
Method Article
Cartographie des relations structure-fonction des facteurs de transcription oncogènes désordonnés à l’aide de l’analyse transcriptomique
Dans cet article
Résumé
Les domaines intrinsèquement désordonnés sont importants pour la fonction du facteur de transcription de fusion oncogène. Pour cibler thérapeutiquement ces protéines, une compréhension plus détaillée des mécanismes de régulation employés par ces domaines est nécessaire. Ici, nous utilisons la transcriptomique pour cartographier les caractéristiques structurelles importantes du domaine EWS intrinsèquement désordonné dans le sarcome d’Ewing.
Résumé
De nombreux cancers sont caractérisés par des translocations chromosomiques qui entraînent l’expression de facteurs de transcription de fusion oncogène. Typiquement, ces protéines contiennent un domaine intrinsèquement désordonné (IDD) fusionné avec le domaine de liaison à l’ADN (DBD) d’une autre protéine et orchestrent des changements transcriptionnels généralisés pour favoriser la malignité. Ces fusions sont souvent la seule aberration génomique récurrente dans les cancers qu’elles provoquent, ce qui en fait des cibles thérapeutiques attrayantes. Cependant, le ciblage des facteurs de transcription oncogènes nécessite une meilleure compréhension du rôle mécaniste que jouent les IDD de faible complexité dans leur fonction. Le domaine N-terminal d’EWSR1 est un IDD impliqué dans une variété de facteurs de transcription de fusion oncogénique, y compris EWS / FLI, EWS / ATF et EWS / WT1. Ici, nous utilisons le séquençage de l’ARN pour étudier les caractéristiques structurelles du domaine EWS importantes pour la fonction transcriptionnelle de EWS / FLI dans le sarcome d’Ewing. La première déplétion médiée par l’ARNh de la fusion endogène des cellules du sarcome d’Ewing associée à l’expression ectopique d’une variété de constructions mutantes EWS est effectuée. Ensuite, le séquençage de l’ARN est utilisé pour analyser les transcriptomes des cellules exprimant ces constructions afin de caractériser les déficits fonctionnels associés aux mutations dans le domaine EWS. En intégrant les analyses transcriptomiques aux informations précédemment publiées sur les motifs de liaison à l’ADN EWS / FLI et la localisation génomique, ainsi qu’aux tests fonctionnels pour la capacité de transformation, nous avons pu identifier les caractéristiques structurelles de EWS / FLI importantes pour l’oncogenèse et définir un nouvel ensemble de gènes cibles EWS / FLI essentiels pour le sarcome d’Ewing. Cet article démontre l’utilisation du séquençage de l’ARN comme méthode pour cartographier la relation structure-fonction du domaine intrinsèquement désordonné des facteurs de transcription oncogènes.
Introduction
Un sous-ensemble de cancers, comprenant de nombreuses tumeurs malignes de l’enfance et de l’adolescence, sont caractérisés par des translocations chromosomiques qui génèrentde nouvelles oncogènes de fusion1,2,3,4,5,6. Les protéines de fusion résultantes fonctionnent fréquemment comme des facteurs de transcription oncogènes, orchestrant des changements généralisés dans la régulation transcriptionnelle pour favoriser la tumorigenèse7,8. Les cancers avec ces translocations possèdent généralement un paysage mutationnel par ailleurs calme, avec peu d’aberrations génomiques récurrentes en dehors de la fusion pathognomonique4,9. En tant que tel, cibler directement la protéine de fusion est une stratégie thérapeutique attrayante dans ces maladies. Cependant, ces facteurs de transcription oncogènes consistent généralement en un domaine de faible complexité, intrinsèquement désordonné, activant la transcription fusionné avec un domaine de liaison à l’ADN (DBD)10,11,12,13,14. Les domaines intrinsèquement désordonnés (IDD) et les DBD de ces protéines se sont révélés difficiles à cibler avec les approches pharmacologiques conventionnelles. Le développement de nouvelles approches thérapeutiques nécessite donc une compréhension moléculaire plus détaillée des mécanismes employés par ces fusions pour réguler aberrantement l’expression des gènes.
La partie N-terminale IDD d’EWSR1 est couramment fusionnée à un DBD dans le cancer, y compris EWS / FLI dans le sarcome d’Ewing, EWS / WT1 dans la tumeur diffuse à petites cellules rondes et EWS / ATF1 dans le sarcome à cellules claires des parties molles10. Le rôle mécaniste de l’EWS IDD dans chacune de ces fusions n’est pas bien compris. La famille de fusions EWS/ETS, en particulier EWS/FLI, est la plus fonctionnellement caractérisée à ce jour. EWS/FLI coordonne les changements épigénétiques et transcriptionnels à l’échelle du génome conduisant à l’activation et à la répression de milliers de gènes7,11,15,16. Des études ont montré que l’IDD est important pour le recrutement des co-activateurs transcriptionnels (tels que p300, WDR5 et le complexe BAF), ainsi que des co-répresseurs (tels que le complexe NuRD)11,15,17. La fusion de l’IDD EWS à la partie C-terminale de FLI1 confère une nouvelle spécificité de liaison à l’ADN au DBD ETS de FLI1, de sorte que l’oncoprotéine de fusion (EWS / FLI) se lie aux régions répétitives GGAA-microsatellites du génome en plus du motif ETS consensuel18,19,20. Combinée à la fonction de recrutement de co-activateurs, cette activité émergente de liaison à l’ADN de l’EWS/FLI favorise la formation d’amplificateurs de novo au niveau des microsatellites GGAA distaux aux sites de départ de transcription (TSS) (microsatellites de type amplificateur) et recrute l’ARN polymérase II pour favoriser la transcription au niveau des microsatellites GGAA-microsatellites proximaux au TSS (microsatellites de type promoteur)11,15,16,21.
Prises ensemble, ces données nous ont amenés à émettre l’hypothèse que des éléments discrets au sein du domaine EWS contribuent au recrutement de co-régulateurs distincts pour différents types de sites de liaison EWS/FLI. Cependant, le discernement de ces éléments dans la partie EWS de EWS /FLI, et leur fonctionnement, a été entravé par la nature hautement répétitive et désordonnée du domaine. Ici, nous utilisons un système de sauvetage précédemment publié dans les cellules du sarcome d’Ewing pour cartographier fonctionnellement ces éléments dans l’IDD EWS. Dans ce système, EWS/FLI est épuisé à l’aide d’un shRNA ciblant le 3'UTR du gène FLI1, et l’expression est sauvée avec différentes constructions d’ADNc mutantes EWS/FLI dépourvues du 3'UTR7,17,22. Ces expériences se sont concentrées sur des constructions avec diverses délétions pour cartographier la relation structure-fonction entre l’IDD EWS et d’importants phénotypes oncogènes, y compris l’activation d’une construction de rapporteur GGAA-microsatellite, les essais de formation de colonies et la validation ciblée des gènes activés et réprimés EWS /FLI 7,17,22 . Cependant, ces études n’ont pas réussi à trouver des sous-domaines discrets au sein de l’IDD EWS dans EWS / FLI qui sont particulièrement importants pour l’activation ou la répression. Toutes les constructions testées étaient soit capables d’activer et de réprimer des gènes cibles spécifiques, conduisant à une formation efficace de colonies, soit incapables de réguler l’un des gènes cibles EWS / FLI, entraînant la perte de la formation de colonies7,17,22.
Les analyses transcriptomiques rendues possibles par l’adoption généralisée du séquençage de nouvelle génération sont couramment utilisées pour comparer les signatures d’expression génique dans deux conditions, souvent dans le cadre d’études de dépistage ou descriptives. Nous voulions plutôt tirer parti de la capacité de capturer des données d’expression à l’échelle du génome en utilisant le séquençage de l’ARN (séquençage de l’ARN) pour caractériser les contributions des IDD à la fonction du facteur de transcription. Dans ce cas, RNA-seq est associé au système knockdown-rescue pour explorer la relation structure-fonction du domaine EWS. Cette approche est applicable à d’autres facteurs de transcription de fusion, y compris d’autres fusions EWS ou facteurs de transcription de type sauvage avec une fonction mal comprise, et présente de multiples avantages par rapport aux autres tests utilisés pour les études de cartographie fonctionnelle, tels que les tests rapporteurs ou la qRT-PCR ciblée. Il s’agit notamment de tester les déterminants structurels de la fonction dans le contexte pertinent de la chromatine, la capacité de tester plusieurs types d’éléments de réponse dans un seul essai (c.-à-d. activé et réprimé, microsatellite GGAA et non microsatellite, etc.), et la capacité qui en résulte de mieux détecter la fonction partielle.
La mise en œuvre réussie de cette approche dépend d’un système cellulaire qui capture les phénotypes d’intérêt (dans ce cas, les cellules A673 avec épuisement EWS/FLI médié par l’ARNh), et d’un panel de constructions mutantes dans un vecteur d’expression approprié pour le système cellulaire (dans ce cas, pMSCV-hygro avec divers mutants EWS/FLI marqués 3x-FLAG à délivrer par transduction rétrovirale). La transduction virale des constructions d’épuisement basées sur CRISPR, des constructions d’épuisement basées sur shRNA et des constructions d’expression de l’ADNc avec une sélection appropriée pour générer des lignées cellulaires stables est recommandée par transfection transitoire. L’interprétation en aval des résultats est renforcée lorsque les données transcriptomiques peuvent être associées à d’autres données liées à la localisation du facteur de transcription et à d’autres lectures phénotypiques, le cas échéant.
Dans cet article, nous appliquons cette approche pour caractériser l’activité du mutant DAF d’EWS/FLI14. Le mutant DAF présente 17 mutations tyrosine à alanine dans les régions répétitives de l’IDD EWS de EWS/FLI14. Ce mutant EWS particulier avait déjà été signalé et est incapable d’activer l’expression du gène rapporteur lorsqu’il est fusionné à l’ATF1 DBD14. Cependant, les données préliminaires de qRT-PCR suggèrent que ce mutant était capable d’activer la transcription de la cible EWS/FLI NR0B123. L’approche transcriptomique décrite ici a permis de détecter avec succès la fonction partielle du mutant DAF. En associant ces données transcriptomiques à des informations sur les motifs de liaison et de reconnaissance EWS/FLI, nous montrons en outre que le mutant DAF conserve sa fonction lors des répétitions de microsatellites GGAA. Ces résultats identifient le DAF comme le premier mutant EWS/FLI partiellement fonctionnel et mettent en évidence la fonction des gènes non microsatellites comme étant important pour l’oncogenèse (comme indiqué23). Cela démontre la puissance de cette approche de cartographie transcriptomique structure-fonction pour fournir un aperçu de la fonction des facteurs de transcription oncogènes.
Protocole
1. Mettre en place un panel in vitro de constructions
REMARQUE: Cette étape variera en fonction de la protéine spécifique à analyser.
- Préparer les aliquotes du virus pour l’épuisement et les constructions d’expression si nécessaire.
- Ensemencez un plat de culture tissulaire de 10 cm avec 3-5 x10 6 cellules HEK293-EBNA ou HEK293T pour chaque construction nécessaire à la transduction virale. Laissez les cellules adhérer pendant la nuit dans le milieu Eagle Media modifié (DMEM) de Dulbecco complété par 10% de sérum bovin fœtal (FBS), de pénicilline / streptomycine / glutamine (P / S / Q) et 0,3 mg / mL G418.
REMARQUE: Les cellules HEK293-EBNA et HEK293T sont recommandées pour la production virale car elles sont faciles à cultiver, ont une efficacité de transfection élevée et expriment efficacement les protéines recombinantes à partir de plasmides épisomiques. Les cellules doivent être confluentes à 50-70% le jour de la transfection. - Préparez un mélange de transfection pour chaque construction de transduction virale. Combiner 2 mL de milieux sériques réduits avec 90 μL de réactif de transfection.
REMARQUE: Il est recommandé de préchauffer les milieux sériques réduits. - Ajoutez 10 μg chacun d’un plasmide d’emballage viral (p. ex., gag-pol), d’un plasmide d’enveloppe virale (p. ex., VSV-G) et l’un des plasmides d’épuisement basés sur CRISPR, de l’épuisement à base d’ARNh ou de la construction de l’expression de l’ADNc (p. ex., pMKO ou pMSCV) au mélange de transfection. Bien mélanger par pipetage doux.
- Laissez reposer le mélange de transfection pendant 20 minutes à température ambiante. Retirer le milieu de croissance HEK293-EBNA des boîtes de culture tissulaire et ajouter 3 mL de DMEM complété par 10 % de FBS, P/S/Q et 10 mM de pyruvate de sodium. À chaque plat, ajouter 2 mL de mélange de transfection goutte à goutte. Laisser les cellules reposer dans un milieu de transfection pendant la nuit dans un incubateur à 37 °C et 5 % de CO2.
- Le lendemain matin, ajoutez 20 mL de milieu DMEM avec 10 % de FBS, une supplémentation en P/S/Q et 10 mM de pyruvate de sodium. Incuber les cellules à 37 °C et 5 % de CO2 pendant la nuit.
- Le lendemain matin, remplacez les milieux par des milieux de collecte virale (VCM) de 5 mL (DMEM complété par 10 % de FBS inactivés par la chaleur, P/S/Q et 20 mM HEPES).
- Après 4 h, prélever le VCM sur les plaques et le conserver dans un tube conique de 50 mL sur de la glace à 4 °C. Remplacer par 5 mL de VCM frais.
- Après 4 h, prélever le VCM sur les plaques dans le même tube conique de 50 mL et le stocker sur de la glace à 4 °C. Remplacer par 8 mL de VCM frais pour la collecte de nuit.
- Le matin, récupérez le VCM des plaques et conservez-le dans le tube conique de 50 mL sur de la glace à 4 °C. Remplacer par 5 mL de VCM frais.
- Après 4 h, prélever le VCM dans les plaques et le conserver dans le tube conique de 50 mL sur de la glace à 4°C. Remplacer par 5 mL de VCM frais. Après 4 h, prélever le VCM dans les plaques et l’ajouter au tube conique de 50 mL.
- Collections d’aliquotes à partir d’un tube de 50 mL en cryotubes (2 mL par aliquote) après filtration à travers un filtre de 0,45 μm. Conserver les aliquotes virales à -80 °C jusqu’à leur utilisation.
REMARQUE: Le protocole peut être mis en pause ici, et les aliquotes virales peuvent être stockées jusqu’à ce qu’elles soient prêtes à l’emploi.
- Ensemencez un plat de culture tissulaire de 10 cm avec 3-5 x10 6 cellules HEK293-EBNA ou HEK293T pour chaque construction nécessaire à la transduction virale. Laissez les cellules adhérer pendant la nuit dans le milieu Eagle Media modifié (DMEM) de Dulbecco complété par 10% de sérum bovin fœtal (FBS), de pénicilline / streptomycine / glutamine (P / S / Q) et 0,3 mg / mL G418.
- Ensemencez des cellules à la densité appropriée dans un plat de culture tissulaire de 10 cm. Ciblez 50 % de confluence. Laisser les cellules adhérer pendant la nuit en les plaçant dans l’incubateur à 37 °C contenant 5% de CO2.
REMARQUE: Pour les cellules A673, il s’agit de 5 x10 6 cellules dans 10 mL de milieu DMEM avec 10% de FBS, une supplémentation en P / S / Q et 10 mM de pyruvate de sodium. Ces conditions peuvent varier en fonction du taux de croissance des cellules utilisées. - Épuiser le facteur d’intérêt endogène. Si les cellules n’ont pas besoin d’avoir la protéine endogène d’intérêt épuisée, passez à l’étape 1.4.
- Dégel de l’aliquote virale pour la transduction de shRNA ou de la construction CRISPR ciblant la protéine d’intérêt. Décongeler rapidement les aliquotes congelées au bain-marie à 37 °C.
- Ajouter 2,5 μL de 8 mg/mL de polybrene à chaque aliquote virale et mélanger par pipetage doux. Retirer le milieu des plaques de cellules et ajouter doucement l’aliquote virale à la plaque de 10 cm en pipetant le long du côté de la plaque. Secouez la plaque pour répandre les 2 mL d’aliquote virale.
- Incuber à 37 °C dans l’incubateur de culture tissulaire pendant 2 h. Secouez la plaque toutes les 30 minutes pour éviter que certaines zones de la plaque ne se dessèchent.
- Ajouter 5 mL de milieu DMEM avec 10 % de FBS, une supplémentation en P/S/Q et 10 mM de pyruvate de sodium, avec 5 μL de polybrene de 8 mg/mL. Laissez les cellules incuber pendant la nuit.
- Le matin, retirez les milieux des cellules et les passez dans les milieux complétés par un réactif de sélection. Lors du passage des cellules, ensemencez-les de manière à leur permettre de croître pendant 48 à 72 h et d’atteindre 50% de confluence.
REMARQUE: Pour les cellules A673 avec pSRP-iEF-2, les cellules sont ensemencées dans une division 1:5 et sélectionnées pendant 72 h avec 2 μg / mL de puromycine.
- Transduire les constructions d’expression de l’ADNc.
- Vérifiez les cellules pour confirmer une confluence de 50 à 70 %.
- Dégel des aliquotes virales pour la transduction de construction(s) d’ADNc d’intérêt. Décongeler rapidement les aliquotes congelées au bain-marie à 37 °C. Ajouter 2,5 μL de 8 mg/mL de polybrene à chaque aliquote virale et mélanger en piquant doucement.
- Retirer le milieu des cellules plaquées et ajouter doucement l’aliquote virale à la plaque de 10 cm en pipetant le long du côté de la plaque. Secouez la plaque pour répandre les 2 mL d’aliquote virale.
- Incuber à 37 °C dans l’incubateur de culture tissulaire pendant 2 h. Secouez la plaque toutes les 30 minutes pour éviter que certaines zones de la plaque ne se dessèchent.
- Ajouter 5 mL de milieu DMEM avec 10 % de FBS, une supplémentation en P/S/Q et 10 mM de pyruvate de sodium, avec 5 μL de polybrene de 8 mg/mL. Laissez les cellules incuber pendant la nuit.
- Le matin, retirez les milieux des cellules et les cellules de passage dans des milieux à double sélection. Cultiver et passer des cellules au besoin pendant 7 à 10 jours pour permettre une double sélection et expression de la construction de l’ADNc.
REMARQUE: Cette division de ce passage peut nécessiter une optimisation pour différentes lignées cellulaires. Pour les cellules A673 avec pSRP-iEF-2 et une construction pMSCV-hygro, les cellules sont passées sans se diviser en 2 μg/mL de puromycine et 100 μg/mL d’hygromycine.
2. Collecter des cellules, valider l’expression des constructions et mettre en place des tests phénotypiques corrélatifs
- Après 7 à 10 jours de double sélection, collectez les cellules dans un tube conique de 15 mL. Compter les cellules prélevées avec un hémocytomètre. Aliquot a collecté des cellules pour le séquençage de l’ARN et pour valider l’expression des constructions d’ADNc.
REMARQUE : Mettre en place tous les tests phénotypiques corrélatifs requis par la question de recherche étudiée. Les essais de formation de colonies sont un exemple de test phénotypique corrélatif qui sont utilisés ici.- Recueillir entre 5 x 105 et 1 x 106 cellules pour le séquençage de l’ARN et 2 x 106 cellules pour l’extraction des protéines. Cellules à granulés par centrifugation à 1 000 x g à 4 °C pendant 5 min et éliminent le surnageant.
- Lavez la pastille avec 1 mL de PBS froid. Granulés par centrifugation à 1 000 x g à 4 °C pendant 5 min et éliminer le surnageant. Congeler les pastilles dans de l’azote liquide et les conserver à -80 °C.
- Mettez en place des tests corrélatifs avec les cellules restantes.
REMARQUE: Le protocole peut être mis en pause ici avec des échantillons collectés stockés dans le congélateur à -80 ° C.
- Valider l’élimination de la protéine d’intérêt (si elle est utilisée) et l’expression du panel de constructions.
- Décongeler les granulés de cellules pour l’extraction des protéines sur la glace. Remettre en suspension des cellules dans un tampon d’extraction nucléaire glacé de 500 μL (20 mM HEPES pH 7,9, 140 mM NaCl, 10% glycérol, 1,5 mM MgCl2,1 mM EDTA, 1 mM DTT, 1% IGEPAL) avec inhibiteur de protéase. Laissez reposer pendant 5 min sur la glace.
- Noyaux de granulés par centrifugation à 1 000 x g à 4 °C pendant 5 min et éliminer le surnageant. Laver les noyaux dans un tampon d’extraction nucléaire glacé de 500 μL (20 mM HEPES pH 7,9, 140 mM NaCl, 10% glycérol, 1,5 mM MgCl2,1 mM EDTA, 1 mM DTT, 1% IGEPAL) avec un inhibiteur de protéase.
- Noyaux de granulés par centrifugation à 1 000 x g à 4 °C pendant 5 min et éliminer le surnageant. Remettre en suspension les noyaux dans un tampon RIPA froid de 200 μL avec inhibiteur de protéase (ajuster le volume du tampon RIPA en fonction de la taille de la pastille). Laissez-le reposer sur la glace pendant 45 à 60 minutes avec un vortex vigoureux toutes les 15 minutes.
- Débris de cellules granulées par centrifugation à 16 000 x g à 4 °C pendant 45-60 min. Conservez le surnageant et transférez-le dans un tube froid frais
- Préparer les échantillons pour l’électrophorèse SDS-PAGE en faisant bouillir 5 à 10 μg de protéine avec 1x tampon de charge pendant 5 min. Exécutez un gel SDS-PAGE au besoin pour la protéine d’intérêt.
- Transférer dans une membrane de nitrocellulose ou de PVDF au besoin pour la protéine d’intérêt. Bloquer et éponger avec les anticorps primaires et secondaires appropriés pour confirmer l’élimination de la protéine endogène (si elle est utilisée) et l’expression ectopique de la construction de l’ADNc.
REMARQUE: Le protocole peut être mis en pause ici.
- Extraire l’ARN. Évaluer la qualité et la quantité d’ARN.
- Décongeler les granulés de cellules sur la glace. Extraire l’ARN total à l’aide d’un kit d’extraction à base de colonne de spin de silice conformément aux instructions du fabricant.
- En bref, lysez les cellules à l’aide du tampon de lyse du kit. Appliquez le lysat sur une colonne de spin de silice avec un bref spin à >13000 tr/min pendant 30 à 60 secondes ou retirez l’ADNg en appliquant le lysat à une colonne d’élimination de l’ADNg avec un bref tour à >13000 tr/min pendant 30 à 60 secondes.
- Effectuer une digestion de l’ADN sur colonne si le lysat a été directement appliqué sur une colonne de spin de silice. Si vous utilisez une colonne d’élimination de l’ADNg, appliquez l’éluat sur une colonne de spin de silice avec un bref spin à >13000 tr/min pendant 30 à 60 s.
- Lavez l’ARN sur la colonne selon les instructions du fabricant. Eluter l’ARN dans 30 μL de tampon d’élution.
- Évaluer la qualité et la quantité d’ARN à l’aide d’un fluoromètre ou de tout autre instrument comparable. Assurez-vous que le rapport 260/280 est proche de 2 et qu’il y a au moins 2,5 μg d’ARN à soumettre au séquençage.
REMARQUE: Au fur et à mesure que les réplicats sont rassemblés, chaque réplicat doit être traité avec le même protocole d’extraction d’ARN. - Utilisez une petite aliquote d’ARN pour confirmer l’élimination stable de la protéine d’intérêt, si nécessaire, par qRT-PCR. Conserver l’échantillon d’ARN restant à -80 °C.
- Recueillez les répliques biologiques en répétant les étapes 1 à 2 jusqu’à ce que 3 à 4 ensembles complets d’ARN aient été collectés. Assurez-vous que chaque réplique affiche une expression adéquate des constructions d’ADNc et un renversement stable de la protéine endogène (si elle est utilisée).
3. Séquençage de nouvelle génération
- Soumettre l’ARN extrait à séquencer à l’aide d’une plate-forme de séquençage de nouvelle génération avec une cible de 50 millions de lectures d’extrémité appariées de 150 paires de bases (pb). Suivez les instructions de l’installation qui traite les échantillons. Sélectionnez pour les ARN poly-adénylés et le séquençage spécifique au brin.
4. Pipeline d’alignement et de comptage des transcriptions
Remarque : Ce protocole suppose qu’après la soumission et le traitement de l’exemple, un ensemble de fichiers FASTQ appariés est renvoyé pour chaque échantillon. Ces fichiers sont fréquemment compressés avec un suffixe de « fastq.gz ». Une analyse plus approfondie de ces fichiers FASTQ nécessitera l’accès à une installation de calcul haute performance (HPC) exécutant un système d’exploitation Linux.
- Transférer des fichiers
- Ouvrez un terminal dans l’environnement HPC avec PuTTY. Créez un répertoire pour l’analyse appelé « projet ».
- Accédez au répertoire « path_to/project » et créez un nouveau répertoire pour les fichiers fastq.gz bruts compressés appelés « fastq ». Créez également un répertoire appelé « trimmed ». Ceci est illustré à la figure S1A-C.
- Transférez les fichiers fastq.gz bruts compressés du stockage local vers le répertoire « path_to/project/fastq/ » à l’aide de WinSCP ou d’un programme similaire. Vérifiez qu’il existe un fichier « R1 » et un fichier « R2 » pour chaque échantillon, comme illustré à la figure S1B.
- Facultatif : Si nécessaire, installez TrimGalore. Définissez le répertoire contenant le fichier exécutable trim_galore dans la variable d’environnement PATH sous Linux.
REMARQUE: Les lectures et les adaptateurs de faible qualité sont coupés avec TrimGalore. TrimGalore est disponible sur https://github.com/FelixKrueger/TrimGalore. - Facultatif : Accédez au répertoire des progiciels téléchargés (c.-à-d. « path_to/logiciel »). Téléchargez le dernier package TrimGalore à l’aide de la commande « curl -fsSL https://github.com/FelixKrueger/TrimGalore/archive/[version].tar.gz -o trim_galore-[version].tar.gz ».
- Facultatif : décompressez le fichier tar.gz. Utilisez la commande « tar -xvzf trim_galore-[version_number].tar.gz ».
- Facultatif : Rendre TrimGalore exécutable. Utilisez la commande « chmod a+x path_to/software/TrimGalore-[version]/trim_galore ». Assurez-vous que ce nouveau répertoire se trouve dans le PATH. Utilisez la commande « export PATH=path_to/software/TrimGalore-[version]:$PATH ».
- Accédez à path_to/project/fastq/. Utilisez TrimGalore pour découper les lectures de faible qualité des fichiers fastq.gz à l’aide de la commande illustrée à la figure S1C.
REMARQUE: Des indicateurs supplémentaires pour cette commande peuvent être pertinents et peuvent être trouvés ici: https://github.com/FelixKrueger/TrimGalore/blob/master/Docs/
Trim_Galore_User_Guide.md - Recherchez les fichiers fastq.gz découpés dans le répertoire path_to/project/trimmed. Assurez-vous qu’ils sont appelés sample1_R1_val_1.fq.gz et sample1_R2_val_2.fq.gz
- Alignez les fichiers FASTQ découpés avec STAR et générez le nombre de transcriptions.
REMARQUE: STAR est disponible à https://github.com/alexdobin/STAR)- Facultatif : Installez STAR version 2.6 ou ultérieure. Définissez l’exécutable STAR dans le chemin d’accès.
- Facultatif : Accédez au répertoire des progiciels téléchargés (c.-à-d. « path_to/logiciel »).
- Facultatif : Téléchargez le package STAR à l’aide de la commande « curl -SLO https://github.com/alexdobin/STAR/archive/[version].tar.gz ». Décompressez le fichier tar.gz.
- Facultatif : Utilisez la commande « tar -xzf [version].tar.gz ». Rendez STAR exécutable. Utilisez la commande « chmod a+x path_to/software/STAR-[version]/bin ».
- Facultatif : Assurez-vous que ce nouveau répertoire se trouve dans le chemin d’accès. Utilisez la commande « export PATH=path_to/software/STAR-[version_number]/bin/linux_x86_64_static:$PATH ».
REMARQUE: Le manuel STAR est disponible à l’adresse suivante: (https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf). - Assurez-vous qu’il y a un indice du génome à utiliser avec STAR. Placez-le dans un répertoire distinct du répertoire path_to/project/. Si un index a déjà été généré pour des expériences antérieures, utilisez-le. Vous pouvez également utiliser un index prégénéré approprié si disponible ici : http://refgenomes.databio.org/. Sinon, construisez un nouvel index à l’aide de la commande « STAR--runMode genomeGenerate » en utilisant les instructions du manuel STAR.
REMARQUE : Pour le reste de ce protocole, le chemin d’accès à l’index STAR sera appelé « path_to/STAR_index ». - Accédez au répertoire path_to/project/. Créez un nouveau répertoire appelé « STAR_output », comme illustré à la figure S1D.
- Accédez au répertoire path_to/project/trimmed/. Utilisez la commande illustrée à la figure S1D pour exécuter STAR afin d’aligner les fichiers fastq.gz découpés.
REMARQUE: Cette étape est la plus intensive en calcul et il est recommandé de l’effectuer sur un cluster HPC avec plusieurs threads (c’est-à-dire >16) désignés pour la tâche d’alignement. Selon le nombre d’échantillons et les ressources de calcul disponibles, cette étape peut prendre plusieurs heures à plusieurs jours. - Trouvez la sortie requise pour les étapes suivantes qui contiennent le nombre de transcriptions à l’emplacement suivant : path_to/project/STAR_output/sampleN_ReadsPerGene.out.tab.
Remarque : Dans le fichier ReadsPerGene.out.tab colonne 1 contient des informations sur la fonctionnalité en cours de comptage. La colonne 2 contient les nombres de lectures non transférées, la colonne 3 contient les comptes de lectures échouées vers l’avant et la colonne 4 contient les comptes de lectures échouées inverses. Les quatre premières lignes de ce fichier contiendront des informations sur les lectures alignées qui ne se sont pas alignées sur un seul gène. Ce protocole nécessite le nombre de lectures non transférées. - Utilisez RStudio (préférable) ou R dans l’environnement HPC pour compiler les données de la ligne 5 et des lignes inférieures pour les colonnes 1 et 2 de chaque exemple. Définissez le répertoire de travail sur « projet » dans R.
- Lisez dans chaque fichier ReadsPerGene.out.tab à l’aide de la commande de la figure S2A. Pour la première colonne, ne prenez que les caractères avant le « . » dans la colonne « ENsembl gene ID » pour faciliter le traitement en aval.
- Compilez les comptes de tous les échantillons dans une trame de données appelée « totcts » à l’aide des commandes de la figure S2B. Enregistrez cette nouvelle table de données de comptage brutes sous la forme d’un fichier .txt délimité par des tabulations, c’est-à-dire sample_counts.txt, si vous le souhaitez, à l’aide de la commande « write.table ».
REMARQUE : L’ordre de l’ID du gène Ensembl est le même pour chaque fichier ReadsPerGene.out.tab dans les échantillons.
5. Expression différentielle et analyse en aval
- Normaliser les effets de lot entre les échantillons avec ComBat.
REMARQUE: Il y a deux variables possibles qui expliquent les changements dans l’expression des gènes, la première étant la construction utilisée (c.-à-d. l’échantillon) et la seconde étant des facteurs externes associés au passage des cellules dans le temps (c.-à-d. le lot). Une étape pour normaliser les échantillons pour la variation de lot à lot avec le R-package ComBat est recommandée.- Installez si nécessaire et chargez les bibliothèques pour sva, DESeq2, AnnotationDBI, org. Hs.eg.db, pheatmap, RColorBrewer, genefilter, Cairo, ggplot2, ggbiplot, rgl et reshape2 comme illustré à la figure S2C. Pour l’installation, utilisez la commande « install.packages » ou Bioconductor selon la documentation de chaque paquet.
- Filtrez d’abord les données uniquement sur les gènes qui ont au moins un nombre par lecture. Enregistrez ce nouveau tableau pour indiquer le filtrage, comme illustré à la figure S2D.
REMARQUE: Fréquemment, de nombreux gènes auront un nombre de lectures très faible ou nul. - Préparez une deuxième table pour la normalisation par lots appelée « vars » comme illustré à la figure S2E. Définissez les noms de ligne sur les noms uniques de chaque exemple. Définissez les noms des colonnes sur « sample », « batch » et « construct ».
- Attribuez à tous les échantillons un numéro unique dans la colonne « échantillon » de 1 à n, n étant le nombre d’échantillons. Attribuez des numéros de lot à tous les échantillons de la colonne « lot » de manière à ce que les a_1 condition et les b_1 de condition se voient attribuer 1, et que les a_2 conditionnelle et b_2 de condition se voient attribuer 2. Attribuez toutes les désignations de condition à tous les échantillons de la colonne « construction » de sorte que les échantillons de condition a soient tous « A » et que les échantillons de condition b soient tous « B ».
- Définissez également la variable batch et une matrice de modèle NULL spécifique pour ComBat, comme illustré à la figure S2F. Exécutez ComBat avec la commande définie à la figure S2F.
- Organisez davantage les données en arrondissant à l’entier le plus proche. Supprimez également les gènes ayant une valeur négative. Utilisez les commandes illustrées à la figure S3A.
REMARQUE: La sortie de la normalisation par lots aura des nombres de lecture non entiers et certains gènes avec des valeurs négatives. Cette étape est nécessaire car l’analyse d’expression différentielle en aval ne prend pas en charge le nombre de lectures négatives. - Définissez le profil d’expression différentielle pour chaque construction à l’aide de DESeq2.
- Entrez le plan d’expérience pour DESeq2 comme illustré à la figure S3B. Construisez un DESeqDataSet (dds) à l’aide de la fonction DESeqDataSetFromMatrix, estimez les facteurs de taille et exécutez DESEq2, comme illustré à la figure S3B.
REMARQUE : Il est impératif que les données de colonne saisies pour " condition " soient dans le même ordre que la colonne dans la matrice de comptage. - Afin d’évaluer la qualité de l’analyse, extraire les comptages normalisés par rlog utilisés par DESeq2 comme le montre la figure S3B.
REMARQUE: Au cours de l’analyse, DESeq2 transforme les comptes avec un « journal régularisé », rlog, transformation pour réduire les différences d’échantillon à échantillon pour les gènes à faible nombre (faible information) afin de préserver les différences dans les gènes avec des comptes plus élevés entre les échantillons (information élevée). - Lors de l’extraction des résultats pour chaque profil transcriptionnel à partir des résultats de DESeq2, effectuez des comparaisons par paires en référence à la condition de knockdown ou au vecteur vide de base, comme illustré à la figure S3C. Modifiez davantage ces résultats avec les symboles du gène HGNC comme le montre la figure S3D.
- Comme le montre la figure S3E,extraire les données des résultats de DESeq2. Exportez en tant que fichier unique avec l’ID du gène Ensembl, le symbole HGNC, l’expression moyenne de base et les données d’expression différentielle pour toutes les constructions avec log2FoldChange et les valeurs p brutes et ajustées.
REMARQUE : L’utilisation d’une valeur de p ajustée < 0,05 est le seuil recommandé pour l’expression différentielle. - Évaluer la normalisation réussie des lots et la similitude intra-échantillon. Vérifiez le regroupement d’échantillons avec des diagrammes de distance d’échantillon à ÉCHANTILLON et d’échantillon à échantillon à l’aide des comptages normalisés rlog à l’aide du code athe illustré dans les figures S4A-B.
- Entrez le plan d’expérience pour DESeq2 comme illustré à la figure S3B. Construisez un DESeqDataSet (dds) à l’aide de la fonction DESeqDataSetFromMatrix, estimez les facteurs de taille et exécutez DESEq2, comme illustré à la figure S3B.
- Utilisez les profils d’expression différentielle pour générer des diagrammes de volcans à l’aide du code de la figure S4C. Évaluer les changements dans l’expression des gènes à travers les constructions.
- Utilisez les comptages normalisés rlog et le regroupement hiérarchique pour identifier les signatures de gènes uniques aux différentes constructions. Utilisez le code illustré à la figure S4D.
- Extrayez les 1000 gènes les plus variables de toutes les constructions d’une matrice. Utilisez pheatmap pour effectuer un regroupement hiérarchique non supervisé de vos échantillons en fonction de ces gènes.
- Extrayez les grappes d’intérêt du dendrogramme en décidant à quel niveau des grappes d’intérêt du dendrogramme apparaissent. Définissez « k » comme étant égal au nombre de clusters à ce niveau. Relotez la carte thermique ordonnée par cluster pour déterminer quels clusters sont d’intérêt comme le montre la figure S5.
- Exportez la liste des gènes associés à chaque cluster comme illustré dans le tableau S1. Utilisez ces informations pour déterminer les gènes dans les groupes d’intérêt.
- Identifier les rôles biologiques des différents groupes de gènes identifiés et comparer entre les classes. Cela peut être effectué à l’aide d’une variété d’outils bioinformatiques. ToppGene24 est utilisé ici et est disponible gratuitement en ligne.
REMARQUE: Il existe de nombreux outils gratuits qui nécessitent simplement une liste de gènes à copier et coller dans un champ sur un site Web. Choisissez les outils d’analyse les plus appropriés pour les questions de recherche étudiées. - Éventuellement, s’il existe des données disponibles sur la liaison génomique qui déterminent la sortie transcriptionnelle pour le facteur de transcription d’intérêt, comparez la réponse transcriptionnelle aux gènes associés à différents éléments de liaison pour évaluer davantage la fonction mutante.
6. Comparaison avec les phénotypes pertinents
- Comparez les phénotypes corrélatifs avec les données de profil transcriptomique générées et interprétez-les le cas échéant.
Résultats
Les données préliminaires de qRT-PCR ont suggéré qu’un mutant EWS/FLI appelé DAF, avec des mutations spécifiques de tyrosine à alanine dans la région répétitive et désordonnée de EWS, maintenait la capacité d’activer les gènes cibles EWS/FLI, mais ne parvenait pas à réprimer les gènes cibles critiques23. Afin de mieux comprendre la relation entre ces résidus dans le domaine EWS et la fonction EWS/FLI, le protocole décrit ci-dessus et décrit à la figure 1 a été utilisé. Les cellules du sarcome d’Ewing A673 ont été transduites viralement avec un shRNA ciblant le 3'UTR de FLI1,ce qui a entraîné l’épuisement des EWS/FLI endogènes. Après quatre jours de sélection, la fonction EWS /FLI a été sauvée avec la transduction virale de différentes constructions mutantes EWS/FLI marquées 3XFLAG, avec un vecteur vide comme contrôle de l’absence de sauvetage. Un mutant non fonctionnel dépourvu du domaine EWS, appelé Δ22, a été utilisé comme témoin négatif et un EWS/FLI de type sauvage, appelé wtEF, a été utilisé comme témoin positif(Figure 2A). DAF a été utilisé comme construction de test, bien que plus d’une construction de test puisse être utilisée si vous le souhaitez. Les cellules ont été sélectionnées pendant 10 jours supplémentaires pour permettre à l’expression de la construction de se stabiliser, puis collectées pour l’ARN (avec une étape d’élimination de l’ADNg), les tests de formation de protéines et de colonies. Quatre répliques ont été recueillies et des qRT-PCR représentatives et des transferts occidentaux montrant un renversement et un sauvetage efficaces sont représentés à la figure 2B-D. Il convient de noter que les cellules sauvées par le DAF n’ont pas réussi à former des colonies, comme le montre la figure 2E,ce qui suggère une altération de la transformation oncogénique.
Après l’achèvement de la validation répliquée et des tests phénotypiques, l’ARN a été soumis à l’Institut de médecine génomique du Nationwide Children’s Hospital pour la préparation de la bibliothèque et le séquençage de nouvelle génération avec environ 50 millions de lectures appariées de 150 pb collectées. Les données ont été renvoyées sous forme de fichiers fastq.gz. Les lectures de mauvaise qualité ont été coupées à partir de ces fichiers avec TrimGalore et STAR a été utilisé pour aligner les lectures sur le génome humain hg19 et compter les lectures par gène. hg19 a été utilisé à des fins de compatibilité avec les autres ensembles de données organisés pour EWS/FLI utilisés dans l’analyse en aval. Ces comptages de lecture ont été combinés en une seule matrice de comptage pour tous les échantillons, dont les 6 premières lignes sont illustrées à la figure 3.
Les comptages ont d’abord été effectués à travers DESeq2 sans normalisation de lot, cependant, l’inspection visuelle de la distance d’échantillon à échantillon a montré des effets de lot potentiellement confondants, comme le montrent des flèches rouges à la figure 4A. Cela est probablement dû à la variabilité biologique introduite par le passage des cellules en culture et aux différences dans le traitement de chaque lot. La normalisation des effets de lot a été effectuée avec ComBat et est généralement recommandée. Les distances d’échantillon à échantillon des données normalisées par lots sont illustrées à la figure 4B. Après la normalisation par lots, DESeq2 a été utilisé pour générer des profils transcriptionnels pour les trois constructions (wtEF, Δ22 et DAF) par rapport à la ligne de base. Notez que bien que les cellules A673 « parentales » (simulation de renversement et de sauvetage simulé, appelées « iLuc » ici) aient été incluses dans l’analyse différentielle, la référence pour cette expérience sont les cellules avec des cellules appauvries en EWS / FLI, appelées cellules iEF. Le profil transcriptionnel peut être généré pour la protéine endogène ici en comparant l’échantillon iLuc à l’iEF, ce qui peut être intéressant pour comprendre le fonctionnement du système de sauvetage, mais ce n’est pas le but de cette analyse particulière. Les profils transcriptionnels générés pour les mutants comprennent des contrôles positifs (wtEF) et négatifs (Δ22), en ce qui concerne l’iEF, de sorte que ceux-ci devraient servir de repères pour d’autres mutants. Ceci est important, car le contrôle positif dans cet exemple n’a pas complètement récapitulé la fonction de l’EWS/FLI endogène comme discutéailleurs 7,23.
L’analyse en composantes principales (ACP) de la figure 5 suggère que le profil transcriptionnel du DAF est intermédiaire entre wtEF et Δ22, confirmant la fonction partielle. De plus, le regroupement hiérarchique des 1000 gènes les plus variables sur les échantillons a montré que le DAF n’a pas réussi à réprimer les gènes cibles EWS/FLI et n’a que partiellement conservé l’activité d’activation des gènes, comme le montrent les figures 6A et S5. L’analyse de ToppGene a suggéré que les classes de gènes que DAF active sont fonctionnellement distinctes des cibles activées par EWS / FLI où DAF n’est pas fonctionnel (Figure 6B). Fait intéressant, la fonction des gènes activés sauvés par wtEF, mais pas par DAF, semble être liée au contrôle transcriptionnel et à la régulation de la chromatine. Sur la base des résultats des essais de formation de colonies, les gènes de cette signature génétique de base devraient être analysés plus en détail pour leur rôle dans l’oncogenèse médiée par EWS / FLI. L’importance de la répression génique médiée par EWS/FLI a déjà été décrite17.
On sait que EWS/FLI possède une affinité de liaison unique pour les éléments répétitifs GGAA-microsatellites19,22, et que la liaison à ces éléments entraîne la régulation génique en aval11,15,18,20,22. Ces microsatellites ont été caractérisés comme étant soit associés à l’activation ou à la répression, et soit proximaux au TSS (< 5 kb), soit distaux au (> 5 kb) TSS25. En outre, il existe des gènes régulés EWS / FLI avec des motifs ETS à haute affinité (HA) proximaux à TSS23. Afin d’analyser davantage les caractéristiques de la fonction DAF et les types de gènes activés par EWS / FLI que DAF a pu sauver, l’expression différentielle des gènes associés à ces différentes classes a été analysée. Fait intéressant, DAF a été le plus en mesure de sauver les gènes activés par microsatellite GGAA, mais incapable de sauver les gènes activés près d’un site HA comme le montre la figure 7. Comme on l’a vu avec le regroupement hiérarchique, DAF ne parvient pas à sauver la répression médiée par EWS / FLI à travers les classes de motifs. Ces données suggèrent que le DAF conserve suffisamment de caractéristiques structurelles de l’EWS pour se lier et s’activer à partir des microsatellites GGAA, à la fois proximaux et distaux au TSS. Cela provient probablement du domaine SYGQ intact considéré comme important pour l’activité EWS / FLI à GGAA répète11. Ces données suggèrent également que les tyrosines spécifiques mutées dans le DAF jouent des rôles importants, mais mal compris, dans la régulation des gènes médiée par EWS / FLI à partir des sites HA, ainsi que dans la répression des gènes, mettant en évidence un domaine important d’investigation supplémentaire.

Figure 1 : Flux de travail. Représentation de la procédure étape par étape pour effectuer un mappage structure-fonction par transcriptomique. Les cellules ont d’abord été préparées pour exprimer la suite de constructions requises pour la cartographie structure-fonction. Après l’expression, les cellules ont été récoltées pour l’ARN et les protéines et testées pour les phénotypes corrélatifs. L’expression des constructions a été validée, et ce processus a été répété 3-4 fois pour recueillir des répliques biologiques indépendantes. L’ARN a ensuite été soumis au séquençage de nouvelle génération (NGS). Lorsque les données ont été reçues, les données ont été ajustées pour la qualité, alignées et le nombre de transcriptions a été calculé. Les effets des lots ont été contrôlés et les signatures transcriptomiques et l’expression différentielle ont été déterminées à l’aide de DESeq2. Le clustering hiérarchique et l’analyse en aval intégrant d’autres ensembles de données -omiques et différentes analyses de chemin ou fonctionnelles peuvent être incorporés. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Validation de l’expression de construction et des essais corrélatifs. (A) Schéma représentant les constructions testées dans cet exemple. (B) Validation de l’élimination des EWS/FLI endogènes et expression des constructions marquées 3X-FLAG par immunoblot. (C,D) Validation de l’activité de construction d’un gène cible activé EWS/FLI(C), NR0B1,et(D)gène cible réprimé, TGFBR2,par qRT-PCR. Les données sont présentées sous forme d’écart-type moyen +/-. Les valeurs de P ont été calculées avec un test de signification honnête de Tukey. * p < 0,05, ** p < 0,01, *** p < 0,005 (E) Nombre de colonies à partir d’essais de gélose molle effectués pour évaluer l’activité de transformation des constructions. Les valeurs de P ont été calculées avec un test de signification honnête de Tukey. * p < 0,05, ** p < 0,01, *** p < 0,005. Cette figure est adaptée de Theisen, et al.23Veuillez cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 3 : Données finales de comptage rassemblées pour analyse. Capture d’écran des 6 premières lignes du fichier de comptage avec les numérations de gènes pour tous les échantillons à normaliser et à analyser par lots. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Cartes thermiques de distance d’échantillon à échantillon. (A) Graphique de distance d’échantillon à échantillon montrant le regroupement d’échantillons des données de comptage brutes. Les échantillons qui se regroupent à la fois par lot et par échantillon sont signalés par des flèches rouges. (B) Diagramme de distance échantillon à échantillon suivant la normalisation du lot avec ComBat. Ici, les échantillons de toutes les répliques se regroupent ensemble, indépendamment du lot. Veuillez cliquer ici pour voir une version agrandie de cette figure.
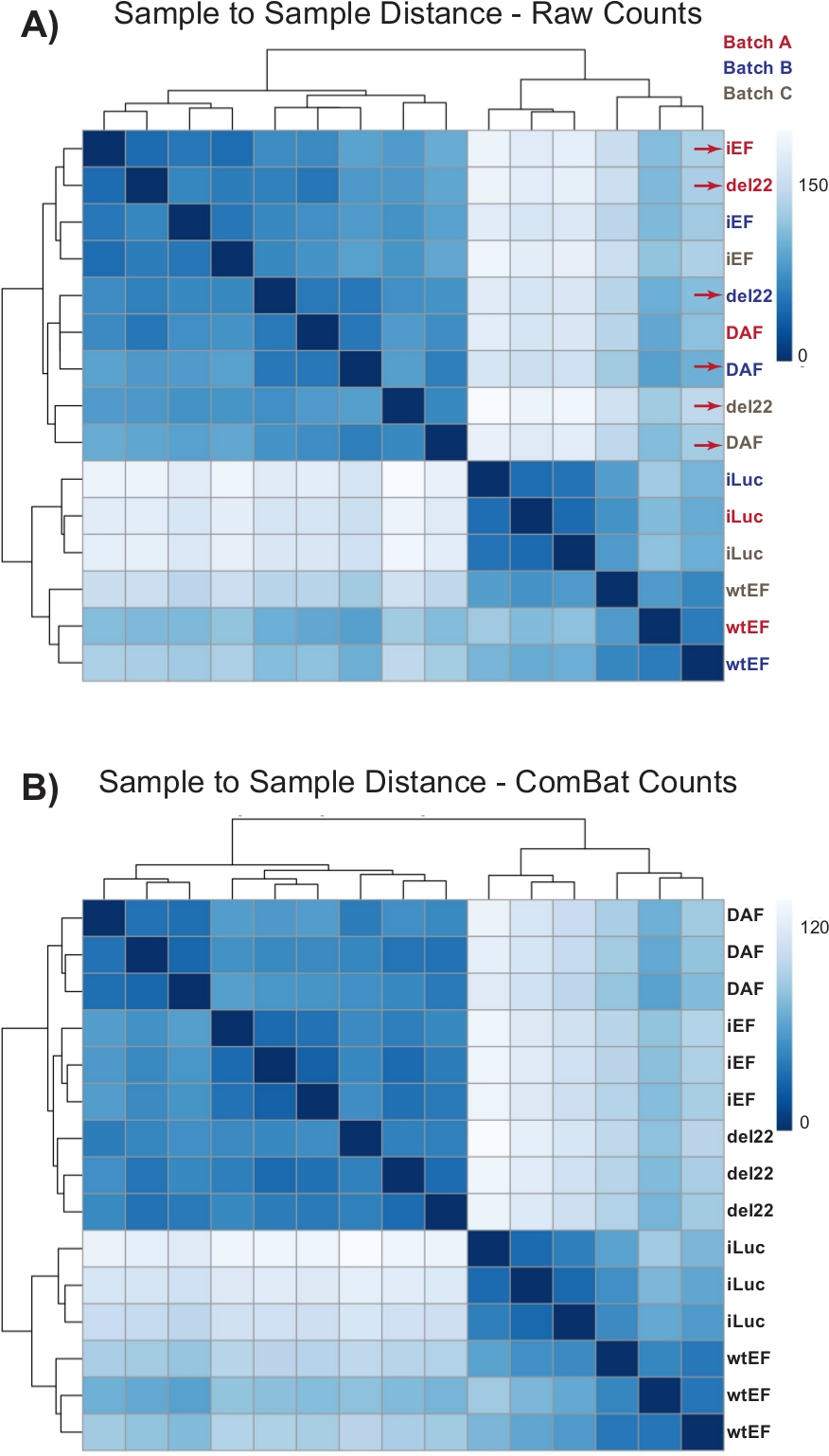{kind=link}

Figure 5 : Résultats de l’analyse de l’expression différentielle. (A) Le diagramme d’analyse composante principale (APC) des signatures transcriptomiques générées pour tous les échantillons montre un fort regroupement intra-échantillon et démontre que le DAF est intermédié entre les témoins positifs (wtEF) et négatifs (Δ22). (B) Diagrammes de volcan montrant le -log(p-value) tracé par rapport au log2FoldChange pour les gènes dans chaque construction. Gènes avec une valeur de p ajustée < 0,05 et une |log2(FoldChange)| > 1 sont considérés comme significatifs et sont indiqués en rouge. Le panneau 5B est adapté de Theisen, et al.23Veuillez cliquer ici pour voir une version plus grande de cette figure.
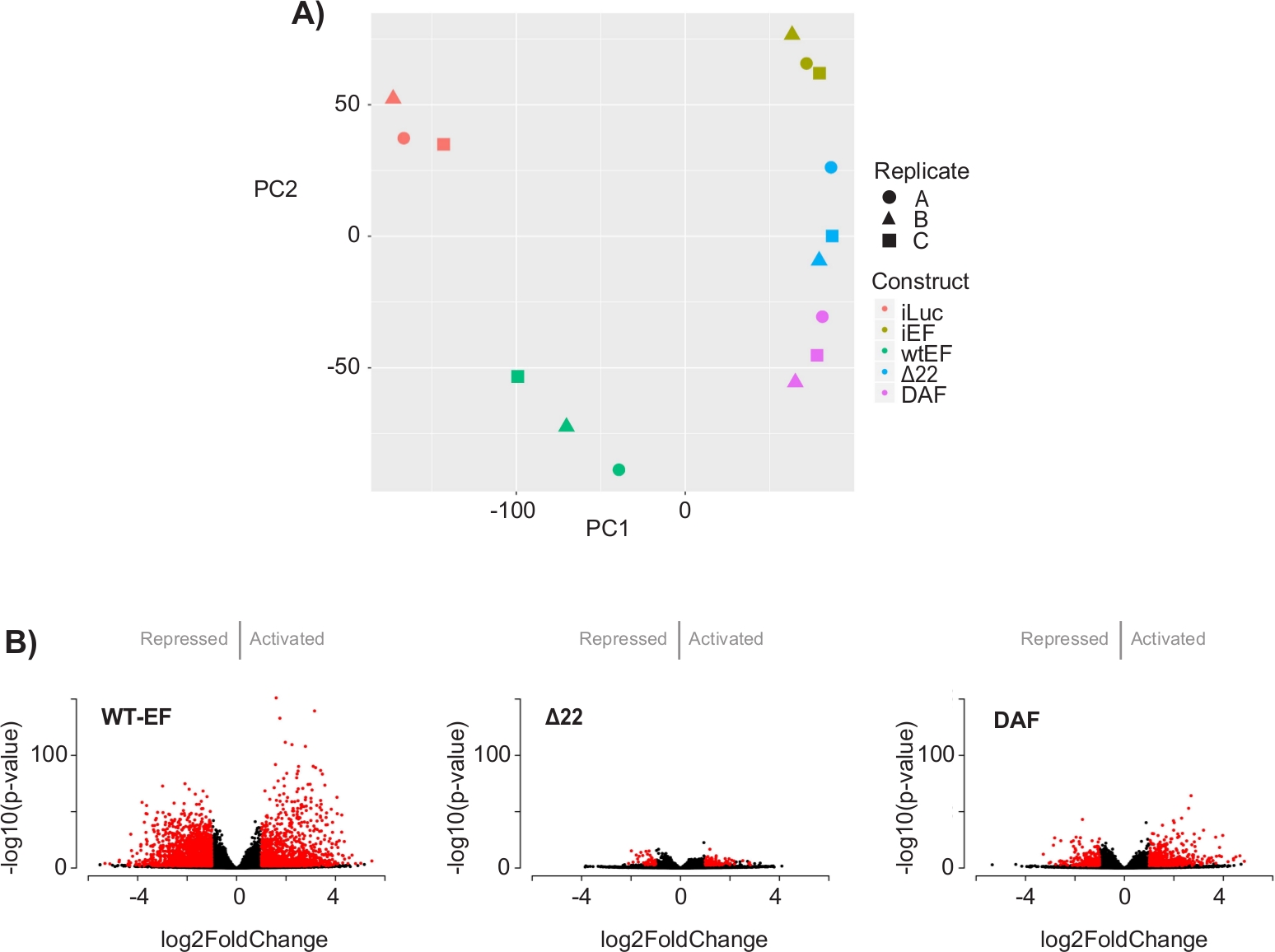{kind=link}

Figure 6 : Regroupement hiérarchique pour identifier les classes de gènes. (A) Le regroupement hiérarchique des 1000 gènes les plus variables dans toutes les constructions et la ligne de base, iEF, montre que DAF sauve partiellement l’activation génique médiée par EWS / FLI. (B) Résultats de l’ontologie génique (fonction moléculaire) de ToppGene montrant l’enrichissement fonctionnel des gènes activés par EWS / FLI qui sont sauvés ou non sauvés par DAF. Le panneau 6B est adapté de Theisen, et al.23Veuillez cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 7: Analyse détaillée de différents éléments de réponse du facteur de transcription à différentes constructions: (A) Schéma illustrant le traitement des données utilisé pour générer des panels (B) et (C) en incorporant d’autres ensembles de données disponibles avec les profils transcriptomiques ici. (B,C) Compilation montrant le sauvetage de différentes classes de cibles directes EWS/FLI- (B) activées et (C) réprimées. Les gènes inclus n’étaient que les gènes dont l’expression différentielle était détectable par EWS/FLI endogène. Dans chaque diagramme à secteurs, le gris représente la partie des gènes qui ne sont pas sauvés par la construction. Le rouge représente la partie des gènes qui sont activés différentiellement, et le bleu représente la partie des gènes qui sont réprimés différentiellement. Cette figure est adaptée de Theisen, et al.23Veuillez cliquer ici pour voir une version plus grande de cette figure.
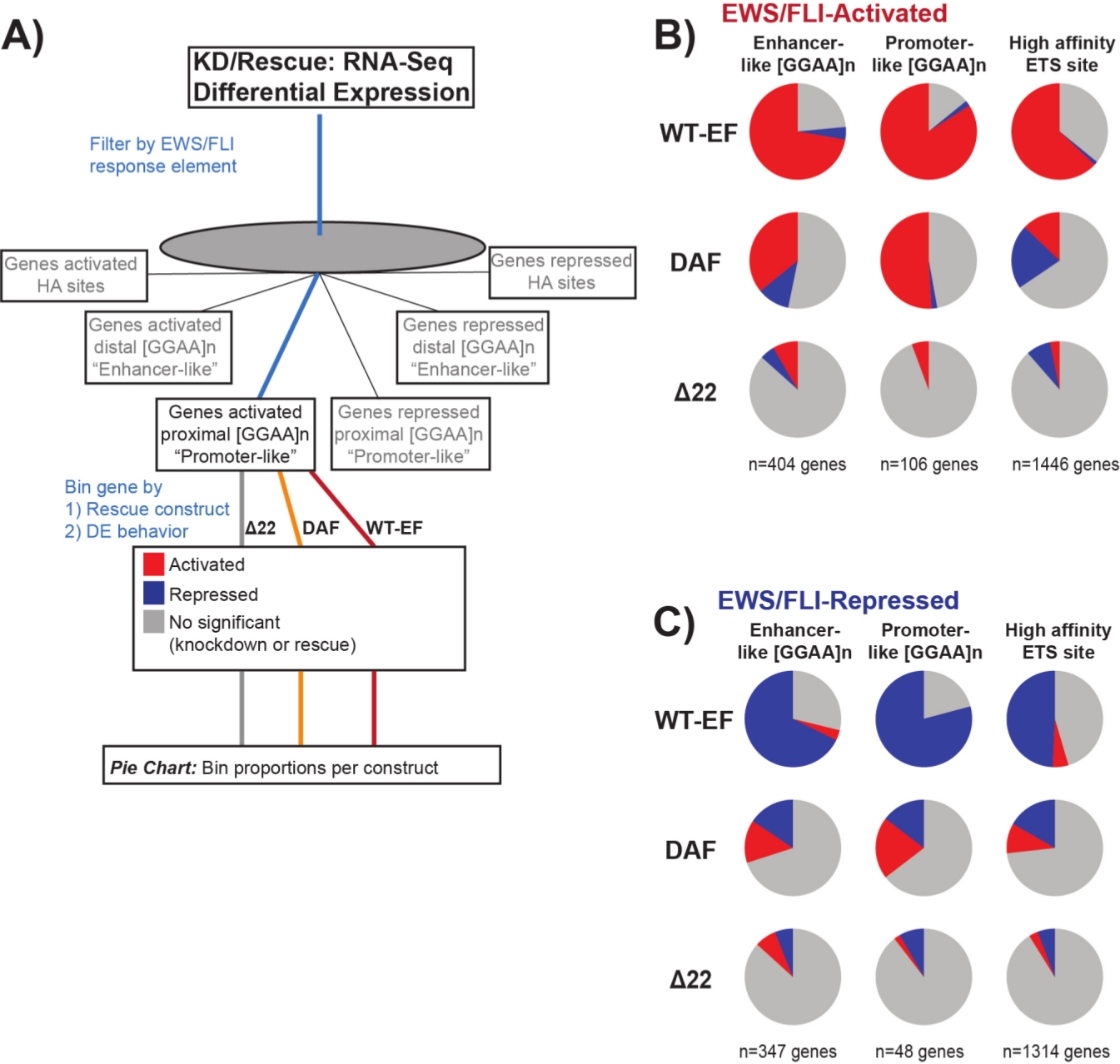{kind=link}
Figure S1 : Chargement des fichiers fastq.gz dans l’environnement HPC, rognage et alignement. Veuillez cliquer ici pour télécharger cette figure.
{kind=link}
Figure S2 : Regroupement des nombres de lectures entre les échantillons et exécution de la normalisation des lots avec ComBat. Veuillez cliquer ici pour télécharger cette figure.
{kind=link}
Figure S3 : Exécution de DESeq2 et extraction des résultats de l’analyse de l’expression différentielle. Veuillez cliquer ici pour télécharger cette figure.
{kind=link}
Figure S4 : Analyse de la sortie. Veuillez cliquer ici pour télécharger cette figure.
{kind=link}
Figure S5 : Regroupement hiérarchique pour identifier les classes de gènes : Regroupement hiérarchique des 1000 gènes les plus variables dans toutes les constructions et la base de référence, iEF, triée en k clusters. Dans ce cas, k= 7, mais ce paramètre est défini par l’utilisateur comme illustré à la figure S4D. Veuillez cliquer ici pour télécharger cette figure.
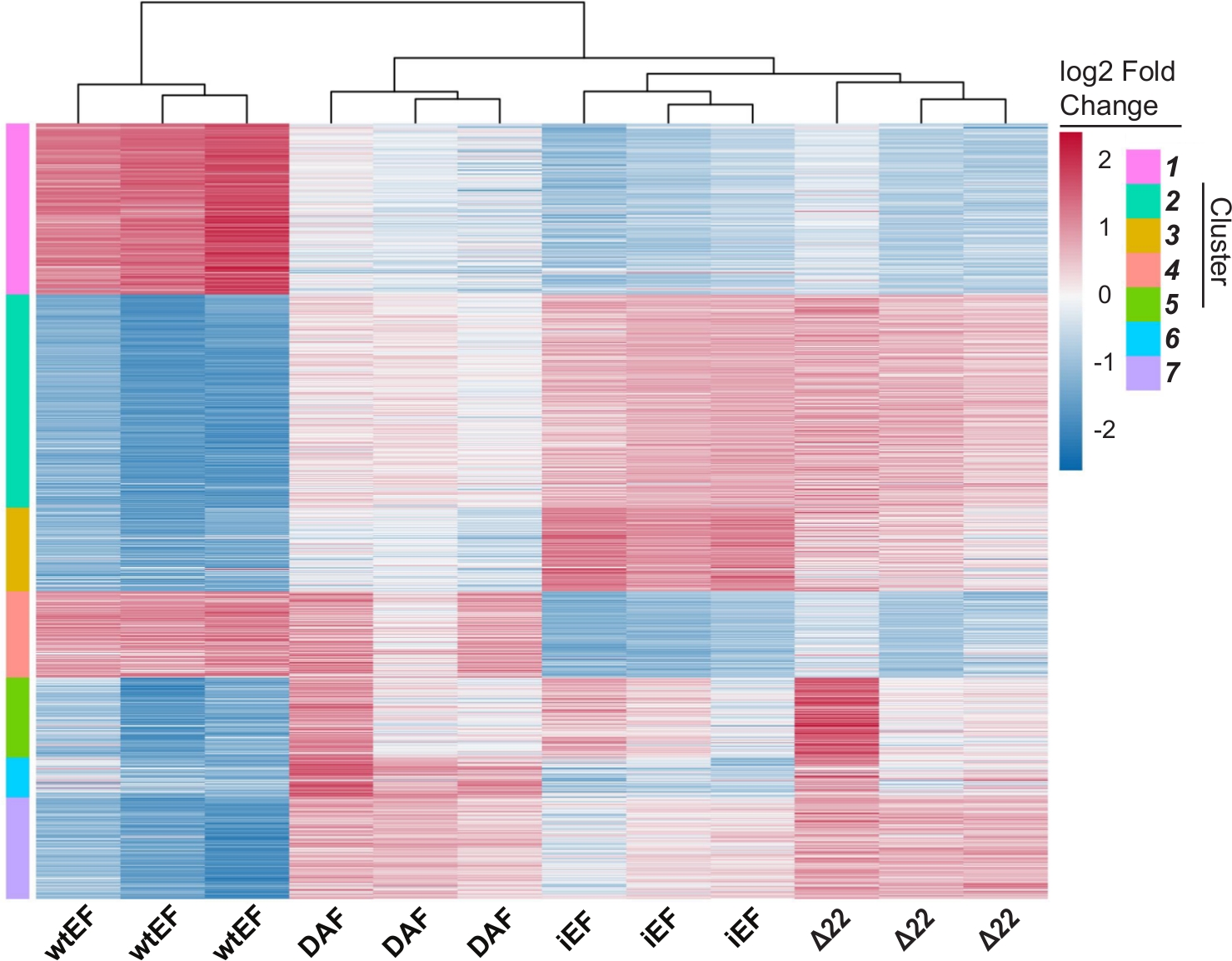{kind=link}
Tableau S1 : Liste des gènes (ID du gène Ensembl) avec annotation de cluster. Veuillez cliquer ici pour télécharger ce tableau.
Discussion
L’étude des mécanismes biochimiques des facteurs de transcription oncogéniques est d’une importance cruciale pour comprendre les maladies qu’ils causent et pour concevoir de nouvelles stratégies thérapeutiques. Cela est particulièrement vrai dans les tumeurs malignes caractérisées par des translocations chromosomiques entraînant des facteurs de transcription de fusion. Les domaines inclus dans ces protéines chimériques peuvent manquer d’interactions significatives avec les domaines régulateurs présents dans les protéines de type sauvage, ce qui complique la capacité d’interpréter les informations structure-fonction dans le contexte de la fusion26,27,28. De plus, beaucoup de ces fusions oncogènes sont caractérisées par des domaines intrinsèquement désordonnés de faible complexité10,13,29,30.
Le domaine EWS est un exemple d’un tel domaine intrinsèquement désordonné qui est impliqué dans une variété de fusions oncogènes10. La nature intrinsèquement désordonnée et répétitive a entravé les efforts de compréhension des mécanismes moléculaires employés par le domaine EWS. Les efforts antérieurs pour étudier la structure-fonction ont largement eu recours à l’utilisation de différents mutants dans le contexte de tests de gènes rapporteurs ou dans des arrière-plans cellulaires qui ne parviennent pas à récapituler le contexte cellulaire pertinent, ou qui n’ont aucune variation structurelle produisant une fonction partielle significative11,17,25. La méthode présentée ici aborde ces questions. La cartographie structure-fonction est effectuée dans un contexte cellulaire pertinent pour la maladie et le séquençage de nouvelle génération permet le profilage transcriptomique pour évaluer la fonction du facteur de transcription dans le contexte de la chromatine native. Dans le cas spécifique du mutant DAF d’EWS/FLI, il a été rapporté que le DAF montrait peu d’activité dans les essais rapporteurs utilisant des éléments de réponse isolés, mais qu’il montrait une activité dans le contexte du promoteur complet du gène, soit dans un test rapporteur, soit dans la chromatine native, suggérant un phénotype23intéressant. L’utilisation de la méthode décrite ici résout plus directement la question de savoir quel type d’éléments régulateurs du génome sont les plus réactifs dans le contexte de la maladie. En testant simultanément tous les gènes cibles candidats dans leur contexte natif de chromatine, une approche transcriptomique est plus susceptible d’identifier des constructions à fonction partielle.
La force inhérente à l’utilisation d’un fond cellulaire pertinent pour la maladie est peut-être la plus grande limitation de cette technique. L’un des facteurs les plus importants est le choix du système cellulaire approprié pour ces expériences. De nombreuses lignées cellulaires dérivées de tumeurs malignes avec des facteurs de transcription pathognomoniques ne tolèrent pas facilement l’élimination de ce facteur de transcription et, dans de nombreux cas, en particulier pour les cancers pédiatriques, la véritable cellule d’origine reste controversée et l’expression de l’oncogène dans d’autres milieux cellulaires est prohibitivement toxique31,32 . Dans ces cas, il peut être utile d’effectuer des expériences dans un contexte cellulaire différent, à condition que le chercheur fasse preuve de prudence dans l’interprétation des résultats et valide de manière appropriée tout résultat pertinent dans un type de cellule plus pertinent pour la maladie.
Il est extrêmement important de valider soigneusement la stabilité et les conséquences phénotypiques de l’expression de l’oncogène et de ne soumettre que des échantillons pour le séquençage qui répondent à des critères stricts. Ici, cela comprenait le transfert western pour confirmer l’élimination et le sauvetage, et la qRT-PCR d’un petit nombre de gènes cibles connus pour valider le témoin positif(Figure 2). Il est également crucial de réduire autant que possible la variabilité des lots en effectuant soigneusement les préparations de cellules et d’ARN de la même manière que possible dans chaque lot.
La méthode décrite ici devient particulièrement puissante lorsqu’elle est associée à d’autres types de données génomiques qui parlent de la fonction pangénomique du facteur de transcription à l’étude. Les orientations futures de ce type d’analyse structure-fonction s’étendraient pour inclure ChIP-seq et ATAC-seq afin de déterminer la liaison du facteur de transcription et tout changement induit dans l’accessibilité de la chromatine. En tant que suite, ce type de données peut indiquer où différents composants structurels d’un facteur de transcription oncogénique contribuent à différents aspects de la fonction (c.-à-d. liaison à l’ADN vs modification de la chromatine vs recrutement co-régulateur). Dans l’ensemble, l’utilisation d’approches basées sur le NGS pour cartographier les relations structure-fonction des facteurs de transcription de fusion peut révéler de nouvelles connaissances sur les déterminants biochimiques de la fonction oncogène de ces protéines. Ceci est important pour approfondir notre compréhension des maladies qu’ils causent et pour permettre le développement de nouvelles stratégies thérapeutiques.
Déclarations de divulgation
SLL déclare un conflit d’intérêts en tant que membre du conseil consultatif et actionnaire de Salarius Pharmaceuticals. SLL est également un inventeur répertorié sur les brevets américains No. US 7 393 253 B2, « Méthodes et compositions pour le diagnostic et le traitement du sarcome d’Ewing », et US 8 557 532, « Diagnostic et traitement du sarcome d’Ewing pharmacorésistant ». Cela ne modifie pas notre adhésion aux politiques de JoVE sur le partage de données et de matériaux.
Remerciements
Cette recherche a été soutenue par le High Performance Computing Facility de l’Abigail Wexner Research Institute du Nationwide Children’s Hospital. Ce travail a été soutenu par le National Institute of Health National Cancer Institute [U54 CA231641 à SLL, R01 CA183776 à SLL]; Alex’s Lemonade Stand Foundation [Prix du jeune chercheur à ERT]; Pelotonia [Bourse à l’ERT]; et la bourse biomédicale CJ Martin Overseas biomedical du Conseil national de la santé et de la recherche médicale [APP1111032 à KIP].
matériels
| Name | Company | Catalog Number | Comments |
| Wet Lab Reagents | |||
| anti-FLI rabbit pAb | Abcam | ab15289 | 1:500 |
| anti-lamin B1 rabbit pAb | Abcam | ab16048 | 1:2000 |
| Cell-based system for introduction of mutant constructs | Determined by cell system used | ||
| Cryotubes | For viral aliquots | ||
| DMEM | Corning Cellgro | 10-013-CV | For viral production |
| Fetal bovine serum | Gibco | 16000-044 | For viral production |
| G418 | ThermoFisher | 10131027 | For viral production |
| HEK293-EBNAs | ATCC | CRL-10852 | For viral production |
| HEPES | Gibco | 15630106 | |
| Hygromycin B | ThermoFisher | 10687010 | |
| M2 anti-FLAG mouse mAb | Sigma | F3165 | 1:2000 |
| Near IR-secondary antibodies | Li-Cor | ||
| Optimem | Gibco | 31985062 | For viral production |
| Penicillin/Streptomycin/Glutamine | Gibco | 10378-016 | For viral production |
| Polybrene | Sigma | TR-1003-G | For viral transduction |
| Puromycin | Sigma | P8833 | Stored at 2 mg/mL stock |
| RNeasy Plus kit | Qiagen | 74136 | Has gDNA removal columns |
| Selection reagents | As dictated by cell system used | ||
| Sodium Pyruvate | Gibco | 11360-070 | For viral production |
| Tissue culture media | Determined by cell system used | ||
| TransIT-LT1 | Mirus | MIR 2304 | For viral production |
| Software | |||
| Access to HPC environment | |||
| AnnotationDbi | 1.38.2 | ||
| Cairo | 1.5-10 | ||
| DESeq2 | 1.16.1 | ||
| genefilter | 1.58.1 | ||
| ggbiplot | 0.55 | ||
| ggplot2 | 3.1.1 | ||
| org.Hs.eg.db | 3.4.1 | ||
| pheatmap | 1.0.12 | ||
| PuTTY | |||
| R | 3.4.0 | ||
| RColorBrewer | 1.1-2 | ||
| reshape2 | 1.4.3 | ||
| rgl | 0.100.19 | ||
| R-studio | |||
| STAR | Version 2.6 or later | ||
| sva | 3.24.4 | ||
| TrimGalore! | |||
| WinSCP |
Références
- Miettinen, M., et al. New fusion sarcomas: histopathology and clinical significance of selected entities. Human Pathology. 86, 57-65 (2019).
- Knott, M. M. L., et al. Targeting the undruggable: exploiting neomorphic features of fusion oncoproteins in childhood sarcomas for innovative therapies. Cancer and Metastasis Reviews. 38, 625-642 (2019).
- Yoshihara, K., et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 34, 4845-4854 (2015).
- Duesberg, P. H. Cancer genes generated by rare chromosomal rearrangements rather than activation of oncogenes. Medical Oncology and Tumor Pharmacotherapy. 4, 163-175 (1987).
- Dupain, C., Harttrampf, A. C., Urbinati, G., Geoerger, B., Massaad-Massade, L. Relevance of Fusion Genes in Pediatric Cancers: Toward Precision Medicine. Molecular Therapy - Nucleic Acids. 6, 315-326 (2017).
- Mitelman, F., Johansson, B., Mertens, F. The impact of translocations and gene fusions on cancer causation. Nature Reviews Cancer. 7, 233-245 (2007).
- Smith, R., et al. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing's sarcoma. Cancer Cell. 9, 405-416 (2006).
- Davicioni, E., et al. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Research. 66, 6936-6946 (2006).
- Gröbner, S. N., et al. The landscape of genomic alterations across childhood cancers. Nature. 555, 321-327 (2018).
- Kim, J., Pelletier, J. Molecular genetics of chromosome translocations involving EWS and related family members. Physiological Genomics. 1, 127-138 (1999).
- Boulay, G., et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell. 171, 163-178 (2017).
- Lessnick, S. L., Braun, B. S., Denny, C. T., May, W. A. Multiple domains mediate transformation by the Ewing’s sarcoma EWS/FLI-1 fusion gene. Oncogene. 10, 423-431 (1995).
- Leach, B. I., et al. Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure. 21, 176-183 (2013).
- Ng, K. P., et al. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proceedings of the National Academy of Sciences U.S.A. 104, 479-484 (2007).
- Riggi, N., et al. EWS-FLI1 Divergent Chromatin Remodeling Mechanisms to Directly Activate or Repress Enhancer Elements in Ewing Sarcoma. Cancer Cell. 26, 668-681 (2014).
- Tomazou, E. M., et al. Epigenome Mapping Reveals Distinct Modes of Gene Regulation and Widespread Enhancer Reprogramming by the Oncogenic Fusion Protein EWS-FLI1. Cell Reports. 10, 1082-1095 (2015).
- Sankar, S., et al. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene. 32, 5089-5100 (2013).
- Gangwal, K., et al. Microsatellites as EWS/FLI response elements in Ewing's sarcoma. Proceedings of the National Academy of Sciences U.S.A. 105, 10149-10154 (2008).
- Gangwal, K., Close, D., Enriquez, C. A., Hill, C. P., Lessnick, S. L. Emergent Properties of EWS/FLI Regulation via GGAA Microsatellites in Ewing's Sarcoma. Genes & Cancer. 1, 177-187 (2010).
- Guillon, N., et al. The Oncogenic EWS-FLI1 Protein Binds In Vivo GGAA Microsatellite Sequences with Potential Transcriptional Activation Function. PLoS One. 4, 4932(2009).
- Chong, S., et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 361, (2018).
- Johnson, K. M., et al. Role for the EWS domain of EWS/FLI in binding GGAA-microsatellites required for Ewing sarcoma anchorage independent growth. Proceedings of the National Academy of Sciences U.S.A. 114, 9870-9875 (2017).
- Theisen, E. R., et al. Transcriptomic analysis functionally maps the intrinsically disordered domain of EWS/FLI and reveals novel transcriptional dependencies for oncogenesis. Genes & Cancer. 10, 21-38 (2019).
- Chen, J., Bardes, E. E., Aronow, B. J., Jegga, A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Research. 37, 305-311 (2009).
- Johnson, K. M., Taslim, C., Saund, R. S., Lessnick, S. L. Identification of two types of GGAA-microsatellites and their roles in EWS/FLI binding and gene regulation in Ewing sarcoma. PLOS One. 12, 0186275(2017).
- Kim, P., Ballester, L. Y., Zhao, Z. Domain retention in transcription factor fusion genes and its biological and clinical implications: a pan-cancer study. Oncotarget. 8, 110103-110117 (2017).
- de Mendíbil, I. O., Vizmanos, J. L., Novo, F. J. Signatures of Selection in Fusion Transcripts Resulting from Chromosomal Translocations in Human Cancer. PLOS One. 4, 4805(2009).
- Frenkel-Morgenstern, M., Valencia, A. Novel domain combinations in proteins encoded by chimeric transcripts. Bioinformatics. 28, 67-74 (2012).
- Hegyi, H., Buday, L., Tompa, P. Intrinsic Structural Disorder Confers Cellular Viability on Oncogenic Fusion Proteins. PLoS Computational Biology. 5, 1000552(2009).
- Latysheva, N. S., Babu, M. M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Research. 44, 4487-4503 (2016).
- Deneen, B., Denny, C. T. Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene. 20, 6731-6741 (2001).
- Kendall, G. C., et al. PAX3-FOXO1 transgenic zebrafish models identify HES3 as a mediator of rhabdomyosarcoma tumorigenesis. eLife. 7, 33800(2018).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.