
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Permettre une compensation en temps réel dans les oxydations photochimiques rapides des protéines pour la détermination des changements de topographie des protéines
Dans cet article
Résumé
L’oxydation photochimique rapide des protéines est une technique émergente pour la caractérisation structurelle des protéines. Différents additifs et ligands de solvant ont des propriétés de récupération radicales hydroxyles variées. Pour comparer la structure protéique dans différentes conditions, une compensation en temps réel des radicaux hydroxyles générés dans la réaction est nécessaire pour normaliser les conditions de réaction.
Résumé
L’oxydation photochimique rapide des protéines (FPOP) est une technique de biologie structurale basée sur la spectrométrie de masse qui sonde la surface des protéines accessible aux solvants. Cette technique repose sur la réaction des chaînes latérales d’acides aminés avec des radicaux hydroxyles librement diffusant dans la solution. FPOP génère ces radicaux in situ par photolyse laser du peroxyde d’hydrogène, créant une explosion de radicaux hydroxyles qui est épuisé sur l’ordre d’une microseconde. Lorsque ces radicaux hydroxyles réagissent avec une chaîne latérale d’acides aminés accessibles aux solvants, les produits de réaction présentent un changement de masse qui peut être mesuré et quantifié par spectrométrie de masse. Étant donné que le taux de réaction d’un acide aminé dépend en partie de la surface moyenne accessible au solvant de cet acide aminé, les changements mesurés dans la quantité d’oxydation d’une région donnée d’une protéine peuvent être directement corrélés aux changements dans l’accessibilité des solvants de cette région entre les différentes conformations (p. ex., ligand-bound versus ligand-free, monomère vs agrégat, etc.) FPOP a été appliqué dans un certain nombre de problèmes en biologie, y compris les interactions protéines-protéines, les changements conformationnels des protéines, et la liaison protéine-ligand. Comme la concentration disponible de radicaux hydroxyles varie en fonction de nombreuses conditions expérimentales dans l’expérience FPOP, il est important de surveiller la dose radicale efficace à laquelle l’analyte de protéine est exposé. Cette surveillance est efficacement réalisée en incorporant un dosimètre en ligne pour mesurer le signal de la réaction FPOP, avec fluence laser ajusté en temps réel pour atteindre la quantité désirée d’oxydation. Avec cette compensation, les changements dans la topographie des protéines reflétant les changements conformationnels, les surfaces de liaison de ligand, et/ou les interfaces d’interaction protéine-protéine peuvent être déterminés dans des échantillons hétérogènes utilisant des quantités relativement faibles d’échantillon.
Introduction
L’oxydation photochimique rapide des protéines (FPOP) est une technique émergente pour la détermination des changements topographiques protéiques par une modification covalente ultra-rapide de la surface exposée aux solvants des protéines suivie de la détection par LC-MS1. FPOP génère une forte concentration de radicaux hydroxyles in situ par photolyse laser uv flash du peroxyde d’hydrogène. Ces radicaux hydroxyles sont très réactifs et de courte durée, consommés sur une période d’environ une microseconde dans les conditions FPOP2. Ces radicaux hydroxyles diffusent à travers l’eau et oxydent divers composants organiques en solution à des vitesses cinétiques allant généralement de rapide (~106 M-1 s-1) à la diffusion contrôlée3. Lorsque le radical hydroxyle rencontre une surface protéique, le radical oxyde les chaînes latérales d’acides aminés sur la surface des protéines, ce qui entraîne un déplacement de masse de cet acide aminé (le plus souvent l’ajout net d’un atome d’oxygène)4. Le taux de réaction d’oxydation à n’importe quel acide aminé dépend de deux facteurs : la réactivité inhérente de cet acide aminé (qui dépend de la chaîne latérale et du contexte de la séquence)4,5 et l’accessibilité de cette chaîne latérale au radical hydroxyle diffusant, qui est étroitement corrélée à la surface moyenne accessible au solvant6,7. Tous les acides aminés standard, à l’exception de la glycine, ont été observés comme étant étiquetés par ces radicaux hydroxyles hautement réactifs dans les expériences FPOP, bien qu’à des rendements très différents; dans la pratique, Ser, Thr, Asn et Ala sont rarement considérés comme oxydés dans la plupart des échantillons, sauf sous des doses radicales élevées et identifiés par une fragmentation etd ciblée attentive et sensible8,9. Après l’oxydation, les échantillons sont éteints pour enlever le peroxyde d’hydrogène et les oxydants secondaires (superoxyde, oxygène singlet, hydroperoxydes peptidyl, etc.) Les échantillons éteints sont ensuite digérés protéolytiquement pour générer des mélanges de peptides oxydés, oùl’informationstructurelle est congelée comme un « instantané » chimique dans les modèles de produits d’oxydation des divers peptides ( Figure 1 ). La chromatographie liquide couplée à la spectrométrie de masse (LC-MS) est utilisée pour mesurer la quantité d’oxydation des acides aminés dans un peptide protéolytique donné basé sur les intensités relatives des versions oxydées et non oxydées de ce peptide. En comparant cette empreinte oxydative de la même protéine obtenue dans différentes conditions conformationnelles (p. ex., ligand-bound versus ligand-free), les différences dans la quantité d’oxydation d’une région donnée de la protéine peuvent être directement corrélées avec les différences dans la surface accessible aux solvants de cette région6,7. La capacité de fournir des informations topographiques protéiques fait de fpop une technologie attrayante pour la détermination de la structure de plus haut niveau des protéines, y compris dans la découverte thérapeutique des protéines et le développement10,11.

Figure 1 : Vue d’ensemble de la FPOP. La surface de la protéine est codévalentement modifiée par des radicaux hydroxyles hautement réactifs. Les radicaux hydroxyles réagiront avec les chaînes latérales d’acides aminés de la protéine à un rythme fortement influencé par l’accessibilité des solvants de la chaîne latérale. Les changements topographiques (par exemple, en raison de la liaison d’un ligand comme indiqué ci-dessus) protégeront les acides aminés dans la région de l’interaction contre la réaction avec les radicaux hydroxyles, ce qui entraînera une diminution de l’intensité du peptide modifié dans le signal LC-MS. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
Les différents constituants présents dans la solution FPOP (p. ex., ligands, excipients, tampons) ont une activité de nettoyage différente vers les radicaux hydroxyles générés lors de la photolyse laser du peroxyde d’hydrogène3. De même, un petit changement dans la concentration de peroxyde, la fluence laser, et la composition tampon peut changer la dose radicale efficace, ce qui rend la reproduction des données FPOP difficile à travers les échantillons et entre les différents laboratoires. Par conséquent, il est important de pouvoir comparer la dose de radicaux hydroxyles disponibles pour réagir avec des protéines dans chaque échantillon à l’aide de l’un des dosimètres radicaux hydroxyle disponibles12,13,14,15,16. Les dosimètres radicaux hydroxyles agissent en rivalisant avec l’analyte (et avec tous les charognards en solution) pour le pool de radicaux hydroxyles; la dose efficace de radicaux hydroxyles est mesurée en mesurant la quantité d’oxydation du dosimètre. Notez que la « dose hydroxyle radicale efficace » est fonction à la fois de la concentration initiale de radicaux hydroxyles générés et de la demi-vie du radical. Ces deux paramètres dépendent en partie l’un de l’autre, ce qui rend la modélisation cinétique théorique quelque peu complexe (figure 2). Deux échantillons pourraient avoir des demi-vies radicales initiales très différentes tout en maintenant la même dose radicale efficace en modifiant la concentration initiale de radicaux hydroxyles formés; ils généreront toujours des empreintes identiques17. L’adénine13 et le Tris12 sont des dosimètres radicaux hydroxyles pratiques parce que leur niveau d’oxydation peut être mesuré par spectroscopie UV en temps réel, permettant aux chercheurs d’identifier rapidement quand il y a un problème avec la dose radicale efficace d’hydroxyle et de résoudre leur problème. Pour résoudre ce problème, un dosimètre en ligne situé dans le système de flux directement après le site d’irradiation qui peut surveiller le signal des changements d’absorption d’adénine en temps réel est important. Cela aide à mener des expériences FPOP dans des tampons ou tout autre excipient avec des niveaux très différents de la capacité de récupération hydroxyle radical17. Cette compensation de dosage radicale peut être exécutée en temps réel, donnant des résultats statistiquement indiscernables pour le même conformer en ajustant la dose radicale efficace.
Dans ce protocole, nous avons des procédures détaillées pour effectuer une expérience FPOP typique avec la compensation de dosage radical utilisant l’adénine comme dosimètre radical optique interne. Cette méthode permet aux chercheurs de comparer les empreintes de pas dans les conditions FPOP qui ont une capacité de récupération différente en effectuant une indemnisation en temps réel.

Figure 2 : Simulation cinétique de la rémunération basée sur la dosimétrie. 1 mM adénine dosimeter réponse est mesurée en 5 μM lysozyme analyte avec une concentration initiale de 1 mM hydroxyle radical (▪OH t1/2=53 ns), et fixé comme réponse dosimètre cible (noir). Lors de l’ajout de 1 mM de l’histidine excipient de charognard, la réponse dosimètre (bleu) diminue avec la quantité d’oxydation de protéine d’une manière proportionnelle (cyan). La demi-vie du radical hydroxyle diminue également (▪OH t1/2=39 ns). Lorsque la quantité de radicaux hydroxyl générés est augmentée pour donner un rendement équivalent de dosimètre oxydé dans l’échantillon avec 1 mM histidine charognard comme atteint avec 1 mM hydroxyl radical en l’absence de charognard (rouge), la quantité d’oxydation des protéines qui se produit de même devient identique (magenta), tandis que l’hydroxyle radical demi-vie diminue encore plus loin (▪OH t1/2=29 ns). Adapté avec la permission de Sharp J.S., Am Pharmaceut Rev 22, 50-55, 2019. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
Protocole
1. Préparer le banc optique et le capillaire pour FPOP
ATTENTION : Les lasers excimères KrF sont des dangers oculaires extrêmes, et la lumière directe ou réfléchie peut causer des dommages oculaires permanents. Portez toujours une protection oculaire appropriée, évitez la présence d’objets réfléchissants près du chemin du faisceau lorsque cela est possible, et utilisez des contrôles techniques pour empêcher l’accès non autorisé à un laser actif et pour retenir toute réflexion errante.
- Préparer le banc optique FPOP.
- Allumez le laser pour vous réchauffer. Réglez le laser sur déclencheur externe, énergie constante, pas de remplacement de gaz. Réglez l’énergie laser par impulsion (généralement entre 80-120 mJ/impulsion).
- Installez le banc optique avec l’objectif plano-convexe (30 mm Dia. x 120 mm FL non couché) directement dans le chemin du faisceau laser et un backstop non réfléchissant pour absorber la lumière comme le montre la figure 3A.

Figure 3 : Banc optique pour l’expérience FPOP. (A) L’échantillon est mélangé avec H2O2, dosimètre radical d’adénine, et charognard de glutamine et chargé dans la seringue. L’échantillon est poussé à travers le capillaire de silice fusionné à travers la trajectoire de faisceau concentré d’un laser UV excimère KrF. La lumière UV photolyzes H2O2 dans les radicaux hydroxyles, qui oxyde la protéine et le dosimètre d’adénine. Le flux de seringue pousse l’échantillon lumineux hors de la trajectoire du laser avant la prochaine impulsion laser, avec un volume d’exclusion non illuminé entre les régions éclairées. Immédiatement après l’oxydation, l’échantillon est passé par un spectrophotomètre UV inline, qui mesure l’absorption UV de l’adénine à 265 nm. L’échantillon est ensuite déposé dans un tampon d’extinction pour éliminer les oxydants H2O2 et secondaires restants. (B) La taille de tache est mesurée après irradier une note collante colorée fixée derrière le capillaire avec le laser à 248 nm. La largeur de l’endroit est utilisée pour calculer le débit de l’échantillon, et la silhouette du capillaire au centre de l’endroit est utilisée pour aligner le banc optique. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
- Couper une longueur appropriée du capillaire de silice fusionné (360 μm de diamètre extérieur et 100 μm de diamètre intérieur) et à l’aide d’un manchon, se connecter à la seringue étanche au gaz à l’aide d’un connecteur à faible volume mort.
- Brûlez doucement le revêtement en polyimide du capillaire à l’aide d’une torche de butane à l’endroit où le dosimètre en ligne lit le signal d’absorption à 265 nm après l’exposition au laser des échantillons. Essuyez les débris sur le capillaire doucement à l’aide de méthanol sur un essuie-tout sans peluche. Le revêtement en polyimide à l’emplacement de l’incidence laser peut être brûlé de la même façon avec la torche butane ou brûlé avec le laser excimer tir à faible puissance.
REMARQUE : Attendez que le capillaire refroidisse car il s’agit d’un risque d’incendie pour utiliser le méthanol sur le capillaire chaud. - Placez ce capillaire à travers le chemin de faisceau du laser et dans le dosimètre en ligne.
- Appuyez sur le levier sur le dessus du dosimètre en ligne pour ouvrir la charnière. Retirez les supports magnétiques. Placez le capillaire dans la rainure usinée du dosimètre en ligne, en utilisant les supports magnétiques pour maintenir le capillaire en place. Fermez la charnière dosimétrique sur le capillaire, en appuyant sur elle jusqu’à ce que le levier se verrouille en place.
- À l’aide du logiciel de dosimétrie, cliquez sur le bouton Démarrer flash pour commencer à tirer le laser excimer. Définissez la puissance laser prédéfinie entre 50-100 mJ/pulse sur le logiciel de contrôle laser lui-même, et définissez le taux de répétition prédéfini entre 10-20 Hz dans l’onglet Paramètres du logiciel de dosimétrie.
- Concentrez le faisceau laser à l’aide d’une lentille convexe plano montée sur une scène motorisée linéaire. Mesurer la largeur et la hauteur de la tache laser à la position du capillaire sur une note collante précisément à l’aide d’un étrier pour calculer la fluence incidente (mJ/mm2) comme indiqué à la figure 3B.
- Placez une ouverture opaque près du capillaire pour assurer une largeur lumineuse constante du capillaire indépendamment des changements dans la taille du faisceau en raison du mouvement de la lentille ou de changer l’énergie par impulsion du laser18.
- Avec le tir au laser, déplacez la scène motorisée à travers sa gamme de mouvement. Assurez-vous que le faisceau reste centré sur l’ouverture et la silhouette du capillaire peut être observée tout au long. Le diamètre de l’ouverture doit être plus petit que la largeur du faisceau focalisant à chaque point de la portée de l’étape motorisée.
- Faire couler de l’eau dans le capillaire à 20 μL/min pendant au moins une minute pour laver le capillaire.
- Cliquez sur le bouton Démarrer les données + AutoZero sur le logiciel dosimètre pour réduire le dosimètre à l’eau et commencer la collecte de données.
REMARQUE : Si le système tampon pour FPOP a une absorption UV significative à 265 nm, le système FPOP doit être mis à zéro sur la mémoire tampon, et non sur l’eau.
- Cliquez sur le bouton Démarrer les données + AutoZero sur le logiciel dosimètre pour réduire le dosimètre à l’eau et commencer la collecte de données.
- Réglez le débit calculé sur la pompe à seringues.
- Le débit de l’échantillon de protéines dépend du volume irradié par coup (VIrr), du nombre de tirs laser par seconde (R),et de la fraction de volume d’exclusion non irradiée souhaitée (FEx) pour corriger les effets de flux laminaires et la diffusion de l’échantillon (0,15-0,30 recommandé)2,19,20. Calculer le VIrr (en μL) en fonction du diamètre intérieur du capillaire en mm (d) et de la largeur de la tache laser empiétant sur le capillaire (c.-à-d. la largeur de l’ouverture) en mm (w) en utilisant l’équation suivante:
VIrr = π(d/2)2w - Calculer le débit souhaité (en μL/min) en fonction de l’équation suivante :
Flux = 60R[VIrr (1 + FEx)]
- Le débit de l’échantillon de protéines dépend du volume irradié par coup (VIrr), du nombre de tirs laser par seconde (R),et de la fraction de volume d’exclusion non irradiée souhaitée (FEx) pour corriger les effets de flux laminaires et la diffusion de l’échantillon (0,15-0,30 recommandé)2,19,20. Calculer le VIrr (en μL) en fonction du diamètre intérieur du capillaire en mm (d) et de la largeur de la tache laser empiétant sur le capillaire (c.-à-d. la largeur de l’ouverture) en mm (w) en utilisant l’équation suivante:
2. Préparation de la solution protéique pour FPOP
- Préparer la protéine dans les deux conditions ou plus différentes à comparer (p. ex., sans ligand et sans ligand; agrégat et monomère; seul et avec un partenaire de liaison protéine-protéines; etc.) pour détecter les changements de conformation.
- Définissez le volume total utilisé pour FPOP en consélez les besoins de l’expérience. La limite minimale dépend habituellement du volume du capillaire de l’irradiation et du matériau requis pour une détection robuste et une quantification relative, et varie en grande partie selon le système LC-MS/MS utilisé et la méthode de traitement de l’échantillon post-étiquetage. Le volume total des solutions FPOP couramment utilisées dans notre groupe est de 20 μL après l’ajout de peroxyde d’hydrogène. La concentration finale de la protéine est généralement de 1-10 μM, avec 17 mM de glutamine (pour limiter la durée de vie du radical hydroxyle), 1 mM adénine (pour agir comme un dosimètre radical)13,,17 et 10 mM tampon de phosphate (un tampon qui est un pauvre charognard de radicaux hydroxyle). Les échantillons sont généralement préparés avec plusieurs répliques pour permettre une modélisation statistique des résultats.
- Pour la plupart des fins générales, préparer des échantillons en triple dans les deux états, plus au moins un échantillon à utiliser comme un contrôle sans laser pour mesurer l’oxydation de fond. Préparer 18 μL de ce mélange de solutions FPOP.
REMARQUE : De nombreux tampons et additifs couramment utilisés en biochimie sont des charognards radicaux hydroxyles. Ces additifs et tampons peuvent être utilisés; cependant, des réductions de l’oxydation dues au nettoyage radical d’hydroxyle du tampon peuvent se produire. En général, gardez tous les additifs au minimum requis par le système biologique pour maximiser le rendement d’oxydation des protéines. Le sulfoxyde de diméthyle devrait être évité en raison de la propension à générer des radicaux secondaires ; diméthylformamide a été une alternative utile dans nos mains. Lors de l’utilisation de tampons qui sont de solides charognards radicaux hydroxyles, la glutamine peut souvent être exclue du mélange de solutions FPOP.
- Pour la plupart des fins générales, préparer des échantillons en triple dans les deux états, plus au moins un échantillon à utiliser comme un contrôle sans laser pour mesurer l’oxydation de fond. Préparer 18 μL de ce mélange de solutions FPOP.
- Préparer 1 M de peroxyde d’hydrogène immédiatement avant l’expérience FPOP.
REMARQUE : 30 % de peroxyde d’hydrogène vendu couramment par les fournisseurs comprend un stabilisateur, ce qui augmente la durée de conservation. Une fois dilué, le peroxyde d’hydrogène doit être utilisé rapidement, certainement dans la même journée. Le peroxyde d’hydrogène devrait également être régulièrement testé pour la décomposition par FPOP à l’aide d’un dosimètre radical hydroxyle. - Préparer des tubes de microcentrifuge contenant 25 μL de solution d’extinction de 0,5 μg/μL d’amide de méthionine et de 0,5 μg/μL de catalase. Si un volume d’échantillon supérieur à 20 μL est utilisé pour le FPOP, augmentez proportionnellement le volume de la solution d’extinction.
3. Effectuer l’expérience FPOP
- Ajouter 2 μL de peroxyde d’hydrogène dans les 18 μL du mélange de solutions FPOP. Mélanger le contenu doucement avec une pipette et faire tourner rapidement la solution vers le bas des tubes de microcentrifuge. Collectez immédiatement à l’aide d’une seringue étanche au gaz et chargez-la dans la pompe à seringues.
- Démarrez le débit sur la pompe à seringues avec le débit déterminé à l’étape 1.8.1 (généralement entre 8-16 μL/min) en cliquant sur le bouton Démarrer la pompe sur le logiciel dosimètre.
- Surveillez la lecture en temps réel de l’adénine à l’aide du dosimètre en ligne (voir tableau des matériaux)et collectez l’échantillon dans les déchets. Attendez que le signal Abs265 se stabilise.
- Cliquez sur le bouton Démarrer flash dans le logiciel dosimètre pour commencer à tirer le laser au taux de répétition prédéfini et de l’énergie.
- Surveiller la lecture en temps réel de l’adénine à l’aide d’un dosimètre en ligne (voir tableau des matériaux); la différence dans Abs265 avec le laser éteint et le laser sur est la lecture ΔAbs265.
NOTE: L’apparition de lectures abs265 très instables lors de la mise à feu du laser en présence de peroxyde d’hydrogène est due à la génération de bulles en solution. Réduire la fluidité du laser et/ou la concentration de peroxyde d’hydrogène pour éliminer les bulles.
4. Effectuer une indemnisation
REMARQUE : Différents ligands, tampons, etc. peuvent avoir une capacité de récupération différente vers les radicaux hydroxyles. Il est important de s’assurer que des doses hydroxyles radicales efficaces comparables sont disponibles pour réagir avec des protéines à travers différents échantillons. Ceci est accompli en assurant la réponse égale de dosimètre radical d’hydroxyle entre les échantillons. À l’aide de la dosimétrie adénine, le changement d’absorption UV à 265 nm (ΔAbs265) reflète la dose hydroxyle radicale efficace; plus le ΔAbs265est grand, plus la dose de radical hydroxyle efficace est élevée.
- Comparez la lecture ΔAbs265 obtenue avec le dosimètre en ligne avec la lecture ΔAbs265 souhaitée obtenue par des expériences ou des contrôles antérieurs. Une lecture ΔAbs265 inférieure à la lecture souhaitée indique une dose efficace insuffisante de radicaux hydroxyles; une lecture de ΔAbs265 indique une dose radicale efficace qui est trop élevée. Si la lecture ΔAbs265 est au niveau souhaité, prélever l’échantillon immédiatement après l’irradiation laser dans la mémoire tampond’extinction 17.
- Compenser la dose radicale efficace pour égaliser les ΔAbs265. Cette compensation peut être effectuée de trois façons : modifier la concentration de peroxyde d’hydrogène, augmenter la fluence laser en changeant l’énergie laser par impulsion, ou augmenter la fluence laser en changeant le plan focal de la lentille de mise au point.
- Pour effectuer un grand changement (>10 mAU) en lecture ΔAbs265, refaire l’échantillon avec plus ou moins de peroxyde d’hydrogène et réexécuter l’échantillon selon la section 3.
- Pour effectuer un petit changement dans la lecture ΔAbs265 en temps réel, ajuster le plan focal du faisceau d’incident en ajustant la position de la lentille de mise au point à l’aide de l’étape motorisée de 50 mm. Rapprocher le plan focal de la position du capillaire augmentera la lecture de ΔAbs265; amener le plan focal plus loin de la position du capillaire diminuera la lecture ΔAbs265.
- Surveiller l’adénine ΔAbs265 pour mesurer la quantité effective de radicaux hydroxyles présents dans l’échantillon après irradiation laser13. La surveillance en temps réel avec un détecteur capillaire UV inline permet une compensation en temps réel comme décrit dans 4.2.2; ajuster la position de la lentille à l’aide de l’étape motorisée jusqu’à ce que la lecture ΔAbs265 soit égale à la lecture souhaitée. Les mesures d’absorption post-expérimentales avec un spectrophotomètre UV sont également précises, mais nécessitent l’utilisation de nouveaux échantillons pour chaque dose radicale efficace.
5. Digérer les échantillons de protéines
REMARQUE : La trypsine est le plus couramment utilisée pour digérer les échantillons de protéines pour le FPOP, et est la protéase utilisée dans ce protocole. Il s’agit d’une protéase fiable qui génère des peptides avec des sites de base à la fois au terminus N et C, favorisant multiplier les ions peptides chargés dans la SP. En outre, il se fend après la lysine et l’arginine, deux acides aminés qui ne sont que modérément réactifs aux radicaux hydroxyles; par conséquent, les changements dans le modèle de digestion dus à l’oxydation d’analyte est rare. D’autres protéases ont été utilisées avec succès avec FPOP21, mais il faut veiller à ce que les habitudes de digestion soient comparables entre les échantillons non oxydés et oxydés.
- Mesurer le volume final de l’échantillon FPOP éteint. Ajouter 500 mM Tris, pH 8.0 avec 10 mM CaCl2 contenant 50 mM dithiothreitol (TNT) à la solution protéique après l’extinction à une concentration finale de 50 mM Tris, 1 mM CaCl2 et 5 mM DTT.
- Chauffer l’échantillon de protéines à 95 °C pendant 15 minutes.
- Refroidir immédiatement l’échantillon sur la glace pendant 2 min.
- Ajouter le rapport poids trypsine/protéine 1:20 aux échantillons.
- Digère la protéine toute la nuit à 37 °C avec le mélange.
- Arrêter la réaction de digestion par l’ajout de 0,1% d’acide formique et/ou le chauffage de l’échantillon à 95 °C pendant 10 min.
- Ajouter 2 mM TNT aux échantillons et chauffer à 60 °C pendant 15 min immédiatement avant LC-MS/MS.
NOTE : Alors que d’autres groupes ont rapporté l’alkylation des thiols dans des expériences de FPOP, dans nos mains nous avons noté des produits secondaires sur l’alkylation des protéines oxydées (probablement due à la réaction avec des carbonyls nucléophilic formés comme produit d’oxydation mineur). Par conséquent, nous choisissons d’éviter l’alkylation des thiols lorsque c’est possible.
6. Effectuer la spectrométrie de masse chromatographie liquide-tandem (LC-MS/MS)
- Préparer la phase mobile A composée d’eau contenant 0,1 % d’acide formique et de phase B mobile composée d’acétonitrule avec 0,1 % d’acide formique.
- Chargez d’abord l’échantillon sur une colonne de piège C18 (300 μm I.D. x 5 mm 100 Å de taille de pores, 5 μm de taille de particule) piégeant la cartouche et lavez-la avec 2% de solvant B pendant 3 minutes à un débit de 5,0 μL/min pour enlever les sels et les petites molécules hydrophiles.
- Séparez ensuite les peptides sur le nanocolumn C18 (0,75 mm x 150 mm, 2 μm de taille de particules, 100 Å de taille de pores) à un débit de 300 nL/min. Le gradient se compose d’une augmentation linéaire de 2 à 35% solvant B sur 22 min, ramped à 95% solvant B sur 5 min et maintenu pendant 3 min pour laver la colonne, puis retourné à 2% B sur 3 min et maintenu pendant 9 min pour ré-équilibrer la colonne.
REMARQUE : Ce gradient est suffisant pour la LC-MS/MS de la plupart des mélanges FPOP à une et deux protéines qui cherchent à faire de la quantification au niveau du peptide. Le pourcentage de solvant B peut avoir besoin d’être modifié pour augmenter la résolution de peptides dans de rares cas où les peptides interfèrent les uns avec les autres en raison de temps de rétention similaires et des valeurs m/z. Proteome-échelle FPOP22 ou des conceptions expérimentales cherchant à séparer les isomères de produit d’oxydation de peptide1,23,24,25 peuvent exiger des gradients plus longs de LC et sont au-delà de la portée du présent rapport. - Elute les peptides directement dans la source nanospray d’un spectromètre de masse à haute résolution à l’aide d’un émetteur de nanospray conducteur.
- Acquérir les données en mode ion positif. Réglez la tension de pulvérisation à 2400 V et la température du tube de transfert d’ions à 300 °C.
- Acquérir les balayages complets de SP de m/z 250 à 2000 à une résolution nominale à m/z 200 de 60 000 suivi de huit balayages de MS/MS de piège linéaire dépendant des données suivantes sur les huit ions peptides les plus abondants utilisant la dissociation induite par la collision à 35 % d’énergie normalisée pour identifier les peptides. Fragmenter les peptides jusqu’à cinq fois dans les 30 s, puis transférer vers une liste d’exclusion pour 60 s.
7. Traitement et calcul des données d’oxydation moyenne des peptides
- Déterminer la couverture de la séquence des valeurs protéiques, des valeurs m/z et des temps de rétention des peptides non oxydés à l’aide du moteur de recherche protéomique MS/MS.
- Réglez la tolérance de masse précurseur à 10 ppm et prévoyez jusqu’à deux sites de clivage manqués pour les échantillons digérés par la trypsine, en utilisant la spécificité standard du clivage trypsine.
- Réglez la tolérance de masse de fragment de peptide à 0.4 Daltons.
- Sur la base du rapport m/z des peptides non modifiés détectés et des changements de masse connus des principaux produits d’oxydation, calculer le m/z des différents produits théoriques d’oxydation de chaque peptide4,26,27,28,29.
- Identifier le chromatogramme d’ions extrait de ces valeurs m/z à l’aide d’un logiciel pour afficher l’exécution spectrométrique de masse (figure 4). Identifiez les produits d’oxydation peptide en fonction de leur m/z, de leur état de charge, et de la similitude dans le temps d’élution avec le peptide non modifié. Dans nos mains, les produits d’oxydation peptide s’élèvent entre 240 secondes avant 180 secondes après le peptide non modifié en utilisant le gradient LC ci-dessus. Comme l’oxydation se traduira souvent par de multiples produits d’oxydation isomérique, il est courant d’observer de multiples pics partiellement résolus dans les chromatogrammes extraits d’ions des produits d’oxydation peptide, comme le montre la figure 4. Les produits d’oxydation peptide sont quantifiés en fonction de la zone du(s) pic(s) dans les chromatogrammes d’ions extraits.

Figure 4 : Chromatogramme d’ions extrait d’un peptide et de ses produits d’oxydation après FPOP. Le m/z des produits d’oxydation du peptide est calculé sur la base du m/z du peptide non oxydé et des produits d’oxydation connus; et les zones de ces produits peptidiques sont déterminées. La zone des produits peptides est ensuite utilisée pour le calcul des événements d’oxydation moyen par peptide. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
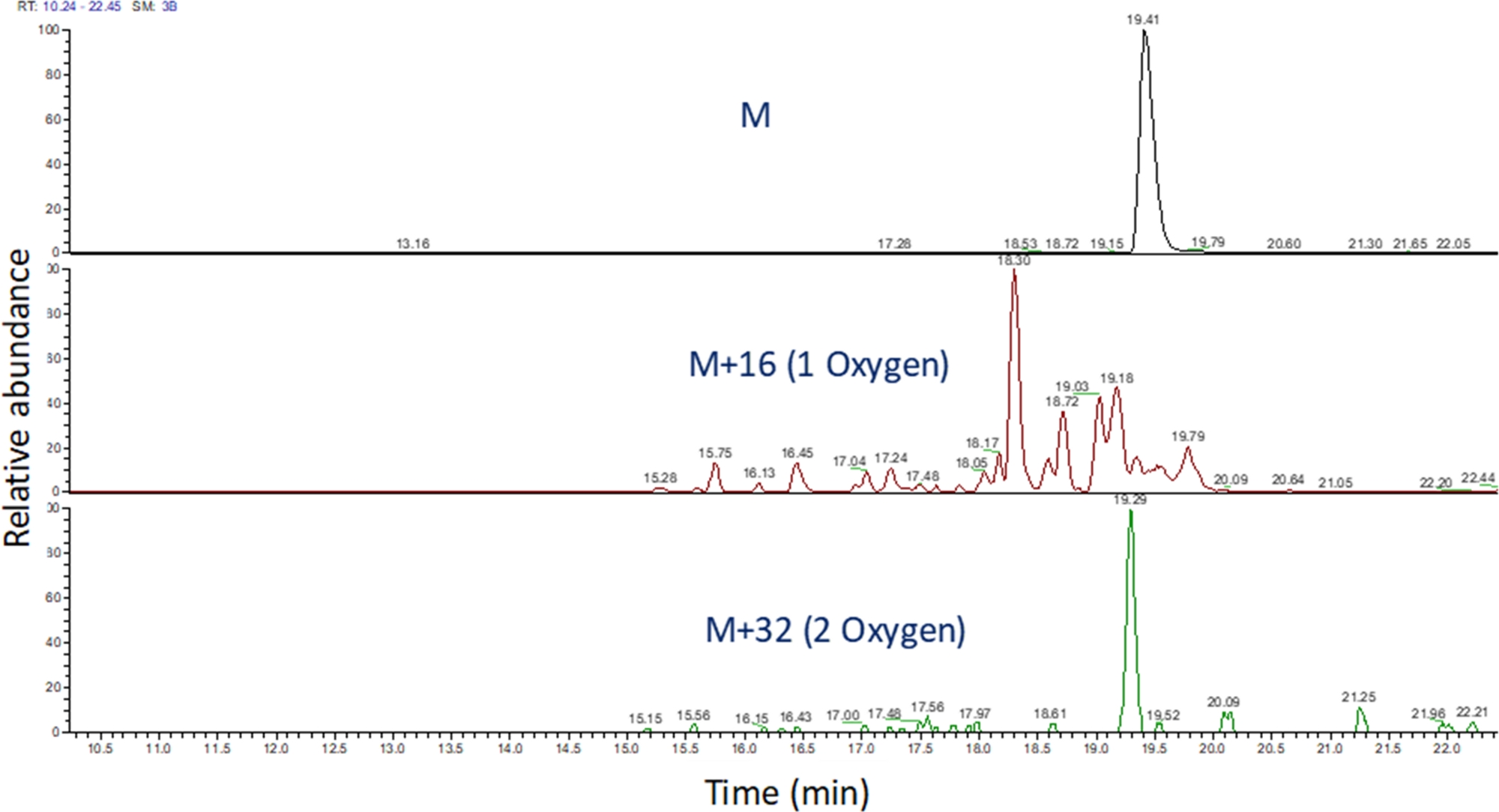{kind=link}
- Calculer l’oxydation moyenne des peptides à l’aide de l’équation suivante.

où P désigne le nombre moyen d’événements d’oxydation par molécule de peptide, et je représente la zone de pointe du peptide non oxydé (Iunoxidized) et le peptide avec des événements d’oxydation n. Notez que je (singly oxydé) comprendrait non seulement des ajouts d’un seul atome d’oxygène, mais aussi d’autres événements d’oxydation unique moins courants que l’investigateur peut choisir de mesurer (p. ex., décarboxylation oxydative, formation de carbonyle, etc.) 4,26,27,28,29.
Résultats
La comparaison de l’empreinte de peptide à chaîne lourde du biosimilaire de l’adalimumab dans le tampon de phosphate et lorsqu’il est chauffé à 55 °C pendant 1 h montre des résultats intéressants. Le test t de l’élève est utilisé pour l’identification des peptides qui sont considérablement modifiés dans ces deux conditions (p ≤ 0,05). Les peptides 20-38, 99-125, 215-222, 223-252, 260-278, 376-413 et 414-420 présentent une protection significative contre le solvant lorsque la protéine est chauff?...
Discussion
Les techniques structurelles basées sur la spectrométrie de masse, y compris l’échange hydrogène-deutérium, la liaison chimique, l’étiquetage covalent, et la spectrométrie de masse de pulvérisation indigène et la mobilité d’ion ont été rapidement croissantes dans la popularité en raison de leur flexibilité, sensibilité, et la capacité de manipuler des mélanges complexes. FPOP bénéficie de plusieurs avantages qui ont augmenté sa popularité dans le domaine des techniques structurelles à base de ...
Déclarations de divulgation
Joshua S. Sharp révèle un intérêt financier important dans GenNext Technologies, Inc., une petite entreprise qui cherche à commercialiser des technologies pour l’analyse de la structure de haute qualité des protéines, y compris l’empreinte de protéines radicales hydroxyles.
Remerciements
Nous reconnaissons le financement de la recherche de l’Institut national des sciences médicales générales subvention R43GM125420-01pour soutenir le développement commercial d’un dispositif FPOP banc et R01GM127267 pour le développement de protocoles de normalisation et de dosimétrie pour fpop haute énergie.
matériels
| Name | Company | Catalog Number | Comments |
| Adenine | Acros Organics | 147440250 | Soluble in water upto 3.5 mM |
| Aperture | Edmund Optics | 39-905 | 1000 μm Aperture Diameter, Gold-Plated Copper Aperture |
| Aperture holder | Edmund Optics | 53-287 | 25.8mm Outer Diameter, Precision Pinhole Mount |
| Catalse | Sigma Aldrich | C-40 | Catalase from bovine liver, lyophilized powder, ≥10,000 units/mg protein |
| COMPex Pro laser | Coherent | 1113836 | COMPexPRO 102, F-Vversion, KrF laser, No XeCl |
| Dithiotheitol (DTT) | Promega | V3151 | DTT, Molecular Grade (DL-Dithiothreitol) |
| Fraction collector | GenNext Technologies, Inc. | N/A | Automated fraction collector |
| Fused silica capillay | Molex | 1068150023 | Polymicro Flexible Fused Silica Capillary Tubing, Inner Diameter 100 µm, Outer Diameter 375 µm, TSP100375 |
| Glutamine | Acros Organics | 119951000 | L(+)-Glutamine, 99% |
| Holder for lens | Edmund Optics | 03-668 | 53 mm Outer Diameter, Three-Screw Adjustable Ring Mount |
| Hydrogen peroxide | Fisher Scientific | H325-100 | Hydrogen Peroxide, 30% (Certified ACS), Fisher Chemical |
| LC-MS/MS system | Thermo Scientific | IQLAAEGAAPFADBMBCX | Dionex Ultimate 3000 coupled to Orbitap Fusion Tribrid mass spectrometer |
| Mas spec grade Acetonitrile | Fisher Scientific | A955-1 | Acetonitrile, Optima LC/MS Grade, Fisher Chemical |
| Mass spec grade formic acid | Fisher Scientific | A117-50 | Formic Acid, 99.0+%, Optima™ LC/MS Grade, Fisher Chemical |
| Mass spec grade water | Fisher Scientific | W6-4 | Water, Optima LC/MS Grade, Fisher Chemical |
| MES buffer | Sigma Aldrich | M0164 | MES hemisodium salt |
| Methionine amide | Bachem | 4000594.0005 | H-met-NH2.HCl |
| Micro V clamp | Thor Labs | VK250 | Micro V-clamp with stainless steel blades |
| Motorized stage | Edmund Optics | 68-638 | 50mm Travel Motorized Stage System with Manual Control |
| Nano C18 colum | Thermo Scientific | 164534 | Acclaim PepMap 100 C18 HPLC Columns |
| Optical bench | Edmund Optics | 56-935 | 18" x 18" breadboard |
| Pioneer FPOP Module System | GenNext Technologies, Inc. | N/A | Inline FPOP Radical Dosimetry System |
| Post holder | Edmund Optics | 58-979 | 3" Length, ¼-20 Thread, Post Holder |
| Sodium phosphate dibasic | Fisher Scientific | BP331-500 | Sodium Phosphate Dibasic Heptahydrate (Colorless-to-White Crystals), Fisher BioReagents |
| Sodium phosphate monobasic | Fisher Scientific | BP330-500 | Sodium Phosphate Monobasic Monohydrate (Colorless-to-white Crystals), Fisher BioReagents |
| Syringe | Hamilton | 81065 | 100 µL, Model 1710 RN SYR, Small Removable NDL, 22s ga, 2 in, point style 3 |
| Syringe pump | KD Scientific | 788101 | Legato 101 syringe pump |
| Trap C18 column | Thermo Scientific | 160454 | Thermo Scientific Acclaim PepMap 100 C18 HPLC Columns |
| Tris | Sigma Aldrich | 252859 | Tris(hydroxymethyl)aminomethane |
| Trypsin | Promega | V5111 | Sequencing Grade Modified Trypsin |
| UV plano convex lens | Edmund Optics | 84-285 | 30 mm Dia. x 120 mm FL Uncoated, UV Plano-Convex Lens |
Références
- Kaur, P., Kiselar, J., Yang, S., Chance, M. R. Quantitative protein topography analysis and high-resolution structure prediction using hydroxyl radical labeling and tandem-ion mass spectrometry (MS). Molecular & Cellular Proteomics. 14 (4), 1159-1168 (2015).
- Hambly, D. M., Gross, M. L. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. Journal of the American Society for Mass Spectrometry. 16 (12), 2057-2063 (2005).
- Buxton, G. V., Greenstock, C. L., Helman, W. P., Ross, A. B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O- in Aqueous Solution. Journal of Physical and Chemical Reference Data. 17 (2), 513 (1988).
- Xu, G., Chance, M. R. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Analytical Chemistry. 77 (14), 4549-4555 (2005).
- Sharp, J. S., Tomer, K. B. Effects of anion proximity in peptide primary sequence on the rate and mechanism of leucine oxidation. Analytical Chemistry. 78 (14), 4885-4893 (2006).
- Huang, W., Ravikumar, K. M., Chance, M. R., Yang, S. Quantitative mapping of protein structure by hydroxyl radical footprinting-mediated structural mass spectrometry: a protection factor analysis. Biophysical Journal. 108 (1), 107-115 (2015).
- Xie, B., Sood, A., Woods, R. J., Sharp, J. S. Quantitative protein topography measurements by high resolution hydroxyl radical protein footprinting enable accurate molecular model selection. Scientific Reports. 7 (1), 4552 (2017).
- Li, Z., et al. High structural resolution hydroxyl radical protein footprinting reveals an extended Robo1-heparin binding interface. Journal of Biological Chemistry. 290 (17), 10729-10740 (2015).
- Li, X., et al. Structural analysis of the glycosylated intact HIV-1 gp120-b12 antibody complex using hydroxyl radical protein footprinting. Biochemistry. 56 (7), 957-970 (2017).
- Li, K. S., Shi, L., Gross, M. L. Mass spectrometry-based fast photochemical oxidation of proteins (FPOP) for higher order structure characterization. Accounts of Chemical Research. 51 (3), 736-744 (2018).
- Li, J., Chen, G. The use of fast photochemical oxidation of proteins coupled with mass spectrometry in protein therapeutics discovery and development. Drug Discovery Today. 24 (3), 829-834 (2019).
- Roush, A. E., Riaz, M., Misra, S. K., Weinberger, S. R., Sharp, J. S. Intrinsic buffer hydroxyl radical dosimetry using Tris(hydroxymethyl)aminomethane. Journal of the American Society for Mass Spectrometry. 31 (2), 169-172 (2020).
- Xie, B., Sharp, J. S. Hydroxyl radical dosimetry for high flux hydroxyl radical protein footprinting applications using a simple optical detection method. Analytical Chemistry. 87 (21), 10719-10723 (2015).
- Niu, B., Zhang, H., Giblin, D., Rempel, D. L., Gross, M. L. Dosimetry determines the initial OH radical concentration in fast photochemical oxidation of proteins (FPOP). Journal of the American Society for Mass Spectrometry. 26 (5), 843-846 (2015).
- Niu, B., et al. Incorporation of a reporter peptide in FPOP compensates for adventitious scavengers and permits time-dependent measurements. Journal of the American Society for Mass Spectrometry. 28 (2), 389-392 (2017).
- Garcia, N. K., Sreedhara, A., Deperalta, G., Wecksler, A. T. Optimizing hydroxyl radical footprinting analysis of biotherapeutics using internal standard dosimetry. Journal of the American Society for Mass Spectrometry. 31 (7), 1563-1571 (2020).
- Sharp, J. S., Misra, S. K., Persoff, J. J., Egan, R. W., Weinberger, S. R. Real time normalization of fast photochemical oxidation of proteins experiments by inline adenine radical dosimetry. Analytical Chemistry. 90 (21), 12625-12630 (2018).
- Zhang, B., Cheng, M., Rempel, D., Gross, M. L. Implementing fast photochemical oxidation of proteins (FPOP) as a footprinting approach to solve diverse problems in structural biology. Methods. 144, 94-103 (2018).
- Konermann, L., Stocks, B. B., Czarny, T. Laminar flow effects during laser-induced oxidative labeling for protein structural studies by mass spectrometry. Analytical Chemistry. 82 (15), 6667-6674 (2010).
- Gau, B. C., Sharp, J. S., Rempel, D. L., Gross, M. L. Fast photochemical oxidation of protein footprints faster than protein unfolding. Analytical Chemistry. 81 (16), 6563-6571 (2009).
- Li, K. S., et al. Hydrogen-Deuterium exchange and hydroxyl radical footprinting for mapping hydrophobic interactions of human bromodomain with a small molecule Inhibitor. Journal of the American Society for Mass Spectrometry. 30 (12), 2795-2804 (2019).
- Espino, J. A., Jones, L. M. Illuminating biological interactions with in vivo protein footprinting. Analytical Chemistry. 91 (10), 6577-6584 (2019).
- Charvatova, O., et al. Quantifying protein interface footprinting by hydroxyl radical oxidation and molecular dynamics simulation: application to galectin-1. Journal of the American Society for Mass Spectrometry. 19 (11), 1692-1705 (2008).
- Gau, B., Garai, K., Frieden, C., Gross, M. L. Mass spectrometry-based protein footprinting characterizes the structures of oligomeric apolipoprotein E2, E3, and E4. Biochemistry. 50 (38), 8117-8126 (2011).
- Gau, B. C., Chen, J., Gross, M. L. Fast photochemical oxidation of proteins for comparing solvent-accessibility changes accompanying protein folding: Data processing and application to barstar. Biochimica et Biophysica Acta. 1834 (6), 1230-1238 (2013).
- Garrison, W. M. Reaction mechanisms in the radiolysis of peptides, polypeptides, and proteins. Chemical Reviews. 87 (2), 381-398 (1987).
- Xu, G., Chance, M. R. Radiolytic modification of sulfur-containing amino acid residues in model peptides: fundamental studies for protein footprinting. Analytical Chemistry. 77 (8), 2437-2449 (2005).
- Xu, G., Chance, M. R. Radiolytic modification of acidic amino acid residues in peptides: probes for examining protein-protein interactions. Analytical Chemistry. 76 (5), 1213-1221 (2004).
- Xu, G., Takamoto, K., Chance, M. R. Radiolytic modification of basic amino acid residues in peptides: probes for examining protein-protein interactions. Analytical Chemistry. 75 (24), 6995-7007 (2003).
- Misra, S. K., Orlando, R., Weinberger, S. R., Sharp, J. S. Compensated hydroxyl radical protein footprinting measures buffer and excipient effects on conformation and aggregation in an adalimumab biosimilar. AAPS Journal. 21 (5), 87 (2019).
- Simmons, D. A., Konermann, L. Characterization of transient protein folding intermediates during myoglobin reconstitution by time-resolved electrospray mass spectrometry with on-line isotopic pulse labeling. Biochemistry. 41 (6), 1906-1914 (2002).
- Vahidi, S., Konermann, L. Probing the time scale of FPOP (fast photochemical oxidation of proteins): radical reactions extend over tens of milliseconds. Journal of the American Society for Mass Spectrometry. 27 (7), 1156-1164 (2016).
- Chance, M. R. Unfolding of apomyoglobin examined by synchrotron footprinting. Biochemical and Biophysical Research Communications. 287 (3), 614-621 (2001).
- Xu, G., Chance, M. R. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chemical Reviews. 107 (8), 3514-3543 (2007).
- Zhang, Y., Rempel, D. L., Zhang, H., Gross, M. L. An improved fast photochemical oxidation of proteins (FPOP) platform for protein therapeutics. Journal of the American Society for Mass Spectrometry. 26 (3), 526-529 (2015).
- Cornwell, O., Radford, S. E., Ashcroft, A. E., Ault, J. R. Comparing hydrogen deuterium exchange and fast photochemical oxidation of proteins: a structural characterisation of wild-type and ΔN6 β(2)-microglobulin. Journal of the American Society for Mass Spectrometry. 29 (2), 2413-2426 (2018).
- Xie, B., Sharp, J. S. Relative Quantification of sites of peptide and protein modification using size exclusion chromatography coupled with electron transfer dissociation. Journal of the American Society for Mass Spectrometry. 27 (8), 1322-1327 (2016).
- Srikanth, R., Wilson, J., Vachet, R. W. Correct identification of oxidized histidine residues using electron-transfer dissociation. Journal of Mass Spectrometry. 44 (5), 755-762 (2009).
- Li, X., Li, Z., Xie, B., Sharp, J. S. Improved identification and relative quantification of sites of peptide and protein oxidation for hydroxyl radical footprinting. Journal of the American Society for Mass Spectrometry. 24 (11), 1767-1776 (2013).
- Li, X., Li, Z., Xie, B., Sharp, J. S. Supercharging by m-NBA Improves ETD-Based Quantification of Hydroxyl Radical Protein Footprinting. Journal of the American Society for Mass Spectrometry. 26 (8), 1424-1427 (2015).
- Khaje, N. A., Sharp, J. S. Rapid quantification of peptide oxidation isomers from complex mixtures. Analytical Chemistry. 92 (5), 3834-3843 (2020).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.