
Method Article
Applications in situ de l’hybridation in situ de l’ARN viral et de l’ADN de nouvelle génération dans la recherche sur le virus de l’immunodéficience humaine et le virus de l’immunodéficience simienne
Dans cet article
Résumé
Nous présentons ici un test d’hybridation in situ de nouvelle génération pour identifier des séquences spécifiques d’ARN ou d’ADN viral dans des tissus inclus dans la paraffine fixée au formol (FFPE). Cette approche permet de visualiser de faibles copies d’ARN et d’ADN en moins de 24 h avec une sensibilité et une spécificité très élevées.
Résumé
L’hybridation in situ est une technique puissante pour identifier des séquences d’ARN ou d’ADN spécifiques dans des cellules individuelles dans des coupes de tissus, fournissant des informations importantes sur les processus physiologiques et la pathogenèse des maladies. L’hybridation in situ (ISH) est utilisée depuis de nombreuses années pour évaluer l’emplacement des cellules infectées par des virus, mais récemment, une approche ISH de nouvelle génération a été développée avec une stratégie de conception de sonde unique qui permet l’amplification du signal et la suppression simultanée du bruit de fond pour obtenir une visualisation d’une seule molécule tout en préservant la morphologie des tissus. Cette ISH de nouvelle génération est basée sur une approche similaire à la PCR ramifiée, mais réalisée in situ et est plus facile, sensible et reproductible que les méthodes ISH classiques ou les approches PCR in situ pour détecter régulièrement l’ARN ou l’ADN dans les tissus inclus dans la paraffine fixée au formol (FFPE). Au cours des dernières années, notre laboratoire a utilisé cette plateforme ISH pour la détection de cellules positives à ARN viral (ARNv) et/ou à ADN viral (ADNv) dans une multitude de tissus FFPE positifs pour le déficit humain (VIH) et l’immunodéficience simienne (SIV). Avec ce manuscrit technique détaillé, nous aimerions partager nos connaissances et nos conseils avec toutes les personnes intéressées à utiliser l’ISH de nouvelle génération dans leurs recherches.
Introduction
L’ISH est l’approche expérimentale utilisée pour cibler et visualiser des brins d’ADN, d’ARN ou d’acides nucléiques modifiés complémentaires (c’est-à-dire des sondes) à des séquences d’ADN ou d’ARN spécifiques au sein d’une cellule ou d’une section de tissu. L’ISH permet la localisation et la visualisation spécifiques de séquences nucléiques spécifiques dans les tissus, ce qui est important pour comprendre le niveau d’expression, l’organisation, la distribution et les interactions entre la cible et son environnement cellulaire, ce qui constitue une information précieuse qui ne peut être obtenue avec l’utilisation d’autres techniques populaires, telles que la qPCR. Jusqu’à récemment, l’ISH était couramment réalisée avec un ADN complémentaire marqué ou un ARN complémentaire (riboprobe). Ces sondes ont été soit conjuguées directement avec des bases marquées par radio, par fluorescence ou par antigène (par exemple, 35 S, FITCet digoxigénine), puis localisées et quantifiées dans le tissu à l’aide d’approches d’autoradiographie, de microscopie à fluorescence ou d’immunohistochimie, respectivement. Bien que ces technologies in situ continuent d’être des approches précieuses, il y a encore beaucoup à faire pour développer des approches moins exigeantes en main-d’œuvre, plus simples, plus rapides, sensibles et spécifiques.
Une autre approche commerciale d’ISH de nouvelle génération (p. ex., le test RNAscope), décrite pour la première fois en 2012, pour la détection de l’ARN messager de l’hôte (ARNm) est basée sur la PCR ramifiée. La détection de l’ARNm est effectuée dans les cellules et les tissus FFPE, avec une sensibilité proche de la visualisation de molécules d’ARN unique dans des cellules individuelles1. La spécificité de cette approche est obtenue à la condition unique que deux sondes cibles double-Z se lient de manière contiguë à leurs séquences d’ARN (ou d’ADN) complémentaires respectives pour qu’un préamplificateur de signal se lie séquentiellement à1. Cela permet d’initier une cascade d’amplification du signal via des étapes d’hybridation ultérieures similaires à l’ADN ramifié (ADNb)1,2. De plus, cette approche est remarquablement rapide et facile, avec des résultats obtenus en seulement 1 jour (<8 h), un avantage significatif par rapport à jusqu’à 4 semaines avec des techniques alternatives, y compris Radio-ISH 1,2. Cette ISH de nouvelle génération a ouvert de nouvelles perspectives et opportunités pour la recherche sur le VIH/SIV. Les principaux obstacles à la guérison du VIH sont les réservoirs cellulaires et tissulaires qui s’établissent au cours des premiers stades de la maladie 3,4. L’objectif global de cette technique est d’identifier, de localiser et, en fin de compte, de comprendre les principaux compartiments tissulaires qui agissent comme un réservoir viral et qui persistent dans un hôte infecté. Cela contribuera à son tour à l’élaboration de stratégies de traitement efficaces contre le VIH.
Dans ce manuscrit, nous expliquons en détail notre protocole ISH multiplex ARN/ADN duplex de nouvelle génération (par exemple, RNAscope/DNAscope) et expliquons comment nous avons modifié le protocole ISH RNA existant pour optimiser l’ISH de nouvelle génération pour nos échantillons et cibles spécifiques. Ce protocole permet la visualisation, la localisation et la quantification de l’ARN viral et de l’ADN viral du VIH/VIS dans des coupes de tissus de 5 μm. La visualisation simultanée de l’ARNv et de l’ADNv est réalisée en combinant deux ensembles de sondes personnalisées : l’une détective, ciblant le brin codant de l’ADNv (sonde C1 SIVmac239 Gag-Pol-Sense [416141-C1]), et l’autre antisens, ciblant les transcrits d’ARNv (sonde C2 SIVmac239 Vif-Env-Nef-Tar-Anti-Sense [416131-C2]) couvrant différentes régions du génome viral (Tableau 1), à l’aide de deux canaux de visualisation différents, C1 et C2. Dans ce protocole, les canaux C1 et C2 nous permettent de visualiser les signaux de différentes couleurs (c’est-à-dire AP en rouge et HRP en marron) et de détecter les sondes avec différentes approches. À l’exclusion du traitement de la fixation des tissus et de la coupe, ce test prend 2 jours. Voici le protocole d’hybridation in situ de l’ARNv duplex et de l’ADNv qui peut être réalisé sur des cuillers cellulaires ou des coupes de tissus.
Protocole
1. Préparation des sections et des lames
- Coupez les blocs de paraffine et utilisez un microtome pour couper 5 sections de +/-1 μm. Montez les coupes ou les pastilles de cellules sur des lames de microscope chargées dans un bain d’eau sans RNase à 40-45 °C. Sécher les diapositives à l’air libre pendant la nuit à 37 °C ou RT.
REMARQUE : Les lames peuvent être stockées jusqu’à 3 mois à température ambiante (RT) et 6 mois à 4 °C. - Déparaffiniser les lames FFPE.
- Faites cuire les lames dans un four sec pendant 1 h à 60 °C.
- Dans une hotte, remplissez deux plats de coloration de lames avec ~200 mL de xylène frais, et deux plats de coloration supplémentaires avec ~200 mL d’éthanol frais à 100 %. Couvrez les récipients avec des couvercles.
- Placez les lames sur une grille et plongez-les dans le premier plat contenant du xylène. Incuber pendant 5 à 10 min à RT avec agitation.
- Placez les lames dans le deuxième plat contenant du xylène et incubez pendant 5 à 10 minutes à RT en agitant.
- Placez immédiatement les lames dans le plat contenant 100 % d’éthanol. Incuber les lames pendant 5 à 10 minutes à RT avec agitation.
- Placez immédiatement les lames dans le deuxième plat contenant 100 % d’éthanol et incubez pendant 5 à 10 min à RT avec agitation.
- Retirez la grille de l’éthanol, tapotez doucement le côté de la grille pour éliminer l’excès d’éthanol et rincez à l’eau libre RNase pendant 5 à 15 minutes.
2. Préparation au four
- Allumez le four d’hybridation et réglez la température à 40 °C.
- Placez un chiffon ou une serviette en papier absorbante solide dans un plateau et mouillez complètement avec de l’eau doublement distillée pour permettre le contrôle de l’humidité.
- Insérez la plaque couverte dans le four et fermez la porte du four. Réchauffez la plaque pendant au moins 30 min à 40 °C avant utilisation. Conservez la plaque dans le four lorsqu’elle n’est pas utilisée.
3. Récupération d’épitopes induite par la chaleur
- Préparez un tampon de récupération de cible d’hybridation ISH à base de citrate 0,5x (10 nmol/L, pH = 6, voir le tableau des matériaux). Portez-le à ébullition dans un bécher sur la plaque chauffante.
- Effectuez la récupération d’épitopes induite par la chaleur en plaçant les lames dans le tampon de récupération de la cible d’ébullition pendant 30 min.
- Retirez les lames du tampon de récupération de la cible et lavez-les immédiatement à l’eau bidistillée. Déshydrater dans de l’éthanol à 100 % pendant 5 min avant de sécher à l’air.
- Une fois que les lames ont séché à l’air, appliquez un stylo barrière hydrophobe pour entourer la section de tissu sur la lame. Assurez-vous de laisser la barrière hydrophobe sécher complètement à l’air.
4. Prétraitement de la protéase
- Placez les lames séchées sur un support à lames verrouillable, puis préparez les réactifs de prétraitement de la protéase (solution de digestion de la protéase, 2,5 μg/mL) en les diluant avec du PBS stérile et froid dans un rapport de 1:5. Bien mélanger.
REMARQUE : Trois réactifs de protéase différents avec des concentrations différentes sont fournis dans le kit disponible dans le commerce. Protéase III (standard), Protéase IV (forte) et Protéase Plus (légère). Testez empiriquement le temps de digestion et la dilution de la protéase avant de la mettre en œuvre dans une étude, car les conditions optimales varient en fonction du type de tissu, de la fixation et de l’épaisseur (voir Discussion). - Distribuez la solution de protéase diluée sur les lames pour couvrir complètement les sections de tissu. Incuber immédiatement les lames pendant 20 min à 40 °C dans un four (préparé à l’étape 1.4), en veillant à ce que les lames soient scellées dans le bac d’hybridation humide. Ne laissez pas les coupes de tissus se dessécher pour le reste du protocole.
- Rincez immédiatement 3 fois en immergeant le support coulissant verrouillable dans un bac de lavage rempli d’eau doublement distillée.
- Effectuez le bloc endogène de la peroxydase en déposant la solution de peroxydase sur chaque section de tissu pour la recouvrir complètement. Incuber les lames pendant 10 min à RT. Une fois cela fait, rincez les sections 3x dans de l’eau doublement distillée.
5. Hybridation de sonde et amplification du signal
REMARQUE : Pour éviter l’évaporation, assurez-vous que le plateau à humidité contrôlée se ferme correctement afin que les tissus ne se dessèchent pas pendant les étapes d’incubation. Remettez la chambre d’humidité dans le four pendant l’étape de lavage pour qu’elle reste à 40 °C.
- Mélangez la sonde C2 et la sonde C1 dans un rapport de 1:50 en pipetant 1 volume de sonde C2 à 50 volumes de sonde C1 dans un tube comme suggéré par le fabricant. Retournez le tube plusieurs fois. Préchauffez le mélange de sonde cible dans un four à 40 °C pendant ~10 min pour dissoudre toute précipitation avant utilisation.
REMARQUE : Les sondes cibles mixtes peuvent être stockées à 4 °C jusqu’à 6 mois. - Retirez les lames de l’eau. Rincez et tapotez ou effleurez les lames pour éliminer l’excès d’eau des sections de tissus. Distribuez immédiatement la sonde sur les lames, en vous assurant que chaque section de tissu est complètement recouverte sans bulles d’air. Incuber le mélange de sonde dans la chambre d’humidité pendant une nuit à 40 °C.
- Le lendemain, sortez les lames du four et placez-les dans le bac de lavage contenant 0,5x tampon de lavage pendant 5 min à RT. Répétez l’étape de lavage une fois de plus.
- Retirez les lames du tampon de lavage. Rincez et tapotez ou effleurez les lames pour enlever l’excès de tampon de lavage des sections de tissu.
- Distribuer commercialement le réactif AMP 1 prêt à l’emploi (2 nmol/L) dans le tampon d’hybridation B (20 % de formamide, 5x SSC, 0,3 % de sulfate de lithiumdodécyle, 10 % de sulfate de dextran, réactifs bloquants) sur les lames, en assurant une couverture complète de la section tissulaire sans aucune bulle d’air. Incuber pendant 30 min à 40 °C dans la chambre d’humidité. Répétez les étapes 1.6.3-1.6.4 en effectuant le lavage pendant 2 minutes chacune.
- Distribution disponible dans le commerce AMP 2. Assurez-vous que la section du tissu est complètement recouverte sans bulles d’air. Incuber les lames dans la chambre d’humidité pendant 15 min à 40 °C. Répétez les étapes 5.3-5.4, en effectuant le lavage pendant 2 minutes chacune.
- Distribution disponible dans le commerce AMP 3. Assurez-vous que la section du tissu est complètement recouverte sans bulles d’air. Incuber les lames dans la chambre d’humidité pendant 30 min à 40 °C. Répétez les étapes 5.3-5.4, en effectuant le lavage pendant 2 minutes chacune.
- Distribution disponible dans le commerce AMP 4. Assurez-vous que la section du tissu est complètement recouverte sans bulles d’air. Incuber les lames dans la chambre d’humidité pendant 15 min à 40 °C. Répétez les étapes 5.3-5.4, en effectuant le lavage pendant 2 minutes chacune.
- Distribution disponible dans le commerce AMP 5. Assurez-vous que la section du tissu est complètement recouverte sans bulles d’air. Incuber les lames dans la chambre d’humidité pendant 30 min à 40 °C. Répétez les étapes 5.3-5.4 en effectuant le lavage pendant 2 minutes chacun.
- Distribution disponible dans le commerce AMP 6. Assurez-vous que la section du tissu est complètement recouverte sans bulles d’air. Incuber les lames dans la chambre d’humidité pendant 15 min à 40 °C. Répétez les étapes 5.3-5.4 en effectuant le lavage pendant 2 minutes chacun.
- Avant la détection, rincez les lames une fois dans 1x TBS-Tween 20 (0,05 % v/v). Retirez les lames du tampon de lavage, rincez et tapotez ou effleurez les lames pour éliminer l’excès de tampon de lavage des sections de tissu. Placez immédiatement dans le bac de lavage rempli de 1x tampon TBS-Tween.
6. Détection du signal cible du canal 1 (C1)
REMARQUE : Ceci est effectué à l’aide de la phosphatase alcaline rouge et de l’amplification chromogène rouge rapide 6 à partir de kits de détection 2-plex (voir le tableau des matériaux) contenant des marqueurs de phosphatase alcaline et la détection chromogène. Il utilise le rouge rapide comme substrat pour générer un signal rouge.
- Préparez une solution de travail rapide en utilisant une dilution de 1:60 de Fast RED-B en Fast RED-A. Bien mélanger. Pour réduire le précipité et obtenir un signal plus propre, filtrez la solution chromogène à travers une membrane MCE de 0,45 μm à l’aide d’une seringue.
REMARQUE : Utilisez la solution Fast RED-B dans les 5 min. Ne pas exposer à la lumière directe du soleil ou à la lumière UV. - Retirez les lames du TBS-Tween, rincez-les et tapotez ou effleurez les lames pour éliminer l’excès de tampon des sections de tissus.
- Versez une solution ignifuge mélangée et filtrée sur chaque section de tissu, en vous assurant que chaque section est complètement recouverte. Incuber à RT pendant 6-8 min. Observer au microscope.
- Rincez les lames dans un tampon de lavage 0,5x 2x. Retirez les lames du tampon de lavage, rincez et tapotez ou effleurez les lames pour éliminer l’excès de tampon de lavage des sections de tissu.
- Distribution disponible dans le commerce AMP 7. Assurez-vous que la section du tissu est complètement recouverte sans bulles d’air. Incuber les lames dans la chambre d’humidité pendant 10 min à 40 °C. Répétez les étapes 5.3-5.4 en effectuant le lavage pendant 2 minutes chacun.
- Distribution disponible dans le commerce AMP 8. Assurez-vous que la section du tissu est complètement recouverte sans bulles d’air. Incuber les lames dans la chambre d’humidité pendant 15 min à 40 °C. Répétez les étapes 5.3-5.4 en effectuant le lavage pendant 2 minutes chacun.
- Distribution disponible dans le commerce AMP 9. Assurez-vous que la section du tissu est complètement recouverte sans bulles d’air. Incuber les lames dans la chambre d’humidité pendant 30 min à 40 °C. Répétez les étapes 5.3-5.4 en effectuant le lavage pendant 2 minutes chacun.
- Distribution disponible dans le commerce AMP 10. Assurez-vous que la section du tissu est complètement recouverte sans bulles d’air. Incuber les lames dans la chambre d’humidité pendant 30 min à 40 °C. Répétez les étapes 5.3-5.4 en effectuant le lavage pendant 2 minutes chacun.
7. Détection du signal cible du canal 2 (C2)
REMARQUE : Ceci est effectué à l’aide de kits chromogènes Brown HRP et DAB disponibles dans le commerce (voir le tableau des matériaux). L’amplification 10 de la détection 2-plex contient des marqueurs de peroxydase de raifort, et la détection chromogène est effectuée à l’aide de DAB pour générer un signal brun.
- Pour une détection optimale du signal DAB, utilisez des kits disponibles dans le commerce et suivez les instructions du fabricant (voir la table des matériaux). Observez au microscope.
ATTENTION : Le DAB est toxique. Suivez les précautions appropriées et les directives de sécurité lors de la manipulation et de l’élimination de ce produit chimique.
8. Contre-coloration et montage
- Contre-colorez les lames avec de l’hématoxyline.
- Contre-colorez les lames pendant 30 s avec 50 % d’hématoxyline fraîche filtrée tout en agitant les lames. Les diapositives apparaîtront en violet. Rincez immédiatement à l’eau courante tout en agitant les lames de haut en bas jusqu’à ce que l’eau soit claire. Les sections de tissu resteront violettes.
- Pour obtenir un meilleur contraste, placez les lames contre-colorées dans de l’eau distillée saturée de carbonate de lithium pendant 1 min. Rincez abondamment à l’eau courante au moins 3 fois tout en agitant les lames. Utilisez de l’eau bi-distillée pour le rinçage final.
- Étant donné que le FR est sensible aux solvants organiques, les lames colorées au FR devront être recouvertes d’un support de montage à base d’eau et séchées pendant la nuit à RT.
- Montez les diapositives.
- Assurez-vous que les sections de tissu recouvertes d’un support de montage à base d’eau sont sèches.
- Tremper les lames dans du xylène avant de glisser le couvercle à l’aide du réactif de montage. Assurez-vous d’éviter ou d’éliminer toute bulle d’air entre la lamelle et la section du tissu et laissez sécher pendant 16 heures à RT.
9. Protocole d’analyse d’images quantitatives pour RNAscope à l’aide de CellProfiler5
- En bref, assurez-vous que le logiciel séparera les colorants d’hématoxyline et de FR en images distinctes. Identifier et mesurer les objets d’intérêt : noyaux, virions, cellules positives, et coloration positive FR agrégée. Enregistrez les mesures dans un fichier CSV et enregistrez l’image analysée.
- Sélectionnez l’option « Démélanger les couleurs », ce qui sépare les taches, divisant la région d’intérêt (ROI) d’origine en images distinctes d’hématoxyline et de FR.
- Si l’hémosidérine, le tatouage ou des caractéristiques similaires interfèrent avec l’analyse, ajoutez une deuxième étape de « démélange » avec de l’hématoxyline, du FR et du DAB. Utilisez la deuxième image FR pour trouver les pixels FR les plus intenses. Cette deuxième image sera utilisée comme masque pour une véritable coloration ignifuge.
- Si vous le souhaitez, lissez les images tachées avant de les seuiller. Il s’agit d’une décision empirique qui n’est pas toujours nécessaire. Seuilz les images colorées individuelles à l’aide de « IdentifyPrimaryObjects » pour sélectionner les pixels positifs.
- Identifier les trois différents types d’objets (virion, agrégats de virions, cellule productive).
- Assurez-vous que les noyaux sont des objets de 4 à 100 pixels (px) de diamètre colorés à l’hématoxyline. Délumper et remplir les trous après le seuillage. Les objets « IntenseFastRed » ont un diamètre de 4 à 100 px et comprennent des virions, des cellules positives et une coloration positive agrégée, comme on le voit sur les cellules dendritiques folliculaires (FDC) dans les follicules des cellules B (BCF). Cette image est utilisée pour filtrer les faux positifs (par exemple, l’hémosidérine).
- Assurez-vous que les petits positifs FR sont des objets de 2 à 12 pixels de diamètre. Cette mesure comprend les virions et les cellules vDNA+. Jetez tous les objets en dehors de cette plage. Délumper et remplir les trous après le seuillage.
- Assurez-vous que les objets de 9 à 100 pixels de diamètre sont de grands positifs. Cette mesure comprend les cellules positives à l’ARNv+ et la coloration positive agrégée. Dégroupage après seuil. La taille des petits et des grands objets FR positifs peut se chevaucher. Ils seront séparés dans une étape ultérieure.
REMARQUE : Le logiciel (par exemple, Cellprofiler) utilise des pixels pour la taille des objets, et ceux utilisés ici sont dérivés de diapositives numérisées à 0,2510 μm/pixel (40x).
- Définir et extraire les résultats.
- Une fois les objets identifiés, définissez et extrayez les résultats pour le nombre de virions, de cellules infectées productives et de noyaux.
- Identifiez les virions en masquant FastRedSmallPositives à l’aide de l’objet IntenseFastRed configuré pour supprimer les faux positifs (par exemple, l’hémosidérine).
- Ensuite, identifiez les cellules positives et agrégez la coloration positive FR. Supprimez les faux positifs et les virions de FastRedLargePositives en le masquant avec IntenseFastRed et avec l’objet virion défini.
- Extrayez les cellules positives du FastRedLargePositives raffiné en masquant à nouveau avec des noyaux. Divisez les objets qui ne se touchent plus et filtrez les résultats par zone d’objet, en supprimant les petits objets (≤6 px). Cela permet d’éliminer les taches créées en masquant le chevauchement des noyaux. Le résultat est les cellules positives.
- Enfin, définissez les résultats positifs globaux des FR. Cette étape utilise IdentifyTertiaryObjects, qui recherche les objets contenus dans un objet parent plus grand. Dans ce cas, l’ensemble d’objets FastRedLargePositives affiné est le parent et les cellules positives sont soustraites.
- Comptez le nombre de virions et de cellules positives.
- Mesurez la surface positive globale et convertissez-la de pixels en mm2. Optionnellement, enregistrer la zone occupée par les virions et les cellules positives en mm2 si l’analyse nécessite l’utilisation d’une cellule standard et de tailles de virions au lieu de comptages directs.
- Superposez les objets positifs sur l’image d’origine et enregistrez le résultat.
REMARQUE : Les données ISH sont rapportées en fonction du nombre de virions par 106 noyaux (cellules) et du nombre de cellules vRNA+ infectées de manière productive par 106 noyaux (cellules) pour une meilleure compréhension et pour faciliter la comparaison avec les données de qPCR, mais les résultats pourraient également être rapportés par zone de tissu en mm2.
Résultats
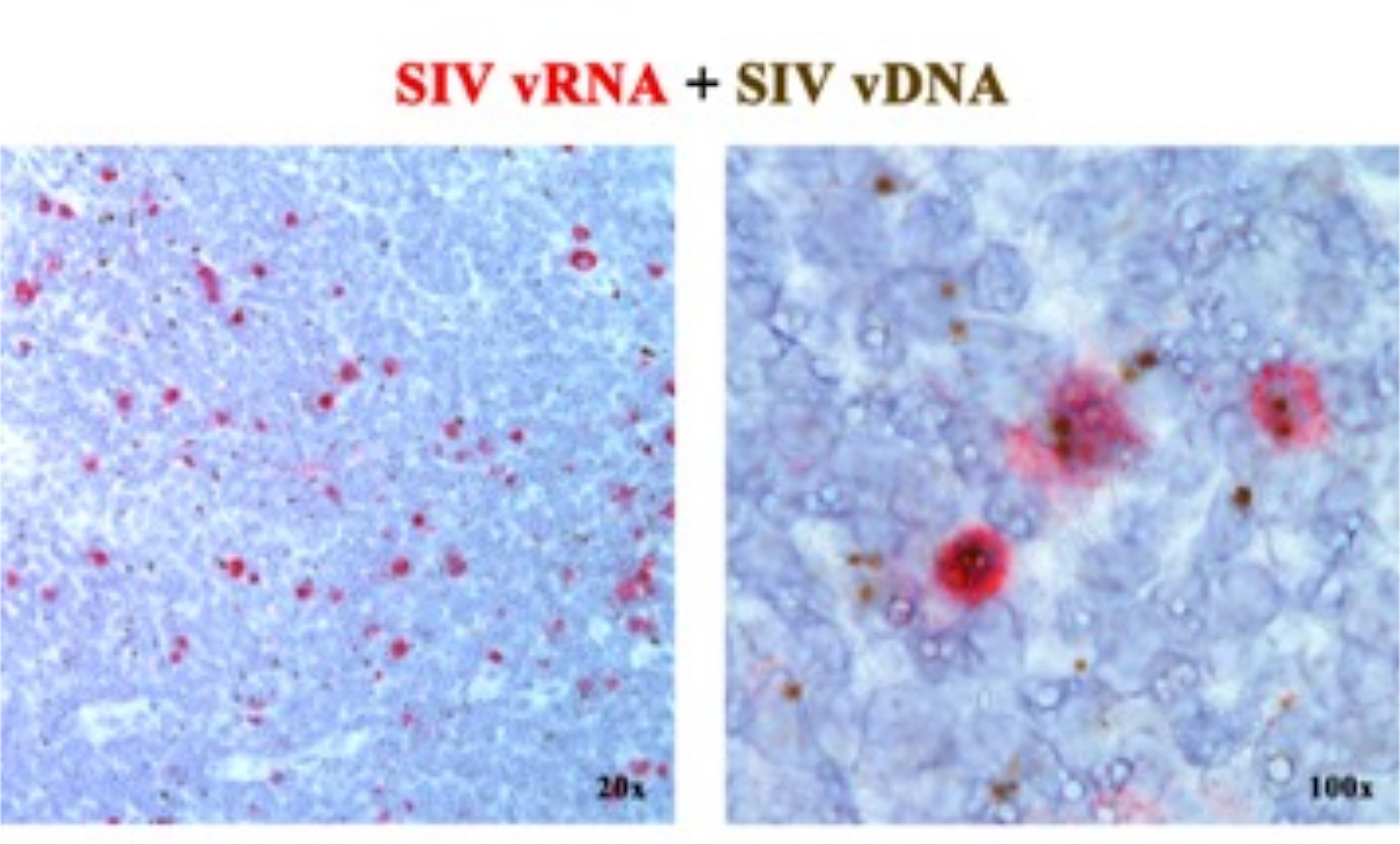
Dans un manuscrit précédent 2,6,7,8,9,10,11, nous avons rapporté que les plateformes ISH de nouvelle génération détectant soit l’ARNv, soit l’ADNv, peuvent être combinées, à l’aide de sondes sensorielles (ADNv) ciblant la partie 5' gag-pol du génome SIV/VIH et de sondes antisens (ARNv) ciblant les gènes de la moitié 3' du génome (vif, vpx, vpr, tat, env et nef) ainsi que l’élément TAR dans le génome 5' (tableau 1). Cette approche distingue les cellules transcriptionnellement actives (vRNA+, vDNA+) des cellules infectées transcriptionnellement inactives (présumées latentes) ou des cellules hébergeant des provirus transcriptionnellement incompétents (vRNA-, vDNA+) dans la même sectiontissulaire 2 (Figure 1).

Figure 1 : Détection de l’ARN viral et de l’ADNv dans la même section de tissu. Combinaison d’un test d’hybridation d’ARN (rouge) et d’hybridation d’ADN (brun) dans un ganglion lymphatique RM infecté par le SIV aiguë, démontrant la capacité de détecter l’ARNv et l’ADNv dans la même section de tissu et fournissant une approche puissante pour identifier in situ les cellules d’ADNv+ vRNA+ transcriptionnellement silencieuses. Cette figure a été modifiée de Deleage C. et al.2. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
| Ensembles de sondes ARN/DNAscope Plex simples | |||
| Nom | Catalogue ACD # | Nombre de ZZ | Description |
| SIVmac239 (Anti-Sens) | 312811 | 83 | Ciblage de la sonde antisens entre 1251 et 9420 pb de D01065.1 (gag, pol, vif, vpx, vpr, tat, env et nef) |
| SIVmac239 (Sense) | 314071 | 83 | Sonde Sense ciblant le brin inverse entre 1251 et 9420 pb de D01065.1 (gag, pol, vif, vpx, vpr, tat, env et nef) |
| V-HIV1-Clade A (anti-sens) | 416101 | 80 | Sonde antisens ciblant entre 879 et 7629 pb du consensus du clade A du VIH-1 (gag, pol, vif, vpr, tat, rev, vpu, env et nef) |
| V-HIV1-Clade A (Sens) | 426341 | 80 | Sonde sensorielle ciblant le brin inverse dans un rayon de 879 à 7629 pb du consensus du clade A du VIH-1 (gag, pol, vif, vpr, tat, rev, vpu, env et nef) |
| V-HIV1-Clade B (Anti-Sens) | 416111 | 78 | Sonde antisens ciblant entre 854 et 8291 pb de AF324493.2, VIH-1 clade B NL4-3 (gag, pol, vif, vpr, tat, rev, vpu, env et nef) |
| V-HIV1-Clade B (Sens) | 425531 | 78 | Sonde Sense ciblant le brin inverse entre 854 et 8291 pb de AF324493.2, VIH-1 clade B NL4-3 (gag, pol, vif, vpr, tat, rev, vpu, env et nef) |
| V-HIV1-Clade D (anti-sens) | 416121 | 76 | Sonde antisens ciblant dans les 894-7697 pb du consensus du clade D du VIH-1 (gag, pol, vif, vpr, tat, rev, vpu, env, nef) |
| V-HIV1-Clade D (Sense) | 426351 | 76 | Sonde sensorielle ciblant le brin inverse dans un rayon de 894 à 7697 pb du consensus du clade D du VIH-1 (gag, pol, vif, vpr, tat, rev, vpu, env, nef) |
| Ensembles de sondes ARN/DNAscope Multi-Plex | |||
| Nom | Catalogue ACD # | Nombre de ZZ | Description |
| V-SIVmac239-gag-pol-Sense-C1 | Référence 416141-C1 | 40 | Sonde Sense ciblant le brin inverse entre 1251 et 4093 pb de D01065.1 (gag et pol) |
| V-SIVmac239-vif-env-nef-tar-C2 (Anti-sens) | Réf. 416131-C2 | 47 | Ciblage de la sonde antisens entre 5381 et 10257 pb de D01065.1 (vif, vpx, vpr, tat, env, nef et l’élément TAR) |
| V-HIV1-Clade_B-gag-pol-sense-C1 | Référence 444051-C1 | 40 | Sonde Sense ciblant le brin inverse entre 854 et 3940 pb de AF324493,2, VIH-1 clade B NL4-3 (gag et pol) |
| V-HIV1-Clade_B-vif-vpr-tat-rev-vpu-env-nef-tar-C2 (Anti-sens) | Référence 444061-C2 | 40 | Sonde antisens ciblant entre 5042 et 9673 pb de AF324493.2, VIH-1 clade B NL4-3 (vif, vpr, tat, env, nef et l’élément TAR) |
| V-HIV1-Clade_C-gag-pol-sense-C1 | Référence 444021-C1 | 48 | Sonde sensorielle ciblant le brin inverse entre 888 et 5032 pb de la séquence consensuelle du clade C du VIH-1 (gag et pol) |
| V-HIV1-Clade_C-vif-vpr- rev-vpu-env-nef-tar-C2 (Anti-sens) | Référence 444041-C2 | 49 | Ciblage de la sonde antisens entre 5078 et 9698 pb de la séquence consensuelle du clade C du VIH-1 (vif, vpr, tat, env, nef et l’élément TAR) |
| V-HIV1-Clade_AE-gag-pol-sense-C1 | Référence 444011-C1 | 55 | Sonde Sense ciblant le brin inverse entre 890 et 4812 pb de AF259954.1, EA du clade du VIH-1 (gag et pol) |
| V-HIV1-Clade_AE-vif-vpr-tat-rev-vpu-env-nef-tar-C2 (Antisens) | Réf. 444031-C2 | 57 | Sonde antisens ciblant entre 5052 et 9694 pb de AF259954,1, EA du clade du VIH-1 (vif, vpr, tat, env, nef et l’élément TAR) |
Tableau 1 : Liste des sondes pour cibler l’ARNv et l’ADNv du VIH-1 et du VIS.
Discussion
L’hybridation in situ est un test méticuleux qui nécessite de la rigueur et des connaissances de base en chimie des acides nucléiques, en biologie cellulaire et en histologie pour pouvoir adapter chaque étape critique pour localiser une cible dans un environnement bien conservé. Dans cette discussion, nous aimerions mettre en évidence les étapes critiques où le dépannage est crucial pour obtenir des résultats précis et interprétables.
La fixation et le traitement des tissus sont essentiels et doivent être abordés dès le départ pour s’assurer que le test peut donner les meilleurs résultats. Le fixateur PFA à tampon neutre (4 % fraîchement préparé) est optimal pour le dosage recto-verso. Cependant, le test peut également être effectué sur des tissus congelés (OCT) avec les conditions de fixation post-cryosectionnement appropriées.
Le prétraitement des coupes de tissus est une étape cruciale. Il y a deux étapes de prétraitement dans ce test : la première est une récupération d’épitopes induite par la chaleur (HIER). Cette étape est importante pour l’inversion des réticulations des ponts méthylènes et la restauration des structures protéiques, ce qui est nécessaire dans les tissus fixés. L’efficacité de ce traitement dépend du temps, de la température, du type de tampon de récupération et du pH. Le deuxième prétraitement est une récupération d’épitopes induite par la protéase (PIER). Cette étape clive les peptides, exposant l’antigène ou les nucléotides, et utilise des enzymes telles que la protéinase K, la trypsine et la pepsine. Il s’agit d’une étape extrêmement sensible qui pourrait potentiellement endommager à la fois la morphologie des tissus et la cible d’intérêt. La concentration de l’enzyme, ainsi que le temps et la température d’incubation sont essentiels dans ce processus. La surdigestion entraîne une mauvaise démarcation des noyaux et des difficultés dans les étapes de quantification. Il est essentiel de trouver un équilibre entre un accès optimal à la cible ARN/ADN et des conditions de prétraitement qui n’endommagent pas le tissu ou la cible d’intérêt. Chaque type de tissu a un niveau de sensibilité différent à chacun de ces prétraitements et chaque paramètre (concentration enzymatique, temps, température) doit être testé empiriquement.
La rigueur du tampon de lavage est basée sur trois paramètres principaux : la température, la concentration des sels et du détergent et le temps. Le tampon de lavage est un tampon salin de citrate de sodium (SSC), et la concentration en sel à l’intérieur du tampon contrôle la rigueur pendant les étapes de lavage. Dans son protocole, ACD conseille d’utiliser le tampon de lavage à une concentration finale de 0,1x SSC, 0,03 % de dodécylsulfate de lithium. En travaillant sur l’optimisation du DNAscope et du multiplexage, nous avons déterminé que l’utilisation du tampon de lavage à une concentration finale de 0,05x SSC nous donnait de meilleurs résultats pour visualiser le signal de l’ADN et contribuait considérablement à réduire l’hybridation hors cible non spécifique résultant de l’incubation nocturne de la sonde sensorielle.
Le choix de l’approche de détection, chromogène (rouge ou brun) ou fluorescence, doit être réfléchi en fonction du type de tissu et de l’objectif avant de commencer le test. L’approche chromogène rouge donnera un joli contraste, car le rouge ne se trouve pas naturellement dans les tissus. Le chromogène brun donnera des résultats similaires au chromogène rouge. Cependant, il est important de garder à l’esprit que certains produits de dégradation du sang présents dans le tissu ont une couleur similaire, et l’encre de tatouage sera difficile à séparer du signal brun lors de la quantification. Une approche de détection par fluorescence permettra une distinction claire des différents marqueurs cellulaires et le multiplexage offrira un test parfait pour phénotyper les cellules hébergeant l’ARNv et/ou l’ADNv.
De multiples contrôles sont nécessaires pour garantir la spécificité des sondes et la qualité du dosage. Chaque sonde nouvellement conçue doit être testée sur des tissus de contrôle positifs et négatifs connus ou sur des pastilles cellulaires. Nous générons souvent des plasmides contenant la séquence que nous avons ciblée et effectuons la transfection dans des lignées cellulaires pour générer des contrôles positifs. Pour chaque passage, nous ajoutons un tissu négatif connu (VIH ou SIV négatif), un témoin sans sonde contenant uniquement le diluant de la sonde, et un témoin traité par RNase pour garantir la qualité et la spécificité du test.
La quantification est une étape extrêmement importante et doit être effectuée à l’aide des outils et de l’algorithme appropriés en fonction de la question posée. Dans ce manuscrit, nous avons présenté un logiciel d’analyse d’images (par exemple, Cellprofiler), que nous avons choisi après évaluation de plusieurs options. Nous avons estimé que ce logiciel était le meilleur logiciel pour nos besoins, mais il existe de nombreux logiciels d’analyse d’images qui pourraient être utilisés.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Ce projet a été financé en totalité par des fonds fédéraux de l’Institut national du cancer, National Institutes of Health, dans le cadre du contrat n°. HHSN261200800001E et par l’Oregon National Primate Research Center NIH grant P51OD011092 (J.D.E). Le contenu de cette publication ne reflète pas nécessairement les opinions ou les politiques du ministère de la Santé et des Services sociaux, et la mention de noms commerciaux, de produits commerciaux ou d’organisations n’implique pas l’approbation du gouvernement américain. Le duplex a été développé à l’aide d’Advanced Cell Diagnostics.
matériels
| Name | Company | Catalog Number | Comments |
| ACD HybEZII Hybridization system (110V) with ACD EZ-Batch Slides system | ACD | 321710 | Hybridization oven |
| CAT Hematoxylin | Biocare medical | CATHE-GAL | colorstain |
| Clear-Mount | ELECTRON MICROSCOPY SCIENCES | 17985-15 | mounting reagent for red chromogen |
| Immpact DAB Peroxidase Kit | Vector | SK-4105 | Used to reveal HRP - DAB (Brown) to replace the DAB coming in the ACD kit |
| lithium carbonate | Fisher chemical | L119-500 | bluing solution |
| paraformaldehyde | ELECTRON MICROSCOPY SCIENCES | 15714-S | for tissue fixation (4%) |
| PBS | life technology | 14190-136 | |
| Permount Mounting Medium | ThermoFisher Scientific | SP15-100 | mounting regaent for brown chromogen |
| Prolong Gold | ThermoFisher Scientific | P36930 | mounting regaent for fluorescence |
| ribonucleases A | ThermoFisher Scientific | 12091039 | for RNAse treatment in DNAscope protocol |
| ribonucleases T1 | Roche | R1003 | for RNAse treatment in DNAscope protocol |
| RNAscope 2.5, 2-plex detection reagent | ACD | 322430 | Brown and red kit chromogen detection |
| RNAscope Target Retrieval Reagents | ACD | 322000 | retrieval buffer |
| SuperFrost Plus Glass Slides | ThermoFisher Scientific | 12-550-17 | |
| TBS | BOSTON BIOPRODUCTS | BM-301-4L | for washes |
| TSA Plus Fluorescence palette kit (Cy3, Cy5, TMR, Fluorescein) | Perkin elmer | NEL760001KT | HRP Fluorescence detection |
| Tween 20 | SIGMA | P1379-1L | for washes |
| XYLENE 20LT | ThermoFisher Scientific | AC422680200 |
Références
- Wang, F., et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. Journal of Molecular Diagnosis. 14 (1), 22-29 (2012).
- Deleage, C., et al. Defining HIV and SIV Reservoirs in Lymphoid Tissues. Pathogens and Immunity. 1 (1), 68-106 (2016).
- Sengupta, S., Siliciano, R. F. Targeting the Latent Reservoir for HIV-1. Immunity. 48 (5), 872-895 (2018).
- Churchill, M. J., Deeks, S. G., Margolis, D. M., Siliciano, R. F., Swanstrom, R. HIV reservoirs: what, where and how to target them. Nature Reviews Microbiology. 14 (1), 55-60 (2016).
- McQuin, C., et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biology. 16 (7), e2005970 (2018).
- Deleage, C., Chan, C. N., Busman-Sahay, K., Estes, J. D. Next-generation in situ hybridization approaches to define and quantify HIV and SIV reservoirs in tissue microenvironments. Retrovirology. 15 (1), 4 (2018).
- Deleage, C., Turkbey, B., Estes, J. D. Imaging lymphoid tissues in nonhuman primates to understand SIV pathogenesis and persistence. Current Opinion in Virology. 19, 77-84 (2016).
- Estes, J. D., et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nature Medicine. 23 (11), 1271-1276 (2017).
- Mavigner, M., et al. Simian Immunodeficiency Virus Persistence in Cellular and Anatomic Reservoirs in Antiretroviral Therapy-Suppressed Infant Rhesus Macaques. Journal of Virology. 92 (18), (2018).
- Peterson, C. W., et al. Differential impact of transplantation on peripheral and tissue-associated viral reservoirs: Implications for HIV gene therapy. PLoS Pathogen. 14 (4), e1006956 (2018).
- Deleage, C., et al. Impact of early cART in the gut during acute HIV infection. Journal of Clinical Investigation Insight. 1 (10), e87065 (2016).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.