
Method Article
Préparation de l’échantillon TMT pour la soumission des installations de protéomique et l’analyse subséquente des données
Dans cet article
Résumé
Nous présentons un protocole d’étiquetage optimisé de l’étiquette de masse en tandem (TMT) qui comprend des informations détaillées pour chacune des étapes suivantes : extraction de protéines, quantification, précipitations, digestion, étiquetage, soumission à une installation de protéomique et analyses de données.
Résumé
Les technologies protéomiques sont des méthodologies puissantes qui peuvent nous aider à comprendre les mécanismes d’action dans les systèmes biologiques en fournissant une vision globale de l’impact d’une maladie, d’un traitement ou d’une autre condition sur le protégé dans son ensemble. Ce rapport fournit un protocole détaillé pour l’extraction, la quantification, les précipitations, la digestion, l’étiquetage et l’analyse subséquente des données des échantillons de protéines. Notre protocole d’étiquetage TMT optimisé nécessite une concentration plus faible d’étiquettes et permet d’obtenir des données fiables. Nous avons utilisé ce protocole pour évaluer les profils d’expression protéique dans une variété de tissus de souris (c.-à-d., le cœur, le muscle squelettique et le cerveau) ainsi que les cellules cultivées in vitro. En outre, nous démontrons comment évaluer des milliers de protéines à partir de l’ensemble de données résultant.
Introduction
Le terme « protéomique » a d’abord été défini comme la caractérisation à grande échelle de l’ensemble du complément protéique d’une cellule, d’un tissu ou d’un organisme1. Les analyses protéomiques permettent d’enquêter sur les mécanismes et les processus cellulaires impliqués dans le développement de la maladie, les voies thérapeutiques et les systèmes sains utilisant des techniques pour effectuer la quantitation relative des niveaux d’expression des protéines2. Les descriptions initiales de ces études ont été publiées en 1975 et ont démontré l’utilisation de l’électrophoresis en gel polyacrylamide bidimensionnel (2D-PAGE) à cette fin1,3. La méthode 2D sépare les protéines basées sur la charge (mise au point isoélectrique, IEF) et la masse moléculaire (sodium dodecyl sulfate polyacrylamide gel electrophoresis, ou SDS-PAGE)4. Pendant des années, la combinaison de 2D-PAGE et de spectrométrie de masse tandem ultérieure effectuée sur chaque composant de gel a été la technique d’analyse non ciblée la plus commune d’expression de protéine exécutée et a identifié de nombreux profils d’expression de protéine précédemment inconnus5,6. Les inconvénients généraux de l’approche 2D-PAGE sont qu’il prend beaucoup de temps, ne fonctionne pas bien pour les protéines hydrophobes, et il ya des limites dans le nombre total de protéines évaluées en raison de la faible sensibilité7,8.
L’étiquetage des isotopes stables par les acides aminés dans la méthode de culture cellulaire (SILAC) est devenu la prochaine approche populaire pour identifier et quantifier l’abondance des protéines dans les échantillons9. Il se compose de l’étiquetage métabolique des cellules qui sont incubées dans le milieu dépourvu d’un acide aminé essentiel standard et complétée par une version isocéocéocée de cet acide aminé spécifique10. L’avantage de cette technique est son efficacité et son étiquetage précis9. La principale limite à l’approche SILAC est principalement la réduction du taux de croissance cellulaire causée par l’incorporation d’étiquettes isotopiques, qui peut être particulièrement difficile dans les lignées cellulaires relativement sensibles modélisant les maladies humaines11.
En 2003, une technique de protéomique nouvelle et robuste impliquant des étiquettes isobaric tandem (TMT) a été introduite sur le terrain12. L’étiquetage TMT est une méthode puissante en raison de sa sensibilité accrue pour détecter les niveaux relatifs d’expression des protéines et les modifications posttranslationnelles13. À la date de publication, des kits TMT ont été développés qui peuvent simultanément étiqueter 6, 10, 11 ou 16 échantillons. En conséquence, il est possible de mesurer l’abondance de peptides dans de multiples conditions avec des répliques biologiques en même temps14,15,16. Nous avons récemment utilisé TMT pour caractériser le profil protéomique cardiaque d’un modèle de souris du syndrome de Barth (BTHS)17. Ce faisant, nous avons pu démontrer une amélioration généralisée des profils cardiaques des souris BTHS traitées avec la thérapie génique et identifier de nouvelles protéines touchées par BTHS qui ont révélé de nouvelles voies thérapeutiques impliquées dans les cardiomyopathies.
Ici, nous décrivons une méthode détaillée pour effectuer des analyses quantitatives de protéomique de TMT multiples utilisant des échantillons de tissu ou des granules cellulaires. Il peut être avantageux d’effectuer la préparation et l’étiquetage de l’échantillon avant la soumission à un noyau parce que les peptides tryptiques étiquetés sont plus stables que les échantillons congelés bruts, pas tous les noyaux ont l’expérience de traiter tous les types d’échantillons, et la préparation des échantillons dans un laboratoire peut gagner du temps pour les noyaux, qui ont souvent de longs arriérés. Pour des descriptions détaillées de la partie de spectroscopie de masse de ce processus s’il vous plaît voir Kirshenbaum et al. et Perumal et al.18,19.
Le protocole de préparation de l’échantillon se compose des étapes majeures suivantes : extraction, quantification, précipitations, digestion et étiquetage. Les principaux avantages de ce protocole optimisé sont qu’il réduit les coûts d’étiquetage, améliore l’extraction des protéines et génère constamment des données de haute qualité. En outre, nous décrivons comment analyser les données TMT pour dépister des milliers de protéines dans un court laps de temps. Nous espérons que ce protocole encouragera d’autres groupes de recherche à envisager d’intégrer cette méthodologie puissante dans leurs études.
Protocole
Le Comité institutionnel de soins et d’utilisation des animaux de l’Université de Floride a approuvé toutes les études sur les animaux.
1. Préparation des réactifs
- Préparer CHAPS Lysis Buffer (150 mM KCl, 50 mM HEPES pH - 7,4, 0,1% CHAPS, et 1 comprimé de cocktail inhibiteur de protéase par 50 ml de tampon). Le tampon sans inhibiteurs de la protéase peut être stocké à 4 oC pendant 6 mois ou tampon avec inhibiteur de protéase stocké à -20 oC jusqu’à 1 an.
- Préparer le bicarbonate de triéthylammonium de 100 mM (TEAB) : Ajouter 500 L de 1 M TEAB à 4,5 ml d’eau ultrapure.
- Préparer 200 mM tris (2-carboxyethyl) phosphine hydrochlorure (TCEP): Ajouter 70 L de 0,5 M TCEP, un réactif dénaturant, à 70 L d’eau ultrapure. Ajouter ensuite 35 L des 1 M TEAB.
- Préparer 5 % d’hydroxylamine : Ajouter 50 ll de 50 % d’hydroxylamine à 450 L de 100 mM TEAB.
2. Extraction de protéines
- Isolez le muscle quadriceps d’une souris euthanasiée selon un protocole approuvé par l’IACUC. Congeler et maintenir à -80 oC ou continuer avec le protocole pour une utilisation immédiate.
- Couper pour isoler environ 10 mg de tissu de souris quadriceps frais ou congelé. Fibres séparées à l’aide de pincettes lorsque vous travaillez avec le muscle squelettique. Alternativement, si vous travaillez avec les cultures cellulaires, réutilisez 3 x 106 cellules dans 300 l de tampon de lyse CHAPS et sautez à l’étape 2.4.
- Homogénéisez les tissus à l’aide d’un perturbateur perlé à l’aide de tubes de 2 ml remplis d’environ 200 ll de perles de zirconia/silice de 1 mm et de 500 lil de tampon de lyse CHAPS. Échelle vers le haut ou vers le bas, le cas échéant (p. ex., 5 mg de tissu dans 250 l de tampon de lyse CHAPS).
- Effectuer la sonication (10x pour 10 s chacun avec 50% d’amplitude et 30 s intervalles sur la glace) pour libérer des protéines liées à l’ADN. Les mêmes résultats de dégradation de l’ADN peuvent être obtenus soit avec la lyse de seringue en passant le lysate 10x par une aiguille de 21 G attachée à une seringue de 1 mL, soit par incubation de benzonase (44 U/mL) à 37 oC pendant 30 min.
- Centrifuge le lysate à 16.000 x g pendant 10 min à 4 oC et transférer le supernatant dans un nouveau tube de centrifugeuse.
3. Mesure des protéines
- Déterminer la concentration de protéines du supernatant à l’aide de protocoles établis (voir Tableau des matériaux).
REMARQUE : Il est préférable d’utiliser des échantillons à 2 g/l, mais des échantillons moins concentrés peuvent également être utilisés. Si un échantillon moins concentré est utilisé, il sera nécessaire d’ajuster de façon appropriée les volumes des réactifs de réduction/alkylating dans l’étape 5.1. - Préparer une dilution de courbe standard BSA à l’aide d’un tampon chapS lysis.
- Suivez les instructions du fabricant, et après 15 min, lisez l’absorption à 750 nm.
4. Réduire/alkylating traitement de réactif
- Transférer 200 g de protéines par condition dans un nouveau tube de centrifugeuse et ajuster à un volume final de 100 ll à l’aide d’un tampon de lyse CHAPS. Il est possible d’étendre jusqu’à 200 ll lorsque la concentration de protéines est trop faible, mais n’oubliez pas d’ajuster de manière appropriée le volume de réduction / alkylating réactif.
- Ajouter 5 ll des échantillons de 200 mM TCEP et incuber des échantillons à 55 oC pour 1 h.
- Immédiatement avant utilisation, préparer 375 mM d’iodoacetamide en dissolvant un tube d’iodoacetamide (c.-à-d. 9 mg) en 132 ll de 100 mM TEAB. Protégez cette solution de la lumière.
- Ajouter 5 L d’iodoacetamide de 375 mM à l’échantillon et incuber pendant 30 minutes à température ambiante (RT) à l’abri de la lumière.
5. Précipitations du méthanol/chloroforme20
- Ajoutez 400 L de méthanol à chaque 100 L de protéines et d’échantillons de vortex brièvement.
- Centrifugeuse à 9.000 x g pour 10 s à RT. Il s’agit d’incorporer des liquides déposés sur les côtés du tube de l’échantillon.
- Ajouter 100 L de chloroforme au mélange et brièvement vortex. Utilisez 200 L de chloroforme si l’échantillon a une forte concentration de phospholipides.
- Centrifugeuse à 9.000 x g pour 10 s à RT. Il s’agit d’incorporer des liquides déposés sur les côtés du tube de l’échantillon.
- Ajouter 300 L d’eau et de vortex vigoureusement. Il est important d’obtenir une solution homogène.
- Centrifugeuse à 9 000 x g pendant 1 min à RT. Soyez extrêmement prudent pour éviter de déranger les couches lors du transfert du tube sur une grille.
REMARQUE : Le tube doit maintenant contenir trois phases : 1) Couche supérieure (c.-à-d. supernatant), un mélange d’eau et de méthanol; 2) Couche moyenne (c.-à-d. interphase), protéine blanche précipitée; et 3) Couche inférieure (c.-à-d. phase inférieure), chloroforme. - Retirez soigneusement le supernatant.
- Ajouter 300 L de méthanol à l’interphase et à la phase inférieure restantes. Vortex vigoureusement.
- Centrifugeuse à 9 000 x g pendant 2 minutes à RT. Soyez extrêmement prudent pour éviter de déranger les couches lors du transfert du tube sur une grille.
- Retirez soigneusement le supernatant.
- Aspirez doucement autant de liquide que possible sous un flux d’air (p. ex., à l’aide d’un concentrateur sous vide) à RT jusqu’à ce que la boulette soit juste un peu humide (10 min). Comme le temps nécessaire peut être différent pour chaque échantillon, vérifiez toutes les 2 minutes à évaluer. Conserver la granulé à -80 oC jusqu’à nouvel traitement.
6. Digestion des protéines
- Réutilisez la granule de protéines précipitée dans un tampon de lyse TEAB de 100 ll.
REMARQUE : Il est facultatif de mesurer la concentration de protéines à cette étape. - Immédiatement avant l’utilisation, préparer la trypsine de 1 g/L en ajoutant 100 l de la solution de stockage de trypsin (50 mM d’acide acétique) au fond de la fiole de verre à trypsine de 100 g et incuber pendant 5 min au RT. Store restant réactif à doses à usage unique à -80 oC.
- Ajouter 2,5 l de trypsine par 100 g de protéines. Digérer l’échantillon pendant la nuit à 37 oC. Cette étape est cruciale pour la solubilisation complète de la protéine; ne modifient pas ces conditions. Après la digestion, il est facultatif de mesurer la concentration de protéines à l’aide d’essais de protéines standard.
7. Étiquetage Peptide
- Immédiatement avant l’utilisation, équilibrez les réactifs du kit d’étiquette TMT à RT.
- Dissoudre chacune des flacons d’étiquette TMT de 0,8 mg par l’ajout de 41 l d’acétonitrile anhydre à chaque tube. Incuber le réactif pendant 5 min à RT avec vortexing occasionnel. Brièvement centrifugeuse les tubes.
REMARQUE : Une concentration de 0,8 mg d’étiquette TMT est généralement suffisante pour étiqueter deux ensembles. D’autres chercheurs, cependant, ont démontré que cette concentration peut être réduite davantage et encore produire des données fiables15. - Ajoutez soigneusement 41 L du réactif de l’étiquette TMT à chaque échantillon de 100 L.
- Incuber la réaction pour 1 h à RT.
- Ajouter 8 ll d’hydroxylamine de 5 % à l’échantillon et incuber pendant 15 min pour étancher la réaction.
- Diviser les échantillons en quantités égales dans un nouveau tube de centrifugeuse et stocker à -80 oC.
REMARQUE : Dans cette étape, les échantillons sont stables et peuvent être soumis pour la spectroscopie de masse. Il est facultatif de mesurer la concentration à ce stade à l’aide d’essais de protéines standard.
8. Spectroscopie de masse
- Soumettez les échantillons à une installation de protéomique (cette étude a utilisé l’installation UF ICBR Proteomics Core), où tous les échantillons sont combinés et purifiés à l’aide de colonnes de spin C18.
REMARQUE : Discutez avec l’installation principale comment soumettre les échantillons avant de les préparer à confirmer les étapes exactes qu’ils préfèrent pour les soumissions. - Demandez les procédures suivantes par échantillon multiplex combiné : extraction de phase solide, HPLC (SCX, SE), pointe zippée, et LC-MS/MS (gradient de 2 h pour ID protéique, si 'gt;10QE Plus).
- Une fois les données recueillies, l’installation de base traitera les fichiers RAW à l’aide d’un logiciel fourni par le fournisseur pour l’identification des protéines.
9. Analyse des données
- Les données sont généralement livrées à partir du noyau vers l’utilisateur dans le format 7z, qui peut nécessiter environ 16 Go d’espace disque par chaque jeu de données (dans ce cas 11 échantillons). Pour le traitement des données, assurez-vous qu’un ordinateur est disponible qui est d’au moins 3,4 GHz.
- Extraire les fichiers à l’aide de 7-Zip File Manager. Ces fichiers extraits contiennent des données RAW, un fichier de format pdStudy et un fichier format pdResultView. Enregistrez tous les fichiers pour d’autres analyses.
- Fichier ouvert à l’aide du logiciel Proteome Discovery 2.2.
REMARQUE : Le format du fichier est « Nom de fichier.pdStudy ». Si le « Nom de fichier.pdResultView » est ouvert, il n’est pas possible de sélectionner l’échantillon de contrôle. - Sélectionnez des échantillons de contrôle sur le panneau «Échantillons».
- Résultat ouvert en sélectionnant l’ID sur le panneau «Résultats d’analyse».
- Exportation vers les logiciels de feuilles de calcul.
- Enregistrer les données brutes (toutes les protéines identifiées).
- Ouvrez le fichier logiciel de la feuille de calcul. Cela contiendra toutes les protéines qui ont été identifiées.
- Dans le fichier logiciel de feuille de calcul utiliser le "Filtre" fonction pour filtrer "Protein FDR Confidence: Combined" en haut (colonne B), "#Unique Peptides" plus haut que 2 (colonne K), et l’un des " Ratiod’abondance" blanc exclusivement(colonne S jusqu’à W).
- Insérer une colonne pour le calcul «p-value» avec la fonction
TTEST (groupe témoin, groupe expérimental, queues, type) - Insérez une colonne pour "Signification Statistique" avec la fonction
IF (p-value-lt;0.05, « Significance »,"NS ») - Utilisez la fonction «Filtre» pour filtrer «Signification statistique» montrant «Signification». Le résultat montre les protéines analysées avec la signification statistique dans le groupe témoin et le groupe expérimental.
- Déterminer l’abondance d’expression protéique significativement plus élevée ou inférieure dans le groupe expérimental par rapport au groupe témoin, insérer une colonne pour "Régulation" avec la fonction
'IF(AVERAGE(groupe de contrôle)'gt;AVERAGE(groupe expérimental),"Upregulated »,"Downregulated »)
10. Méthodes pour évaluer les coups significatifs
- Pour identifier les interactions protéines-protéines entre les succès significatifs identifiés dans les études TMT, utilisez Search Tool pour la récupération de la version 11.021: https://string-db.org/
- Pour classer par groupes (c.-à-d. la fonction moléculaire, les processus biologiques et les classes de protéines) utiliser l’analyse des protéines par le biais du logiciel de classification de l’ontologie des relations évolutives(PANTHER) 22: http://www.pantherdb.org/
- Pour identifier les interactions protéiques dans une variété de voies, utilisez le logiciel d’analyse des voies23.
11. Téléchargement de données protéomiques dans une banque de dépôts
- Pour soumettre des données protéomiques à la base de données d’identification protéomique (PRIDE) ou à l’environnement virtuel interactif de spectrométrie de masse (MassIVE) inclure les informations suivantes : les fichiers de liste de pointe (fichiers de spectre de masse traités dans un format standard tel que mzXML, mzML, ou MGF), fichiers de résultats (identifications de spectre dans un format standard tel que mzIdentML ou mzTab), et les fichiers de spectre brut (fichiers de spectre de masse brute dans un format non standard ou spécifique aux instruments tels que . Fichiers RAW ou . Fichiers WIFF).
- Pour soumettre, créez un compte et incluez des informations telles que les détails de l’affiliation et du projet. Ensuite, sélectionnez les fichiers énumérés dans l’étape 11.1 et téléchargez-les.
- Pour créer un jeu de données officiel, exécutez un flux de soumission sur ces fichiers téléchargés.
REMARQUE : Après la soumission, l’ensemble de données sera privé dans la banque de dépôt. Avec l’option privée, les données ne sont disponibles que pour les utilisateurs autorisés. Il existe deux options supplémentaires : 1) Jeu de données partagé, qui donne accès aux critiques de revues et aux collaborateurs; ou 2) Jeu de données publics, qui s’affichera dans les recherches publiques de dataset. Une autre caractéristique importante de ces dépôts est la possibilité de mettre à jour les données téléchargées et d’associer les publications ultérieures avec le jeu de données existant.
Résultats
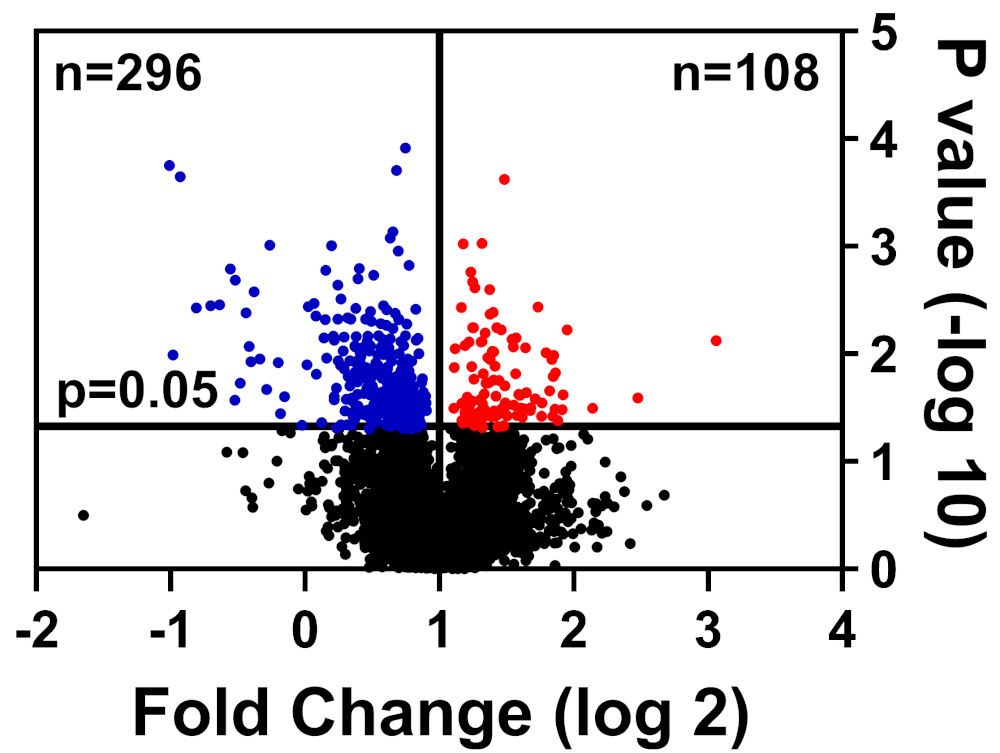
Les cellules saines et malades ont été analysées dans le tampon CHAPS, préparées comme détaillées dans notre méthode d’étiquetage TMT, et soumises au Centre interdisciplinaire de l’Université de Floride pour la recherche en biotechnologie (UF-ICBR) Proteomics Core for Liquid Chromatography avec spectrométrie de masse tandem. À la suite de l’acquisition et de la livraison de données à partir du noyau, l’ensemble de données a été ouvert dans les logiciels fournis par le fournisseur et les filtres de coupure suivants ont été appliqués : 2 peptides uniques, ions reporter pour chaque échantillon de protéines présents dans tous les canaux, et ne comprennent que des protéines modifiées de manière significative (p - 0,05). Le tableau 1 résume les données : 39 653 peptides au total, dont 7 211 ont un nombre égal ou supérieur à deux peptides uniques, et 3 829 comprennent des ions de journaliste pour tous les canaux. Les valeurs p pour ces 3 829 peptides ont été calculées par le test t de l’étudiant et p 0,05 a été considérée comme significative. De plus, une coupure de changement a été utilisée pour déterminer la répartition relative des protéines provenant de cellules malades par rapport aux cellules saines : régulées (bleues) ou régulées (rouge)(figure 1).
La liste de l’expression significativement dysrégulée de protéine a été évaluée utilisant le système de classification d’ontologie de PANTHER et les analyses de STRING. Les analyses de panthères ont montré une liste classée de protéines basée sur une abondance significativement plus faible(figure 2A) ou une abondance plus élevée dans les cellules malades en fonction de la fonction moléculaire(figure 2B). Les analyses de chaînes de protéines de façon significative plus faible (figure 2C) et d’abondance plus élevée(figure 2D) ont permis d’identifier de multiples interactions et de fortes associations entre les protéines.

Figure 1 : Parcelle de volcan affichant des protéines dont l’abondance n’a pas été significativement altérée (noir), significativement abaissée (bleu), ou augmentée significativement (rouge) dans les cellules de contrôle malades vs saines. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : Évaluations représentatives des coups dysrégulés significativement identifiés par PANTHER (A, B) et String (C, D) de protéines d’abondance significativement plus faibles ou plus élevées. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
| Peptides totaux | Total identifié | 2 peptides uniques | Protéines quantifiées | Protéines considérablement altérées | |
| Faible | Haute | ||||
| 39653 | 7211 | 4457 | 3829 | 296 | 108 |
Tableau 1 : Tableau représentatif des protéines quantifiées par analyse de jeu de données.
Discussion
Pour préparer avec succès des échantillons pour l’analyse protéomique à l’aide de méthodologies d’étiquetage isobarique stable isobarique à base de TMT, il est crucial d’effectuer des extractions de protéines très soigneusement à 4 oC et d’utiliser un tampon de lyse qui contient un cocktail inhibiteur de la protéase24,25. Le cocktail inhibiteur de la protéase est un réactif crucial pour éviter une dégradation inattendue des protéines pendant la digestion des protéines. Une différence clé entre notre protocole et le présent tel que le fournisseur est que nous recommandons fortement l’utilisation de tampons chapS lysis en fonction de notre expérience avec les cellules et les tissus de mammifères. Nous suggérons également d’utiliser une approche de précipitations de protéines de méthanol/chloroforme pour les granulés cellulaires et les tissus.
Idéalement, l’extraction de protéines, la mesure, la réduction/l’alkylating des traitements de réactif, et les précipitations de méthanol/chloroforme sont tous exécutés le même jour. Suite à cette recommandation, des concentrations de protéines plus précises pour l’étiquetage ultérieur se traduiront. L’étape de précipitation des protéines est importante pour l’élimination des réactifs qui interfèrent avec la spectrométrie de masse tandem. L’inclusion de l’étape de précipitations améliore considérablement la résolution de TMT26. En somme, les principaux avantages de notre protocole TMT sont l’efficacité élevée d’étiquetage pour différents types d’échantillons, sa reproductibilité, et les données fiables acquises.
Alors que la nature multiplexe de cette stratégie de protéomique non ciblée TMT continue de s’étendre, elle améliorera progressivement la capacité des chercheurs dans une grande variété de domaines à faire de nouvelles découvertes. Plus précisément dans le domaine biomédical, nous et d’autres avons trouvé cette technologie de plus en plus instructive dans les études explorant de nouveaux mécanismes d’action dans la maladie et les impacts relatifs de diverses thérapies. Pour toutes ces raisons, cette technologie puissante complète le répertoire d’autres approches OMICS utilisées dans les études de recherche modernes et fournit des informations clés qui peuvent guider davantage le développement thérapeutique.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous tenons à remercier l’installation de protéomique UF-ICBR pour leur traitement de nos échantillons. Ces travaux ont été soutenus en partie par les National Institutes of Health R01 HL136759-01A1 (CAP).
matériels
| Name | Company | Catalog Number | Comments |
| 1 M Triethylammonium bicarbonate (TEAB), 50 mL | Thermo Fisher | 90114 | Reagent for protein labeling |
| 50% Hydroxylamine, 5 mL | Thermo Fisher | 90115 | Reagent for protein labeling |
| Acetic acid | Sigma | A6283 | Reagent for protein digestion |
| Anhydrous acetonitrile, LC-MS Grade | Thermo Fisher | 51101 | Reagent for protein labeling |
| Benzonaze nuclease | Sigma-Aldrich | E1014 | DNA shearing |
| Bond-Breaker TCEP solution, 5 mL | Thermo Fisher | 77720 | Reagent for protein labeling |
| BSA standard | Thermo | 23209 | Reagent for protein measurement |
| CHAPS | Thermo Fisher | 28300 | Reagent for protein extraction |
| Chloroform | Fisher | BP1145-1 | Reagent for protein precipitation |
| cOmplete, EDTA-free Protease Inhibitor Cocktail Tablet | Roche | 4693132001 | Reagent for protein extraction |
| DC Protein Assay | BioRad | 500-0116 | Reagent for protein measurement |
| Excel | Microsoft Office | Software for data analyses | |
| Heat block | VWR analog | 12621-104 | Equipment for protein digestion incubation |
| HEPES | Sigma | RDD002 | Reagent for protein extraction |
| Methanol | Fisher | A452-4 | Reagent for protein precipitation |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher | 90058 | Reagent for protein digestion |
| Potassium chloride | Sigma | 46436 | Reagent for protein extraction |
| Sigma Plot 14.0 | Sigma Plot 14.0 | Software for data analyses | |
| Sonicator | Fisher Scientific | FB120 | DNA shearing |
| Spectra Max i3x Multi-Mode Detection Platform | Molecular Devices | Plate reader for protein measurement | |
| Thermo Scientific Pierce Quantitative Colorimetric Peptide Assay | Thermo Fisher | 23275 | Reagent for protein measurement |
| Thermo Scientific Pierce Quantitative Fluorescent Peptide Assay | Thermo Fisher | 23290 | Reagent for protein measurement |
| Thermo Scientific Proteome Discoverer Software | Thermo Fisher | OPTON-30945 | Software for data analyses |
| TMT 10plex Isobaric Label Reagent Set 0.8 mg, sufficient reagents for one 10plex isobaric experiment | Thermo Fisher | 90110 | Reagent for protein labeling |
| TMT11-131C Label Reagent 5 mg | Thermo Fisher | A34807 | Reagent for protein labeling |
| Water, LC-MS Grade | Thermo Fisher | 51140 | Reagent for protein extraction |
Références
- Graves, P. R., Haystead, T. A. Molecular biologist's guide to proteomics. Microbiology and Molecular Biology Reviews. 66 (1), 39-63 (2002).
- Erdjument-Bromage, H., Huang, F. K., Neubert, T. A. Sample Preparation for Relative Quantitation of Proteins Using Tandem Mass Tags (TMT) and Mass Spectrometry (MS). Methods in Molecular Biology. 1741, 135-149 (2018).
- O'Farrell, P. H. High resolution two-dimensional electrophoresis of proteins. Journal of Biological Chemistry. 250 (10), 4007-4021 (1975).
- Rabilloud, T., Lelong, C. Two-dimensional gel electrophoresis in proteomics: a tutorial. Journal of Proteomics. 74 (10), 1829-1841 (2011).
- Ong, S. E., et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Molecular & Cellular Proteomics. 1 (5), 376-386 (2002).
- Anderson, N. G., Anderson, N. L. Twenty years of two-dimensional electrophoresis: Past, present and future. Electrophoresis. 17 (3), 443-453 (1996).
- Haynes, P. A., Yates, J. R. Proteome profiling-pitfalls and progress. Yeast. 17 (2), 81-87 (2000).
- Bunai, K., Yamane, K. Effectiveness and limitation of two-dimensional gel electrophoresis in bacterial membrane protein proteomics and perspectives. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences. 815 (1-2), 227-236 (2005).
- Sury, M. D., Chen, J. X., Selbach, M. The SILAC fly allows for accurate protein quantification in vivo. Molecular & Cellular Proteomics. 9 (10), 2173-2183 (2010).
- Zhang, G., Neubert, T. A. Use of stable isotope labeling by amino acids in cell culture (SILAC) for phosphotyrosine protein identification and quantitation. Methods in Molecular Biology. 527, 79-92 (2009).
- Wang, X., et al. SILAC-based quantitative MS approach for real-time recording protein-mediated cell-cell interactions. Scientific Reports. 8 (1), 8441 (2018).
- Thompson, A., et al. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Analytical Chemistry. 75, 1895-1904 (2003).
- Cheng, L., Pisitkun, T., Knepper, M. A., Hoffert, J. D. Peptide Labeling Using Isobaric Tagging Reagents for Quantitative Phosphoproteomics. Methods in Molecular Biology. 1355, 53-70 (2016).
- Navarrete-Perea, J., Yu, Q., Gygi, S. P., Paulo, J. A. Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. Journal of Proteome Research. 17 (6), 2226-2236 (2018).
- Zecha, J., et al. TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Molecular & Cellular Proteomics. 18 (7), 1468-1478 (2019).
- Bachor, R., Waliczek, M., Stefanowicz, P., Szewczuk, Z. Trends in the Design of New Isobaric Labeling Reagents for Quantitative Proteomics. Molecules. 24 (4), E701 (2019).
- Suzuki-Hatano, S., et al. AAV9-TAZ Gene Replacement Ameliorates Cardiac TMT Proteomic Profiles in a Mouse Model of Barth Syndrome. Molecular Therapy - Methods & Clinical Development. 13, 167-179 (2019).
- Kirshenbaum, N., Michaelevski, I., Sharon, M. Analyzing large protein complexes by structural mass spectrometry. Journal of Visualized Experiments. (40), e1954 (2010).
- Perumal, N., et al. Sample Preparation for Mass-spectrometry-based Proteomics Analysis of Ocular Microvessels. Journal of Visualized Experiments. (144), e59140 (2019).
- Wessel, D., Flügge, U. I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Analytical Biochemistry. 138, 141-143 (1984).
- Jensen, L. J., et al. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Research. 37, D412-D416 (2009).
- Mi, H., Muruganujan, A., Casagrande, J. T., Thomas, P. D. Large-scale gene function analysis with the PANTHER classification system. Nature Protocols. 8 (8), 1551-1566 (2013).
- Cirillo, E., Parnell, L. D., Evelo, C. T. A Review of Pathway-Based Analysis Tools That Visualize Genetic Variants. Frontiers in Genetics. 8, 174 (2017).
- Plaxton, W. C. Avoiding Proteolysis during the Extraction and Purification of Active Plant Enzymes. Plant and Cell Physiology. 60 (4), 715-724 (2019).
- Ryan, B. J., Henehan, G. T., Walls, D., Loughran, S. T. . Protein Chromatography: Methods and Protocols. , 53-69 (2017).
- Fic, E., Kedracka-Krok, S., Jankowska, U., Pirog, A., Dziedzicka-Wasylewska, M. Comparison of protein precipitation methods for various rat brain structures prior to proteomic analysis. Electrophoresis. 31 (21), 3573-3579 (2010).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.