
Method Article
Profilage cellulaire à haut débit de composés de dégradation de protéines ciblés à l’aide de lignées cellulaires HiBiT CRISPR
Dans cet article
Résumé
Ce protocole décrit la détection luminescente quantitative de la cinétique de dégradation des protéines dans les cellules vivantes qui ont été modifiées à l’aide de CRISPR / Cas9 pour exprimer l’étiquette de détection de protéine endogène sans anticorps fusionnée à une protéine cible. Des instructions détaillées pour calculer et obtenir les paramètres de dégradation quantitatifs, le taux, Dmax, DC 50 et Dmax50 sont incluses.
Résumé
Les composés de dégradation des protéines ciblés, y compris les colles moléculaires ou la protéolyse ciblant les chimères, constituent une nouvelle modalité thérapeutique passionnante dans la découverte de médicaments à petites molécules. Cette classe de composés induit la dégradation des protéines en rapprochant la protéine cible et les protéines de machinerie de la ligase E3 nécessaires pour ubiquitiner et finalement dégrader la protéine cible par la voie ubiquitine-protéasomique (UPP). Le profilage de la dégradation des protéines cibles à haut débit reste toutefois très difficile compte tenu de la complexité des voies cellulaires nécessaires pour parvenir à la dégradation. Nous présentons ici un protocole et une stratégie de dépistage basés sur l’utilisation du marquage endogène CRISPR / Cas9 des protéines cibles avec le marqueur HiBiT de 11 acides aminés qui complète avec une forte affinité pour la protéine LgBiT, pour produire une protéine luminescente. Ces lignées cellulaires ciblées CRISPR avec des marqueurs endogènes peuvent être utilisées pour mesurer la dégradation induite par le composé en temps réel, en mode cellule vivante cinétique ou en mode lytique final en surveillant le signal luminescent à l’aide d’un lecteur à plaque luminescente. Nous décrivons ici les protocoles de criblage recommandés pour les différents formats, ainsi que le calcul des principaux paramètres de dégradation de la vitesse, Dmax, DC 50, Dmax50, ainsi que le multiplexage avec des tests de viabilité cellulaire. Ces approches permettent la découverte et le triage rapides de composés à un stade précoce tout en maintenant l’expression endogène et la régulation des protéines cibles dans les milieux cellulaires pertinents, permettant une optimisation efficace des composés thérapeutiques principaux.
Introduction
La dégradation ciblée des protéines est devenue l’un des domaines à la croissance la plus rapide dans la découverte de médicaments à petites molécules, grandement renforcée par le succès thérapeutique des composés de colle moléculaire immunomodulateurs (par exemple, IMiD) pour le traitement du cancer, et les données prometteuses des premiers essais cliniques de Protéolyse ciblant les composés chimères 1,2,3,4,5,6,7,8, 9,10,11,12. Les composés de dégradation des protéines ciblés fonctionnent en rapprochant une protéine cible des protéines de machinerie E3 ligase 1,2,3,4,5,6,7,8,9,10,11,12 . Ce recrutement induit par le composé de la protéine cible dans la ligase E3 conduit à l’ubiquitination et à la dégradation de la protéine cible via la voie protéasomale de l’ubiquitine (UPP)1,2,3,4,5,6,7,8,9,10,11,12 . Historiquement, les programmes de criblage de la découverte de médicaments à petites molécules se sont appuyés sur des tests biochimiques initiaux pour évaluer l’activité et classer les composés par ordre. Ceci, cependant, a présenté un défi important pour les dégradateurs de protéines ciblés dont l’activité ultime, la dégradation via le protéasome, dépend d’une cascade d’événements cellulaires 1,2,4,5,6,11,12,13,14,15,16,17, 18. Les multiples voies et la complexité des complexes protéiques nécessaires à la dégradation réussie de la cible nécessitent des approches d’essai cellulaire pour le criblage précoce et le triage des composés initiaux. Actuellement, la disponibilité des technologies permettant de surveiller la dégradation des protéines cibles à haut débit dans le contexte de l’environnement cellulaire fait cruellement défaut14. Ici, nous présenterons des protocoles pour l’évaluation en temps réel de l’activité de dégradation lytique des cellules vivantes cinétiques ou des critères finaux à l’aide des lignées cellulaires cibles HiBiT marquées de manière endogèneCRISPR / Cas9 18,19,20 pour surveiller la perte de la protéine cible via une mesure luminescente après traitement avec des composés dégradateurs10,11,18,19.
Pour réussir la dégradation des cibles thérapeutiques et étendre le protéome médicamentable, de nombreuses approches et types de dégradateurs ont émergé qui peuvent cibler un large éventail de protéines pour la destruction, y compris celles localisées au niveau ou dans la membrane plasmique, les lysosomes, les membranes mitochondriales, le cytoplasme et le noyau21-57. Les deux principales classes de composés les plus étudiées sont les colles moléculaires et les protéines ciblant les cimères 2,4,5,6,7,12,26. Les colles moléculaires sont monovalentes, donc généralement de plus petite taille, et facilitent une nouvelle interface d’interaction protéine/protéine avec une protéine cible lors de la liaison à un composant de la ligase E3 2,12,26. Ce sont le plus souvent des dégradateurs qui se lientau composant 2,12,26,55,56,57 du Cereblon (CRBN) E3. Récemment, de nouveaux exemples passionnants utilisant d’autres machines E3 ligase telles que DCAF1558,59,60 et le recrutement CDK / Cyclin à DDB145 montrent le potentiel d’expansion de cette classe de composés. En revanche, les PROTACs sont des molécules bivalentes plus grosses, constituées d’un ligand de liaison cible, le plus souvent un inhibiteur, ponté via un agent de liaison chimique à une poignée de ligase E3 1,3,4,5,7,13. En tant que tels, ces composés sont capables de se lier directement à la fois à la ligase E3 et à la protéine cible 1,3,4,5,7,13. De nombreuses protéines se sont dégradées via ces molécules bivalentes, et les poignées de ligase E3 les plus utilisées recrutent soit CRBN, soit Von Hippel Lindau (VHL)1,3,4,5,7,13. Cependant, le nombre de poignées disponibles pour le recrutement de la ligase E3 chez les chimères ciblant la conception de la protéolyse augmente rapidement, élargissant les capacités de cette classe de composés ayant le potentiel de dégrader diverses classes cibles ainsi que d’améliorer la spécificité des types cellulaires ou tissulaires 24,48,61,62 . Combinés à l’exigence minimale d’engager une protéine cible, même avec une affinité marginale, les composés de dégradation sont prometteurs pour l’expansion du protéome médicamentable.
La caractérisation de la dynamique cellulaire de la perte de protéines, ainsi que la récupération potentielle des protéines après le traitement, est essentielle pour comprendre la fonction et l’efficacité des composés de dégradation. Bien qu’il soit possible d’étudier les changements de niveau de protéines endogènes dans les systèmes cellulaires pertinents à l’aide de tests d’anticorps Western blot ou de spectrométrie de masse, ces approches sont difficiles à adapter aux formats de criblage à haut débit, ont une capacité de quantification limitée ou la capacité de mesurer les changements cinétiques à de nombreux points temporels14. Pour relever ces défis, nous avons développé un système luminescent cellulaire à base de plaques pour surveiller les changements dans les niveaux de protéines endogènes, qui utilise l’insertion génomique via CRISPR / Cas9 de l’étiquette de 11 acides aminés, HiBiT, aux loci de toutes les cibles de dégradation clés18,19,20. Ce peptide complète avec une grande affinité avec son partenaire de liaison, LgBiT, pour produire une luminescence brillante en présence de son substrat 18,19,20,63, rendant ainsi ces protéines endogènes marquées luminescentes dans les cellules ou les lysats 18,19,20,63 . Les unités de lumière relative (RLU) mesurées avec un instrument luminomètre sont directement proportionnelles aux niveaux de protéines cibles marquées 18,19,20,63. Avec le développement de substrats stabilisés de luciférase, des mesures en temps réel du niveau de protéines cinétiques sur des périodes de 24 à 48 heures sont possibles 18,53,64. Cela permet de déterminer un profil de dégradation complet pour une cible donnée à une concentration de composé donnée, y compris une analyse quantitative du taux de dégradation initial, du maximum de dégradation (Dmax) et de la récupération après traitement du composé18,53. Cependant, si vous criblez de grandes banques de composés de dégradation, l’analyse des paramètres peut également être facilement effectuée au format 384 puits à diverses concentrations de médicaments et à des moments désignés.
Les protocoles présentés dans ce manuscrit représentent des stratégies de criblage cellulaire pour les composés de dégradation des protéines ciblés, applicables à tous les types de dégradateurs. L’utilisation de lignées cellulaires HiBiT CRISPR avec ces protocoles ne se limite cependant pas à la dégradation des protéines, mais plutôt à des outils généraux pour surveiller tout niveau de protéine cible endogène qui pourrait être modulé post-traitement pour étudier l’impact des composés ou même des mécanismes de résistance 20,65,66. Une condition préalable à ces méthodes de détection luminescentes est une lignée cellulaire cible HiBiT marquée de manière endogène CRISPR, ce qui est essentiel car elle permet une détection luminescente sensible, tout en maintenant l’expression de la cible endogène et la régulation du promoteur natif18,19,20. Des progrès significatifs ont été réalisés dans l’utilisation de CRISRP / Cas9 pour l’insertion de balises génomiques, en particulier dans l’évolutivité 20 et avec la haute sensibilité de détection, dans divers formats, y compris les pools CRISPR ou les clones avec insertions alléliques hétérozygotes ou homozygotes18,19,20. L’utilisation d’une expression exogène de HiBiT ou d’autres fusions rapporteures dans les cellules au lieu d’un marquage endogène est possible, mais des précautions importantes doivent être prises en utilisant des systèmes présentant une surexpression protéique14,18. Ceux-ci peuvent conduire à des artefacts dans la compréhension de la véritable puissance des composés et de la dynamique de récupération des protéines14,18, y compris des boucles de rétroaction transcriptionnelles potentielles activées après la dégradation de la cible. En outre, les composés à un stade précoce à faible puissance pourraient être manqués et se présenter comme des faux négatifs lors du dépistage. Comme la perte de protéines pourrait résulter d’une toxicité induite par un composé et de la mort cellulaire, les protocoles décrits ici contiennent des tests luminescents ou fluorescents hautement recommandés, mais facultatifs, associés au protocole de dégradation. Le protocole comporte deux sections principales, le critère d’évaluation lytique et le criblage cinétique des cellules vivantes. Dans chacune de ces sections, des options sont incluses pour les mesures de viabilité des cellules multiplexées dans les formats final ou cinétique. La surveillance des changements de la protéine endogène marquée nécessite une complémentation avec LgBiT dans les cellules. Par conséquent, la section de criblage cinétique fait référence à des protocoles importants pour l’introduction de celui-ci, qui peut être réalisé par expression transitoire ou stable et est essentiel pour effectuer les mesures luminescentes sur cellules vivantes. Toutes les approches présentées ici permettent un classement rapide et une évaluation de l’activité des composés, ce qui permet des efforts de criblage précoce des composés et une identification plus rapide des dégradateurs de plomb.
Ce protocole est conçu pour l’étude des composés de dégradation en conjonction avec une lignée cellulaire HiBiT CRISPR. Les protocoles de génération d’insertions HiBiT CRISPR pour de nombreuses cibles ont été décrits dans plusieurs publications récentes18,19,20.
Protocole
1. Études de dégradation des critères d’évaluation avec des protéines cibles HiBiT CRISPR au format lytique avec analyse de fluorescence de viabilité cellulaire facultative
- Préparation et placage de lignée cellulaire adhérente ou en suspension de mammifères
- Ajuster la densité cellulaire à 2,22 x 105/mL par dilution dans un milieu cellulaire approprié utilisé pour le passage et la croissance cellulaire.
- Distribuer les cellules dans des plaques avec un minimum de 3 puits par condition expérimentale et témoin. Distribuer 90 μL (20 000 cellules) par puits de suspension cellulaire dans des plaques blanches à 96 puits. Pour le format 384 puits, distribuer 36 μL (8 000 cellules) par puits de suspension cellulaire dans des plaques blanches de 384 puits.
- Préparation et addition de composés
- Préparer des plaques PROTAC ou des plaques de composés d’essai de dégradateur diluées en série à une concentration finale de 1 000 fois dans du DMSO à 100 %. Ensuite, diluez-le à 10x la concentration finale dans le milieu de culture cellulaire. Ajouter un volume égal de DMSO au support, à utiliser comme contrôle DMSO non composé.
- Pour le format 96 puits, ajouter 10 μL de 10x composés et solutions témoins à 90 μL de cellules. Pour un format 384 puits, ajoutez 4 μL de solutions composées et témoins 10x à 36 μL de cellules.
- Incuber les plaques dans un incubateur à 37 °C et 5% de CO2 pendant la durée souhaitée ou dans des conditions optimales pour leur croissance.
NOTA: Comme il s’agit d’un essai sur les paramètres finaux, l’essai de plusieurs points temporels nécessitera la préparation de plaques de dégradation distinctes pour chaque point temporel, comme décrit à l’étape 1.1.2 ci-dessus. Les temps d’incubation pour détecter la dégradation médiée par les composés sont très variables et dépendent probablement de la concentration du composé. Les points de temps initiaux suggérés seraient 6 h et 24 h. - Si vous mesurez la détection luminescente du point final sans la mesure facultative de la viabilité de la cellule, passez directement à l’étape 1.3 ci-dessous. Si vous effectuez un multiplexage avec mesure de viabilité de cellule, passez à la section 1.4 suivante ci-dessous.
- Mesure lytique des cellules
- Immédiatement avant les mesures lytiques HiBiT, préparer 2x réactif de détection lytique en ajoutant 20 μL de substrat lytique et 10 μL de protéine LgBiT par 1 mL de tampon lytique. Préparer suffisamment de réactif de détection 2x pour le nombre de puits à analyser, y compris un volume supplémentaire pour tenir compte de l’erreur de pipetage (c.-à-d. nombre de puits + 10%).
- Ajouter un réactif de détection lytique préparé aux cellules. Pour le format à 96 puits, ajouter 100 μL de 2x réactif de détection lytique à chaque puits contenant 100 μL de cellules. Pour un format à 384 puits, ajouter 40 μL de 2x réactif de détection lytique à chaque puits contenant 40 μL de cellules. Mélanger la plaque sur un mélangeur vortex à microplaques pendant 10-20 min à 350 tr / min.
- Mesurer la luminescence sur un luminomètre capable de lire la luminescence dans une plaque de 96 ou 384 puits.
- Multiplexage de viabilité cellulaire en option
REMARQUE: Cette étape est effectuée à l’aide d’un kit CellTiter-Fluor (CTF) disponible dans le commerce (voir le tableau des matériaux).- 30 à 40 minutes avant la mesure souhaitée, préparer une solution de réactif de détection de viabilité cellulaire 6x en ajoutant 10 μL du substrat à 2 mL du tampon de dosage. Préparer suffisamment de réactif 6x pour chaque puits à analyser, y compris le volume supplémentaire pour l’erreur de pipetage (c.-à-d. nombre de puits + 10%).
- Ajouter le réactif préparé aux puits. Pour le format 96 puits, ajoutez 20 μL de réactif 6x à chaque puits contenant déjà un volume de 100 μL. Pour un format 384 puits, ajoutez 8 μL de réactif 6x à chaque puits contenant 40 μL de cellules. Mélanger brièvement sur un mélangeur vortex à microplaques, puis incuber la plaque pendant 30 min dans un incubateur à 37 °C.
- Au point de terminaison souhaité de la mesure (c.-à-d. 6 ou 24 heures après le traitement, étape 1.2.3), mesurer la fluorescence sur un instrument capable de lire la fluorescence (380-400 nmEx/505 nmEm) au format 96 ou 384 puits.
- Préparer 2x réactif de détection lytique en ajoutant 20 μL de substrat lytique et 10 μL de protéine LgBiT par 1 mL de tampon lytique. Préparer suffisamment de réactif de détection 2x pour le nombre de puits à analyser, y compris le volume supplémentaire pour tenir compte de l’erreur de pipetage (p. ex., nombre de puits + 10 %).
- Ajouter le réactif de détection lytique préparé aux puits. Pour le format 96 puits, ajouter 120 μL de 2x réactif de détection lytique à chaque puits contenant déjà un volume de 120 μL. Pour un format 384 puits, ajoutez 48 μL de 2x réactif de détection lytique à chaque puits contenant déjà un volume de 48 μL. Mélanger la plaque sur un mélangeur vortex à microplaques pendant 10-20 min.
- Mesurer la luminescence sur un luminomètre capable de lire la luminescence dans des plaques de 96 ou 384 puits.
- Quantification de la dégradation et de la viabilité cellulaire
- Faites la moyenne des unités d’éclairage relatif (RLU) du contrôle DMSO au point de temps mesuré. Utilisez cette valeur comme niveau protéique de base de la cible pour calculer la dégradation fractionnée en normalisant tous les autres traitements testés au même moment indiquent cette valeur. Par exemple, si le RLU moyen pour les puits témoins de DMSO à 6 h était de 10 000 et que le RLU pour un traitement composé donné à 6 h était de 5 000, la dégradation fractionnelle serait calculée comme suit : 5 000 ÷ 10 000 = 0,5 (équation 1).
Équation 1 :
- Déterminez le pourcentage de dégradation à partir de la RLU fractionnaire :
Équation 2 :
- Tracer l’UFR fractionnaire ou le % de dégradation à des moments précis pour classer l’activité des composés.
- Vous pouvez éventuellement analyser les données de l’unité de fluorescence relative (UFR) pour la mesure du test de viabilité cellulaire en comparant les valeurs de tous les traitements au contrôle DMSO. Si une baisse significative de l’UFR est observée pour tout traitement relatif au contrôle du DMSO, les données de dégradation peuvent également être normalisées aux données de l’essai de viabilité cellulaire pour déterminer les changements dans le niveau de protéines par rapport aux pertes de viabilité cellulaire.
- Faites la moyenne des unités d’éclairage relatif (RLU) du contrôle DMSO au point de temps mesuré. Utilisez cette valeur comme niveau protéique de base de la cible pour calculer la dégradation fractionnée en normalisant tous les autres traitements testés au même moment indiquent cette valeur. Par exemple, si le RLU moyen pour les puits témoins de DMSO à 6 h était de 10 000 et que le RLU pour un traitement composé donné à 6 h était de 5 000, la dégradation fractionnelle serait calculée comme suit : 5 000 ÷ 10 000 = 0,5 (équation 1).
2. Dégradation cinétique en temps réel des protéines cibles HiBiT CRISPR et test de luminescence optionnel de viabilité cellulaire
NOTE: La capacité d’effectuer le criblage cinétique et la dégradation nécessite une co-expression de la protéine LgBiT dans la cellule, qui a été décrite précédemment18,19,63. Cela peut être réalisé par transfection transitoire d’un vecteur LgBiT, utilisation de BacMam LgBiT, ou en effectuant l’insertion de HiBiT CRISPR dans une lignée cellulaire stable LgBiT.
- Placage de lignées cellulaires adhérentes.
- Retirer le milieu de la fiole cellulaire par aspiration, laver les cellules avec du DPBS, dissocier les cellules avec 0,05% de trypsine-EDTA et permettre aux cellules de se dissocier du fond de la fiole. Pour les lignées cellulaires en suspension, passez à la section 2.2.
- Neutraliser la trypsine à l’aide d’un milieu de culture cellulaire contenant du sérum, mélanger pour recueillir et remettre en suspension les cellules et transférer la suspension cellulaire dans un tube conique.
- Faites tourner les cellules à 125 x g pendant 5 min. Jeter le milieu de culture cellulaire et remettre en suspension dans un volume égal de milieu de culture cellulaire frais.
- Cellules en plaques dans des plaques d’essai avec un minimum de puits triples par condition expérimentale et témoin. Pour le dénombrement du format de 96 puits afin d’estimer la densité cellulaire, ajuster la densité à 2 x 105 cellules/ml dans le milieu d’essai et distribuer 100 μL (20 000 cellules) par puits dans une plaque de 96 puits. Pour le dénombrement du format de 384 puits afin d’estimer la densité cellulaire, ajuster la densité à 4,44 x 105 cellules/ml dans le milieu d’essai et distribuer 18 μL (8 000 cellules) par puits.
- Incuber les plaques à 37 °C, 5% de CO2 pendant la nuit ou dans des conditions optimales pour leur croissance.
- Placage de cellules en suspension
- Ajuster la densité cellulaire à 2,22 x 105 cellules/ml dans un milieu indépendant du CO2 supplémenté avec 10 % de FBS et 1 x endurazine (dilution de 1:100 du réactif mère).
- Cellules de plaque dans des plaques d’essai avec un minimum de 3 puits par condition expérimentale et témoin. Pour le format 96 puits, distribuer 90 μL (20 000 cellules) par puits. Pour le format 384 puits, distribuer 36 μL (8 000 cellules) par puits.
REMARQUE: Pour les lignées cellulaires en suspension qui ont une faible luminescence du signal au fond (S: B), par exemple, lorsque vous travaillez avec des pools CRISPR plutôt que des clones, il est possible d’augmenter la luminescence en augmentant le nombre de cellules plaquées, jusqu’à 100 000 cellules / puits au format 96 puits, ou 40 000 cellules / puits au format 384 puits.
- Tests de dégradation cinétique utilisant des cellules HiBiT CRISPR exprimant LgBiT
- Pour les cellules en suspension contenant déjà de l’endurazine, qui a été incluse à l’étape de placage au point 2.2., passer directement à l’étape 2.3.3. Pour les lignées cellulaires adhérentes, préparez la solution d’endurazine Nano-Glo. Pour le format 96 puits, préparer une solution 1x d’endurazine en diluant le réactif mère 1:100 dans un milieu indépendant du CO2 complété par 10% de FBS. Pour le format 384 puits, préparer une solution 2x d’endurazine en diluant le réactif mère 1:50 dans un milieu indépendant du CO2 complété par 10% de FBS.
- Ajouter la solution d’endurazine à chaque puits de cellules adhérentes. Pour le milieu d’aspiration au format 96 puits, ajouter 90 μL de 1x solution d’endurazine. Pour un format à 384 puits, ajouter 18 μL de solution d’endurazine 2x à 18 μL de cellules. Ne pas aspirer le milieu car l’essai de dégradation est effectué dans un mélange 50:50 de milieu de culture et de milieu indépendant du CO2 dans un format 384 puits.
- Incuber des plaques cellulaires en suspension ou adhérentes contenant de l’endurazine pendant 2,5 h dans un incubateur à 37 °C et 5% de CO2 pour permettre à la luminescence de s’équilibrer.
- Préparer une concentration de 10x de titrage PROTAC d’essai dans un milieu indépendant du CO2 et ajouter 10 μL à chaque puits de plaque de 96 puits ou 4 μL pour une plaque de 384 puits. Pour les composés dont l’efficacité est inconnue, une concentration finale de 1 à 10 μM au point le plus élevé est recommandée comme point de départ.
- Recueillir des mesures cinétiques de la luminescence dans un luminomètre prééquilibré à 37 °C pendant une période comprise entre 0 et 48 h. Les incréments de temps de mesure peuvent être personnalisés pour chaque expérience, mais une expérience initiale recommandée serait des mesures de luminescence toutes les 5-15 minutes pendant 24 h ou la durée souhaitée.
- Analyse multiplex de même puits sur la viabilité cellulaire en option après la mesure cinétique finale
REMARQUE : Ce test est effectué avec un kit CellTiter-Glo (CTG) disponible dans le commerce (voir le tableau des matériaux).- Équilibrer le réactif CTG à température ambiante.
- Après la mesure de la dégradation au dernier point temporel de l’analyse cinétique, ajouter 100 μL (plaque de 96 puits) ou 40 μL (plaque de 384 puits) de réactif par puits de la plaque, et mélanger sur un agitateur à plaque à 500-700 tr/min (plaque de 96 puits) ou un mélangeur vortex à microplaques (plaque de 384 puits) pendant 5 min.
- Incuber la plaque à température ambiante pendant 30 minutes pour permettre la lyse cellulaire et l’extinction du signal HiBiT.
- Mesurer la luminescence totale sur un luminomètre en suivant les recommandations du fabricant.
- Quantification des profils de dégradation cinétique
- À l’aide des mesures de luminescence cinétique recueillies, normaliser les RLU brutes pour chaque concentration de PROTAC à la condition moyenne de DMSO répliquée à chaque point temporel pour tenir compte des changements dans la concentration de furimazine libre au fil du temps. Calculez le RLU fractionnaire à l’aide de l’équation 1.
Équation 1 : - À partir des courbes de dégradation, ajustez un modèle de décroissance exponentielle à composante unique utilisant l’équation 2 à la partie de dégradation initiale de chaque courbe jusqu’au point où les données atteignent un plateau.
NOTA: Il peut être utile d’exclure de l’ajustement les premiers points de données car il peut y avoir un bref décalage avant que la dégradation ne soit observée.
Équation 2 :
- À partir de l’équation 2, déterminez le paramètre ƛ, qui représente la constante de vitesse de dégradation et le plateau, qui représente la plus faible quantité de protéines restantes.
- Calculez Dmax, qui est la quantité fractionnaire maximale de protéines dégradées et est calculée comme 1-Plateau.
- Tracer Dmax pour chaque concentration de PROTAC afin de déterminer une courbe de pouvoir de dégradation indépendante du temps.
- Déterminer la valeur Dmax50 pour la parcelle en 2.3.5 pour analyser l’efficacité des composés.
REMARQUE : Pour déterminer un courant continu50 à un moment précis, tracez le pourcentage de dégradation calculé pour chaque concentration au moment choisi. Cela peut être spécifié comme DC 50 t = 4 h ou DC50 t = 12 h.
- À l’aide des mesures de luminescence cinétique recueillies, normaliser les RLU brutes pour chaque concentration de PROTAC à la condition moyenne de DMSO répliquée à chaque point temporel pour tenir compte des changements dans la concentration de furimazine libre au fil du temps. Calculez le RLU fractionnaire à l’aide de l’équation 1.
Résultats
Pour démontrer l’analyse de la dégradation lytique à concentration unique, plusieurs protéines cibles CDK; CDK2, CDK4, CDK7 et CDK10 ont été marqués de manière endogène avec HiBiT à leur extrémité C dans des cellules HEK293 et traités avec une concentration de 1 μM du PROTAC pankinase à base de Cereblon, TL12-18654 (Figure 1A). Le niveau de protéine CDK a été mesuré à différents moments et le RLU fractionnaire par rapport au témoin DMSO a été déterminé (Figure 1A). Chaque protéine CDK a montré différents degrés de dégradation en réponse au traitement composé et aux différents points temporels (Figure 1A). Pour comprendre comment les protéines CDK se comparent directement les unes aux autres en termes de perte de protéines, les RLU fractionnaires de la figure 1A ont été calculées en % de dégradation totale et tracées pour chaque point temporel de la figure 1B. Cela montre que même à des points temporels précoces, 2 ou 4 h, certains membres de la famille CDK présentent des niveaux élevés de dégradation qui continuent à augmenter au fil du temps (Figure 1B).
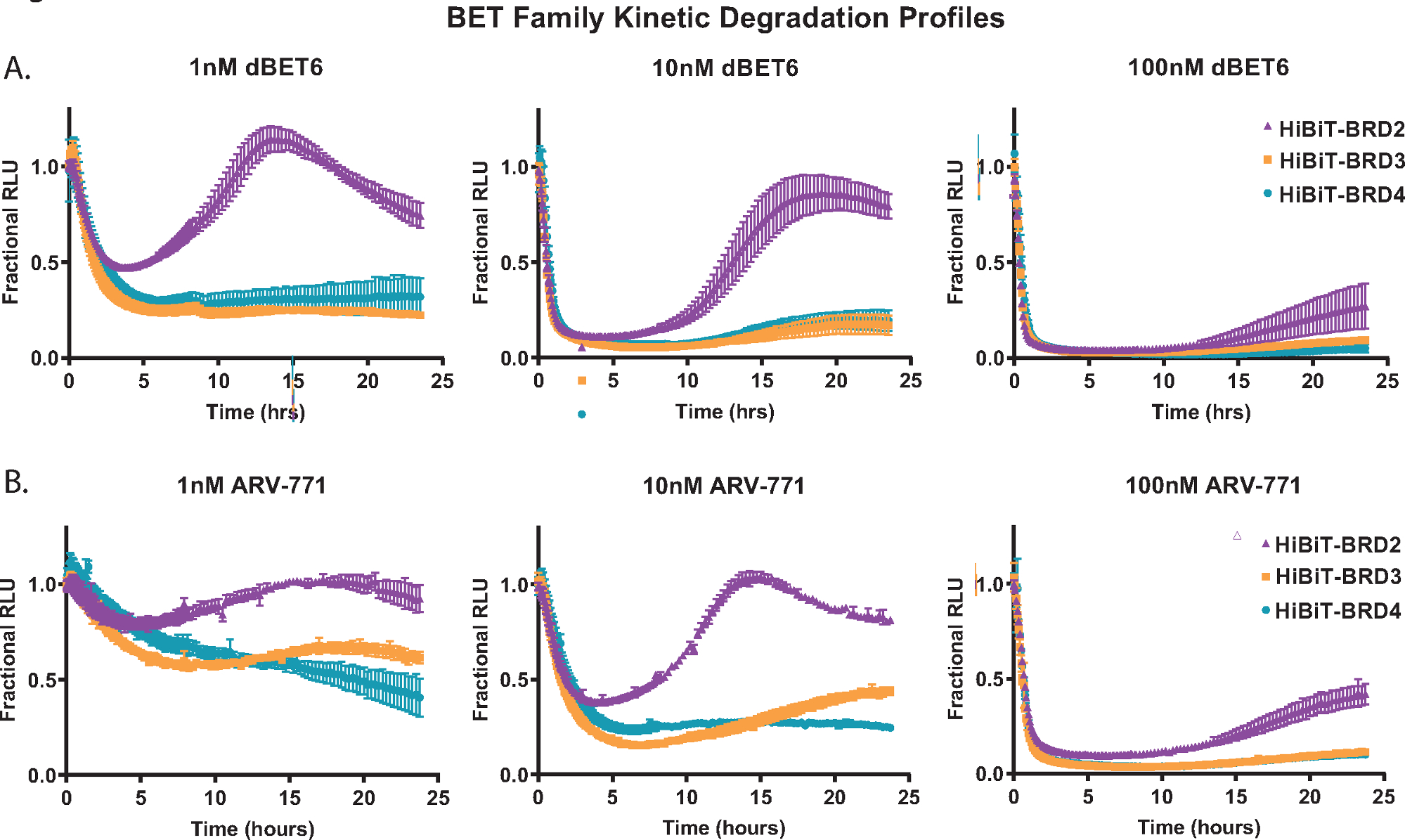
Démontrer l’analyse de dégradation cinétique, chacune des protéines membres de la famille BET; BRD2, BRD3 et BRD4 ont été marqués de manière endogène avec HiBiT à leur extrémité N dans les cellules HEK293 exprimant de manière stable la protéine LgBiT18. Ceux-ci ont ensuite été traités avec trois concentrations différentes des PROTAC pan-BET; le dBET650 à base de Cereblon (Figure 2A) et l’ARV-77141 à base de VHL (Figure 2B). Les mesures cinétiques ont été recueillies sur une période de 24 heures et, à partir des profils à chaque concentration, les différences dans la réponse des membres de la famille BET sont évidentes. La capacité de BRD2 à amorcer une réponse de récupération plus rapide après un traitement composé de dégradation (figure 2A,B) a déjà été observée avec d’autres PROTAC pan-BET et est probablement due à une réponse de rétroaction transcriptionnelle compétitive au processus de dégradation18.
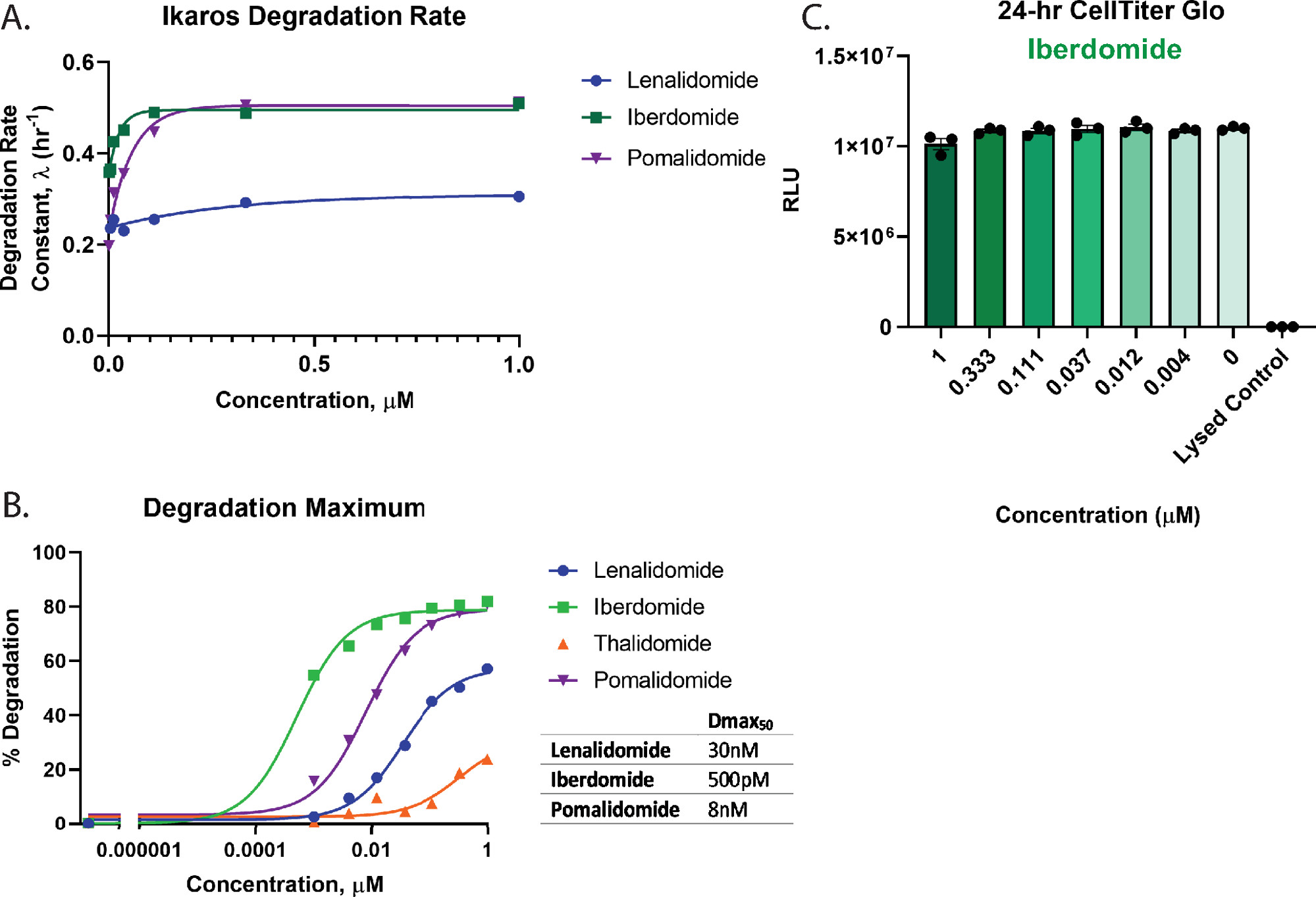
L’analyse des paramètres et de la cinétique peut être effectuée avec des traitements dose-réponse composés complets. La figure 3 présente les profils de dégradation de la relation dose-réponse cinétique du traitement des cellules Ikaros/IKZF1-HiBiT CRISPR Jurkat exprimant de manière stable la protéine LgBiT avec quatre composés moléculaires de colle 2,26,55,57; lénalidomide (figure 3A), iberdomide (CC-220) (figure 3B), thalidomide (figure 3C) et pomalidomide (figure 3D). Ces dégradateurs montrent des différences significatives dans la réponse à la dégradation entre les composés ainsi qu’entre les séries de concentrations (Figure 3).
Pour évaluer quantitativement la dégradation et classer les composés de la figure 3, les profils dose-réponse ont été utilisés pour calculer les principaux paramètres de dégradation, y compris le taux de dégradation (figure 4A), les valeurs Dmax (figure 4B) et Dmax50 (figure 4B). Ces analyses montrent que l’iberdomide (CC-220) et le pomalidomide ont des taux de dégradation initiale rapide très similaires (figure 4A), mais que l’iberdomide (CC-220) a la puissance la plus élevée comme on l’a déjà vu dans des études orthogonales55,57 (figure 4B). Étant donné que l’iberdomide présente une puissance élevée et que toutes les concentrations testées montrent une dégradation supérieure à 50%, la valeur Dmax50 obtenue pour l’iberdomide représente une estimation basée sur la limitation de l’ajustement précis des données. D’après les graphiques des figures 3C, D et 4B, ni le lénalidomide ni la thalidomide ne dégradent la cible d’Ikaros/IKZF1 jusqu’à son achèvement aux concentrations les plus élevées testées. En raison de la très faible dégradation observée avec la thalidomide, les traces de dégradation n’ont pas pu être ajustées avec précision à un modèle de désintégration exponentielle, par conséquent, le taux de dégradation n’a pas été quantifié pour ce traitement. Pour le dégradateur le plus puissant, l’iberdomide (CC-220)55,57 (figure 4B). Les essais multiplex de viabilité cellulaire n’ont montré aucune perte de viabilité cellulaire pour les concentrations testées (figure 4C).

Figure 1 : Dégradation et toxicité du critère d’évaluation CDK avec la pankinase PROTAC, TL12-18654. (A) Sélectionner un panel de protéines cibles CDK endogènes fusionnées avec HiBiT sur l’extrémité C via CRISPR/Cas9 et évalué pour la dégradation avec 1 μM TL12-186 PROTAC 54 à 2 h, 4 h, 8 h et24 h de traitement. Les valeurs sont représentées sous forme de RLU fractionnaire par rapport à un contrôle DMSO mesuré à chaque point temporel. Les barres d’erreur représentent l’écart-type de la moyenne de 3 répétitions techniques. (B) Pourcentage de dégradation du panel de protéines cibles CDK calculé à partir de (A) représentant la quantité de dégradation de chaque membre de la famille observée à 2, 4, 8 et 24 h de temps. Les barres d’erreur représentent l’écart-type de la moyenne de 3 répétitions techniques. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Profilage de la sélectivité de dégradation cinétique des membres de la famille BET avec des dégradateurs BET, dBET650 et ARV-77141. Profils de dégradation cinétique des membres endogènes de la famille BET, BRD2, BRD3 et BRD4, marqués avec HiBiT sur l’extrémité N via CRISPR / Cas9 avec traitement de concentrations uniques de 1 nM (gauche), 10 nM (milieu) ou 100 nM (droite) dBET650 (A) ou ARV-77141 (B) PROTACs. Les valeurs sont représentées sous forme de RLU fractionnaire calculée à partir d’un contrôle DMSO à chaque point de temps cinétique. Les barres d’erreur représentent l’écart-type de la moyenne de 4 répétitions techniques. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Profils dose-réponse de dégradation cinétique des cellules vivantes d’Ikaros/IKZF1-HiBiT avec un panneau de colle moléculaire 2,26,55,57. Les cellules Jurkat exprimant de manière stable la protéine LgBiT ont été modifiées à l’aide de CRISPR / Cas9 pour marquer l’extrémité C d’Ikaros / IKZF1 avec le peptide HiBiT. Les cellules ont été traitées avec une série de concentrations dose-réponse de 8 points, y compris le DMSO, de quatre composés moléculaires de colle 2,26,55,57: (A) lénalidomide, (B) iberdomide (CC-220), (C) thalidomide ou (D) pomalidomide. La luminescence a été mesurée toutes les 5 minutes pour un total de 19,5 h. Les données relatives aux unités lumineuses (UGR) de (A-D) ont été converties en UFR fractionnelles comme décrit à l’étape 2.4.1 et représentées graphiquement en fonction du temps. Les barres d’erreur représentent SD de 3 répétitions techniques. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Calcul du taux de dégradation et de Dmax50 pour Ikaros/IKZF1-HiBiT, et tests de santé cellulaire multiplexage. Les données de dégradation cinétique de la figure 3 ont été utilisées pour calculer les paramètres quantitatifs de dégradation. (A) Les taux de dégradation et (B) les valeurs maximales de dégradation (Dmax) sont représentés graphiquement à chaque concentration de médicament pour les composés de colle moléculaireindiqués 2,26,55,57. (B) Les valeurs Dmax50 pour chaque composé ont été calculées à l’aide d’un modèle dose-réponse avec une pente de Hill contrainte de 1, qui peut être utilisé pour classer les composés de dégradation par ordre pour une cible. (C) Des essais de viabilité cellulaire avec l’iberdomide(CC-220)55,57 dose-réponse à la dégradation de la figure 3B ont été effectués comme mesure du paramètre final une fois les mesures de dégradation cinétique terminées. Les barres d’erreur représentent SD de 3 répétitions techniques. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Discussion
Nous présentons ici deux méthodes de criblage de l’activité des composés de dégradation sous forme de point final lytique ou de mode cinétique de cellule vivante. Ces approches sont basées sur les mêmes principes de mesure luminescente, mais offrent différents niveaux de détail et de compréhension. Le choix de l’une ou l’autre approche dépendra probablement des objectifs de criblage et de la taille de la bibliothèque de composés. Pour que les grands criblages de composés ou les criblages primaires observent toute dégradation détectable, le criblage lytique des paramètres offre une compatibilité sensible et efficace à haut débit lorsque d’autres approches de point final, telles que le transfert Western ou la spectrométrie de masse, peuvent être peu pratiques ou difficilesà adapter 14. Un point de départ pour ces écrans pourrait être effectué avec un nombre limité de concentrations et de points temporels. Les concentrations initiales recommandées pour l’essai sont comprises entre 100 nM-10 μM, pour tenir compte des dégradateurs initiaux ayant une faible puissance, une faible perméabilité ou, dans certains cas, des composés très puissants, la présence d’un effet crochet. Il est en outre recommandé d’essayer au moins deux points temporels différents pour établir une dégradation précoce à 4-6 h et une dégradation latente ou soutenue à 18-24 h. Les composés qui présentent un pouvoir de dégradation élevé et un mécanisme sur la cible sont facilement observés dans un délai de 4 à 6 heures, alors que la dégradation ou la perte apparente de protéines observée seulement à des moments ultérieurs pourrait être due à divers mécanismes. Il est fortement recommandé de surveiller la viabilité cellulaire à des moments précoces et tardifs, de sorte que la perte de protéines puisse être dissociée de la perte due à la mort cellulaire. Comme pour tout type d’essai luminescent ou fluorescent, il est possible que les composés dans les bibliothèques interfèrent ou inhibent le signal, par conséquent, des expériences de suivi orthogonales avec des composés principaux utilisant des fusions non apparentées ou des approches alternatives pour surveiller le niveau de protéines seront importantes pour évaluer que la perte de RLU dans ces tests est directement associée à la dégradation des protéines cibles.
La capacité de cribler dans un format cinétique de cellule vivante sur de longues périodes de temps dépend fortement du signal de test au bruit de fond (S: B). Les facteurs qui contribuent à S:B comprennent le niveau d’expression de la protéine cible elle-même, qui peut couvrir plusieurs ordres de grandeur, l’efficacité de l’expression de LgBiT dans la lignée cellulaire choisie pour l’insertion peptidique, et la disponibilité de la cible étiquetée pour la complémentarité dans ses divers complexes natifs. Nous avons établi une exigence générale de coupure consistant en un S:B de 15 pour mesurer avec succès la dégradation en mode cinétique avec l’endurazine ou la vivazine. Le S:B est déterminé en mesurant le signal de base des cellules éditées par HiBiT co-exprimant LgBiT par rapport aux cellules parentales non éditées exprimant LgBiT seule en présence de substrats de cellules vivantes d’endurazine ou de vivazine. La vivazine produira un signal luminescent plus élevé, mais se désintégrera plus rapidement que l’endurazine et pourrait limiter l’acquisition du signal à 24 heures ou moins. En outre, S:B peut également dépendre fortement de l’utilisation de pools ou de clones CRISPR. Pour les cibles dans les lignées cellulaires qui sont plus faciles et ont une efficacité élevée pour l’ingénierie CRISPR / Cas9, une population hétérogène de cellules modifiées CRISPR peut avoir suffisamment de S: B pour l’analyse cinétique. Pour les cibles dans des lignées cellulaires plus difficiles où une intégration génomique moins efficace via CRISPR entraîne des pools à faible S:B, il pourrait être nécessaire d’isoler les clones de CRISPR pour enrichir les populations éditées et atteindre un S:B suffisamment élevé pour l’analyse cinétique. Pour l’un ou l’autre de ces scénarios, si S:B est inférieur à 15 avec des substrats d’endurazine ou de virazidine, un dépistage lytique final est conseillé.
Pour une meilleure compréhension et caractérisation des composés, y compris la détermination d’un profil de dégradation avec des paramètres quantitatifs, l’analyse cinétique en temps réel dans les cellules vivantes est l’approche de dépistage recommandée14,18. À l’instar de l’analyse des paramètres décrite ci-dessus, le criblage cinétique initial peut être effectué avec un nombre limité de concentrations comprises entre 100 nM et 10 μM à haut débit. Dans un format de 384 puits, plus de 100 composés peuvent facilement être criblés en triple à une concentration sur une seule plaque. Les profils de dégradation qui en résulteront fourniront des indications non seulement sur l’étendue de la dégradation observée, mais aussi sur le taux de dégradation, la durée de la dégradation et la récupération potentielle de la protéine14,18 (figure 2 et figure 3). Les formes du profil de dégradation fournissent également des informations précieuses. Les dégradateurs spécifiques et puissants montrent souvent une perte rapide initiale de la protéine cible à un plateau en quelques heures18,53, alors que d’autres mécanismes tels que la rétroaction transcriptionnelle ou la toxicité du composé entraînent généralement une perte plus linéaire de la protéine au fil du temps. Ces détails et nuances sont manqués avec l’analyse lytique des points finaux, et avec l’analyse en temps réel sur 24-48 heures, il n’est pas nécessaire de prédire le temps nécessaire pour capturer le vrai Dmax dans des ensembles de composés nouveaux ou inconnus.
La cinétique en temps réel permet également un dépistage efficace de la relation dose-réponse pour mieux comprendre l’efficacité des composés, l’impact de la concentration des composés sur le taux de dégradation initial et offre la possibilité de classer les composés en fonction de plus d’un paramètre. Les mesures classiques du pouvoir de dégradation impliquent des calculs de DC50 à un moment précis en fonction des maxima de dégradation apparents. En revanche, notre approche cinétique pour évaluer la puissance intègre le véritable maximum de dégradation à chaque concentration, quel que soit le moment où elle se produit dans le temps18. Nous appelons cette mesure de la puissance de dégradation cinétique, le Dmax5018. L’analyse de cette manière tient compte des composés qui peuvent initier la dégradation plus lentement à des concentrations plus faibles et donc prendre plus de temps après le traitement pour atteindre leur Dmax. Il peut être particulièrement instructif de classer les composés à la fois sur le taux de dégradation et sur Dmax. Pour les dégradateurs les plus puissants, cela différenciera davantage les dégradateurs lents mais puissants de ceux qui sont à la fois rapides et puissants. Ensemble, le criblage cinétique lytique et cellulaire vivant utilisant les lignées cellulaires HiBiT CRISPR sont des approches puissantes qui donnent une image plus complète de la dégradation ciblée des protéines, de la fonction des composés et permettent le processus de criblage de l’évaluation initiale de l’activité à l’optimisation chimique en aval grâce à l’amélioration des paramètres clés de dégradation.
Déclarations de divulgation
Promega Corporation est le propriétaire commercial par cession de brevets des technologies et applications HiBiT et NanoLuc.
Remerciements
K.M.R, S.D.M, M.U. et D.L.D sont tous des employés de Promega Corporation
matériels
| Name | Company | Catalog Number | Comments |
| CellTiter-Glo 2.0 reagent | Promega | G9241 | Cell Viability luminescent assay |
| CellTiter-Fluor Cell Viability Assay | Promega | G6080 | Cell Viability fluorescent assay |
| CO2-independent medium | ThermoFisher | 18045-088 | Cell culture |
| DMSO | Sigma Aldrich | D2650 | For compound dilution and control |
| DPBS | Gibco | 14190 | Cell culture |
| Fetal Bovine Serum | Seradigm | 89510-194 | Cell culture |
| HEK293 LgBiT stable cell line | Promega | N2672 | For complementation with HiBiT to generate luminescence |
| HiBiT CRISPR mammalian cell line | Promega | https://www.promega.com/crispr-tpd | |
| Hygromycin B solution | Gibco | 10-687-010 | Cell culture |
| LgBiT BacMam | Promega | CS1956C01 | For complementation with HiBiT to generate luminescence |
| LgBiT Expression Vector | Promega | N2681 | For complementation with HiBiT to generate luminescence |
| Luminometer Plate Reader | Luminomenter capable of measuring luminescence and fluorescence (e.g. GloMax Discover System, Promega GM3000) | ||
| NanoGlo Endurazine live cell substrate | Promega | N2570 | Kinetic HiBiT reagent |
| NanoGlo Vivazine live cell substrate | Promega | N2580 | Kinetic HiBiT reagent |
| NanoGlo HiBiT Lytic Detection system | Promega | N3030 | Enpoint lytic HiBiT reagent |
| Opti-MEM Reduced Serum Medium, no phenol red (ThermoFisher) | ThermoFisher | 11058-021 | Cell culture |
| Tissue culture plates, white, 96 well plate | Costar | 3917 | Cell culture |
| Tissue culture plates, white, 384 well plate | Corning | 3570 | Cell culture |
| Trypsin/EDTA | Gibco | 25300 | Cell culture |
Références
- Burslem, G. M., Crews, C. M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. 181 (1), 102-114 (2020).
- Chamberlain, P. P., Hamann, L. G. Development of targeted protein degradation therapeutics. Nature Chemical Biology. 15 (10), 937-944 (2019).
- Churcher, I. Protac-induced protein degradation in drug discovery: Breaking the rules or just making new ones. Journal of Medicinal Chemistry. 61 (2), 444-452 (2018).
- Ciulli, A., Farnaby, W. Protein degradation for drug discovery. Drug Discovery Today: Technologies. 31, 1-3 (2019).
- Crews, C. M. Inducing protein degradation as a therapeutic strategy. Journal of Medicinal Chemistry. 61 (2), 403-404 (2018).
- Cromm, P. M., Crews, C. M. Targeted protein degradation: from chemical biology to Drug Discovery. Cell Chemical Biology. 24 (9), 1181-1190 (2017).
- Deshaies, R. J. Protein degradation: Prime time for PROTACs. Nature Chemical Biology. 11 (9), 634-635 (2015).
- Lai, A. C., Crews, C. M. Induced protein degradation: an emerging drug discovery paradigm. Nature Reviews Drug Discovery. 16 (2), 101-114 (2017).
- Ottis, P., Crews, C. M. Proteolysis-targeting chimeras: Induced protein degradation as a therapeutic strategy. ACS Chemical Biology. 12 (4), 892-898 (2017).
- Wu, T., et al. Targeted protein degradation as a powerful research tool in basic biology and drug target discovery. Nature Structural and Molecular Biology. 27, 605-614 (2020).
- Hanan, E. J., et al. Monomeric targeted protein degraders. Journal of Medicinal Chemistry. , (2020).
- Collins, I., Wang, H., Caldwell, J. J., Chopra, R. Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway. Biochemical Journal. 474 (7), 1127-1147 (2017).
- Carmony, K. C., Kim, K. B. PROTAC-induced proteolytic targeting. Methods in Molecular Biology. 832, 627-638 (2012).
- Daniels, D. L., Riching, K. M., Urh, M. Monitoring and deciphering protein degradation pathways inside cells. Drug Discovery Today: Technologies. 31, 61-68 (2019).
- Gu, S., Cui, D., Chen, X., Xiong, X., Zhao, Y. PROTACs: An emerging targeting technique for protein degradation in drug discovery. Bioessays. 40 (4), 1700247 (2018).
- Neklesa, T. K., Winkler, J. D., Crews, C. M. Targeted protein degradation by PROTACs. Pharmacology and Therapy. 174, 138-144 (2017).
- Raina, K., Crews, C. M. Targeted protein knockdown using small molecule degraders. Current Opinion in Chemical Biology. 39, 46-53 (2017).
- Riching, K. M., et al. Quantitative live-cell kinetic degradation and mechanistic profiling of PROTAC mode of action. ACS Chemical Biology. 13 (9), 2758-2770 (2018).
- Schwinn, M. K., et al. CRISPR-mediated tagging of endogenous proteins with a luminescent peptide. ACS Chemical Biology. 13 (2), 467-474 (2018).
- Schwinn, M. K., Steffen, L. S., Zimmerman, K., Wood, K. V., Machleidt, T. A simple and scalable strategy for analysis of endogenous protein dynamics. Science Reports. 10 (1), 8953 (2020).
- Bensimon, A., et al. Targeted degradation of SLC transporters reveals amenability of multi-pass transmembrane proteins to ligand-induced proteolysis. Cell Chemical Biology. 27 (6), 728-739 (2020).
- Bondeson, D. P., et al. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chemical Biology. 25 (1), 78-87 (2018).
- Buckley, D. L., et al. HaloPROTACS: Use of small molecule PROTACs to induce degradation of HaloTag fusion proteins. ACS Chemical Biology. 10 (8), 1831-1837 (2015).
- Bulatov, E., Ciulli, A. Targeting Cullin-RING E3 ubiquitin ligases for drug discovery: structure, assembly and small-molecule modulation. Biochemical Journal. 467 (3), 365-386 (2015).
- Burslem, G. M., et al. The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chemical Biology. 25 (1), 67-77 (2018).
- Chamberlain, P. P., et al. Evolution of cereblon-mediated protein degradation as a therapeutic modality. ACS Medicinal Chemistry Letters. 10 (12), 1592-1602 (2019).
- Crew, A. P., et al. Identification and characterization of Von Hippel-Lindau-recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. Journal of Medicinal Chemistry. 61 (2), 583-598 (2018).
- DeMars, K. M., Yang, C., Castro-Rivera, C. I., Candelario-Jalil, E. Selective degradation of BET proteins with dBET1, a proteolysis-targeting chimera, potently reduces pro-inflammatory responses in lipopolysaccharide-activated microglia. Biochemical and Biophysical Research Communication. 497 (1), 410-415 (2018).
- Erb, M. A., et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature. 543 (7644), 270-274 (2017).
- Farnaby, W., et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nature Chemical Biology. 15 (7), 672-680 (2019).
- Gadd, M. S., et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nature Chemical Biology. 13 (5), 514-521 (2017).
- Gechijian, L. N., et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nature Chemical Biology. 14 (4), 405-412 (2018).
- Gustafson, J. L., et al. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angewandte Chemie International Edition England. 54 (33), 9659-9662 (2015).
- Kerres, N., et al. Chemically induced degradation of the oncogenic transcription factor BCL6. Cell Reports. 20 (12), 2860-2875 (2017).
- Lohbeck, J., Miller, A. K. Practical synthesis of a phthalimide-based Cereblon ligand to enable PROTAC development. Bioorganic and Medicinal Chemistry Letters. 26 (21), 5260-5262 (2016).
- Lu, J., et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chemical Biology. 22 (6), 755-763 (2015).
- Lu, M., et al. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eurupean Journal of Medicinal Chemistry. 146, 251-259 (2018).
- Nabet, B., et al. The dTAG system for immediate and target-specific protein degradation. Nature Chemical Biology. 14 (5), 431-441 (2018).
- Nowak, R. P., et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nature Chemical Biology. 14, 706-714 (2018).
- Powell, C. E., et al. Chemically induced degradation of Anaplastic Lymphoma Kinase (ALK). Journal of Medicinal Chemistry. 61 (9), 4249-4255 (2018).
- Raina, K., et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proceedings of National Academy of Science U. S. A. 113 (26), 7124-7129 (2016).
- Sakamoto, K. M., et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proceeding National Academy Science. 98 (15), 8554-8559 (2001).
- Sakamoto, K. M., et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Molecular Cell Proteomics. 2 (12), 1350-1358 (2003).
- Schiedel, M., et al. Chemically induced degradation of Sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). Journal of Medicinal Chemistry. 61 (2), 482-491 (2018).
- Slabicki, M., et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature. , (2020).
- Smith, B. E., et al. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nature Communications. 10 (1), 131 (2019).
- Sun, B., et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia. 32 (2), 343-352 (2018).
- Tong, B., et al. A Nimbolide-based kinase degrader preferentially degrades oncogenic BCR-ABL. ACS Chemical Biology. 15 (7), 1788-1794 (2020).
- Winter, G. E., et al. Drug Development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 348 (6241), 1376-1381 (2015).
- Winter, G. E., et al. BET Bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Molecular Cell. 67 (1), 5-18 (2017).
- Zengerle, M., Chan, K. H., Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chemical Biology. 10 (8), 1770-1777 (2015).
- Zhang, C., et al. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK). European Journal of Medicinal Chemistry. 151, 304-314 (2018).
- Zoppi, V., et al. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel-Lindau (VHL) based dual degrader probe of BRD9 and BRD7. Journal of Medicinal Chemistry. 62 (2), 699-726 (2019).
- Huang, H. T., et al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chemical Biology. 25 (1), 88-99 (2018).
- Bjorklund, C. C., et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia. 34 (4), 1197-1201 (2020).
- Matyskiela, M. E., et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nature Chemical Biology. 14 (10), 981-987 (2018).
- Matyskiela, M. E., et al. A cereblon modulator (CC-220) with improved degradation of Ikaros and Aiolos. Journal of Medicinal Chemistry. 61 (2), 535-542 (2018).
- Bussiere, D. E., et al. Structural basis of indisulam-mediated RBM39 recruitment to DCAF15 E3 ligase complex. Nature Chemical Biology. 16 (1), 15-23 (2020).
- Du, X., et al. Structural basis and kinetic pathway of RBM39 recruitment to DCAF15 by a sulfonamide molecular glue E7820. Structure. 27 (11), 1625-1633 (2019).
- Ting, T. C., et al. Aryl sulfonamides degrade RBM39 and RBM23 by recruitment to CRL4-DCAF15. Cell Reports. 29 (6), 1499-1510 (2019).
- Hughes, S. J., Ciulli, A. Molecular recognition of ternary complexes: a new dimension in the structure-guided design of chemical degraders. Essays in Biochemistry. 61 (5), 505-516 (2017).
- Schapira, M., Calabrese, M. F., Bullock, A. N., Crews, C. M. Targeted protein degradation: expanding the toolbox. Nature Review Drug Discovery. 18 (12), 949-963 (2019).
- Dixon, A. S., et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chemical Biology. 11 (2), 400-408 (2016).
- Gilan, O., et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immuno-inflammation. Science. 368 (6489), 387-394 (2020).
- Oh-Hashi, K., Furuta, E., Fujimura, K., Hirata, Y. Application of a novel HiBiT peptide tag for monitoring ATF4 protein expression in Neuro2a cells. Biochemical Biophysical Report. 12, 40-45 (2017).
- Ottis, P., et al. Cellular resistance mechanisms to targeted protein degradation converge toward impairment of the engaged ubiquitin transfer pathway. ACS Chemical Biology. 14 (10), 2215-2223 (2019).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.