger une seringue de 1 mL avec de l’huile fluorés (HFE) contenant un agent de surface 2 % w/w perfluoropolyéthers-polyéthylène glycol (PEG-PFPE), fixez-le avec une aiguille et placez-le dans un pousse-seringue.
-
Générer 25 µm barcode gouttes à l’aide de l’appareil de dropmaking de débit Co.
Remarque : Voir la Figure 3 a pour un dispositif schématique indiquant l’emplacement des entrées de réactif et prise de courant.- Brancher l’entrée de cellules d’un dispositif de dropmaker flux Co avec un petit morceau de plomb.
- Connecter les seringues chargés de HFE et PCR mélanger pour les piquages des dispositifs microfluidiques à l’aide de morceaux de tubes de PE, à l’aide de l’entrée de la Fusion d’Agarose pour le code à barres PCR. Avant d’insérer les tubes dans le dispositif, amorcer la pompe pour enlever l’air de la conduite.
- Raccorder un bout de tube de sortie d’et placer l’extrémité libre dans un tube de PCR de 0,2 mL. Utiliser les taux de débit (recommandé) suivant pour dropmaking : 600 µL/h pour le HFE 2 % w/w PFPE-PEG et 200 µL/h pour le mélange PCR. Recueillir les gouttes dans les tubes PCR avec environ 50 µL de gouttes dans chaque tube.
- Après le dropmaking, retirer délicatement la couche inférieure de l’huile HFE de l’émulsion à l’aide de gel-chargement embouts et remplacez-le par FC-40 fluorés huile contenant un surfactant PFPE-PEG de 5 % w/w. Cycle thermique avec le protocole suivant : 98 ° C pendant 3 min, 40 x (98 ° C pendant 10 s, 62 ° C pendant 20 s, 72 ° C pendant 20 s), 72 ° C pendant 5 min et puis maintenez à 12 ° C.
Remarque : Les gouttelettes sous vélo thermique peuvent être conservés à 4 ° C jusqu'à 1 nuit. -
Vérifier le taux d’amplification et l’encapsulation de code à barres.
- Préparer un acide nucléique de 1 x tache (voir Table des matières) dans HFE avec un tensioactif de PFPE-PEG 2 % w/w ; la tache est marginalement miscible dans HFE et créera une liaison à l’ADN dans les gouttelettes.
- Ajouter 1 µL d’émulsion de code-barres thermique-pédalé à 10 µL de coloration de l’huile. Les incuber pendant 5 min à température ambiante.
- Gouttelettes d’image par microscopie fluorescente (GFP channel, longueurs d’onde de 497/520 nm excitation/émission) à un grossissement X 200 et compte le taux d’encapsulation de code à barres. Notez que le signal sera discret : les gouttelettes contenant les codes à barres amplifié seront réagissent en couleurs vives, alors que les gouttes vides apparaîtra sombres (Figure 4 b).
7. Tagmentation de l’ADN génomique dans les gouttelettes
Remarque : Voir la Figure 2 b.
- Préparer 500 µL de solution de tagmentation à l’aide de réactifs d’une préparation de bibliothèque de séquençage de la prochaine génération de kit (voir Table des matières). Utilisez 7 µL d’enzyme tagmentation, 250 µL de tampon de tagmentation et 243 µL d’eau PCR-grade. Mélangez-les au vortex et actionner le mélange vers le bas pour recueillir. Charger cette solution dans une seringue à huile dos 1 mL HFE et fixez-le à l’aide d’une aiguille.
-
Préparer le microgels pour la réinjection.
- Tournez en bas du tube de microgel pendant 2 min à 2 000 x g et aspirer le surnageant. Transférer 200 µL de gels en haut d’une seringue HFE-soutenu avec une pointe de pipette gel-chargement et sceller la buse avec un petit morceau de ruban adhésif.
- En utilisant un adaptateur 3D-imprimé centrifugeuse (voir supplémentaire fichiers 1 et 2), faire tourner la seringue microgel pendant 3 min à 3 000 x g.
- Retirer le liquid surnageant de la seringue à l’aide d’une pointe de pipette gel-chargement. Poussez la couche microgel à la base de l’embout de la seringue. Adapter la seringue avec une aiguille.
- Charger une seringue de 3 mL avec de l’huile fluorés (HFE) contenant un agent de surface 2 % w/w perfluoropolyéthers-polyéthylène glycol (PEG-PFPE), fixez-le avec une aiguille et placez-le dans un pousse-seringue.
- Ré-encapsuler le microgels en gouttelettes contenant des réactifs tagmentation.
Remarque : Voir la Figure 3 b pour un dispositif schématique indiquant l’emplacement des entrées de réactif et prise de courant.- Connecter les seringues contenant HFE, mélange de tagmentation et microgels pour les piquages des dispositifs microfluidiques à l’aide de morceaux de tubes PE. Avant d’insérer les tubes dans le dispositif, amorcer la pompe pour enlever l’air de la conduite.
- Raccorder un bout de tube de sortie d’et placer l’extrémité libre dans une seringue de 1 mL vide avec le plongeur attiré sur la ligne de 1 mL.
- Utiliser les taux de débit (recommandé) suivant pour dropmaking : 2 000 µL/h pour le HFE 2 % w/w PFPE-PEG 200 µL/h pour le microgels et 500 µL/h pour le mélange de tagmentation.
- Vérifier le taux d’encapsulation microgel sous un microscope optique avec un grossissement de X 400. Environ 80 à 90 % des gouttes doit contenir une microgel, comme illustré à la Figure 4.
- Adapter la seringue contenant les émulsions de tagmentation avec une aiguille et incuber il verticalement dans un bloc chauffant ou au four pendant 1 h à 55 ° C à fragmenter l’ADN génomique.
8. unicellulaires Barcoding de Microfluidic Double fusion
Remarque : Voir la Figure 2.
- Préparer les gouttelettes de codes à barres pour la fusion, en remplaçant la fraction d’huile FC-40 par HFE 2 % w/w PFPE-PEG. Soigneusement transférer ces gouttes dans une seringue de 1 mL, fixez-le à l’aide d’une aiguille et placez-le dans un pousse-seringue.
- Charger la seringue de gouttelettes microgel couvée et tagmented dans un pousse-seringue.
- Préparer 500 µL du mélange PCR. Ajouter les réactifs dans l’ordre suivant pour prévenir la formation de précipités : 140 µL d’eau PCR-grade, 10 µL de P5_DNA primer (10 µM stock, finale de 0,2 µM), 10 µL de P7_BAR primer (10 µM stock, finale de 0,2 µM), 50 µL de PEG 6k (stock 50 % p/v final de 5 %), 50 µL de polysorbate 20 (50 % v/v stock, 5 % final), 250 µL du mélange réactionnel Taq (x 2) (voir la Table des matières), 10 µL de polymérase isotherme (voir Table des matières) et 10 µL de tampon de neutralisation (voir Table des matières). Mélangez-les de pipetage et descendre de 10 x et tournez le mélange en bas de la pour percevoir. Charger cette solution dans une seringue de 1 mL dos HFE, fixez-le à l’aide d’une aiguille et placez-le dans un pousse-seringue.
- Charge trois 3 mL seringues avec huile fluorés (HFE) contenant un 2 % w/w perfluoropolyéthers-polyéthylène glycol (PEG-PFPE) agent tensio-actif, chacun avec une aiguille monter et rangez-les dans pompes à seringue.
- Charger trois seringues de 1 mL avec 2 M NaCl, correspondent chacun avec une aiguille et mettez-les de côté.
-
Fusionner les gouttelettes de codes à barres, gouttelettes de tagmented de génome, et PCR mélanger en utilisant le dispositif de double fusion.
Remarque : Voir la Figure 5 pour un dispositif schématique indiquant l’emplacement du réactif et électrode entrées et sortie.- Connecter les 3 seringues de NaCl à 2 entrées d’électrode et les douves unique inletusing des tubes PE. Pour les électrodes, appuyer sur les seringues manuellement jusqu'à ce que les électrodes sont complètement remplis de solution saline. Après avoir rempli les électrodes, s’appliquent à pression manuelle sur la seringue de douves jusqu'à ce que le fossé est rempli. Branchez l’extrémité libre de la douve avec un petit morceau de plomb.
- Connecter les 3 seringues HFE montés sur les pompes pour les 2 prises d’huile entretoise et dropmaking huile inletusing morceaux de tubes PE. Avant d’insérer les tubes dans le dispositif, amorcer la pompe pour enlever l’air de la conduite.
- Connecter la seringue de mélange PCR, microgel dépose seringue et barcode tombe seringue à leurs entrées respectives à l’aide de tubes PE. Tirer toutes les gouttelettes réinjection du tube avec un pistolet antistatique avant de les raccorder à des aiguilles de seringue ; le traitement antistatique réduit le risque de la coalescence de gouttelettes induite par des charges électrostatiques sur les tubes de PE.
- Raccordez un bout de tube PE à la sortie et placer l’extrémité libre dans un tube de PCR de 0,2 mL.
- Se connecter à l’aiguille de la seringue de l’électrode à un onduleur fluorescent à cathode froide à l’aide d’une pince crocodile. Alimentation de l’onduleur DC la valeur 2 V.
- Exécutez le dispositif de double fusion avec les débits recommandés : 300 µL/h pour les gouttes de microgel tagmented, 100 µL/h pour les gouttes de codes à barres, 1500 µL/h pour le HFE 2 % w/w PFPE-PEG (dropmaking de pétrole), 600 µL/h pour l’amplification mix, 200 µL/h pour les (HFE 2 % w/w PFPE-PEG huile d’entretoise à code à barres) et 700 µL/h pour le HFE 2 % w/w PFPE-PEG (huile de microgel entretoise). Recueillir les gouttes dans des tubes PCR avec environ 50 µL d’émulsion dans chaque tube.
- Avant le cyclage thermique, retirer délicatement la couche inférieure de l’huile HFE de l’émulsion à l’aide de gel-chargement embouts et remplacez-les par FC-40 fluorés huile contenant un surfactant PFPE-PEG de 5 % w/w. Cycle thermique avec le protocole suivant : 65 ° C pendant 5 min, 95 ° C pendant 2 min, 30 x (95 ° C pendant 15 s, 60 ° C pendant 1 min, 72 ° C pendant 1 min), 72 ° C pendant 5 min, puis maintenez à 12 ° C.
-
Récupérer l’ADN avec code à barres de gouttelettes sous vélo thermique.
- Centraliser les gouttelettes dans un tube de microcentrifuge et briser les émulsions à l’aide de 20 µL de PFO. Vortex pour 10 s pour mélanger.
- Tourner le tube à 10 000 x g pendant 1 min de fractionner le mélange en solution aqueuse (en haut) et phases d’huile (en bas). Avec précaution, retirer la couche aqueuse supérieure du tube à l’aide d’une pipette et transférez-le dans un nouveau tube de microcentrifuge. Jeter la phase huileuse.
- Purifier le produit PCR avec code à barres dans une colonne selon le protocole du fabricant et il éluer dans 20 µL de tampon de TE 1 x.
- Passez à la réalisé le séquençage et l’analyse selon les étapes 9 et 10.
9. séquençage et préparation de bibliothèque
- Préparer la bibliothèque de cellules individuelles pour le séquençage en suivant les protocoles du fabricant pour sélection taille de fragment et la quantification.
- Séquence de la bibliothèque de bout jumelé avec chimie par défaut pour lire 1 et 2 de la lecture. Utiliser le primaire de 1 Index personnalisé I7_READ (tableau 2) pour un index de 15-bp lu, correspondant pour le code à barres de cellule unique.
10. analyse des données de cellule unique
Remarque : Les scripts Python personnalisé pour le contrôle de la qualité et l’analyse préliminaire des données de SiC-seq peuvent être téléchargés de https://www.github.com/AbateLab/SiC-seq.
- Exécutez le script « barcodeCleanup.py » pour effectuer un contrôle qualité sur les lectures avec code à barres et d’exporter les données de cellule unique dans une base de données SQLite. Pour une expérience de contrôle, utiliser ce script avec le «-align » indicateur est défini pour aligner les lectures et les génomes de référence connue.
- Analyser la pureté des groupes de codes à barres (pour une expérience de contrôle) en utilisant le script « purity.py » et de confirmer les valeurs de haute pureté conformes à la Figure 6 b.
Résultats
Le flux de travail expérimental de SiC-seq contient 3 Dispositifs microfluidiques PDMS fabriquées à l’aide d’une procédure de lithographie douce (Figure 1). Un flux de co dropmaker (Figure 3 a) génère 25 µm de gouttelettes de code-barres numérique pour le marquage de l’ADN génomique avec un identificateur unique de la cellule unique. Les oligonucléotides barcode consistent en une séquence dégénérée bp 15, flanqué de poignées PCR amplification (tableau 2, BAR amorce). Les codes à barres sont diluées à une concentration de femtomolar pour atteindre l’encapsulation de molécules simples, et toutes les gouttelettes recevoir des fragments de code à barres soit 0 ou 1. Les gouttelettes contenant un code à barres sont amplifiés, ce qui donne de nombreuses copies des amplicons barcode bicaténaire. Une tache d’acide nucléique est utilisée pour vérifier l’amplification réussie et de quantifier le taux d’encapsulation des fragments de code à barres (Figure 4 b). Les microgels sont générés par co coulant une suspension de cellules bactériennes et un gel d’agarose fondus à des débits égaux (Figure 2 a). L’agarose est prête à deux fois la concentration finale souhaitée, que le processus de dropmaking de flux co dilue efficacement les solutions aqueuses d’un facteur 2. L’agarose se refroidissant, elle se solidifie en une 25 µm de diamètre microgel occupant le volume sphérique de la goutte.
Une série d’étapes de lavage et lyse purifie l’ADN génomique de high-molecular-weight dans la microgels (Figure 2 b). Après la rupture de l’émulsion, lavages aqueux sont réalisées en grandes quantités pour diluer la trace des solvants organiques qui peuvent inhiber les traitements enzymatiques en aval. Les microgels lavés sont observés au microscope afin de vérifier le taux d’encapsulation de cellules (Figure 4 a). Un cocktail d’enzymes ayant une activité lytique large est ajouté à la suspension de microgel à digérer les parois cellulaires des bactéries et microbes eucaryotes19. Un second traitement avec la protéinase K et détergent dégrade les protéines et solubilise les débris cellulaires.
Tagmentation de l’ADN purifié est effectuée dans les gouttelettes pour éviter la contamination croisée potentielle résultant de la diffusion des fragments d’ADN de petits tagmented entre les microgels18. Un dispositif d’encapsulation de gouttelettes (Figure 3 b) compartimente chaque microgel avec une enzyme de tampon et tagmentation, qui en même temps des fragments ADN bicaténaire tout en également « tagging » avec un oligonucléotide préchargées20. Les microgels sont chargés dans les gouttelettes sous forme de particules compacte, encapsulation taux approchant 1 microgel pour chaque goutte avec quelques doublets21 (Figure 4).
Dans la dernière étape du flux de travail microfluidique (Figure 2), un appareil effectue une opération de double fusion combinant 1goutte de codes à barres et 1goutte microgel contenant le mélange d’amplification dans un processus en deux étapes contrôlées. Tout d’abord, une gouttelette contenant les réactifs PCR est jumelée et fusionnée avec une goutte de code-barres dans la région indiquée en jaune (Figure 5). Électrodes d’eau salée dans le canal microfluidique produisent un gradient de champ électrique élevé qui déclenche la fusion de gouttelettes. De manière similaire, la première goutte fusionnée est jumelée avec une gouttelette microgel et fusionnée une seconde fois dans la région en rouge. Les gouttelettes sont collectés et thermique vélo hors puce dans une extension simple chevauchement (SOE) PCR. Les extrémités complémentaires qui se chevauchent de la code à barres et l’ADN génomique de tagmented permettent de fusion et amplification exponentielle d’à bon droit les constructions avec code à barres.
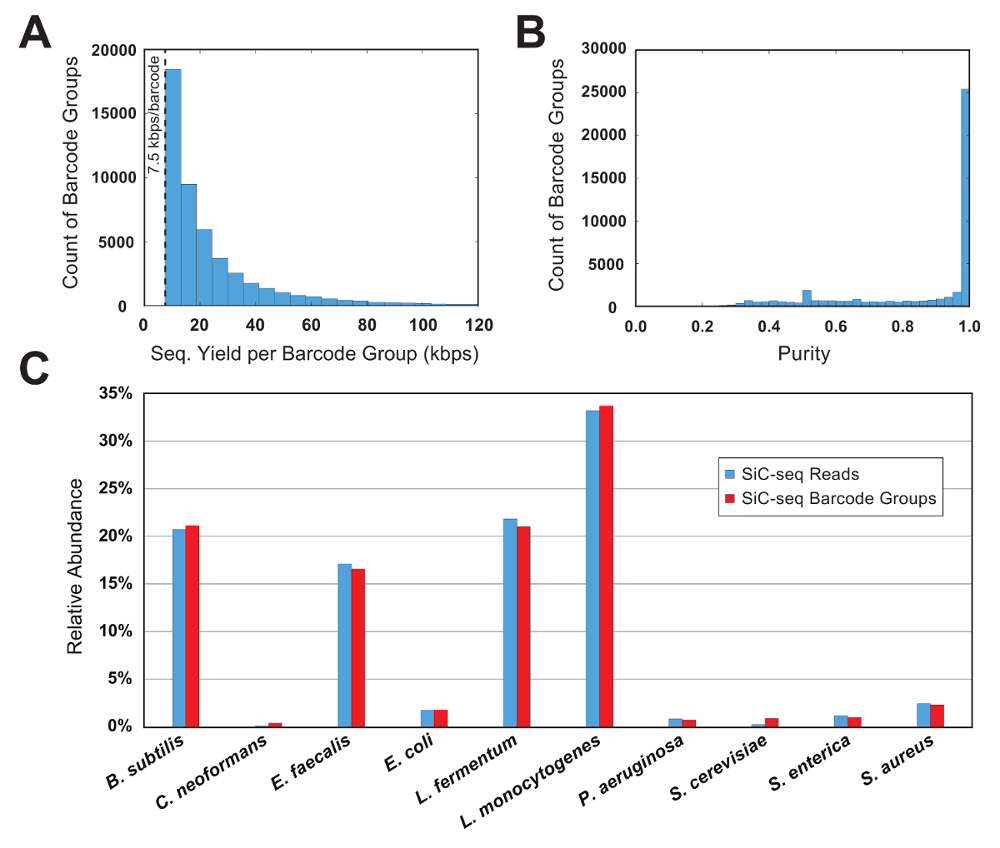
Les données de séquençage sont d’abord filtrées par une qualité de lecture et puis analysées en groupant les lectures selon leur ordre de 15-bp unicellulaires barcode. Pour un groupe de codes à barres soit considérée valide, il doit contenir un nombre minimal de lectures ; ce seuil limite l’analyse aux cellules avec un volume utile de données de séquençage et supprime le code à barres muté PCR « orphelins » de l’objet dataset. Dans cet exemple est exécuté, le minimum est défini sur 7,5 KB/s par groupe (50 lectures de 150 bp chaque). Un histogramme des comtes code-barres par rapport à la taille du groupe montre qu’une partie importante des groupes de code à barres valide est juste au-dessus de la taille de seuil (Figure 6 a).
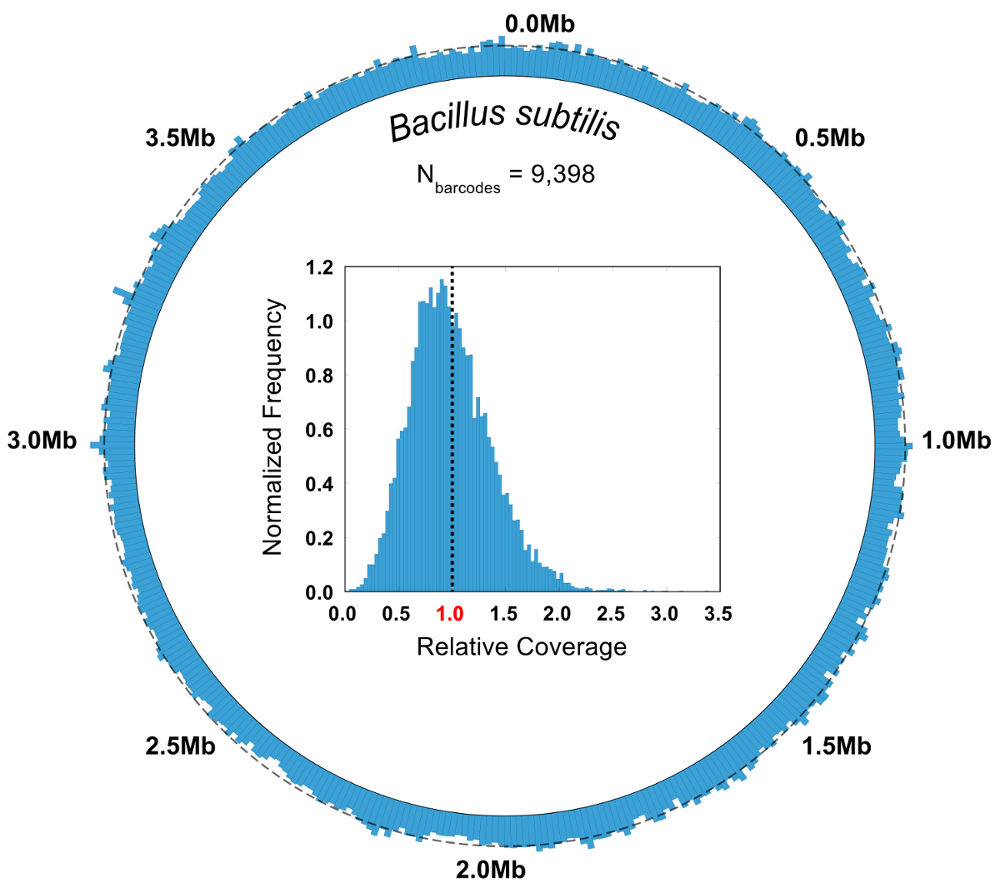
Dans une expérience de contrôle où la composition de la communauté microbienne est connue, la pureté et la métrique de l’abondance relative est utilisés pour évaluer la qualité d’un SiC-seq exécuter. Ici, une communauté de 10 éléments synthétique consistant en 3 bactéries Gram-négatives, 5 bactéries Gram-positives et 2 levures est analysée. La pureté d’un groupe donné de code à barres est définie comme le nombre de lectures de cartographie au génome plus courant dans le groupe divisé par le nombre total de lectures dans le groupe. La grande majorité des groupes de codes à barres ont des degrés de pureté supérieures à 0,95 (Figure 6 b). L’abondance relative des types de cellules est calculé en comptant les lectures brutes et en comptant les groupes de codes à barres, où les groupes reçoivent un type de cellule correspondant au consensus de ses lectures de membre (Figure 6). Suivi de l’abondance des lectures et des groupes de codes à barres dans des proportions à peu près égales, ce qui indique que les populations de cellules sont échantillonnées telle que certaines espèces ne sont pas contenus dans barcode anormalement petits ou grands groupes. Traçant la couverture globale de tous les groupes de codes à barres d’une seule espèce indique un taux de couverture élevé au sein du génome entier, avec peu ou pas les régions d’abandon (Figure 7). L’uniformité de la couverture peut être vérifiée avec une distribution de fréquence des valeurs normalisée de couverture, avec la plupart des valeurs centrées autour de la moyenne (Figure 7, encart).

Figure 1 : Fabrication des dispositifs microfluidiques par photolithographie. (A) maître moules avec une hauteur de fonctionnalité unique sont fabriquées par spin enduit d’une couche de résine photosensible SU-8 sur une plaquette de silicium. La résine photosensible est modelée ensuite avec un masque photolithographique et les rayons UV, réticulation l’exposé SU-8. Enfin, non SU-8 est dissoute dans un bain de développeur. Le moule qui en résulte est utilisé pour effectuer un cast PDMS qui est collé sur une lame de verre pour produire du dispositif microfluidique complet. ()B) d’un dispositif de double-couche, la fabrication commence de la même façon avec mesures de revêtement et exposition de spin. Ces étapes sont répétées puis créer un dispositif de deux couches. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
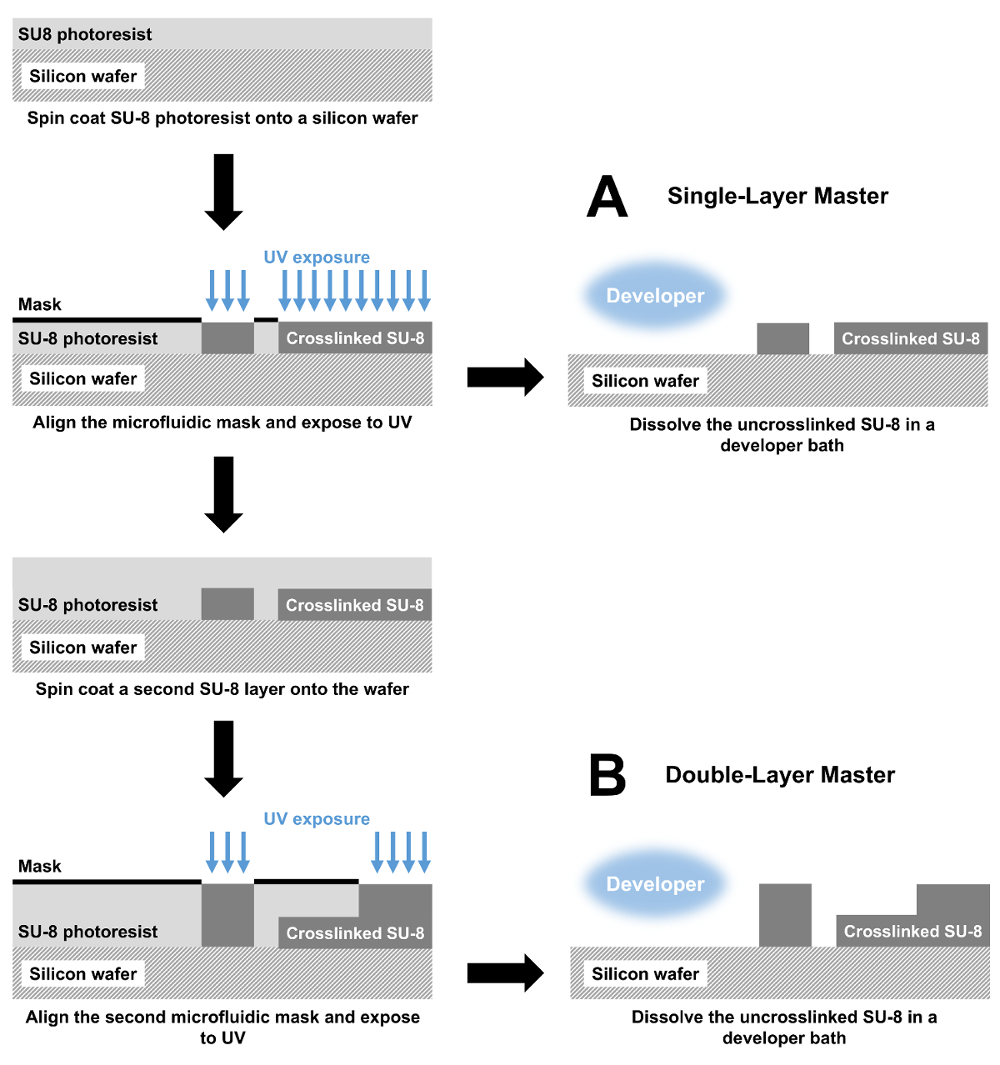{kind=link}

Figure 2 : Vue d’ensemble du workflow SiC-seq. (A) A suspension microbienne est passée conjointement avec agarose fondu dans un dispositif dropmaker pour encapsuler des cellules individuelles en microgels. (B) les microgels sont soumis à une série de lavages pour purifier l’ADN génomique bactérienne. Des enzymes lytiques digèrent les parois cellulaires de levures et de bactéries Gram-positives et détergent solubilise les débris cellulaires. Les microgels sont re-encapsulés en gouttelettes pour la tagmentation de réduire la contamination croisée. (C), la fusion de la microfluidique combine un code-barres numérique de la PCR, un génome de microgel tagmented et un mélange d’amplification à un taux > 1 kHz. Hors puce SOE-PCR épissures un code-barres unique d’unicellulaires sur le génome de tagmented et sélectivement amplifie entièrement construit avec code à barres. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
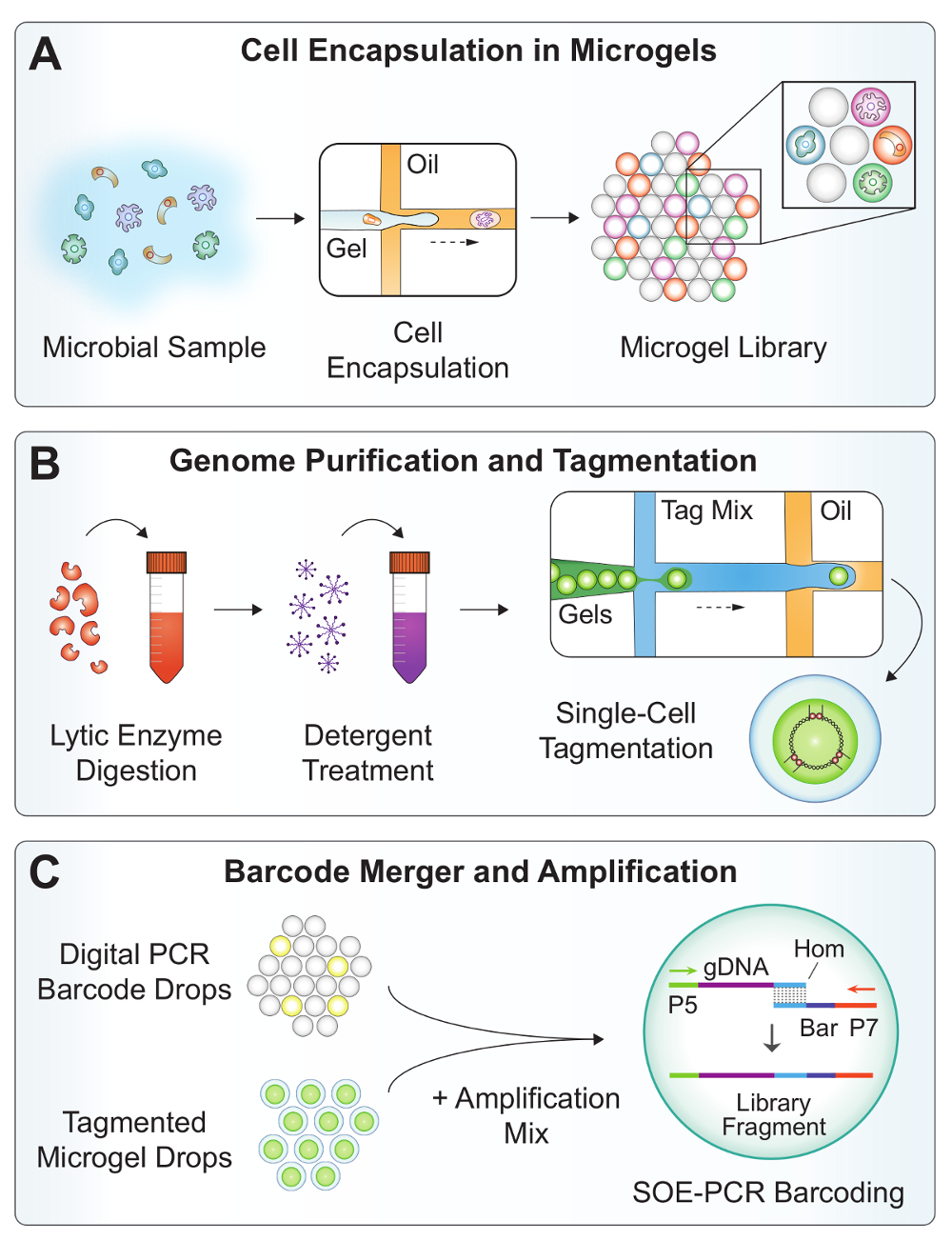{kind=link}

Figure 3 : Dispositifs microfluidiques pour Re-l’encapsulation de dropmaking et microgel. (A), ce panneau indique un écoulement co dropmaker (25 µm de hauteur de l’élément). Cellules et fondu agarose sont introduits dans l’appareil à des débits égaux pour produire 25 µm gouttelettes à la jonction de µm 25 µm x 25. Pour le dropmaking de code-barres numérique, l’entrée de la cellule est branchée, et un mélange PCR est introduit dans l’entrée d’agarose. (B), ce panneau montre un dispositif de Re-encapsulation microgel (25 µm de hauteur de l’élément). Les microgels se jettent dans une baie en forme d’entonnoir pour maintenir leur commande compacte et recevoir un volume de tagmentation mélange avant la Re-encapsulation à un carrefour de µm de 25 µm x 30. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
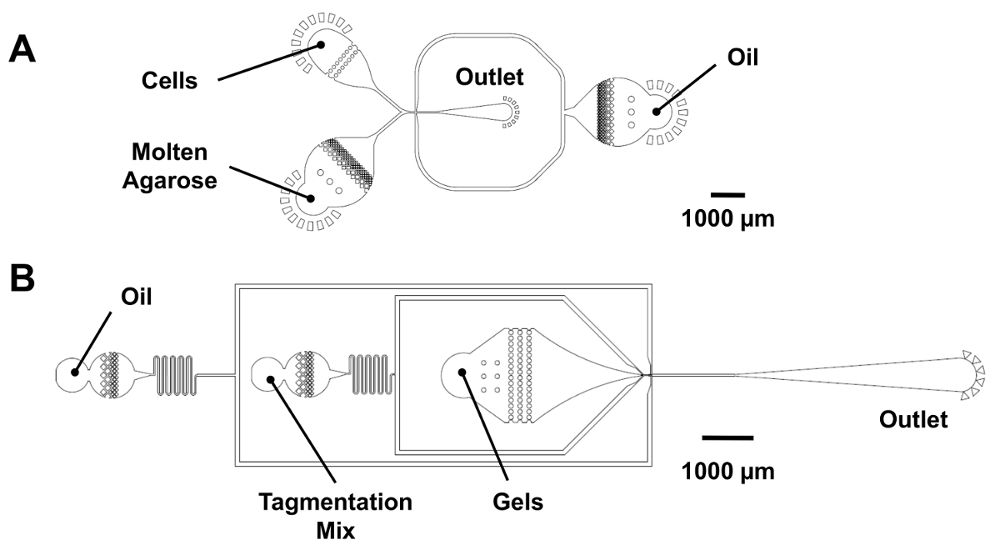{kind=link}

Figure 4 : Micrographies de gouttelettes et microgels. (A), ce panneau affiche lavé 25 µm microgels avant lyse enzymatique. Les bactéries sont colorées fluorescent pour la quantification du taux d’encapsulation. Statistiques de chargement de poisson exigent que les cellules puisse être encapsulé à raison de 1 à 10 gouttes ou moins pour réduire la fréquence des événements multiples-encapsulation. (B) ce panneau montre une image de microscopie de fluorescence de 25 µm code-barres numérique gouttelettes traités avec une tache d’acide nucléique. Les gouttelettes contenant des fragments amplifiés barcode produisent un signal de fluorescence forte. (C) ce panneau montre microgels re-encapsulé dans 50 µm gouttes. La clôture-emballage de la microgels permet des taux d’encapsulation approchant 1 gel par goutte d’eau avec quelques doublets. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 5 : Dispositif de double fusion microfluidique de génome de la cellule unique barcoding. Une opération de fusion en deux étapes des paires des gouttelettes de code-barres avec les génomes de tagmented à un haut débit. Une goutte de mélange PCR est d’abord générée et fusionnée avec une goutte de codes à barres sur la région indiquée en jaune à l’aide d’électrodes de l’eau salées. Ensuite, une gouttelette contenant une microgel est introduite et fusionnée une seconde fois dans la région en rouge. Prises d’huile permettent un contrôle précis de l’espacement entre les gouttelettes réinjectées. La chambre de réinjection de codes à barres et son huile d’espacement sont placés sur la couche plus courte de 25 µm, ombragée en bleu. Toutes les autres fonctionnalités de l’appareil appartenant à la couche plus épaisse avec 45 µm de hauteur totale. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
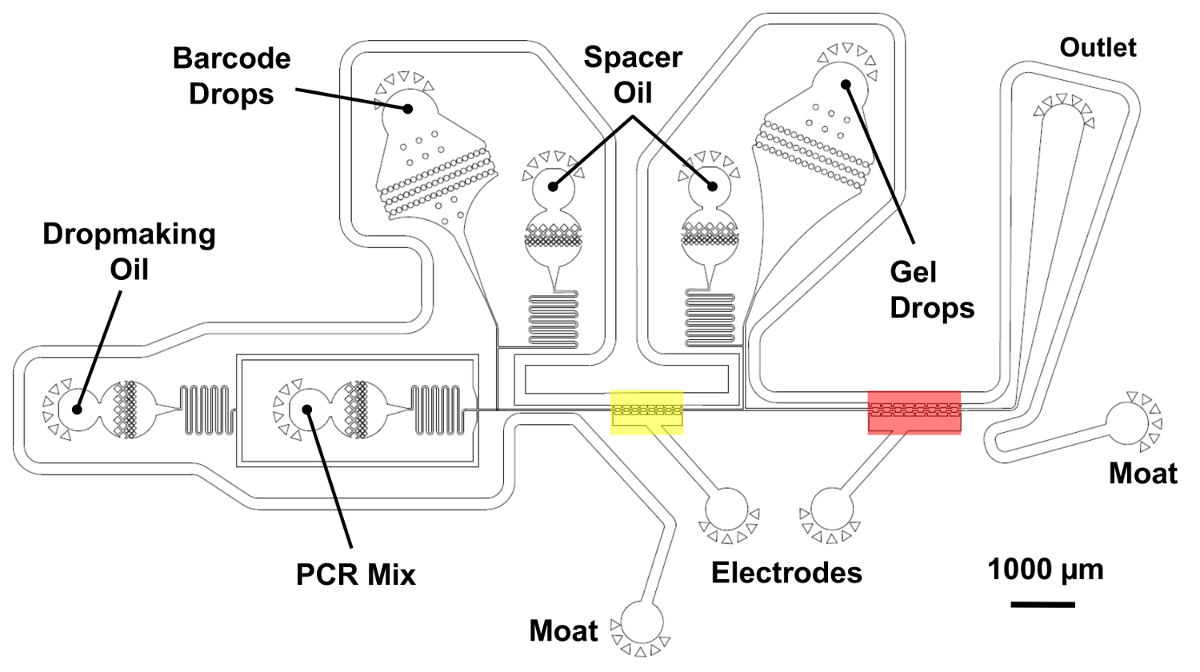{kind=link}

Figure 6 : Mesures de groupe de codes à barres pour une communauté microbienne synthétique de 10 cellules. (A), ce panneau montre la distribution du code barre taille des groupes. Le nombre de groupes d’une taille donnée diminue exponentiellement avec l’augmentation de taille de groupe. Un seuil minimum de 7,5 KB/s par groupe restreint l’analyse aux groupes avec une quantité suffisante de renseignements et supprime la séquence muté PCR « orphelins ». (B), ce panneau montre la répartition des codes à barres puretés de groupe. La grande majorité (> 90 %) des groupes sont de très haute pureté (> 95 %). (C) ce panneau montre l’abondance relative des 10 espèces calculé au niveau du groupe lecture et code à barres. Les 2 méthodes de comptage donnent des résultats similaires, indiquant que les tailles de groupe de codes à barres sont conformes à toutes les espèces. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 7 : Agréger génomique couverture de Bacillus subtilis des groupes de codes à barres. Les lectures de tous les groupes de codes à barres, cartographie pour la bactérie b. subtilis (N = 9 398) sont regroupées et analysées dans leur ensemble. Une carte de couverture circulaire illustre l’uniformité de la couverture de la SiC-seq dispose, avec aucune région d’abandon observables. Une ligne pointillée autour de la circonférence indique la couverture moyenne (5,55 x). L’histogramme de l’encart des fréquences couverture relative montre qu’une grande partie de la base sont couverts à une profondeur près de la moyenne pour l’ensemble du génome, représentée par la ligne pointillée. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
| Appareil | 1ère couche hauteur (µm) | 1ère couche vitesse d’essorage (tr/min) | 2ème couche hauteur (µm) | 2ème couche vitesse d’essorage (tr/min) |
| Flux de co dropmaker | 25 | 4000 | N/A | N/A |
| Re-encapsuleur gel | 25 | 4000 | N/A | N/A |
| Double fusion | 25 | 4000 | 20 | 5000 |
Tableau 1 : paramètres de fabrication de dispositif microfluidique. Ce tableau présente la liste des dispositifs microfluidiques utilisés dans le workflow de SiC-seq avec leurs vitesses requises pour photorésine spin coating (basé sur les spécifications du fabricant pour 3025 SU-8).
| Étiquette | Séquence (5' > 3') | ||||
| BAR | GCAGCTGGCGTAATAGCGAGTACAATCTGCTCTGATGCCGCATAGNNNNNNNNNNNNNNNTAAGCCAGCCCCGACACT | ||||
| DNA_BAR | CTGTCTCTTATACACATCTCCGAGCCCACGAGACGTGTCGGGGCTGGCTTA | ||||
| P7_BAR | CAAGCAGAAGACGGCATACGAGATCAGCTGGCGTAATAGCG | ||||
| P5_DNA | AATGATACGGCGACCACCGAGATCTACACTCGTCGGCAGCGTC | ||||
| I7_READ | GCCCACGAGACGTGTCGGGGCTGGCTTA | ||||
Tableau 2 : Séquences d’amorce.
Fichier supplémentaire 1 : S’il vous plaît cliquez ici pour télécharger ce fichier.
Fichier supplémentaire 2 : S’il vous plaît cliquez ici pour télécharger ce fichier.
Discussion
Le flux de travail microfluidique SiC-seq produit les données de séquençage du génome de la cellule unique parmi des milliers de cellules bactériennes. Codes à barres numériques épissés sur les génomes de cellules encapsulées microgel permettant la déconvolution de silico des données d’end en groupes de lectures avec code à barres, provenant de la même cellule. Une expérience de contrôle avec une communauté microbienne de composition connue, est nécessaire pour évaluer la pureté des groupes de codes à barres. Une grande partie des groupes de faible pureté indique que le taux d’encapsulation de cellules est trop élevé ou qu’il y a contamination croisée important gouttelette se produisant durant les étapes de traitement de la microfluidique. Selon les statistiques de Poisson, les codes-barres et les cellules doivent être isolés à un ratio de 1 particule pour chaque 10 gouttes à limiter le taux de plusieurs événements d’encapsulation à moins de 5 % de toutes les gouttelettes non vide. Un taux d’encapsulation au-dessus de celle-ci augmente les tarifs des doublets exponentiellement, donc la vérification du ratio encapsulation au cours du processus de dropmaking est d’une importance cruciale. Les utilisateurs doivent être particulièrement prudents de l’encapsulation des cellules multiples en un seul microgel parce que les lectures de différentes cellules partage la même séquence de code à barres ne peut pas être séparé de bioinformatically. Dans le cas que 1 cellule reçoit 2 différents codes à barres, la pureté de groupe de codes à barres n’est pas affectée même si les mesures d’abondance sont étalent lors du comptage par séquence de codes à barres.
Contamination croisée gouttelettes peut-être également survenir en raison des conditions sous-optimales fusion. Lors d’une opération réussie, le dispositif de fusion de microfluidique (Figure 5) pouvez contrôlable coupler 1 gouttelette de codes à barres avec 1 microgel et un volume de réactif PCR. Non-idéal débits seront traduira par une goutte d’appariement des ratios incorrect : 1 code à barres peut être couplé avec 2 microgels, par exemple. Tous les débits indiqués dans le protocole sont censés être des estimations et peuvent devoir être ajustée selon les légères variations dans les tailles de géométrie et de gouttelettes de dispositif. Les utilisateurs ayant accès aux caméras avec des capacités d’enregistrement à grande vitesse (> 10 000 images/s) devraient vérifier la fusion de gouttelettes correcte au début et au cours de l’opération de la microfluidique. Les utilisateurs n’ont pas accès à une caméra haute vitesse peuvent recueillir un petit volume de la sortie fusionnée et mesurer manuellement la taille des gouttelettes sous un microscope. La taille des gouttelettes devrait être uniforme : un excès de code à barres non fusionné ou gouttes microgel indique que le taux de réinjection devrait être réduite en conséquence.
Plusieurs générales devraient prendre les précautions nécessaires lors de la manipulation microgels et projetées à préserver leur intégrité. MICROGELS, bien que mécaniquement robuste, il faut refroidir suffisamment avant la rupture et mesures pour s’assurer de gélification complete de lavage. Microgels non-spherical sont une indication que l’agarose n’était pas donné suffisamment de temps pour se solidifier. Lors du lavage microgels, tournez les suspensions en bas à la vitesse nécessaire pour éviter une perte de produit. D’agarose hydrogel a un indice de réfraction correspondant étroitement à celle de l’eau et peut-être être difficile à voir dans un tube22, donc les utilisateurs doivent bien identifier la limite gel-liquide avant l’aspiration. Gouttelettes d’eau dans l’huile sont sensibles à la coalescence de l’accumulation des forces statiques23 sur les tubes et les gants de laboratoire. Pour cette raison, nous vous recommandons de charger les seringues de réinjection de gouttelette à mains nues et de traiter toutes les lignes de réinjection avec un pistolet antistatique avant l’amorçage de la pompe. Grosses gouttelettes coalescentes peuvent être supprimées en tournant lentement les émulsions dans une seringue et en aspirant manuellement les gouttes plus grandes, qui s’accumulent près du sommet en raison de leur plus grande force de flottabilité.
SiC-seq est la première technologie de démontrer le séquençage du génome de cellules individuelles de > 50 000 cellules bactériennes. Cette plate-forme offre des avantages significatifs en matière de rapport aux approches existantes et permet un échantillonnage plus profond des communautés microbiennes hétérogènes. A ce jour, technologies microfluidiques pour le séquençage du génome de cellules individuelles ont employé microchambers micropuits et9 24 pour l’amplification et l’isolement cellulaire, mais avec des débits de l’ordre de seulement quelques dizaines à des centaines de cellules. Le flux tri des cellules uniques en wellplates5,6 ne nécessite aucune instrumentation de microfluidique spécialisés mais possède un débit de même faible. Étant donné que les échantillons de sol et l’eau de l’environnement ont généralement des diversités alpha de > 1 000 espèces de niveau25,26, SiC-seq est très avantageux en raison de sa capacité à déguster un bien plus grand nombre d’organismes. Le workflow de SiC-seq est adaptable aux entrées des cellules de culture en laboratoire, l’environnement naturel ou un hôte vivant. Un échantillon cellulaire doivent seulement être dans une suspension aqueuse et gratuits de grosses particules (> 10 µm) sont adaptés pour l’encapsulation de la microfluidique. Par exemple, la méthode a été appliquée auparavant à un échantillon d’eau de mer à l’aide d’une série de lavage et de filtrage des étapes pour pré-traiter les cellules avant encapsulation17.
Le protocole de SiC-seq génère une quantité relativement clairsemée de données de séquençage de chaque cellule unique et peut ne pas convenir pour toutes les applications. Certains algorithmes de bioinformatique comme assemblage de génome de novo ou variante de nucléotide (SNV) appelant nécessitent des profondeurs de couverture supérieurs à travailler efficacement. Au lieu de cela, des groupes de codes à barres peuvent être ordonné en clusters en silico par taxonomique binning méthodes27 afin que les algorithmes peuvent être appliqués sur les plus grands ensembles de lectures. L’efficacité de codage à barres globale relativement faible du workflow SiC-seq peut également présenter des défis dans les cas où la disponibilité de l’échantillon d’entrée est faible. SiC-seq s’appuie sur une étape d’encapsulation de barcode Poisson-distribué, donc environ 10 % des cellules reçoivent un code-barres moléculaire et sont amplifiés au cours de l’étape de préparation finale de bibliothèque. Tout cela est comparable aux autres régimes de barcoding axée sur les microgouttelettes10, utilisateurs qui travaillent avec des échantillons cellulaires précieux peuvent avoir des difficultés à atteindre le rendement de la bibliothèque adéquate pour le séquençage et devrez peut-être augmenter le nombre de cycles de la PCR en finale étape d’amplification. Une autre solution possible pour les utilisateurs connaissant bien microfluidique est de trier les gouttelettes de barcode positif après l’étape PCR numérique, ce qui porte l’efficacité globale des codes à barres à > 85 %28.
Une orientation future potentielle pour SiC-seq technologie s’adaptant le flux de travail pour une utilisation avec des cellules de mammifères, ouvrant la voie à nouvelles études cliniques de cellule unique. À titre d’exemple, une analyse de la variation numéro de copie entre cancer de simple cellules mai plus loin notre compréhension du rôle de l’hétérogénéité du cancer pathologie2. Alternativement, intégrant les méthodes existantes pour sonder et enrichir les séquences d’ADN d’intérêt29 de SiC-seq permettrait le séquençage unicellulaires ciblé des sous-populations ou rares souches de cellules. Avec des échantillons environnementaux, gènes de dans une voie métabolique connue pourraient être ciblés et analysés contextuellement aux côtés de voisins gènes afin d’identifier de nouveaux îlots génomiques. De dans un environnement hôte humain, des échantillons de bactéries pathogènes bas-titre a peuvent être isolés et séquencé au niveau cellule unique d’examiner de plus près leurs origines génotypiques de la virulence.
Déclarations de divulgation
Brevets se rapportant à ce flux de travail peuvent être autorisé à Mission Bio, de laquelle Adam R. Abate est actionnaire.
Remerciements
Ce travail a été soutenu par la National Science Foundation grâce à une bourse de carrière (numéro de licence DBI-1253293) ; le National Institutes of Health (NIH) (subvention nombres HG007233-01, R01-EB019453-01, 1R21HG007233, DP2-AR068129-01, le R01-HG008978) ; et le Defense Advanced Research projets Agence vivant fonderies Program (numéros de contrat HR0011-12-C-0065, N66001-12-C-4211, HR0011-12-C-0066).
matériels
| Name | Company | Catalog Number | Comments |
| 3" silicon wafers, P type, virgin test grade | University Wafers | 447 | |
| SU-8 3025 photoresist | Microchem | 17030192 | |
| Spin coater | Specialty Coating Systems | G3P-8 | |
| Photomasks | CadArt Servcies | (custom) | See Supplemental Files for mask designs |
| PGMEA developer | Sigma-Aldrich | 484431 | |
| Isopropanol | Sigma-Aldrich | 109827 | |
| Sylgard 184 silicone elastomer kit | Krayden | 4019862 | |
| Degassing chamber | Bel-Art | 42025 | |
| 0.75 mm biopsy punch | World Precision Instruments | 504529 | |
| Glass microscope slides (75 mm x 50 mm) | Corning | 294775X50 | |
| Aquapel (hydrophobic glass treatment) | Pittsburgh Glass Works | 47100 | |
| PE-2 polyethylene tubing | Scientific Commodities | B31695-PE/2 | |
| 1 mL syringes | BD | 309628 | |
| 27 gauge needles | BD | 305109 | |
| Syringe pump | New Era Pump Systems | NE-501 | |
| Novec HFE-7500 fluorinated oil (HFE) | 3M | 98-0212-2928-5 | |
| FC-40 fluorinated oil | Sigma-Aldrich | F9755 | |
| PEG-PFPE surfactant | Ran Biotechnologies | 008-FluoroSurfactant | |
| Space heater | Lasko | CD09250 | |
| Agarose, low gelling temperature | Sigma-Aldrich | a9414 | |
| TE (10X) | Rockland | mb-007 | |
| PBS 1X, pH 7.4 | E&K Scientific Products | EK-65083 | |
| OptiPrep (density gradient medium) | Sigma-Aldrich | d1556 | |
| 1H,1H,2H-Perfluoro-1-Octanol (PFO) | Sigma-Aldrich | 370533 | |
| Span 80 (sorbitane monooleate) | Sigma-Aldrich | s6760 | |
| Hexane | Sigma-Aldrich | 139386 | |
| Tween 20 (polysorbate 20) | Sigma-Aldrich | p2287 | |
| Lysozyme Type IV | MP Biomedicals | 195303 | |
| Mutanolysin | Sigma-Aldrich | M9901 | |
| Zymolyase (yeast lytic enzyme) | Zymo Research | e1004 | |
| Lysostaphin | Sigma-Aldrich | L7386 | |
| Sodium chloride | Sigma-Aldrich | S9888 | |
| EDTA | Sigma-Aldrich | E6758 | |
| Tris-HCl, pH 7.5, 1M | Invitrogen | 15567-027 | |
| Dithiothreitol (DTT) | Teknova | d9750 | |
| Lithium dodecyl sulfate | Sigma-Aldrich | L9781 | |
| Proteinase K | New England Biosciences | P8107S | |
| Ethanol, 200 Proof (100%) | Koptec | V1001 | |
| SYBR Green I (nucleic acid stain) | Invitrogen | S7563 | |
| PEG 6k | Sigma-Aldrich | 81260 | |
| Triton X-100 (octylphenol ethoxylate) | Sigma-Aldrich | t8787 | |
| Nextera DNA Library Prep Kit | Illumina | FC-121-1030 | |
| Phusion Hot Start Flex Master Mix (High-Fidelity Hot Start Master Mix) | New England Biosciences | m05365 | |
| Platinum Multiplex PCR Master Mix (Taq Master Mix) | Applied Biosystems | 4464263 | |
| Warmstart 2.0 Bst Polymerase (isothermal polymerase) | New England Biosciences | m0538m | |
| NT buffer from Nextera XT kit (neutralization buffer) | Illumina | FC-131-1024 | |
| Cold cathode fluorescent inverter | (custom) | (custom) | |
| DC power supply | Mastech | HY1503D | |
| Zerostat 3 anti-static gun | Milty | 5036694022153 | |
| 3D-printed centrifuge syringe holder | (custom) | (custom) | See Supplemental Files for 3D print file |
| Zymo DNA Clean & Concentrator-5 | Zymo Research | D4003 |
Références
- Navin, N., et al. Tumour evolution inferred by single-cell sequencing. Nature. 472 (7341), 90-94 (2011).
- Ni, X., et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proceedings of the National Academy of Sciences of the United States of America. 110 (52), 21083-21088 (2013).
- Schmidt, H., Hensel, M. Pathogenicity islands in bacterial pathogenesis. Clinical Microbiology Reviews. 17, 14-56 (2004).
- Martínez, J. L., Baquero, F. Interactions among strategies associated with bacterial infection: pathogenicity epidemicity, and antibiotic resistance. Clinical Microbiology Reviews. 15 (4), 647-679 (2002).
- Rinke, C., et al. Obtaining genomes from uncultivated environmental microorganisms using FACS-based single-cell genomics. Nature Protocols. 9 (5), 1038-1048 (2014).
- Rinke, C., et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 499 (7459), 431-437 (2013).
- Zhang, H., Liu, K. K. Optical tweezers for single cells. Journal of the Royal Society Interface. 5 (24), 671-690 (2008).
- Xu, L., Brito, I. L., Alm, E. J., Blainey, P. C. Virtual microfluidics for digital quantification and single-cell sequencing. Nature Methods. 13 (9), 759-762 (2016).
- Gawad, C., Koh, W., Quake, S. R. Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics. Proceedings of the National Academy of Sciences of the United States of America. 111 (50), 17947-17952 (2014).
- Macosko, E. Z., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 161 (5), 1202-1214 (2015).
- Klein, A. M., et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 161 (5), 1187-1201 (2015).
- Rotem, A., et al. High-throughput single-cell labeling (Hi-SCL) for RNA-Seq using drop-based microfluidics. PLoS One. 10 (5), 1-14 (2015).
- Amini, S., et al. Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing. Nature Reviews Genetics. 46 (12), 1343-1349 (2014).
- Zheng, G. X. Y., et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nature Biotechnology. 34 (3), 303-311 (2016).
- Lan, F., Haliburton, J. R., Yuan, A., Abate, A. R. Droplet barcoding for massively parallel single-molecule deep sequencing. Nature Communications. 7, 11784 (2016).
- Rotem, A., et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nature Biotechnology. 33 (11), 1165-1172 (2015).
- Lan, F., Demaree, B., Ahmed, N., Abate, A. R. Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. Nature Biotechnology. 35 (7), 640-646 (2017).
- Novak, R., et al. Single-cell multiplex gene detection and sequencing with microfluidically generated agarose emulsions. Angewandte Chemie Internation Edition. 50 (2), 390-395 (2011).
- Gill, C., Van De Wijgert, J. H. H. M., Blow, F., Darby, A. C. Evaluation of lysis methods for the extraction of bacterial DNA for analysis of the vaginal microbiota. PLoS One. 11 (9), 1-16 (2016).
- Picelli, S., et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Research. 24 (12), 2033-2040 (2014).
- Abate, A. R., Chen, C. H., Agresti, J. J., Weitz, D. A. Beating Poisson encapsulation statistics using close-packed ordering. Lab on a Chip. 9 (18), 2628 (2009).
- Jain, A., Yang, A. H. J., Erickson, D. Gel-based optical waveguides with live cell encapsulation and integrated microfluidics. Optic Letters. 37 (9), 1472 (2012).
- Karbaschi, M., Shahi, P., Abate, A. R. Rapid chemical-free breaking of microfluidic emulsions with a hand-held antistatic gun. Biomicrofluidics. 11 (4), 1-6 (2017).
- Gole, J., et al. Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells. Nature Biotechnology. 31 (12), 1126-1132 (2013).
- Chao, Y., et al. Metagenomic analysis reveals significant changes of microbial compositions and protective functions during drinking water treatment. Scientific Reports. 3 (1), 3550 (2013).
- Fierer, N., et al. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proceedings of the National Academy of Sciences of the United States of America. 109 (52), 21390-21395 (2012).
- Mande, S. S., Mohammed, M. H., Ghosh, T. S. Classification of metagenomic sequences: methods and challenges. Briefings in Bioinformatics. 13 (6), 669-681 (2012).
- Eastburn, D. J., et al. Microfluidic droplet enrichment for targeted sequencing. Nucleic Acids Research. 43 (13), e86 (2015).
- Clark, I. C., Abate, A. R. Finding a helix in a haystack: nucleic acid cytometry with droplet microfluidics. Lab on a Chip. 17 (12), 2032-2045 (2017).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.