
Method Article
Une plate-forme de méthyl-Seq de Rat pour identifier les changements épigénétiques liés à l’exposition de Stress
Dans cet article
Résumé
Nous décrivons ici le protocole et la mise en œuvre du méthyl-Seq, une plate-forme épigénomique, utilisant un modèle de rat pour identifier les changements épigénétiques liés à l’exposition de stress chronique. Les résultats démontrent que la plate-forme de méthyl-Seq de rat est capable de détecter des différences de méthylation que résultent d’une exposition de stress chez les rats.
Résumé
Que les génomes d’une plus grande variété d’animaux seront disponibles, il y a un besoin croissant d’outils qui permet de capturer les changements épigénétiques dynamiques dans ces modèles animaux. Le rat est un animal modèle particulier où un outil épigénétique peut compléter les nombreuses études pharmacologiques et comportementales pour fournir des informations perspicaces mécanistes. À cette fin, nous avons adapté le système de Capture SureSelect cible (dénommé méthyl-Seq) chez le rat, qui peut évaluer le taux de méthylation d’ADN au sein du génome du rat. La conception de rat ciblés promoteurs, îles de CpG, rives de l’île et les régions riches en GC de tous les gènes RefSeq.
Pour appliquer le programme sur une expérience de rat, rats Sprague Dawley mâles ont été exposés à un stress chronique variable pendant 3 semaines, après quoi les échantillons de sang ont été prélevés pour l’extraction d’ADN génomique. Méthyl-Seq bibliothèques ont été construits des échantillons d’ADN de rat par cisaillement, ligation de l’adaptateur, enrichissement de la cible, conversion de bisulfite et multiplexage. Bibliothèques ont été séquencés sur une plate-forme de séquençage de prochaine génération et les lectures séquencés ont été analysées afin d’identifier les DMR entre les ADN des rats stressés et non stressés. Meilleur candidat DMR ont été validés indépendamment par pyrosequencing bisulfite pour confirmer la robustesse de la plate-forme.
Les résultats démontrent que la plate-forme de méthyl-Seq de rat est un outil utile et épigénétique qui permet de capturer les changements de méthylation induites par l’exposition au stress.
Introduction
Avancées du séquençage haut-débit ont conduit à une profusion de séquences génomiques pour modèle et organismes non-modèles. La disponibilité de telles séquences a facilité les recherches en génétique, génomique comparative et transcriptomique. Par exemple, les séquences génomiques disponibles sont très utiles pour aligner les données de séquençage de ChIP-Seq expériences qui enrichissent l’ADN issu de sa liaison avec l’histone modifications1ou séquençage de bisulfite, qui mesure la méthylation de l’ADN par détecter l’uracile formé de conversion de bisulfite de cytosines non méthylé2. Toutefois, on a des retards dans la mise en œuvre de plates-formes épigénomique qui incorporent des données de séquençage génomique disponible dans leur conception en raison d’un manque de données annotées des séquences régulatrices spécifiques qui peuvent influencer la fonction du gène.
En particulier, méthylation de l’ADN est l’un des plus largement étudiées modifications épigénétiques sur l’ADN qui peuvent tirer parti des données génomiques disponibles pour la construction d’une plate-forme de méthylomiques. Un tel exemple est une plateforme basée sur la baie pour l’humain méthylome3, qui a été largement utilisé dans les différentes disciplines de l’oncologie de psychiatrie4,5. Malheureusement, les plates-formes similaires pour des modèles animaux non-humains sont rares, qu’il n’y a pratiquement aucune plateforme largement utilisé qui ont profité de la séquence génomique dans leur conception initiale.
Une méthode commune pour évaluer le paysage méthylomiques de modèles animaux non-humains est bisulfite de représentation réduite séquençage (Orr)6. Cette approche permet de surmonter le coût du séquençage du génome entier bisulfite qui, tout en offrant un paysage complet méthylomiques, offre une couverture de lecture-profondeur inférieure en raison de coût et des informations fonctionnelles limitées dans de vastes zones pauvres en gène du génome2 . Obligations à rendement réel implique Sommaire de restriction et de sélection de taille de l’ADN génomique d’enrichir à fortement les séquences riches en GC comme les îles de CpG qui sont communément trouvés près de promoteurs de gènes et joueraient un rôle dans le règlement de gène7. Alors que la méthode Orr a été utilisée dans un certain nombre d’études importantes, sa dépendance sur les enzymes de restriction n’est pas sans limites et défis remarquables. Par exemple, enrichissement des séquences riches en GC dans Orr est entièrement dépendante de la présence de séquences spécifiques reconnus par les enzymes de restriction et sélection taille ultérieure par électrophorèse. Cela signifie que n’importe quel régions génomiques qui ne contiennent pas ces sites de restriction sont exclues lors de la sélection de la taille. En outre, hétérospécifique comparaisons sont difficiles à moins que les mêmes sites de restriction sont présents dans les mêmes locus parmi les différentes espèces.
Une des façons de surmonter les limitations de ces obligations sont d’utiliser une méthode d’enrichissement qui tire profit de la séquence génomique publiée dans la conception de la plate-forme. La plate-forme humaine basée sur la baie utilise des sondes d’apprêt conçus contre GPC spécifique d’allèle spécifique (CG vs TG après conversion du bisulfite) cible recuit et amorce l’extension. Sa conception reflète non seulement la séquence génomique humaine disponible, mais des régions régulatrices vérifiée expérimentalement acquises de multiples champs d’enquête, comme ENCODE et ENSEMBL8. Malgré sa large utilisation dans les enquêtes de méthylomiques humaine, une plate-forme similaire n’existe pas pour les animaux de modèle. En outre, le format tableau met une contrainte importante sur la surface disponible pour positionner la sonde. Dans les dernières années, des efforts ont été faits pour combiner la cible-spécificité offerte par capture sonde design et la fonctionnalité de haut débit de nouvelle génération séquençage. Une telle entreprise a entraîné le système enrichissement cible basée sur le séquençage du génome de la souris (souris méthyl-Seq), qui servait à identifier les différences de cerveau-spécifique ou induite par les glucocorticoïdes de méthylation9,10. Plates-formes similaires pour d’autres modèle et non-modèle animaux sont nécessaires pour faciliter la recherche épigénomique chez ces animaux.
Ici, nous démontrons la mise en œuvre de cette plateforme novatrice pour analyser les méthylomiques sur le rat. Le rat a servi comme modèle animal important en pharmacologie, le métabolisme, neuroendocrinologie et comportement. Par exemple, il y a un besoin croissant de comprendre les mécanismes sous-jacents qui donnent lieu à la toxicité des médicaments, l’obésité, réponse au stress ou la toxicomanie. Une plate-forme haut débit capable de capturer les changements de méthylomiques liés à ces conditions augmenterait notre compréhension des mécanismes. Le génome du rat est toujours sans annotation pour les régions régulatrices, nous constituée de promoteurs non redondant, îles de CpG, île rives11et précédemment identifié les séquences riches en GC dans le rat méthyl-Seq plate-forme12.
Afin d’évaluer la conception et mise en oeuvre de la plate-forme SureSelect cible enrichissement (génériquement appelés méthyl-Seq) pour le génome du rat, nous avons utilisé un modèle de rat de stress chronique de variables (CV)13 à identifier différentiellement méthylés régions entre animaux non accentués et stressés. Notre conception de la plate-forme, protocole et mise en œuvre peuvent être utiles pour les chercheurs qui veulent mener une enquête complète et impartiale d’épigénétique sur un organisme dont la séquence génomique est déjà disponible, mais reste mal annoté.
Protocole
Toutes les expériences ont été achevées en conformité et le respect de toutes les directives réglementaires et institutionnelles pertinentes, y compris le Comité de l’urbanisme à la Johns Hopkins School of Medicine et d’institutionnels animalier.
1. les animaux
- Obtenir des rats Sprague-Dawley adolescents mâles à l’âge de 4 semaines. Loger les animaux dans des cages de rat en polycarbonate dans une température- et pièce à hygrométrie contrôlée sur un 12 h, 12 h dark cycle de lumière avec une apparition lumineuse à 0600 h. fournir les animaux ad libitum accès à l’eau.
- Permettre à des rats s’acclimater pendant 1 semaine réduire le stress liés au transport. Paire-maison les animaux (N = 16) ne permettent pas de stress de l’isolement et à l’âge de 5 semaines, commencer la variable chronique stress régime (CVS) pendant 3 semaines.
2. chronique effort Variable
- Administrer le régime CV une fois le matin (9-11:00) et une fois dans l’après-midi (1-15:00) à des moments irréguliers pour garder la routine imprévisible. Incorporent des stresseurs douces pendant la nuit. Le CVS régime comprend : 1) 3 h dans un cylindre de retenue ; 2) natation 10 min ; cage 3) 3 h incliner 4) 1 h lente secousse plate-forme ; et 5) 1 h dans la chambre froide de 4 ° C.
Remarque : Une nuit agents stressants incluent social encombrement (5 par cage), isolement social, literie humide, restriction alimentaire et lumières-sur. Un service hebdomadaire typique du régime stress est fourni au tableau 1.
3. système endocriniens dosages
-
Déterminer les niveaux de la corticostérone (CORT) à l’aide de prélèvements de sang (environ 50 mL) queue recueillies en même temps (09:00) deux fois par semaine tout au long de l’expérience, avant le régime CVS pour établir les niveaux d’hormone de base (jour 0), une fois au milieu de la CVS hebdomadaire (jours 4,11 et 18), après tous les 7 jours de CVS (jours 7 et 14), ainsi qu’à la fin du CVS (jour 21). Recueillir des échantillons de sang avant le régime de stress quotidien.
- Recueillir un échantillon de sang de tronc final pendant l’euthanasie (25 jours) pour RIA et extraction de l’ADN génomique.
- Centrifuger les échantillons de sang (600 x g, 4 ° C, 10 min) pour séparer le plasma des globules. Pipette de sortir du plasma (liquide surnageant) et stocker les échantillons à-80 ° C.
- Décongeler et utiliser le plasma pour déterminer les niveaux CORT par dosage radio-immunologique (RIA). Veiller à ce que les taux CORT de plasma de 3 semaines sont élevés chez les animaux stressés pour vérifier la solidité du régime stress.
4. comportement
- Après le régime CVS (jours 23 et 24), évaluer chaque animal pour comportement anxieux sur l’élévation plus maze (EPM)14.
- À l’aide d’une caméra vidéo, enregistrement les animaux sur l’appareil EMP pour 300 s et note le temps passé dans le centre, fermé les bras et bras ouverts.
5. conception de la Rat méthyl-Seq
- À l’aide de l’UCSC Genome Browser, obtenir les coordonnées génomiques non redondant (rat Nov 2004 rn4 Assemblée) pour CpG îles et les rivages de l’île (± 1 Ko bordant les îles de CpG), les promoteurs (± 1 Ko de chaque TSS) de chaque gène RefSeq et autres séquences qui peuvent être obtenus auprès documentation pertinente.
Note : Pour le rat méthyl-Seq, séquences riches en GC supplémentaires depuis une plate-forme basée sur la baie de méthylation précédente a été ajouté12. Pour les régions supérieures à 5 Kbits/s, alternance de régions de 500 points de base ont été échantillonnées suivie de 1 kbit/s qui ont été ignorés. La conception de méthyl-Seq rat final se compose de 111 Mbits/s, GPC 2,3 millions ; et une taille moyenne région de 594 bps. Il cible 228 800 locus uniques. - Entrez une liste de coordonnées génomiques dans un logiciel de conception de capture disponibles sur le marché cible pour la conception de la sonde appropriée.
6. construction de la bibliothèque de méthyl-Seq de Rat de l’ADN génomique
Remarque : Pour éliminer les effets du traitement par lots, traiter des échantillons multiples en même temps et intensifier les mélanges maîtres en conséquence. Extraire l’ADN à l’aide d’un kit d’extraction d’ADN disponible dans le commerce. Colonne ou précipitation-méthodes basées sur les deux donnent l’ADN génomique de haute qualité (260/280 ratio ~ 1,8). Utilisation des méthodes axées sur le phénol ne sont pas recommandés. Éluer ou remettre en suspension l’ADN dans un tampon faible TE (10 mM TE, 0,1 mM EDTA, pH 8,0).
- Préparation des échantillons
Remarque : Pour chaque étape à l’aide de la liaison à l’ADN des billes magnétiques, assurez-vous que les perles sont acclimatés à la température ambiante pendant au moins 30 min et bien mélangés avant utilisation.- ADN de cisaillement
- Utilisez un fluorimètre pour déterminer la concentration initiale de l’ADN double-brin de chaque échantillon. Diluer > 1 µg d’ADNg à 50 µL avec tampon de basse TE (TE, 0,1 mM EDTA, pH 8,0 de 10 mM) dans des tubes de microcentrifuge faibles liaison à l’ADN.
- Échantillons en utilisant un sonicateur isotherme de cisaillement (10 % Duty Cycle, intensité 5, 200 Cycles / Burst, 6 cycles de 60 s, balayage de fréquence, 4 ° C).
- Évaluer la qualité de l’ADN à l’aide d’un système basé sur l’électrophorèse qui mesure la quantité et la taille de l’ADN.
Remarque : La quantité d’ADN recommandée 1 µg, soit 3 µg. Si il est limité à partir de matériau, la quantité d’entrée la plus basse doit être > 500 ng, comme des quantités plus faibles n’affectera négativement la quantité et la qualité des bibliothèques générée.
- Réparer les extrémités de l’ADN.
- Le rat méthyl-Seq kit permet de préparer le mélange maître fin-réparation sur la glace. Ajouter 52 µL du mélange dans chaque échantillon et incuber dans un thermocycleur sans couvercle chauffé (20 ° C pendant 30 min, attente de 4 ° C).
Fin-réparation Master Mix (par exemple) :
35,2 µL d’eau
10 µL de tampon de réparation fin (x 10)
1,6 µL de dNTP Mix
1 µL de T4 ADN polymérase
2 µL de Klenow ADN polymérase
2.2 µL de T4 polynucléotide Kinase - Purifier les échantillons à l’aide de 180 µL de billes magnétiques de liaison à l’ADN et de 400 µL d’éthanol à 70 % fraîchement préparée par échantillon. Ajouter 180 µL de perles dans chaque échantillon et incuber pendant 5 min à température ambiante. Perles de granule, éliminer le surnageant et Resuspendre le culot dans 200 µL d’éthanol à 70 %. Retirez l’éthanol et répéter le lavage une fois.
- Utiliser une plaque magnétique pour perles de granule et supprimer autant que possible d’éthanol. Sec dans un 37 ° C bloc chauffant pendant 3 à 5 min jusqu'à ce que la pastille de perle est complètement sec. Resuspendre dans 44 µL d’eau exempte de nucléase et recueillir environ 42 µL de liquide surnageant.
Point d’arrêt : Après la fin de la réparation de l’ADN, échantillons peuvent être scellés et conservés à-20 ° C.
- Le rat méthyl-Seq kit permet de préparer le mélange maître fin-réparation sur la glace. Ajouter 52 µL du mélange dans chaque échantillon et incuber dans un thermocycleur sans couvercle chauffé (20 ° C pendant 30 min, attente de 4 ° C).
- L’adénylate le 3' se termine.
- Préparer adénylation Master Mix sur la glace. Ajouter 9 µL mélange dans chaque échantillon et incuber dans un thermocycleur sans couvercle chauffé (37 ° C pendant 30 min, attente de 4 ° C).
Adénylation Master Mix (par exemple) :
5 µL de tampon de Klenow
1 µL de dATP
3 µL de Klenow ADN polymérase - Purifier les échantillons à l’aide de 90 µL de billes magnétiques de liaison à l’ADN et de 400 µL d’éthanol à 70 % fraîchement préparée par échantillon. Ajouter 90 µL de perles dans chaque échantillon et incuber pendant 5 min à température ambiante. Perles de granule, éliminer le surnageant et Resuspendre le culot dans 200 µL d’éthanol à 70 %. Retirez l’éthanol et répéter le lavage une fois.
- Utiliser une plaque magnétique pour perles de granule et supprimer autant que possible d’éthanol. Sec dans un 37 ° C bloc chauffant pendant 3 à 5 min jusqu'à ce que la pastille de perle est complètement sec. Resuspendre dans 35 µL d’eau exempte de nucléase et recueillir environ 33,5 µL de liquide surnageant.
- Préparer adénylation Master Mix sur la glace. Ajouter 9 µL mélange dans chaque échantillon et incuber dans un thermocycleur sans couvercle chauffé (37 ° C pendant 30 min, attente de 4 ° C).
- Ligaturer l’adaptateur méthylé.
- Préparer la ligature Master Mix sur la glace et ajouter 16,5 µL du mélange dans chaque échantillon. Incuber dans un thermocycleur sans couvercle chauffé (20 ° C pendant 15 min, attente de 4 ° C).
La ligature Master Mix (par exemple) :
2,5 µL d’eau
2,5 µL de méthyl-Seq méthylé adaptateur
10 µL de tampon de T4 DNA Ligase (x 5)
1,5 µL de T4 DNA Ligase - Purifier les échantillons à l’aide de 90 µL de billes magnétiques de liaison à l’ADN et de 400 µL d’éthanol à 70 % fraîchement préparée par échantillon. Ajouter 90 µL de perles dans chaque échantillon et incuber pendant 5 min à température ambiante. Perles de granule, éliminer le surnageant et Resuspendre le culot dans 200 µL d’éthanol à 70 %. Retirez l’éthanol et répéter le lavage une fois.
- Utiliser une plaque magnétique pour perles de granule et supprimer autant que possible d’éthanol. Sec dans un 37 ° C bloc chauffant pendant 3 à 5 min jusqu'à ce que la pastille de perle est complètement sec. Resuspendre dans 22 µL d’eau exempte de nucléase et recueillir environ 22 µL de liquide surnageant. Évaluer la qualité à l’aide d’un bioanalyzer.
Remarque : Si la quantité totale d’ADN est inférieure à 500 ng, cisaillement et processus ADN supplémentaires avant d’effectuer les étapes suivantes. Si la taille moyenne des ADN n’augmente pas de plus de 30 points de base, vérifiez que les réactifs sont nouvelles, comme T4 ADN polymérase, Klenow et/ou T4 ligase peut être vieux.
Point d’arrêt : Après ligature adaptateur méthylé, échantillons peuvent être scellés et conservés à-20 ° C.
- Préparer la ligature Master Mix sur la glace et ajouter 16,5 µL du mélange dans chaque échantillon. Incuber dans un thermocycleur sans couvercle chauffé (20 ° C pendant 15 min, attente de 4 ° C).
- ADN de cisaillement
- Hybridation
- Transférer les échantillons aux tubes de microcentrifuge faibles liaison à l’ADN et utiliser un concentrateur sous vide chauffé pour réduire le volume de l’échantillon à moins de 3,4 µL. reconstituer les échantillons à 3,4 µL.
NOTE : Concentré les échantillons pour environ ~ 3 µL à s’assurer que les échantillons sont retirés de concentrateur sous vide avant tout le liquide s’évapore. - Préparer un tampon d’hybridation à température ambiante et méthyl-Seq bloc Mix sur la glace. Ajouter 5,6 µL du mélange de bloc de méthyl-Seq dans chaque échantillon et incuber dans cycleur thermique (95 ° C pendant 5 min, 65 ° C pendant 2 min, attente de 65 ° C).
Tampon d’hybridation (par exemple) :
6.63 µL de Hyb méthyl-Seq 1
0,27 µL de méthyl-Seq Hyb 2
2.65 µL de méthyl-Seq Hyb 3
3.45 µL de Hyb méthyl-Seq 4
Méthyl-Seq Mix de bloc (par exemple) :
2,5 µL du méthyl-Seq indexation bloc 1
2,5 µL de méthyl-Seq bloc 2
0,6 µL du méthyl-Seq bloc 3 - Préparer la RNase bloc Mix et le mélange de l’hybridation de bibliothèque de Capture. Ajouter 20 µL du mélange de l’hybridation bibliothèque de Capture de chaque échantillon et incuber à 65 ° C pendant au moins 16 h.
RNase Mix de bloc (par exemple) :
0,5 µL de RNase bloc
1,5 µL d’eau
Capturer des Mix de l’hybridation de bibliothèque (par exemple) :
13 µL de tampon d’hybridation
2 µL de RNase bloc Mix
Bibliothèque de Capture de 5 µL de Rat méthyl-Seq
NOTE : garder à 65 ° C, les réactions lors de l’ajout d’hybridation Mix pour empêcher les liaisons non spécifiques. - Aliquote 50µl de billes magnétiques streptavidine par échantillon dans un nouveau tube de bande de 8 puits. Laver les perles avec 200 µL de tampon de liaison méthyl-Seq. Plaque magnétique permet de perles de granule et éliminer le surnageant entre chaque lavage pour un total de 3 lavages. Après le lavage final, resuspendre streptavidine perles dans 200 µL de tampon de liaison méthyl-Seq.
- Ajouter des échantillons à 200 µL de billes magnétiques de streptavidine lavées et incuber à température ambiante pendant 30 min à l’aide d’un mélangeur rotatif. Tout en mélangeant, aliquote à 200 µL de tampon de lavage méthyl-Seq 2 dans trois puits d’une plaque à 96 puits par échantillon et le placer dans un thermocycleur de préchauffage à 65 ° C.
- Après incubation, pellet streptavidine billes magnétiques à l’aide de la plaque magnétique et remettre en suspension les perles dans 200 µL tampon de lavage méthyl-Seq 1. Incuber pendant 15 min à température ambiante. Utiliser une plaque magnétique à pellet et jeter le surnageant.
- Laver les perles 3 fois avec le tampon de lavage méthyl-Seq 2 : resuspendre perle culot dans 200 µL de tampon de lavage 2 (préchauffée à l’étape 6.2.5.), incuber les perles dans le thermocycleur (65 ° C, 10 min) et perles de granule. Jeter le surnageant après chaque lavage à l’aide d’une plaque magnétique.
Remarque : Maintenir les réactions de l’hybridation à 65 ° C lors de l’ajout de tampon de lavage 2 afin d’éviter les liaisons non spécifiques. - Ajouter 20 µL de tampon d’élution méthyl-Seq aux talons lavées et incuber à température ambiante pendant 20 min. Utilisez une plaque magnétique à granulés perles et transfert surnageant dans un nouveau tube de bande. Jeter les perles.
Remarque : Lors de l’incubation, préparer le réactif de conversion de bisulfite.
- Transférer les échantillons aux tubes de microcentrifuge faibles liaison à l’ADN et utiliser un concentrateur sous vide chauffé pour réduire le volume de l’échantillon à moins de 3,4 µL. reconstituer les échantillons à 3,4 µL.
- Conversion de bisulfite
Remarque : Effectuez la conversion de bisulfite de l’ADN simple brin éluée à l’aide de réactifs appropriés et les instructions d’un kit de conversion disponibles sur le marché de bisulfite.- Ajoutez 130 µL de réactif de conversion du bisulfite préparés au surnageant de l’étape précédente. Diviser chacune des 150 réactions µL également dans deux puits. Incuber dans un thermocycleur (64 ° C pendant 2,5 h, cale de 4 ° C).
Remarque : La réaction de 150 µL est également répartie dans deux puits séparés pour assurer une température homogène. Après incubation pendant 2,5 h, procéder immédiatement à l’étape suivante. - Lier des échantillons pour tourner des colonnes en ajoutant 600 µL de tampon de liaison et de laver une fois avec 100 µL de tampon de lavage. Centrifuger les colonnes (15 000 x g, 1 min) entre toutes les étapes de conversion de bisulfite et éliminer les traversent.
- Échantillons de Desulphonate en ajoutant 200 µL de tampon de Desulphonation à colonnes. Incuber à température ambiante pendant 15-20 min. Repeat centrifugation et jeter les traversent.
- Laver les colonnes deux fois avec 200 µL de tampon de lavage. Éluer chaque échantillon en ajoutant 10 µL de tampon d’élution pour la colonne, en incubation pendant 3 min à température ambiante et centrifugation (15 000 x g, 1 min). Répétez l’étape d’élution pour un total de 20 µL.
- Préparer la réaction PCR Master Mix 1 sur la glace. Ajouter 82 µL du mélange dans chaque échantillon. Incuber dans un thermocycleur avec le programme suivant.
PCR réaction Master Mix 1 (par exemple) :
30 µL d’eau
50 µL de méthyl-Seq PCR Master Mix
1 µL de méthyl-Seq PCR1 Primer F
1 µL de méthyl-Seq PCR1 Primer R
Programme du thermocycleur :
Étape 1, 1 cycle : 95 ° C 2 min
Étape 2, 8 cycles : 95 ° C 30 s, 60 ° C, 30 s, 72 ° C 30 s
Étape 3, 1 cycle : 72 ° C 7 min
Étape 4, 1 cycle : 4 ° C Hold - Purifier les échantillons à l’aide de 180 µL de billes magnétiques de liaison à l’ADN et de 400 µL d’éthanol à 70 % fraîchement préparée par échantillon. Ajouter 180 µL de perles dans chaque échantillon et incuber pendant 5 min à température ambiante. Perles de granule, éliminer le surnageant et Resuspendre le culot dans 200 µL d’éthanol à 70 %. Retirez l’éthanol et répéter le lavage une fois.
- Utiliser une plaque magnétique pour perles de granule et supprimer autant que possible d’éthanol. Sec dans un 37 ° C bloc chauffant pendant 3 à 5 min jusqu'à ce que la pastille de perle est complètement sec. Resuspendre dans 21 µL d’eau exempte de nucléase et recueillir environ 19,5 µL de liquide surnageant.
- Ajoutez 130 µL de réactif de conversion du bisulfite préparés au surnageant de l’étape précédente. Diviser chacune des 150 réactions µL également dans deux puits. Incuber dans un thermocycleur (64 ° C pendant 2,5 h, cale de 4 ° C).
- D’indexation
- Préparer la réaction PCR Master Mix 2 sur la glace. Ajouter µL 25,5 Master Mix 2 à chaque échantillon. Ajouter 5 µL de commercial indexation des amorces d’échantillons individuels et incuber dans un thermocycleur.
PCR réaction Master Mix 2 (par exemple) :
25 µL méthyl-Seq PCR Master Mix
Primer d’indexation commun de 0,5 µL méthyl-Seq
Programme du thermocycleur :
Étape 1, 1 cycle : 95 ° C 2 min
Étape 2, 6 cycles : 95 ° C 30 s, 60 ° C, 30 s, 72 ° C 30 s
Étape 3, 1 cycle : 72 ° C 7 min
Étape 4, 1 cycle : 4 ° C Hold
Remarque : Les cycles supplémentaires (2-3) peuvent être nécessaires si la concentration initiale de l’ADN est en dessous des valeurs recommandées. - Purifier les échantillons à l’aide de 90 µL de billes magnétiques de liaison à l’ADN et de 400 µL d’éthanol à 70 % fraîchement préparée par échantillon. Ajouter 90 µL de perles dans chaque échantillon et incuber pendant 5 min à température ambiante. Perles de granule, éliminer le surnageant et Resuspendre le culot dans 200 µL d’éthanol à 70 %. Retirez l’éthanol et répéter le lavage une fois.
- Utiliser une plaque magnétique pour perles de granule et supprimer autant que possible d’éthanol. Sec dans un 37 ° C bloc chauffant pendant 3 à 5 min jusqu'à ce que la pastille de perle est complètement sec. Resuspendre dans 24 µL d’eau exempte de nucléase et recueillir environ 24 µL de liquide surnageant.
- Évaluer la concentration et le dimensionnement de bp en utilisant les réactifs de détection de l’ADN de haute sensibilité sur un bioanalyzer.
Remarque : Si le bioanalyzer ne parvient pas à détecter la présence de la bibliothèque de l’ADN, répétez les étapes de préparation avec l’ADN supplémentaire.
Point d’arrêt : après purification, échantillons indexées peuvent être scellés et conservés à-20 ° C. - Mise en commun des échantillons pour la plate-forme de séquençage de génération approprié utilisée.
- En utilisant les données de concentration de la bioanalyzer, qui détermine la molarité d’ADN basée sur la taille de la bibliothèque et la quantité dans un volume donné, diluer avec un tampon faible TE (6.1.1.1) et combiner tous les échantillons à une concentration finale de 15 h.
Remarque : Une méthode plus sensible de la quantification de la bibliothèque est par quantitative PCR en temps réel en utilisant des amorces ciblant les adaptateurs ligaturés. - Exécuter des échantillons sur le nombre de voies qui suffisent pour 4 échantillons par couloir sur un séquenceur de prochaine génération.
Remarque : par exemple, si 16 exemples de bibliothèque ont été indexés et combinés unique, exécutez les bibliothèques sur 4 voies, équivalant à 4 échantillons par couloir.
- En utilisant les données de concentration de la bioanalyzer, qui détermine la molarité d’ADN basée sur la taille de la bibliothèque et la quantité dans un volume donné, diluer avec un tampon faible TE (6.1.1.1) et combiner tous les échantillons à une concentration finale de 15 h.
- Préparer la réaction PCR Master Mix 2 sur la glace. Ajouter µL 25,5 Master Mix 2 à chaque échantillon. Ajouter 5 µL de commercial indexation des amorces d’échantillons individuels et incuber dans un thermocycleur.
7. séquençage sur un séquenceur de prochaine génération
- Envoyer les échantillons au noyau institutionnel séquençage des clusters de la bibliothèque de méthyl-Seq, suivie par séquençage sur une machine de séquençage de prochaine génération.
8. analyses pour déceler des DMR
- Implémenter le Bismark15, qui appelle noeud papillon 2.0 comme une séquence interne aligneur16,17, pour aligner les lectures d’entrée brutes au bisulfite-convertis, plus-brin de génome. Suite à l’alignement, utilisez le Bismark_methylation_extractor pour réaliser un contrôle de qualité et assigner une valeur estimative de méthylation à chaque CpG.
- Générer une liste de DMR avec le paquet de BS-Seq18 dans Bioconductor. Filtre le DMR basé sur ayant plus de GPC consécutive 3 et P-value < 0,05.
NOTE : Générer une liste DMR qui comprend les coordonnées génomiques, la distance et le gène le plus proche de RefSeq, nombre des GPC au sein de chaque DMR, moyenne % valeur de méthylation de CpG à travers la DMR pour les groupes de deux comparaison (par exemple, a souligné vs non accentué), la P-valeur, et la valeur FDR (taux de fausse découverte). Utilisez la liste DMR, i.e., coordonnées génomiques, pour concevoir des amorces de pyrosequencing pour la validation.
9. validation par Bisulfite Pyrosequencing
-
Conception d’amorce
- Design des amorces de PCR du bisulfite et pyrosequencing. Conception de deux séries d’amorces PCR (extérieurs et imbriquées) afin que la PCR nichée amplifiera 150 – 400 bits/s d’une DMR.
Remarque : en général, amorces sont au moins 24 bases de long avec au moins 4-5 non consécutifs G (C pour l’apprêt inverse) au compte pour réduit la température de perte de la complexité de la séquence de recuit. Une des amorces imbriquées sera biotine-étiquetés et purifié par HPLC. Cependant, les amorces standards doivent être commandés premier à optimiser l’étape de l’ACP en résolvant les réactions sur gel d’agarose.- Conception le pyrosequencing assay apprêt afin qu’il cible le biotinylé complémentaire chapelet juste 1 à 2 bases en amont de la GPC à doser. Concevoir plusieurs amorces pyrosequencing autant que nécessaire, car chaque amorce pyrosequencing peut doser fiable de 30 points de base en aval.
- Pour la Rt1-m4, procédez comme suit :
rRT1M4 à l’extérieur – F TGTAYGATTTTGGTTATYGTAAAT
rRT1M4 à l’extérieur – R AACTTACAAATTTCACCAACTCA
rRT1M4 Nested – F GTGGGTTAYGTGGATAATATATAG
rRT1M4 Nested – R AATCACTTACCATTCTCTCTCTAACTA
rRT1M4 Pyro1 TAYGTGGATAATATATAGAT
rRT1M4 Pyro2 GATAGTTATTTGGYGAGTTAG
rRT1M4 Pyro3 GAGTATTTGGAGGAGTTGAT
rRT1M4 Pyro4 GGATTTTAATATTTGGT
- Design des amorces de PCR du bisulfite et pyrosequencing. Conception de deux séries d’amorces PCR (extérieurs et imbriquées) afin que la PCR nichée amplifiera 150 – 400 bits/s d’une DMR.
-
Utiliser un kit disponible dans le commerce pour la conversion de bisulfite de rat sang ADNg.
Remarque : Les étapes de conversion du bisulfite ont été adaptés à partir du kit disponible dans le commerce avec les modifications suivantes : À l’étape 1, ajouter 50 – 100 ng de sang ADNg et diluer avec de l’eau à 20 µL. À l’étape 9, éluer 20 µL par échantillon.- Préparer le bisulfite réactif de conversion selon le protocole du fabricant et combiner avec ADNg dilué. Incuber dans thermocycleur (64 ° C pendant 2,5 h, cale de 4 ° C).
- Ajouter tampon contraignant à ADNg converti en colonnes à centrifuger et centrifuger (15 000 x g, 1 min). Colonnes de lavage une fois puis ajouter Desulphonation tampon aux colonnes et incuber pendant 15 min à température ambiante. Centrifugeuse (15 000 x g, 1 min).
- Laver la colonne avec le tampon de lavage et centrifuger (15 000 x g, 1 min). Répétez l’étape de lavage avec centrifugation (15 000 x g, 2 min). Ajouter 20 µL tampon d’élution et centrifuger (15 000 x g, 1 min) pour éluer.
-
Amplification par PCR
- Préparer l’extérieur PCR Master Mix. Ajouter 21,5 µL du mélange réactionnel à 3,5 µL convertie par bisulfite ADNg et exécutez programme thermocycleur.
L’extérieur PCR Master Mix :
16.25 µL d’eau
2,5 µL de tampon de polymérisation [10 x]
0,5 µL de dNTP [10 mM]
1 µL d’apprêt avant [0,1 µM]
1 µL d’apprêt inverse [0,1 µM]
0.25 µL de Taq DNA polymérase [5000 U/mL].
Programme du thermocycleur :
Étape 1, 1 cycle : 94 ° C 4 min
Étape 2, 47 cycles : 94 ° C, 1 min, 53 ° C 30 s, 72 ° C 1 min
Étape 3, 1 cycle : 72 ° C, 8 min, 4 ° C Hold - Préparer le mélange réactionnel PCR nichée. Ajouter 23 µL du mélange réactionnel à 2 µL de l’échantillon de PCR externe et répéter le programme extérieur de Thermocycleur PCR. Évaluer la qualité des produits PCR par électrophorèse sur gel (1 x TAE tampon, gel d’agarose à 1 %).
PCR nichée Master Mix :
17,75 µL d’eau
2,5 µL de tampon de polymérisation [10 x]
0,5 µL de dNTP [10 mM]
1 µL d’apprêt avant [0,1 µM]
1 µL d’apprêt inverse [0,1 µM]
0.25 µL de Taq DNA polymérase [5000 U/mL]
Remarque : Pour PCR nichée, soit vers l’avant ou l’amorce de marche arrière doit être biotinylé.
- Préparer l’extérieur PCR Master Mix. Ajouter 21,5 µL du mélange réactionnel à 3,5 µL convertie par bisulfite ADNg et exécutez programme thermocycleur.
-
Pyrosequencing
- Faire un mélange maître contenant 38 µL de tampon de liaison, 35 µL d’eau et 2 µL de streptavidine Sépharose perles par exemple. Dans une plaque à 96 puits, ajoutez 75 µL du mélange maître et 5 µL du produit PCR imbriqué. Agiter dans un agitateur de plaque pour 15-60 min.
- Et en agitant, ajouter 12 µL d’apprêt (0,5 µM, dilué dans tampon recuit) dans les puits d’une plaque de test pyrosequencing.
- Après agitation, effectuez les étapes de lavage à l’aide de tampons de lavage de réaction obligatoire. Placez l’outil vide dans la cuve remplie d’eau puis prélever des échantillons de plaque. Plonger un outil sous vide dans une fosse remplie à moitié, contenant de l’éthanol à 70 %, NaOH (0,2 M) et un tampon Tris acétate (10 mM, pH 7,4). Retirer de l’outil vide vide et place dans HS test plaque pour perles de transfert.
- Placer la plaque sur le bloc chauffant et laisser incuber à 80 ° C pendant 2 min. Allow plaque refroidir pendant 5 min puis commencer le programme de pyro.
Résultats
Une mise en œuvre de la plate-forme de méthyl-Seq rat dépend de plusieurs critères. Figure 1 illustre le flux de travail global de l’étude et met en lumière les mesures spécifiques contrôle qualité (CQ) qui sont nécessaires avant d’avancer. Parmi les premiers facteurs à considérer est la robustesse du modèle animal et le traitement du stress, qui déterminent l’ampleur des changements épigénétiques qui se produisent à travers le méthylome. Étant donné que notre travail animale repose sur notre observation précédente exposition de corticostérone (CORT) pouvant mener à des changements dans la méthylation de l’ADN19,20, notre régime de stress chronique de variable (CVS) devait être de rigueur suffisante pour produire a souligné rats plasmatiques élevés de CORT. Un schéma typique de CVS hebdomadaire est indiqué dans le tableau 1 et se composait des facteurs de stress quotidiens dans le matin, après-midi, et durant la nuit, qui sont constamment modifiés pour empêcher l’accoutumance et diminué la réponse au stress. Dans le traitement de 3 semaines, les animaux stressés présentaient des taux significativement élevés de plasmatique moyenne CORT [jours 4 à 21, contrôlent : 32,7 3,7 ng/mL, Stress : 103,0 11,9 ng/mL (moyenne SEM), P = 2,2 x 10-4, Figure 2 a] au-dessus de ceux d’atones, animaux témoins. Invariablement, ces animaux ont également montré une plus grande anxiété comme comportement sur l’élévation plus labyrinthe (EPM), comme en témoigne le significativement plus de temps passé dans les bras fermés de l’EMP et moins de temps dans les bras ouverts (Figure 2 b). Ces résultats démontrent que l’exposition CVS conduit à endocrines significative et des changements de comportement, qui nous conduit à examiner si ces changements ont été associés à des signatures spécifiques de méthylation de l’ADN.
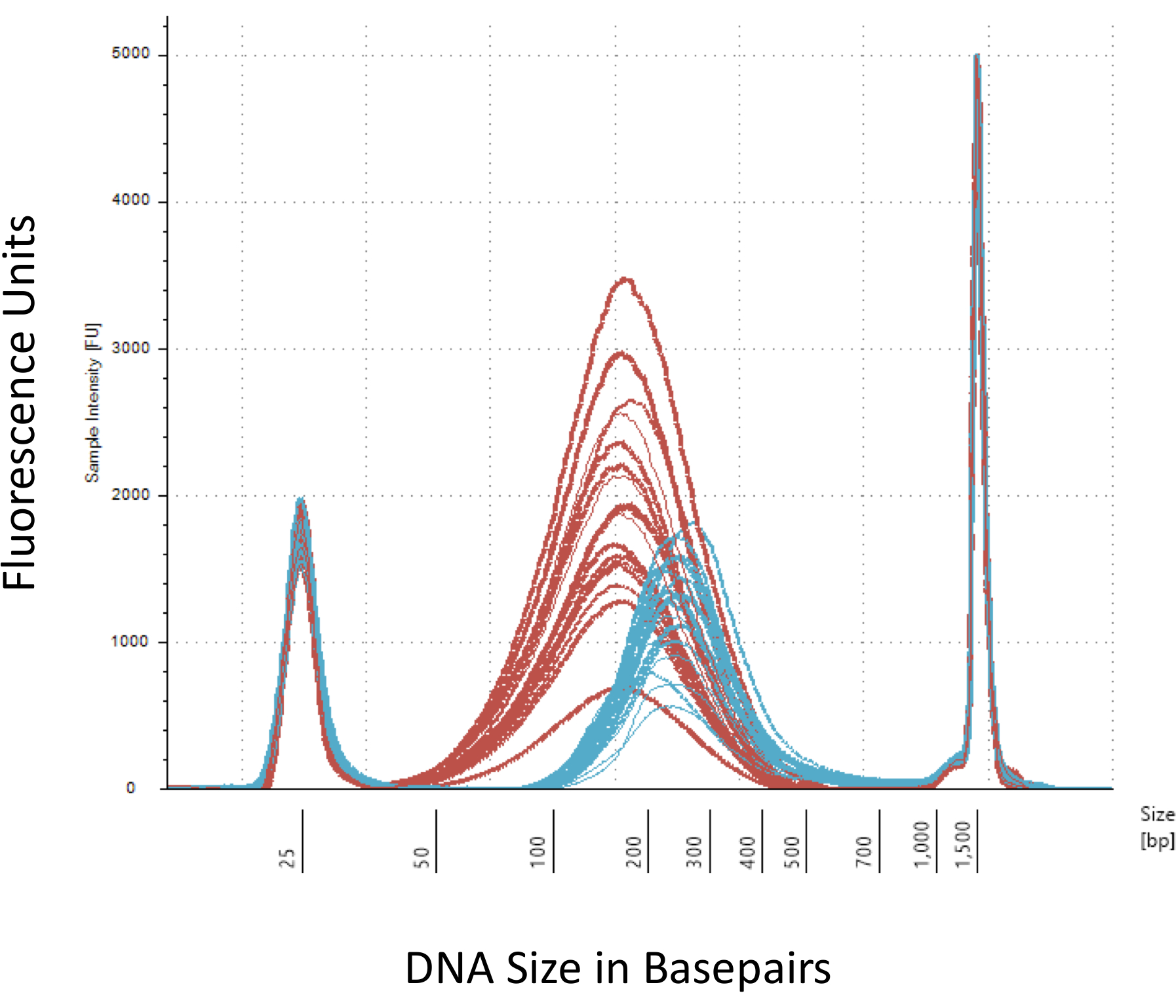
Nous insistons sur plusieurs points de contrôle qui sont cruciaux pour la construction réussie de la bibliothèque de méthyl-Seq. Commençant par une quantité suffisante d’ADN est nécessaire, comme la sonication, multiples lavage/purification, enrichissement de la cible, et les étapes de conversion du bisulfite successivement réduisent la quantité d’ADN dans la bibliothèque de fini. Bien que plusieurs étapes d’amplification PCR atténuer la perte de la matrice d’ADN, un nombre de cycle PCR excessif peut introduire supérieure lit double. Pour la présente étude de rat méthyl-Seq, 2 g de sang ADNg par rat a été utilisé. Nous notons que le méthyl-Seq bibliothèques peuvent être faites avec quantité d’ADN de départ aussi bas que 500 ng. Petit matériel de départ permet aux utilisateurs de générer des bibliothèques de l’ADN isolé de FACS (fluorescence-lancée de cellules tri) ou aiguille à coups de poing, bien qu’il y a un risque accru de produire une quantité insuffisante de librairies pour le séquençage subséquent. QC est interprété par électrophorèse de 1 L de l’échantillon sur un bioanalyzer, qui fournit le poids moléculaire, la quantité et la molarité ADN. Trois étapes essentielles qui nécessitent l’utilisation de la bioanalyzer sont : 1) après l’étape de sonication pour vous assurer un cisaillement suffisante d’ADN (~ 170 bp, rouge, Figure 3) ; 2) après l’étape de ligature adaptateur indiqué par un changement dans la taille moyenne de l’ADN cisaillée (~ 200 bp, bleu, Figure 3) pour assurer leur ultérieure amplification par PCR ; et 3) après l’étape de purification finale bibliothèque afin d’assurer la quantité et la taille de la bibliothèque pour l’ordonnancement.
Les paquets R BSSeq et BSmooth en Bioconductor ont été utilisées pour analyser le bisulfite de séquençage données18. Ils comprennent des outils et des méthodes d’alignement des séquence lectures, effectuer le contrôle de la qualité, et identifiant différentiellement méthylés des régions (DMR). BSmooth logiciel appelle noeud papillon 2.016,17 comme un alignement de séquence interne pour obtenir des résumés de CpG-niveau de mesure, par l’alignement des lectures d’entrée brutes au bisulfite-converti des séquences génomiques. Les lectures alignées sont ensuite filtrés à travers des procédures de contrôle de qualité rigoureux qui cherchent à identifier le séquençage systématique et des erreurs de base d’appel qui peuvent biaiser les analyses en aval. Une série de parcelles sont générés pour aider visuellement dans ce processus de filtrage. Séquençage métriques sont également générés à l’information pertinente de document tels que le nombre de lectures alignés, % cible et par la couverture de CpG, parmi d’autres (tableau 2). Une fois que les données sont filtrées, un algorithme de lissage/normalisation est réalisé, où chaque CpG est attribuée une valeur estimative de méthylation selon QC tous les lit à partir de chaque échantillon et estimations des voisins GPC pour assurer les appels plus précise de la méthylation statut même dans les cas où la couverture de la séquence est faible. Cette valeur fournit une estimation lissée de la probabilité de méthylation sur chaque site de CpG. En comparant la moyenne des estimations lissée de la méthylation de l’échantillon entre les deux groupes de traitement et le classement des régions génomiques du plus significativement différentes au moins, une liste des DMR est générée (tableau 3).
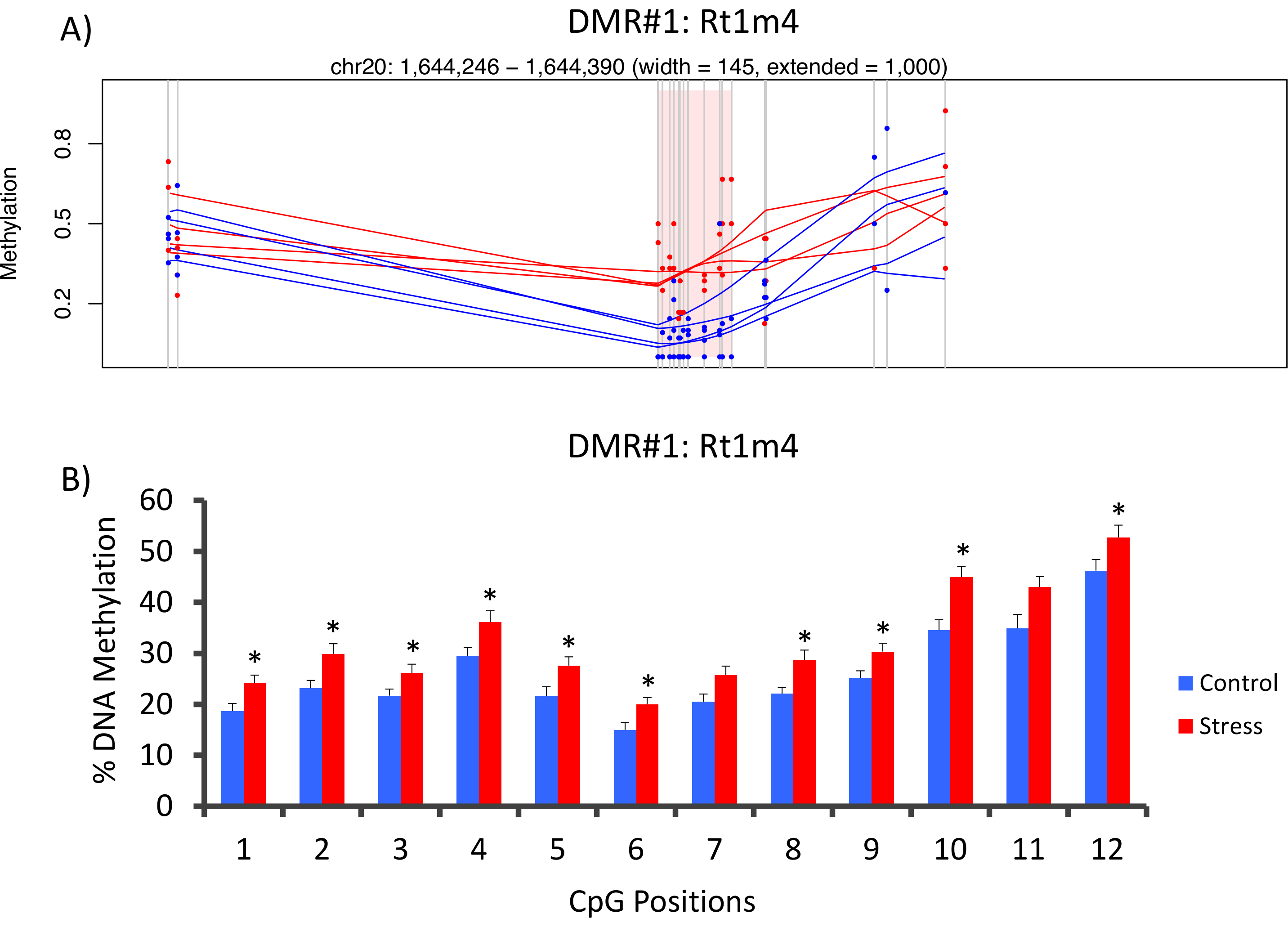
Le haut avec que DMR entre les groupes soumis à une contrainte et non accentués se trouvait dans le promoteur du gène majeur d’histocompatibilité rat Rt1-m4, a insisté sur les animaux présentant des niveaux plus élevés de la méthylation dans l’ensemble de tous les GPC que les animaux non accentué (Figure 4 a). Pour confirmer la mise en œuvre réussie de la plate-forme de méthyl-Seq et l’analyse des données, les amorces ont été conçus contre la DMR et sanguin de méthylation de l’ADN dans l’ensemble de la cohorte d’animaux stressés et non stressés (8 séquencé par méthyl-Seq et 8 non séquencé) ont été évalués par bisulfite pyrosequencing. Les résultats démontrent une augmentation significative de la méthylation de l’ADN à travers 10 hors de la 12 GPC dosés (changement de 5.1 – 10,4 % méthylation, P < 0,037, Figure 4 b). KEGG voie analyse a été effectuée sur l’ensemble de la DMR nominalement important d’identifier les voies associées au stress. Constamment, DMR associées aux voies mis en cause les maladies liés à l’exposition de stress chronique, comme le diabète, les maladies cardiovasculaires et le cancer (tableau 4). 21 , 22 , 23 pour démontrer un lien entre les données de l’épigénétiques et le degré d’exposition au stress, niveaux de méthylation au CpG-10 on a comparé les concentrations moyennes de CORT de 3 semaines pour chaque animal. Les résultats ont montré une corrélation modeste entre les données du système endocrinien et de méthylation (R2= 0,54, P = 0,001, Figure 5).

Figure 1 : ensemble schématique "workflow" pour la plateforme de rat méthyl-Seq. Un g de l’ADN génomique extrait le sang de souligner et de rats témoins est d’abord transformé pour construire les bibliothèques de méthyl-Seq pour séquençage, analyse et identification des cibles. Un autre 100 ng d’ADN est utilisé pour une validation indépendante des cibles identifiées épigénétiques par bisulfite pyrosequencing. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
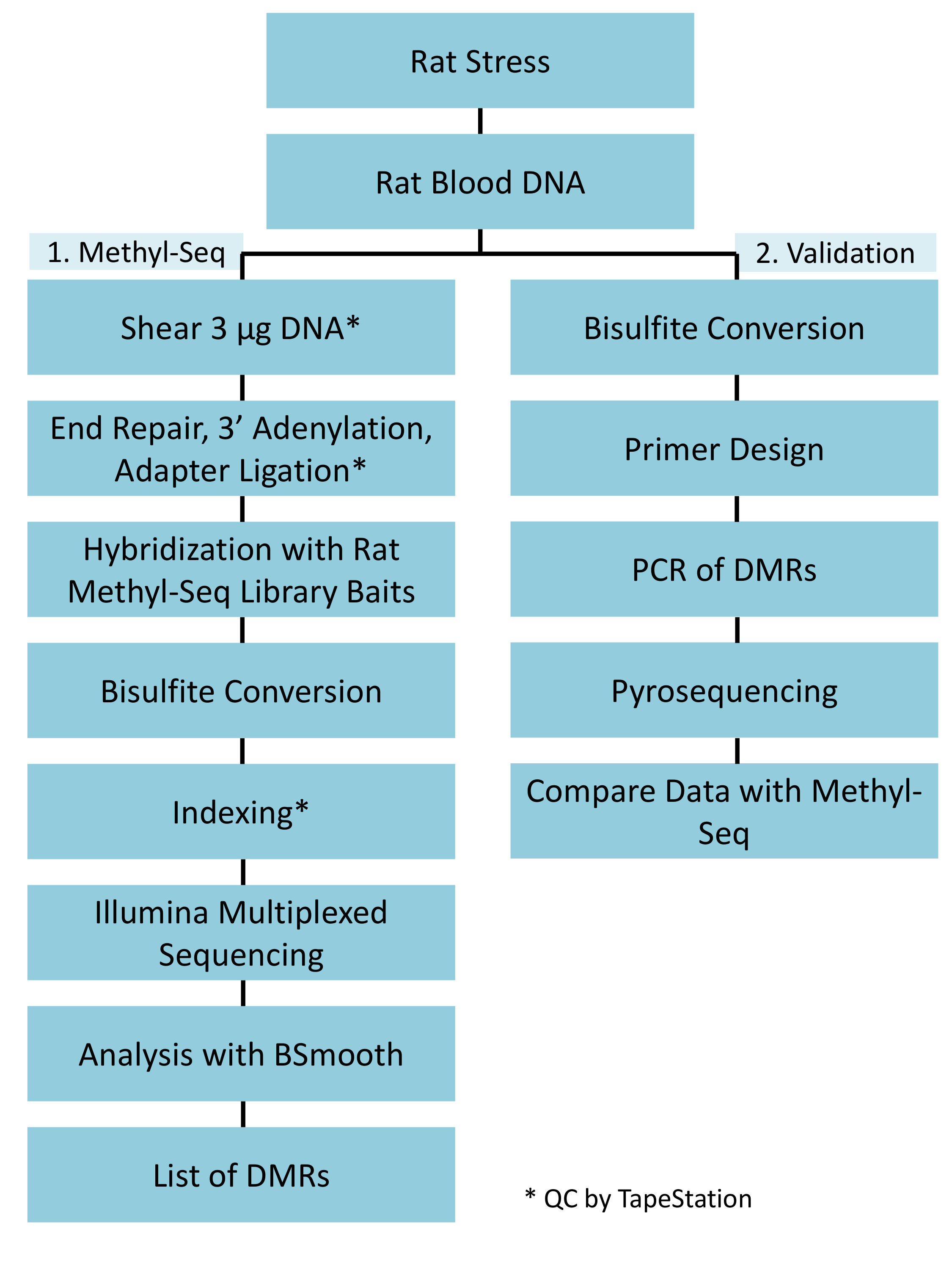{kind=link}

Figure 2 : exposition à un stress chronique variable (CVS) entraîne des changements endocriniens et comportements chez les rats. (A) plusieurs prélèvements de corticostérone (CORT) démontrent la robustesse de la semaine 3 régime CVS. Des échantillons de sang ont été prélevés dans la matinée avant le régime de stress quotidien. (B) a souligné animaux passée plus de temps dans les bras fermés et moins de temps dans les bras ouverts de l’élevé plus de labyrinthe (EPM). Boîtes avec point de données pour chaque animal sont indiqués. Test T de Student a été réalisé pour une signification statistique. * P < 0,05, ** P < 0,01, et *** P < 0,001. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 3 : dosage du rat cisaillé et adaptateur-ligaturé ADN sur un bioanalyzer. Les courbes rouges et bleues montrent la quantité et la taille de l’ADN génomique (rouge) après la tonte en un sonicateur isotherme et la ligature de l’adaptateur, respectivement. Chaque ligne représente un échantillon et le rouge et bleus courbes reflètent les deux perte d’ADN au cours des plusieurs étapes (fin-réparation, 3'-adénylation et le nettoyage de l’échantillon) et augmentent en taille de bp en raison de la ligature des adaptateurs. Forte des pics à 25 bp et bp 1500 sont des marqueurs standards qui ont été ajoutés au tampon de chargement. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 4 : les changements épigénétiques induites par CVS sont détectés par rat méthyl-suiv. (A) analyse du rat méthyl-Seq données impliqués du promoteur du gène Rt1m4 comme une région différentiellement méthylée (DMR) entre stressés (rouge) et les rats témoins (bleu). La sortie graphique pour la Rt1m4 DMR (région ombrée rose) affiche chaque CpG (ligne grise verticale), les quatre échantillons dans chaque groupe (lignes rouges ou bleues) et les niveaux de méthylation % pour chaque animal (point rouge ou bleu). (B) douze GPC au sein de la DMR ont été validés par le bisulfite pyrosequencing. Les graphiques à barres sont représentés en moyenne de SEM, et un test de Student T-a été réalisé pour une signification statistique. * P < 0,05. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 5 : analyse de régression linéaire ont montré une corrélation modeste entre % ADN méthylation au CpG-10 de Rt1m4 et la semaine 3 plasma CORT les concentrations moyennes de tous les deux souligné et contrôlent les animaux (N = 16). Données d’animaux stressés sont représentées par des cercles rouges. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
| Semaine | Jour 1 | Jour 2 | Jour 3 | Jour 4 | Jour 5 | Jour 6 | Jour 7 |
| AM | Dispositif de retenue pour | Nager | Chambre froide | Nager | Dispositif de retenue pour | Shaker | Nager |
| PM | Shaker | Inclinaison de la cage | Dispositif de retenue pour | Shaker | Chambre froide | Dispositif de retenue pour | Chambre froide |
| Pendant la nuit | Restreindre les aliments | Mouiller la literie | Isolement | Lumière sur | Le surpeuplement | Lumière sur | Mouiller la literie |
Tableau 1 : Un horaire hebdomadaire typique du traitement chronique d’effort variable (CVS).
| Séquençage Metrics | Stress,1 | Contrôle1 |
| (n = 4) | (n = 4) | |
| Jumelé fin lectures (PER) | 89,290,397 | 80,165,674 |
| Fin apparié unique mappé lit (UMPER) | 39,200,255 | 35,013,406 |
| Efficacité de taux/cartographie alignement (UMPER / PER) | 44 % | 44 % |
| Double lit (% de UMPER) | 73 % | 65 % |
| UMPER dédupliquée | 10,481,031 | 12,306,018 |
| Moyenne lecture profondeur couverture (x) (ARDC) | x 6 | x 6 |
| GPC (N) | 12,056,878 | 12,056,878 |
| ARDC (x) des GPC | x 2 | x 2 |
| GPC au moins 10 lectures (N) | 481 383 | 595 850 |
| ARDC (X) des GPC au moins 10 lectures | 19 | 19 |
| Sur cible GPC (chevauchement complet avec sonde les régions cibles) | 1 923 872 | 2 007 638 |
| Sur la cible ARDC (x) des GPC | x 7 | 8 x |
| Sur cible GPC au moins 10 lectures (N) | 428 249 | 531 419 |
| Sur cible ARDC (x) des GPC au moins 10 lectures | 18 x | 18 x |
| Sur la cible (p. avec 1 ou plusieurs paires de bases chevauchement avec sonde les régions cibles) (UMPER) | 8 277 715 | 9 369 523 |
| % Sur la cible (de UMPER dédupliquée) | 78 % | 77 % |
| Sur la cible (Total Bases mappées) Mb | 125 Mo | 128 Mo |
| Couverture de profondeur sur cible lecture moyenne (x) (ARDC) | 9 x | x 10 |
| 1 Métriques de séquençage basé sur les moyennes dans l’ensemble de sujets dans chaque groupe |
Tableau 2 : Séquençage paramètres obtenus à partir de la plate-forme de méthyl-Seq de rat.
| Chr | début | fin | gène | distance | areaStat | meanDiff | stress | contrôle | Direction |
| chr20 | 1 644 246 | 1 644 390 | RT1-M4 | in_gene | 93.03 | 0,22 | 0,33 | 0,11 | gain |
| chr5 | 160,361,352 | 160,361,564 | LOC690911 | in_gene | -70.75 | -0,19 | 0,72 | 0,91 | perte |
| CHR3 | 61,138,281 | 61,138,330 | RGD1564319 | 265569 | 61.79 | 0,21 | 0,94 | 0,72 | gain |
| CHR2 | 143,064,811 | 143,065,010 | UFM1 | 8569 | -59.48 | -0,11 | 0,13 | 0,24 | perte |
| chr7 | 30,764,111 | 30,764,284 | Ntn4 | in_gene | 57.04 | 0,21 | 0,94 | 0,73 | gain |
| chr17 | 12,469,112 | 12,469,218 | Idnk | 41996 | -50.91 | -0,13 | 0,74 | 0,88 | perte |
| chr7 | 47,101,725 | 47,101,930 | PAWR | in_gene | -50.54 | -0,12 | 0,64 | 0,76 | perte |
| chr5 | 76,111,248 | 76,111,822 | Txndc8 | 151703 | -50.38 | -0,11 | 0,85 | 0,96 | perte |
| chr11 | 80,640,132 | 80,640,356 | Dgkg | in_gene | -50.07 | -0,16 | 0,73 | 0,89 | perte |
| chr8 | 71,759,248 | 71,759,411 | Mir190 | 210226 | -47.84 | -0,17 | 0,58 | 0,75 | perte |
Tableau 3 : Top 10 méthylé différentiellement régions. Pour chaque DMR, la table de sortie montre de gauche à droite de la colonne de droite : localisation chromosomique (chr), coordonnées (début/fin), le nom de gène, distance de la transcription site, le statistique de la superficie différentielle entre a souligné et les témoins des groupes (areaStat), signifie méthylation différentielle (meanDiff), niveaux de méthylation moyenne sur l’ensemble de chaque DMR pour a souligné et groupes témoins (/ contrôle du stress) et la direction de méthylation changent de contrôles.
| Termes de voie KEGG | Comte de gène | % | P-valeur | Benjamini |
| Diabète | ||||
| Diabète sucré de type II | 12 | 0,1 | 3,6 x 10-4 | 9,8 x 10-3 |
| Maladies cardiovasculaires | ||||
| Contraction du muscle lisse vasculaire | 18 | 0,1 | 1.6 x 10-3 | 3,6 x 10-2 |
| Cardiomyopathie ventriculaire droite arythmogène (CVDA) | 13 | 0,1 | 4,0 x 10-3 | 7.1 x 10-2 |
| Cardiomyopathie dilatée | 14 | 0,1 | 7,6 x 10-3 | 1,2 x 10-1 |
| Fonction neuronale | ||||
| Potentialisation à long terme | 11 | 0,1 | 1.5 x 10-2 | 1. 4 x 10-1 |
| De signalisation | ||||
| Voie de signalisation MAPK | 35 | 0,2 | 2,4 x 10-4 | 9,9 x 10-3 |
| Voie de signalisation calcique | 22 | 0,1 | 1,2 x 10-2 | 1. 4 x 10-1 |
| Voie de signalisation de chimiokine | 21 | 0,1 | 1,2 x 10-2 | 1,3 x 10-1 |
| Cancer | ||||
| Voies du cancer | 42 | 0,3 | 4.1 x 10-5 | 3.4 x 10-3 |
| Gliome | 15 | 0,1 | 4.4 x 10-5 | 2,4 x 10-3 |
| Cancer du poumon non à petites cellules | 10 | 0,1 | 7,9 x 10-3 | 1,1 x 10-1 |
| Cancer colorectal | 13 | 0,1 | 8,4 x 10-3 | 1,1 x 10-1 |
| Leucémie myéloïde chronique | 12 | 0,1 | 1,2 x 10-2 | 1,3 x 10-1 |
Tableau 4 : Analyse KEGG voie de DMR identifié chez le rat méthyl-suiv.
Discussion
Dans cette étude, nous avons conçu et mis en œuvre de la plate-forme de méthyl-Seq pour le génome du rat. En faisant preuve de son utilité avec un modèle de rat du stress, nous avons démontré que le pipeline expérimental et analytique peut fournir des régions différentiellement méthylées entre deux groupes de comparaison.
Afin d’assurer une mise en œuvre de la plate-forme, plusieurs étapes critiques doivent être respectés. Première qualité de l’ADN et la quantité a un impact significatif sur la qualité et la quantité de la bibliothèque de méthyl-Seq finale. Nous avons utilisé un fluorimètre, plutôt que d’un spectrophotomètre, pour s’assurer que nos mesures d’ADN reflète la quantité d’ADN double brin. Le bioanalyzer a été utilisé pour mesurer la taille moléculaire et la quantité d’ADN après la tonte et après ligature de l’adaptateur. Vérifier la taille de la molécule « décalage » entre ces étapes est essentielle confirmer la présence des adaptateurs aux extrémités de chaque fragment d’ADN qui fera l’objet d’adaptateur-mediated PCR dans les étapes ultérieures. La quantité d’ADN à la fin de l’étape de la ligature d’adaptateur est également importante, depuis au moins 100 ng du produit bibliothèque est nécessaire à cette étape afin d’assurer une quantité suffisante est disponible après les étapes de conversion enrichissement et bisulfite de cible. Une mesure de haute sensibilité finale a été réalisée sur la bibliothèque de méthyl-Seq construite afin que la bibliothèque peut être correctement diluée pour le clustering ultérieures sur le séquenceur de prochaine génération. Enfin, bisulfite pyrosequencing travaillait comme une méthode très quantitative, indépendante pour évaluer la précision du pipeline analytique. La validation finale avec les échantillons et réplication à l’aide d’animaux supplémentaires d’origine sont des étapes cruciales pour assurer que l’expérience peut détecter des changements biologiquement significatifs dans la méthylation de l’ADN.
Nous incluons également plusieurs lignes directrices en cas d’écart par rapport au protocole ou si des problèmes sont rencontrés. Tout d’abord, il est possible de perdre trop d’ADN au cours de la fin-réparation, ligature de l’adaptateur ou étapes de purification des billes magnétiques. Par ailleurs, à partir des quantités d’ADN pourrait être petit (< 200 ng) en raison de la disponibilité limitée des tissus/ADN ou mise en œuvre des différentes méthodes d’enrichissement telles que le tri des cellules fluorescence activé. Augmentant le nombre de cycles au cours des étapes de l’amplification de deux bibliothèques peut être en mesure de compenser la perte excessive de l’ADN ou faible à partir de quantité d’ADN dans tout le protocole de construction de bibliothèque. Cependant, pas plus qu’un autre cycles 2 et 3 sont recommandés, comme l’amplification excessive modèle est susceptible d’entraîner une augmentation du nombre de lectures en double en cours de séquençage. Ces doublons sont exclus pendant l’étape de l’alignement afin d’éviter les biais dans le calcul du pourcentage de méthylation. Deuxièmement, si la taille moyenne des ADN n’augmente pas de plus de 30 points de base, vérifiez que les réactifs sont nouvelles, comme T4 ADN polymérase, Klenow et/ou T4 ligase peut être vieux. Réactifs de rechange disponibles dans le commerce peuvent être utilisés.
En outre, il est possible que le DMR prévue ne pourrait pas valider par pyrosequencing, où les différences de méthylation de l’ADN n’existent pas ou sont sensiblement inférieures à celles prédites par analyse. Validation pauvre des régions de candidat est un problème trop commun pour nombre d’analyses Génome-large, par exemple quand pyrosequencing résultats ne confirment pas la méthylation différentielle ou l’ampleur de l’effet est beaucoup plus petite que celle prédite par l’analyse. BSmooth est un paquet analytique qui « lisse » les niveaux de la méthylation dans une fenêtre des GPC multiples. Pour l’expérience en cours, BSmooth mis en cause un DMR dont les niveaux de méthylation ont été validés par le bisulfite pyrosequencing. Cependant, il y aura probablement des différences entre les niveaux de méthylation prévues par BSmooth et celles vérifiées par pyrosequencing. Les écarts résultent de la fonction de lissage qui permet d’estimer les valeurs moyennes de la méthylation dans l’ensemble de la GPC dans un DMR, y compris consécutifs GPC qui peut-être différer dans la méthylation de l’ADN de plus de 50 % ou GPC dont les valeurs de méthylation ont été exclus due à sous seuil lire la profondeur. R-paquets comme MethylKit24 peuvent servir à identifier les petites fenêtres de GPC ou même unique GPC qui le taux de méthylation corréler fortement avec ceux validés par pyrosequencing. Mise en œuvre de différents forfaits et tester leurs régions prévues ou GPC de méthylation différentielle par pyrosequencing assurera la solidité des données. Alternativement, bibliothèques de méthyl-Seq originales peuvent être reséquencées et ajoute les fichiers lus pour augmenter la profondeur. Étant donné que la détermination des niveaux de méthylation sont semi-quantitatifs et dicté par le nombre de lectures [(# of CpGs) / (nombre de TpGs + GPC)], augmentant la profondeur pour un CpG donnée augmentera la précision de sa valeur de pourcentage de méthylation. Dans cette étude, nous avons ne considéré que GPC dont les valeurs de méthylation ont été déterminées par au moins dix lectures et atteint une couverture globale de lecture de 19 x pour chaque CpG.
La plate-forme de méthyl-Seq de rat n’est pas sans ses limites. Bien qu’il soit plus rentable que le séquençage du génome entier bisulfite, c’est beaucoup plus cher que les autres méthodes. Néanmoins, la plupart des coûts a été pour l’achat de voies sur le séquenceur et non pour le système de capture. Selon la profondeur nécessaire, avec des comparaisons de croix-tissus nécessitant moins en raison de différences de grande (25 à 70 %)12 dans la méthylation de l’ADN, le coût peut être réduit par multiplexage plus d’échantillons par voie de circulation et à l’aide d’une plateforme de plus grande capacité. En outre, la préparation de l’échantillon est plus de temps que d’autres méthodes. Bien que similaire aux autres approches pulldown qui incorporent le séquençage de nouvelle génération, les étapes de purification et de conversion de bisulfite supplémentaire ajoutent à la charge de travail. Dans l’ensemble, la plate-forme de méthyl-Seq est une alternative rentable au séquençage du génome entier et offre une résolution paires de bases à plus de 2,3 millions de GPC, qui est beaucoup plus que ceux analysés par microarray-plateformes. À ce jour, l’homme disponibles sur le marché et la souris, méthyl-Seq plates-formes ont été utilisés pour documenter les changements d’alcooliques dans le macaque cerveau25,26, sur le développement neurologique des gènes dans le cerveau de souris9et hémato-encéphalique cibles de glucocorticoïdes10. En outre, la possibilité de cibler des régions spécifiques, indépendamment de la reconnaissance de séquences par des enzymes de restriction rend une plate-forme idéale pour les comparaisons entre les espèces. Pour cette étude, nous avons conçu la plate-forme méthyl-Seq pour le rat, pour laquelle de nombreuses expériences pharmacologiques, métaboliques et comportementales sont effectuées sans le bénéfice d’un outil de Génome-large méthylomiques. Nos résultats montrent qu’il peut être utilisé pour détecter les DMR dans un modèle de rat de stress et autres paramètres physiologiques tels que Global plasmatique CORT en corrélation.
La plateforme de méthyl-Seq est idéale pour des expériences épigénétiques chez les animaux avec des génomes séquencés qui ne disposent pas de suffisamment de preuves expérimental documentant les régions régulatrices. Lorsque ces régions sont mises à disposition, des régions supplémentaires peuvent être personnalisés et attachées à la version actuelle. En outre, la plateforme est idéale pour la génomique comparative, étant donné que l’enrichissement de la cible n’est pas limité par la reconnaissance de l’enzyme de restriction. Par exemple, la région du promoteur d’un gène d’intérêt peut être capturée indépendamment de savoir si elle abrite un site de restriction spécifique. De même, les régions régulatrices, comme celles de la souris ou des humains, qui sont conservées dans le génome d’intérêt peuvent être capturées.
Déclarations de divulgation
Le manuscrit fait partie d’un prix du concours d’Agilent Technologies.
Remerciements
Cette étude a été financée par les NIH grant MH101392 (RSL) et de soutien des prix et fondations suivantes : un NARSAD Young Investigator Award, Margaret Ann prix enquêteur, le James Wah Mood Disorders érudit fonds via le Charles T. Bauer Foundation, Baker Foundation et la projet Match Foundation (RSL).
matériels
| Name | Company | Catalog Number | Comments |
| Radioimmuno assay (RIA) | MP Biomedicals | 7120126 | Corticosterone, 125I labeled |
| Master Pure DNA Purification Kit | Epicentre/Illumina | MC85200 | |
| Thermal-LOK 2-Position Dry Heat Bath | USA Scientific | 2510-1102 | Used with 1.5 mL tubes |
| Vortex Genie 2 | Fisher | 12-812 | Vortex Mixer |
| Ethyl alcohol, Pure | Sigma-Aldrich | E7023 | 100% Ethanol, molecular grade |
| Centrifuge 5424 R | Eppendorf | - | Must be capable of 20,000 x g |
| Qubit 2.0 | ThermoFisher Scientific | Q32866 | Fluorometer |
| Qubit dsDNA BR Assay Kit | ThermoFisher Scientific | Q32850 | |
| Qubit dsDNA HS Assay Kit | ThermoFisher Scientific | Q32851 | High sensitivity DNA detection reagents |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| SureSelectXT Rat Methyl-Seq Reagent Kit | Agilent Technologies | G9651A | Reagents for preparing the Methyl-Seq library |
| SureSelect Rat Methyl-Seq Capture Library | Agilent Technologies | 931143 | RNA baits for enrichment of rat targets |
| IDTE, pH 8.0 | IDT DNA | 11-05-01-09 | 10 mM TE, 0.1 mM EDTA |
| DNA LoBind Tube 1.5 mL | Eppendorf | 22431021 | |
| Covaris E-series or S-series | Covaris | - | Isothermal sonicator |
| microTUBE AFA Fiber Pre-Slit Snap-Cap 6x16mm (25) | Covaris | 520045 | |
| Water, Ultra Pure (Molecular Biology Grade) | Quality Biological | 351-029-721 | |
| Veriti 96 Well-Thermal Cycler | Applied Biosystems | 4375786 | |
| AMPure XP Beads | Beckman Coulter | A63880 | DNA-Binding magnetic beads |
| 96S Super Magnet | ALPAQUA | A001322 | Magnetic plate for purification steps |
| 2200 TapeStation | Agilent Technologies | G2965AA | Electrophresis-based bioanalyzer |
| D1000 ScreenTape | Agilent Technologies | 5067-5582 | |
| D1000 ScreenTape High Sensitivity | Agilent Technologies | 5067-5584 | |
| D1000 Reagents | Agilent Technologies | 5067-5583 | |
| D1000 Reagents High Sensitivity | Agilent Technologies | 5067-5585 | |
| DNA110 SpeedVac | ThermoFisher Scientific | - | Vacuum Concentrator |
| Dynabeads MyOne Streptavidin T1 magnetic beads | Invitrogen | 65601 | Streptavidin magnetic beads |
| Labquake Tube Rotator | ThermoFisher Scientific | 415110Q | Nutator Mixer is also acceptable |
| EZ DNA Methylation-Gold Kit | Zymo Research | D5006 | Bisulfite conversion kit. Contains Binding, Wash, Desulphonation, and Elution buffers |
| Illumina Hi-Seq 2500 | Illumina | - | Next-generation sequencing machine |
| PCR and Pyrosequencing Primers | IDT DNA | Variable | |
| Taq DNA Polymerase with ThermoPol Buffer - 2,000 units | New England BioLabs | M0267L | |
| Deoxynucleotide (dNTP) Solution Set | New England BioLabs | N0446S | |
| Pyromark MD96 | QIAGEN | - | Pyrosequencing machine |
| Ethyl Alcohol 200 Proof | Pharmco-Aaper | 111000200 | 70% Ethanol solution |
| Sodium Hydroxide Pellets | Sigma-Aldrich | 221465 | 0.2 M NaOH denature buffer solution |
| Tris (Base) from J.T. Baker | Fisher Scientific | 02-004-508 | 10 mM Tris Acetate Buffer wash buffer solution |
| PyroMark Gold Q96 Reagents (50x96) | QIAGEN | 972807 | Reagents required for pyrosequencing |
| PyroMark Annealing Buffer | QIAGEN | 979009 | |
| PyroMark Binding Buffer (200 mL) | QIAGEN | 979006 | |
| Streptavidin Sepharose High Performance Beads | GE Healthcare | 17-5113-01 | Streptavidin-coated sepharose beads |
| PyroMark Q96 HS Plate | QIAGEN | 979101 | Pyrosequencing assay plate |
| Eppendorf Thermomixer R | Fisher Scientific | 05-400-205 | Plate mixer. 96-well block sold separately (cat. No 05-400-207) |
| SureDesign Website | Agilent Technologies | - | Target capture design software (https://earray.chem.agilent.com/suredesign/) |
| UCSC Genome Browser | University of California Santa Cruz | - | rat Nov 2004 rn4 assembly |
| Agilent Methyl-Seq Protocol | Agilent Technologies | - | https://www.agilent.com/cs/library/usermanuals/public/G7530-90002.pdf |
Références
- Barski, A., et al. High-resolution profiling of histone methylations in the human genome. Cell. 129 (4), 823-837 (2007).
- Meissner, A., et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 454 (7205), 766-770 (2008).
- Bibikova, M., et al. High density DNA methylation array with single CpG site resolution. Genomics. 98 (4), 288-295 (2011).
- Naumov, V. A., et al. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics. 8 (9), 921-934 (2013).
- Wockner, L. F., et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Translational Psychiatry. 4, e339 (2014).
- Meissner, A., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Research. 33 (18), 5868-5877 (2005).
- Smith, Z. D., Gu, H., Bock, C., Gnirke, A., Meissner, A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 48 (3), 226-232 (2009).
- Slieker, R. C., et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 6 (1), 26 (2013).
- Hing, B., et al. Adaptation of the targeted capture Methyl-Seq platform for the mouse genome identifies novel tissue-specific DNA methylation patterns of genes involved in neurodevelopment. Epigenetics. 10 (7), 581-596 (2015).
- Seifuddin, F., et al. Genome-wide Methyl-Seq analysis of blood-brain targets of glucocorticoid exposure. Epigenetics. 12 (8), 637-652 (2017).
- Irizarry, R. A., et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genetics. 41 (2), 178-186 (2009).
- Lee, R. S., et al. Adaptation of the CHARM DNA methylation platform for the rat genome reveals novel brain region-specific differences. Epigenetics. 6 (11), 1378-1390 (2011).
- Jankord, R., et al. Stress vulnerability during adolescent development in rats. Endocrinology. 152 (2), 629-638 (2011).
- Pellow, S., Chopin, P., File, S. E., Briley, M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. Journal of Neuroscience Methods. 14 (3), 149-167 (1985).
- Krueger, F., Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27 (11), 1571-1572 (2011).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 10 (3), R25 (2009).
- Hansen, K. D., Langmead, B., Irizarry, R. A. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biology. 13 (10), R83 (2012).
- Lee, R. S., et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology. 151 (9), 4332-4343 (2010).
- Lee, R. S., et al. A measure of glucocorticoid load provided by DNA methylation of Fkbp5 in mice. Psychopharmacology. , (2011).
- Bose, M., Olivan, B., Laferrere, B. Stress and obesity: the role of the hypothalamic-pituitary-adrenal axis in metabolic disease. Current Opinion in Endocrinology, Diabetes, and Obesity. 16 (5), 340-346 (2009).
- Brydon, L., Magid, K., Steptoe, A. Platelets, coronary heart disease, and stress. Brain, Behavior, and Immunity. 20 (2), 113-119 (2006).
- McKlveen, J. M., et al. Chronic Stress Increases Prefrontal Inhibition: A Mechanism for Stress-Induced Prefrontal Dysfunction. Biological Psychiatry. 80 (10), 754-764 (2016).
- Akalin, A., et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biology. 13 (10), R87 (2012).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Alcohol-dose-dependent DNA methylation and expression in the nucleus accumbens identifies coordinated regulation of synaptic genes. Translational Psychiatry. 7 (1), e994 (2017).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Genome-wide analysis of the nucleus accumbens identifies DNA methylation signals differentiating low/binge from heavy alcohol drinking. Alcohol. 60, 103-113 (2017).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.