
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
ARN polymérase attachée à l’ADN pour la transcription in vitro programmable et le calcul moléculaire
Dans cet article
Résumé
Nous décrivons l’ingénierie d’une nouvelle ARN polymérase T7 attachée à l’ADN pour réguler les réactions de transcription in vitro. Nous discutons des étapes de la synthèse et de la caractérisation des protéines, validons la régulation transcriptionnelle de la preuve de concept et discutons de ses applications en informatique moléculaire, en diagnostic et en traitement de l’information moléculaire.
Résumé
La nanotechnologie de l’ADN permet l’auto-assemblage programmable des acides nucléiques en formes et dynamiques prescrites par l’utilisateur pour diverses applications. Ces travaux démontrent que les concepts de la nanotechnologie de l’ADN peuvent être utilisés pour programmer l’activité enzymatique de l’ARN polymérase T7 dérivée des phages (RNAP) et construire des réseaux de régulation de gènes synthétiques évolutifs. Tout d’abord, un RNAP T7 attaché à un oligonucléotide est conçu par l’expression d’un RNAP marqué SNAP N-terminal et le couplage chimique ultérieur du marqueur SNAP avec un oligonucléotide modifié par benzylguanine (BG). Ensuite, le déplacement des brins d’acide nucléique est utilisé pour programmer la transcription de la polymérase à la demande. En outre, les assemblages auxiliaires d’acides nucléiques peuvent être utilisés comme « facteurs de transcription artificiels » pour réguler les interactions entre l’ARNP T7 programmé par l’ADN et ses modèles d’ADN. Ce mécanisme de régulation de la transcription in vitro peut mettre en œuvre une variété de comportements de circuit tels que la logique numérique, la rétroaction, la cascade et le multiplexage. La composabilité de cette architecture de régulation des gènes facilite l’abstraction, la normalisation et la mise à l’échelle de la conception. Ces caractéristiques permettront le prototypage rapide de dispositifs génétiques in vitro pour des applications telles que la biodétection, la détection de maladies et le stockage de données.
Introduction
Le calcul de l’ADN utilise un ensemble d’oligonucléotides conçus comme support de calcul. Ces oligonucléotides sont programmés avec des séquences pour s’assembler dynamiquement selon la logique spécifiée par l’utilisateur et répondre à des entrées d’acide nucléique spécifiques. Dans les études de preuve de concept, le résultat du calcul consiste généralement en un ensemble d’oligonucléotides marqués par fluorescence qui peuvent être détectés par électrophorèse sur gel ou lecteurs de plaques de fluorescence. Au cours des 30 dernières années, des circuits de calcul de l’ADN de plus en plus complexes ont été démontrés, tels que diverses cascades logiques numériques, des réseaux de réactions chimiques et des réseaux de neurones1,2,3. Pour aider à la préparation de ces circuits d’ADN, des modèles mathématiques ont été utilisés pour prédire la fonctionnalité des circuits de gènes synthétiques4,5, et des outils informatiques ont été développés pour la conception de séquences d’ADN orthogonales6,7,8,9,10 . Par rapport aux ordinateurs à base de silicium, les avantages des ordinateurs à ADN comprennent leur capacité à s’interfacer directement avec les biomolécules, à fonctionner en solution en l’absence d’alimentation électrique, ainsi que leur compacité et leur stabilité globales. Avec l’avènement du séquençage de nouvelle génération, le coût de la synthèse des ordinateurs à ADN a diminué au cours des deux dernières décennies à un rythme plus rapide que la loi de Moore11. Des applications de ces ordinateurs basés sur l’ADN commencent maintenant à émerger, telles que pour le diagnostic des maladies12,13, pour alimenter la biophysique moléculaire14, et comme plates-formes de stockage de données15.

Figure 1: Mécanisme de déplacement des brins d’ADN médiés par les orteils. L’orteil, δ, est une séquence libre et non liée sur un duplex partiel. Lorsqu’un domaine complémentaire (δ*) est introduit sur un deuxième brin, le domaine δ libre sert de base pour l’hybridation, permettant au reste du brin (ɑ*) de déplacer lentement son concurrent par une réaction réversible de zipping/décompression connue sous le nom de migration de brin. À mesure que la longueur de δ augmente, le ΔG de la réaction vers l’avant diminue et le déplacement se produit plus facilement. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
À ce jour, la majorité des ordinateurs à ADN utilisent un motif bien établi dans le domaine de la nanotechnologie dynamique de l’ADN connu sous le nom de déplacement de brin d’ADN médié par les orteils (TMDSD, Figure 1)16. Ce motif est constitué d’un duplex d’ADN partiellement double brin (dsDNA) affichant de courts surplombs « toehold » (c’est-à-dire 7 à 10 nucléotides (nt)). Les brins d’entrée d’acide nucléique peuvent interagir avec les duplex partiels à travers l’orteil. Cela conduit au déplacement de l’un des brins du duplex partiel, et ce brin libéré peut alors servir d’entrée pour les duplex partiels en aval. Ainsi, TMDSD permet la cascade du signal et le traitement de l’information. En principe, les motifs tmDSD orthogonaux peuvent fonctionner indépendamment dans la solution, ce qui permet un traitement parallèle de l’information. Il y a eu un certain nombre de variations sur la réaction TMDSD, telles que l’échange de brins d’ADN médié par la toehold (TMDSE)17,les orteils « sans fuite » avec des domaines doubles longs18,les orteils dépareillés par séquence19et le déplacement de brin médié par la « poignée »20. Ces principes de conception innovants permettent une énergie et une dynamique TMDSD plus finement ajustées pour améliorer les performances de calcul de l’ADN.
Les circuits de gènes synthétiques, tels que les circuits de gènes transcriptionnels, sont également capables de calculer21,22,23. Ces circuits sont régulés par des facteurs de transcription des protéines, qui activent ou répriment la transcription d’un gène en se liant à des éléments d’ADN régulateurs spécifiques. Par rapport aux circuits à base d’ADN, les circuits transcriptionnels présentent plusieurs avantages. Premièrement, la transcription enzymatique a un taux de rotation beaucoup plus élevé que les circuits d’ADN catalytiques existants, générant ainsi plus de copies de sortie par copie unique d’entrée et fournissant un moyen plus efficace d’amplification du signal. En outre, les circuits transcriptionnels peuvent produire différentes molécules fonctionnelles, telles que des aptamères ou des ARN messagers (ARNm) codant pour des protéines thérapeutiques, en tant que sorties de calcul, qui peuvent être exploitées pour différentes applications. Cependant, une limitation majeure des circuits transcriptionnels actuels est leur manque d’évolutivité. En effet, il existe un ensemble très limité de facteurs de transcription basés sur des protéines orthogonales, et la conception de novo de nouveaux facteurs de transcription protéique reste techniquement difficile et prend beaucoup de temps.

Figure 2: Abstraction et mécanisme du complexe de polymérase « attache » et « cage ». (A et B) Une téther oligonucléotidique est marquée enzymatiquement en une polymérase T7 par la réaction SNAP-tag. Une cage constituée d’un « faux » promoteur T7 avec un porte-à-faux attache-complément lui permet de s’hybrider à l’attache et de bloquer l’activité transcriptionnelle. (C) Lorsque l’opérateur (a*b*) est présent, il se lie à l’orteil sur l’attache oligonucléotidique (ab) et déplace la région b* de la cage, permettant la transcription. Cette figure a été modifiée à partir de Chou et Shih27. Abréviations : RNAP = ARN polymérase. Veuillez cliquer ici pour voir une version agrandie de cette figure.
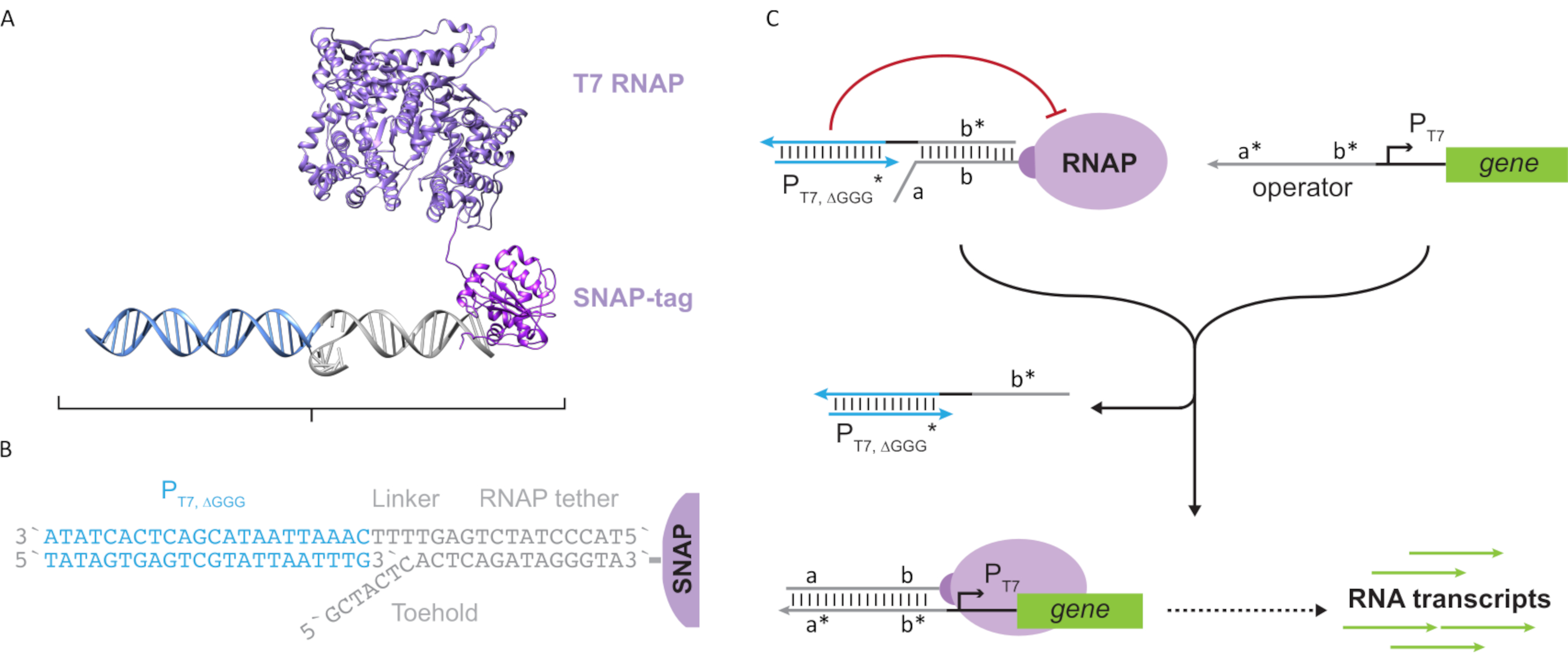{kind=link}
Cet article présente un nouveau bloc de construction pour l’informatique moléculaire qui combine les fonctionnalités des circuits transcriptionnels avec l’évolutivité des circuits à base d’ADN. Ce bloc de construction est un RNAP T7 attaché de manière covalente avec une attache d’ADN simple brin(Figure 2A). Pour synthétiser ce RNAP T7 attaché à l’ADN, la polymérase a été fusionnée à un marqueur SNAP N-terminal24 et exprimée de manière recombinante dans Escherichia coli. Le SNAP-tag a ensuite réagi avec un oligonucléotide fonctionnalisé avec le substrat BG. L’attache oligonucléotidique permet le positionnement d’invités moléculaires à proximité de la polymérase via l’hybridation de l’ADN. L’un de ces invités était un bloqueur transcriptionnel compétitif appelé « cage », qui consiste en un « faux » duplex d’ADN promoteur T7 sans gène en aval(Figure 2B). Lorsqu’elle est liée à l’ARNP via son attache oligonucléotidique, la cage bloque l’activité de la polymérase en surpassant les autres modèles d’ADN pour la liaison À l’ARNP, rendant l’ARNP dans un état « OFF »(Figure 2C).
Pour activer la polymérase à un état « ON », des modèles d’ADN T7 avec des domaines « opérateurs » monocaténaires en amont du promoteur T7 du gène ont été conçus. Le domaine de l’opérateur (c’est-à-dire le domaine a*b* Figure 2C) peut être conçu pour déplacer la cage de l’ARNP via TMDSD et positionner le RNAP proximal au promoteur T7 du gène, initiant ainsi la transcription. Alternativement, des modèles d’ADN ont également été conçus lorsque la séquence de l’opérateur était complémentaire aux brins auxiliaires d’acide nucléique appelés « facteurs de transcription artificiels » (c.-à-d. les brins TFA et TFB à la figure 3A). Lorsque les deux brins sont introduits dans la réaction, ils s’assemblent sur le site de l’opérateur, créant un nouveau domaine pseudo-contigu a*b*. Ce domaine peut ensuite déplacer la cage via TMDSD pour initier la transcription(Figure 3B). Ces brins peuvent être fournis de manière exogène ou produits.

Figure 3: Programmation sélective de l’activité de la polymérase à l’aide d’un activateur de commutation à trois composants. (A) Lorsque les facteurs de transcription (TFA et TFB)sont présents, ils se lient au domaine opérateur en amont du promoteur, formant une pseudo séquence monocaténaire (a*b*)capable de déplacer la cage par déplacement d’ADN médié par l’orteil. (B) Ce domaine a*b* peut déplacer la cage via TMDSD pour initier la transcription. Cette figure a été modifiée à partir de Chou et Shih27. Abréviations : TF = facteur de transcription ; RNAP = ARN polymérase; TMDSD = déplacement de brin d’ADN médié par les orteils. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
L’utilisation de facteurs de transcription à base d’acides nucléiques pour la régulation transcriptionnelle in vitro permet la mise en œuvre évolutive de comportements de circuit sophistiqués tels que la logique numérique, la rétroaction et la cascade de signaux. Par exemple, on peut construire des cascades de portes logiques en concevant des séquences d’acides nucléiques telles que les transcriptions d’un gène en amont activent un gène en aval. Une application qui exploite la cascade et le multiplexage rendus possibles par cette technologie proposée est le développement de circuits informatiques moléculaires plus sophistiqués pour le diagnostic portable et le traitement des données moléculaires. En outre, l’intégration des capacités de calcul moléculaire et de synthèse d’ARN de novo peut permettre de nouvelles applications. Par exemple, un circuit moléculaire peut être conçu pour détecter un ou une combinaison d’ARN définis par l’utilisateur en tant qu’ENTRÉES ET SORTIES D’ARN thérapeutiques ou d’ARNm codant pour des peptides ou des protéines fonctionnels pour des applications médicales au point de service.
Protocole
1. Préparation du tampon
REMARQUE: La préparation du tampon de purification des protéines peut se produire n’importe quel jour; ici, cela a été fait avant de commencer les expériences.
- Préparer un tampon d’lyse/équilibrage contenant 50 mM de tris(hydroxyméthyl)aminométhane (Tris), 300 mM de chlorure de sodium (NaCl), 5 % de glycérol et 5 mM de β-mercaptoéthanol (BME), pH 8. Ajouter 1,5 mL de Tris 1M, 1,8 mL de NaCl 5M, 1,5 mL de glycérol, 25,2 mL d’eau désionisée (ddH2O) dans un tube centrifuge de 50 mL et ajouter 10,5 μL de 14,2 M BME juste avant utilisation.
REMARQUE: Tris peut causer une toxicité aiguë; par conséquent, évitez de respirer sa poussière et évitez le contact avec la peau et les yeux. Le BME est toxique et ne doit être utilisé que dans une hotte aspirante. Il est important d’ajouter le BME en dernier, juste avant la remise en suspension et la lyse cellulaire. Voir le tableau 1 pour la formule tampon de lyse. - Préparer un tampon de lavage (pH 8) contenant 50 mM de Tris, 800 mM de NaCl, 5 % de glycérol, 5 mM de BME et 20 mM d’imidazole. Ajouter 1,5 mL de Tris de 1 M, 4,8 mL de NaCl de 5 M, 1,5 mL de glycérol et 22,2 mL de ddH2O dans un tube de centrifugeuse de 50 mL. Juste avant utilisation, ajouter 7 μL de 14,2 M BME et 200 μL de 2 M d’imidazole à 20 mL de la solution ci-dessus.
REMARQUE: Pour prévenir la toxicité aiguë due à l’imidazole, utilisez un équipement de protection individuelle. Il est important d’ajouter le BME et l’imidazole en dernier, juste avant de laver la protéine hors de la colonne. Voir le tableau 2 pour la formule tampon de lavage. - Préparer un tampon d’élution (pH8) contenant 50 mM de Tris, 800 mM de NaCl, 5 % de glycérol, 5 mM de BME et 200 mM d’imidazole. Ajouter 0,5 mL de Tris 1 M, 1,6 mL de NaCl 5 M, 0,5 mL de glycérol et 6,4 mL de ddH2O dans un tube centrifuge de 15 mL. Juste avant utilisation, ajouter 3,5 μL de 14,2 M BME et 1 mL de 2 M d’imidazole à 10 mL de la solution ci-dessus.
REMARQUE: Il est important d’ajouter le BME et l’imidazole en dernier, juste avant d’éluer la protéine hors de la colonne. Voir le tableau 3 pour la formule du tampon d’élution. - Préparer 2x tampon de stockage (à mélanger 1:1 avec du glycérol) contenant 100 mM de Tris, 200 mM de NaCl, 40 mM de BME et 2 mM d’acide éthylènediaminetétraacétique (EDTA), soit 0,2 % d’un tensioactif non ionique (voir la Table des matières). Préparer 50 mL du tampon de stockage en ajoutant 5 mL de 1 M Tris, 2 mL de 5 M NaCl, 42,56 mL de ddH2O, 200 μL de 0,5 M EDTA, 100 μL du tensioactif non ionique dans un tube centrifuge de 50 mL. Mélanger jusqu’à ce que la solution soit homogène, filtrer le tampon de stockage à travers un filtre à seringue de 0,2 μm et ajouter 140,8 μL de BME à la solution ci-dessus avant utilisation.
REMARQUE: Pour éviter la toxicité aiguë due à l’EDTA, évitez de respirer sa poussière et évitez le contact cutané et oculaire. Il est important d’ajouter le BME en dernier et de mélanger tout le tampon de stockage 1: 1 avec du glycérol, juste avant de stocker la protéine purifiée. Reportez-vous au tableau 4 pour connaître la formule de la mémoire tampon de stockage.
2. Croissance culturelle du jour au lendemain : Jour 1
- Préparer 1 000x de kanamycine en dissolvant 500 mg de kanamycine dans 10 mL de ddH2O.
REMARQUE: Utilisez un équipement de protection individuelle pour prévenir la toxicité aiguë due à la kanamycine. - Ajouter 20 μL du bouillon de kanamycine 1 000x à 20 mL de bouillon de lysogénie. À l’aide d’une pointe de pipette stérile, piquez un bouillon de glycérol BL21 E. coli transformé, puis inoculez la culture en introduisant la pointe dans le bouillon du milieu de croissance.

Figure 4: Carte plasmidique pour SNAP T7 RNAP. Le plasmide code un RNAP T7 contenant une balise histidine N-terminale (6x His) et un domaine SNAP-tag (SNAP T7 RNAP) sous un répresseur lac (lacI) sur une dorsale pQE-80L. D’autres caractéristiques incluent la résistance à la kanamycine (KanR) et les gènes de résistance au chloramphénicol (CmR). Abréviation : RNAP = ARN polymérase. Veuillez cliquer ici pour voir une version agrandie de cette figure.
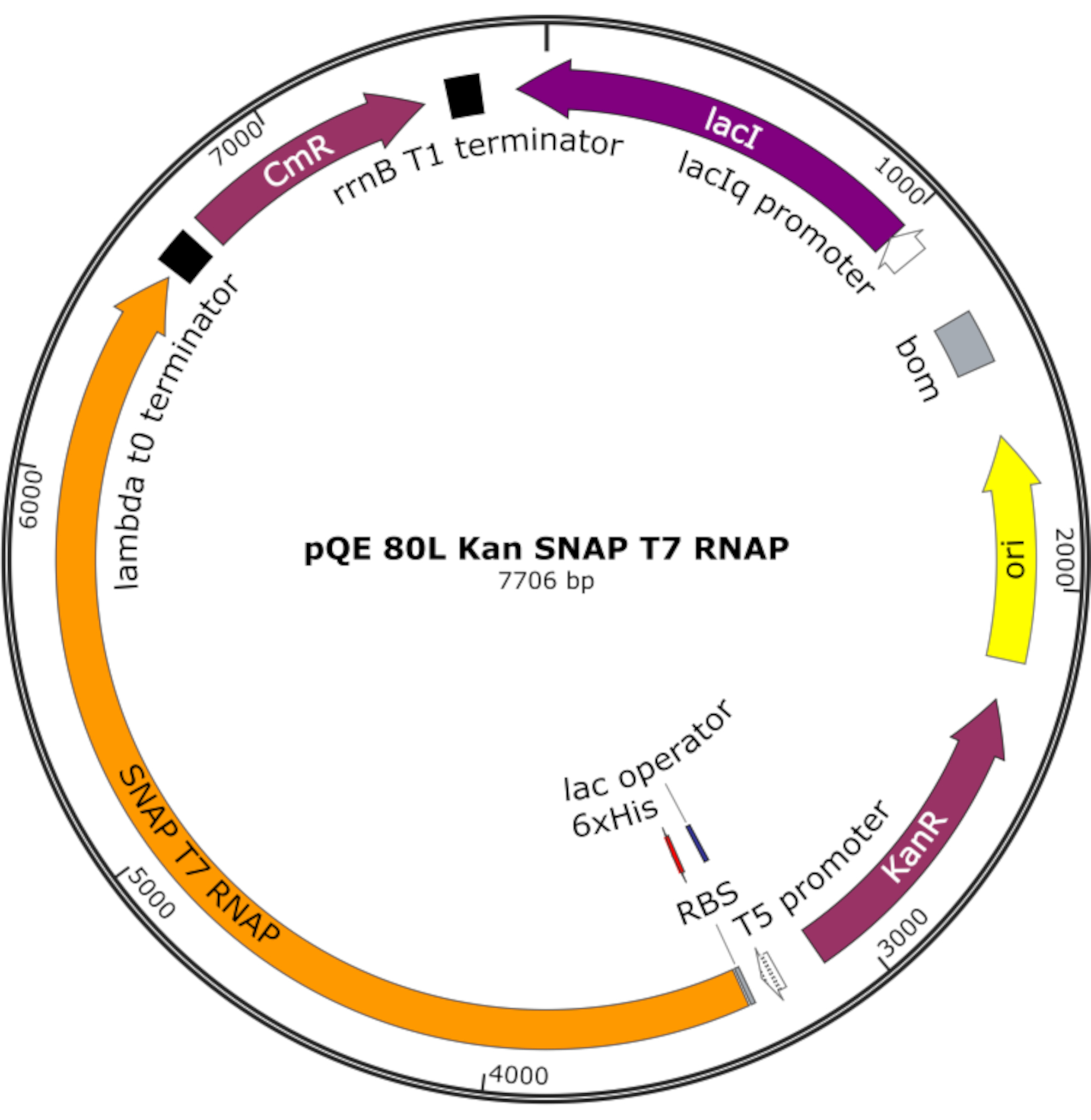{kind=link}
REMARQUE: Le plasmide code un RNAP T7 contenant une étiquette d’histidine N-terminale et un domaine de balise SNAP (SNAP T7 RNAP), ainsi qu’un gène de résistance à la kanamycine sous un squelette pQE-80L (Figure 4)25.
- Encore une fois, ajoutez 20 μL du bouillon de kanamycine 1 000x dans une fiole de culture séparée contenant 20 mL de bouillon de lysogénie et incubez-la comme témoin.
- Incuber les deux échantillons (des étapes 2.2 et 2.3) pendant la nuit pendant 12 à 18 h à 37 °C, tout en tournant à 10 × g.
3. Croissance et induction cellulaires : Jour 2
- Inoculer 400 mL de bouillon de lysogénie contenant 400 μL de bouillon de kanamycine avec 4 mL de culture de croissance pendant la nuit à partir de l’étape 2.4. Incuber les flacons de culture à 37 °C, tout en tournant à 10 × g.
- Une fois que la culture a atteint une densité optique (OD) à 600 nm d’environ 0,5, prélever 1 mL d’échantillon de la fiole de croissance comme témoin. Conserver l’échantillon de contrôle à 4 °C.
- Induire les cellules avec de l’isopropyle β-D-1-thiogalactopyranoside (IPTG) en ajoutant 40 μL de 1M IPTG par 100 mL de culture pour atteindre une concentration finale de 0,4 mM IPTG. Incuber l’échantillon pendant 3 h à 37 °C, en tournant à 10 × g,puis faire tourner la culture induite à 8 000 × g pendant 10 min pour granuler les cellules. Retirez le surnageant et conservez la pastille à -20 °C jusqu’à une utilisation ultérieure.
REMARQUE: Pour éviter la toxicité aiguë due à l’IPTG, évitez de respirer sa poussière et évitez le contact avec la peau et les yeux. Si nécessaire, vous pouvez mettre l’expérience en pause ici et continuer le lendemain.
4. Lyse cellulaire, purification des protéines : Jour 3
- Remettez en suspension la pastille de cellule stockée avec 10 mL de tampon de lyse sur de la glace et tourbillonnez doucement pour vous assurer que la pastille entière est remise en suspension. Ensuite, pipettez 1 mL d’échantillon dans dix tubes de 1,5 mL qui sont conservés sur de la glace.
- Soniquer chaque échantillon à un réglage d’amplitude de « 1 », pulsé pendant 2 s avec un rapport cyclique de 50% sur une période de 30 s. Avant et après chaque échantillon, nettoyez l’embout de sonication avec 70% d’éthanol et ddH2O. Conservez tous les échantillons sur de la glace pendant et après la sonication.
REMARQUE: Gardez 70% d’éthanol à l’abri de la chaleur et de la flamme nue. - Équilibrer une colonne de spin de purification à l’acide nitrilotriacétique chargé de nickel (Ni-NTA) à une température de fonctionnement de 4 °C. Placez/rangez la colonne à 4 °C et conservez-la sur la glace pendant l’utilisation.
- Centrifuger les dix échantillons de 1 mL à 15 000 × g pendant 20 min à 4 °C. Pipettez soigneusement le surnageant contenant le RNAP recombinant sans perturber la pastille. Si nécessaire, utilisez un tampon d’équilibrage supplémentaire pour régler le volume total à ≥ 6 mL.
- Retirez délicatement la languette inférieure de la colonne de rotation Ni-NTA pour permettre l’écoulement dans la colonne. Placez la colonne dans un tube de centrifugeuse et gardez-la sur la glace.
REMARQUE: Utilisez un tube de centrifugeuse de 50 mL avec les colonnes de spin Ni-NTA de 3 mL. - Centrifuger la colonne à 700 × g et 4 °C pendant 2 min pour retirer le tampon de stockage. Équilibrez la colonne en ajoutant 6 mL de tampon d’équilibrage à la colonne. Laissez le tampon pénétrer complètement dans le lit de résine.
- Retirer le tampon d’équilibrage de la colonne par centrifugation à 700 × g et 4 °C pendant 2 min. Avant d’ajouter l’extrait de cellule préparé à la colonne, placez un bouchon inférieur sur la colonne pour éviter de perdre du produit. Ensuite, ajoutez l’extrait de cellule à la colonne et mélangez sur un mélangeur à agitateur orbital pendant 30 min à 4 °C.
- Retirez le bouchon inférieur de la colonne et placez la colonne dans un tube de centrifugeuse de 50 mL étiqueté à travers. Centrifuger la colonne à 700 × g pendant 2 min pour recueillir le débit.
- Ajouter 6 mL de tampon de lavage à la colonne pour laver la résine. Centrifuger la colonne à 700 × g pendant 2 min pour recueillir la fraction dans un nouveau tube de centrifugeuse étiqueté lavage 1. Répétez cette étape deux fois de plus pour un total de 3 fractions distinctes, et collectez les fractions dans des tubes centrifuges séparés(lavage 2 et lavage 3).
- Ajouter 3 mL de tampon d’élution pour éluer les protéines marquées His de la résine. Centrifuger la colonne à 700 × g pendant 2 min pour recueillir la fraction dans un nouveau tube de centrifugeuse étiqueté éluat 1. Répétez cette étape deux fois de plus pour un total de 3 fractions distinctes, et collectez les fractions dans des tubes centrifuges séparés(éluer 2 et éluer 3).
- Mélanger les éluats et effectuer le dessalement pour éliminer les sels de la solution protéique.
- Pipette 15 mL de 0,05 % p/v polysorbate 20 sur une unité de filtration centrifuge de 100 kDa. Centrifuger à 4 000 × g pendant 40 min et jeter le flux.
- Utilisez le filtre revêtu pour concentrer les éluats 1, 2 et 3 (9 mL d’éluat de protéine total + 6 mL de tampon de stockage) à environ 1 500 μL. Centrifugez le filtre à 3 220 × g pendant 20 min et lavez doucement la membrane à la pipette pour éviter les précipitations.
- Diluer l’échantillon à 15 mL avec un tampon de stockage. Effectuez un échange de mémoire tampon à l’aide de la mémoire tampon de stockage 1:1 000 en répétant l’étape 4.11.2 deux fois de plus.
- Quantifier la protéine purifiée en mesurant l’absorbance de la fraction à 280 nm. Vider le spectrophotomètre avec un tampon de stockage (2x tampon de stockage à 4 °C). Mélangez doucement l’échantillon des éluats combinés et mesurez son absorbance.
REMARQUE: Effectuez trois lectures distinctes à des dilutions 1x, 10x et 50x de l’échantillon de protéines pour faire la moyenne et quantifier la protéine. Diluer les échantillons dans un tampon de stockage. - Ajustez les échantillons de protéines à 100 μM à l’aide d’un tampon de stockage 2x. Diluer l’échantillon ajusté 1:1 en volume avec 100% de glycérol. Conserver la solution protéique obtenue à -80 °C.
5. Analyse par électrophorèse sur gel de dodécylsulfate de sodium-polyacrylamide (SDS-PAGE) du produit protéique : Jour 3
- Exécutez un gel SDS-PAGE pour l’analyse des protéines. Mélanger 9 μL de l’échantillon avec 3 μL de 4x colorant de charge protéique de dodécylsulfate de lithium (LDS). Chauffer les échantillons à 95 °C pendant 10 min.
- Chargez les échantillons sur une configuration de gel Bis-Tris SDS-PAGE à 4-12%. Chargez l’échelle protéique dans le puits 1, puis avec des échantillons (de gauche à droite): écoulement, lavage 1, lavage 2, lavage 3, élution 1, élution 2, élution 3 et élution dessalée totale.
REMARQUE : Le tableau 5 contient un exemple de table de chargement pour le gel SDS-PAGE. - Exécutez les échantillons de gel chargés dans un tampon d’acide éthanesulfonique (MES)2(N-morpholino)pendant 35 min à 200 V. Rincez le gel dans un bac propre trois fois pendant 10 min chacun en utilisant 200 mL de ddH2O, avec une agitation douce pour éliminer toute FDS de la matrice de gel.
REMARQUE: Portez un équipement de protection individuelle pour éviter la toxicité aiguë due au MES. - Tacher le gel avec 20 mL de bleu coomassie et incuber le gel pendant la nuit à température ambiante avec une légère agitation. Décolorez le gel deux fois pendant 1 h chacun avec 200 mL de ddH2O avec une légère agitation sur un agitateur orbital.
REMARQUE: Laver le gel pendant une période plus longue ou remplacer fréquemment l’eau augmentera la sensibilité. De plus, placer un tissu d’essuyage plié à tâche délicate dans le récipient pour absorber l’excès de colorant accélérera le processus de décoloration.
6. Vérification fonctionnelle de SNAP T7 RNAP par transcription in vitro
REMARQUE: Ce protocole utilise un modèle d’ADN, qui code pour l’aptamère fluorescent de l’ARN du brocoli et permet l’utilisation de la fluorescence pour surveiller la cinétique de transcription sur un lecteur de plaque de fluorescence.
- Mettre en place trois réactions de transcription in vitro (IVT) pour comparer l’activité du SNAP T7 RNAP avec le RNAP T7 de type sauvage (WT) provenant d’une source commerciale et d’un contrôle tampon uniquement. Réglez le volume de chaque réaction à 20 μL.
- Préparer la réaction SNAP T7 RNAP IVT en mélangeant 2 μL de tampon de transcription 10x, 0,4 μL de mélange de 25 mM de ribonucléoside triphosphate (rNTP), 5 μL de 500 nM de gabarit d’ADN, 2 μL de 500 nM SNAP T7 RNAP et 10,6 μL de ddH2O.
- Préparer la réaction WT RNAP IVT en mélangeant 2 μL de tampon de transcription 10x, 0,4 μL de mélange rNTP de 25 mM, 5 μL de modèle d’ADN 500 nM, 2 μL de WT T7 RNAP et 10,6 μL de ddH2O.
- Préparer la réaction IVT tampon uniquement en mélangeant 2 μL de tampon de transcription 10x, 0,4 μL de mélange rNTP de 25 mM, 5 μL de modèle d’ADN 500 nM et 12,6 μL de ddH2O.
REMARQUE: Ajoutez le RNAP en dernier, en gardant les échantillons sur la glace jusqu’à son introduction. Les tableaux 6, 7et 8 contiennent les formules de réaction IVT.
- Surveiller la cinétique de transcription sur un lecteur de plaque de fluorescence pendant 2 h à des intervalles de 2 min à 37 °C en utilisant une longueur d’onde d’excitation de 470 nm et une longueur d’onde d’émission de 512 nm.
7. Préparation d’oligonucléotides modifiés par BG: Jour 1
- Dissoudre l’oligonucléotide avec modification de la3'-amine dans la ddH2 O jusqu’à une concentration finale de 1 mM. Étiquetez ce S1.
- Mélanger 25 μL de 1 M de bicarbonate de sodium (NaHCO3),284 μL de sulfoxyde de diméthyle à 100 % (DMSO), 125 μL de S1 (stock d’oligonucléotides) et 66 μL de 50 mM d’ester BG-N-hydroxysuccinimide (NHS) (BG-GLA-NHS) dilué avec du DMSO, ajuster le volume à 500 μL et incuber toute la nuit à température ambiante à 100 × g.
REMARQUE: Gardez le DMSO à l’écart de la chaleur et des flammes car il s’agit d’un liquide combustible. Le tableau 9 contient la formule de réaction pour la conjugaison BG à l’oligonucléotide.
- Mélanger 25 μL de 1 M de bicarbonate de sodium (NaHCO3),284 μL de sulfoxyde de diméthyle à 100 % (DMSO), 125 μL de S1 (stock d’oligonucléotides) et 66 μL de 50 mM d’ester BG-N-hydroxysuccinimide (NHS) (BG-GLA-NHS) dilué avec du DMSO, ajuster le volume à 500 μL et incuber toute la nuit à température ambiante à 100 × g.
8. Précipitation éthanol/acétone du conjugué BG-oligonucléotide : Jour 2
- Centrifuger le produit de l’étape 7.1.1. à 13 000 × g pendant 5 min. Transférer soigneusement le surnageant dans un tube frais et jeter tout BG précipité. Divisez la réaction en deux aliquotes égales de 250 μL pour éviter tout débordement et effectuez les étapes suivantes sur les deux aliquotes.
- Ajouter 1/10ème du volume d’acétate de sodium 3 M (25 μL), suivi de 2,5x le volume en éthanol à 100% (625 μL). Incuber à -80 °C pendant 1 h.
REMARQUE: Utilisez un équipement de protection individuelle lorsque vous manipulez à la fois de l’acétate de sodium (peut causer une irritation des yeux, de la peau, des voies digestives et respiratoires) et de l’éthanol (extrêmement inflammable, provoque une irritation au contact). Si nécessaire, mettez l’expérience en pause ici et continuez le lendemain. - Placez les tubes dans la centrifugeuse et marquez le bord extérieur. Centrifuger les tubes à 17 000 × g pendant 30 min à 4 °C.
REMARQUE: La pastille d’oligonucléotide apparaîtra sur le bord marqué du tube. - Sans déranger la pastille, jetez le surnageant. Complétez avec 750 μL d’éthanol réfrigéré à 70 % et faites tourner à 17 000 × g pendant 10 min à 4 °C.
- Sans déranger la pastille, jetez le surnageant. Complétez avec 750 μL d’acétone à 100 % et faites tourner à 17 000 × g pendant 10 min à 4 °C.
REMARQUE: Utilisez un équipement de protection individuelle lorsque vous manipulez de l’acétone, car elle est extrêmement inflammable et provoque une irritation au contact. - Avec le couvercle du tube ouvert, sécher à l’air libre pendant 5 min pour éliminer tout excès d’acétone par évaporation. Dissoudre à nouveau l’oligonucléotide dans 250 μL de 1x tampon Tris-EDTA (TE) pour produire une solution d’oligonucléotide BG d’environ 850 μM.
- Répétez les étapes 8.2 à 8.6 et dissolvez à nouveau dans 70 μL de tampon TE 1x. Étiquetez ce S2.
9. Nettoyage des bg-oligonucléotides par chromatographie par filtration sur gel
- Suspendre la matrice en inversant vigoureusement les colonnes plusieurs fois; retirez le capuchon supérieur et enclenchez l’extrémité inférieure de la colonne. Placez la colonne dans un tube de centrifugeuse de 1,5 mL et centrifugez le tube à 1 000 × g pendant 1 min à température ambiante. Jetez le tampon élué et le tube de collecte.
REMARQUE: Il est important d’empêcher la formation de vide. Utilisez immédiatement les colonnes préparées. - Placez les colonnes emballées dans des tubes de centrifugeuse propres de 1,5 mL. Ajouter 300 μL de tampon TE 1x au centre du lit de colonne et centrifuger à 1 000 × g pendant 2 min pour échanger la solution tampon. Encore une fois, jetez le tampon élué et le tube de collecte.
- Placez les colonnes échangeuses tampons dans des tubes centrifuges propres de 1,5 mL. Appliquer jusqu’à 75 μL d’échantillon au centre du lit. Tourner à 1 000 × g pendant 4 min.
REMARQUE: Ne pas déranger le lit ou toucher les côtés de la colonne; le point le plus élevé du support de gel doit pointer vers le rotor extérieur. - Recueillir l’éluat du tube de collecte, car il contient l’acide nucléique purifié. Pour quantifier l’échantillon, mesurer son absorbance à 260 nm; étiquetez ce S3.
REMARQUE: Notez la longueur du chemin utilisé dans la mesure et calculez la concentration en utilisant la loi de Beer-Lambert.
10. Analyse PAGE dénaturante du conjugué BG-oligonucléotide
- Jetez un gel 18% Tris-borate-EDTA (TBE)-Urea PAGE. Dissoudre 4,8 g d’URÉE, 4,5 mL d’acrylamide à 40 % (19:1) et 1 mL de 10x TBE dans 2,8 mL de ddH2O; ajouter 5 μL de tétraméthyléthylènediamine (TEMED) et bien mélanger. Répéter avec 100 μL de persulfate d’ammonium (APS) à 10 %. Versez la solution dans une cassette de gel vide et laissez la polymérisation pendant 40 min.
REMARQUE: Utilisez l’équipement de protection individuelle approprié lors de la manipulation de l’urée (provoque une irritation des yeux et de la peau), de l’acrylamide (toxique et cancérigène) et du TEMED (toxique, inflammable, corrosif). Le tableau 10 contient la formule de réaction d’un gel de polyacrylamide TBE-UREA à 18 %. - Micro-ondes 500 mL de tampon TBE (0,5x) pendant 2 min et 30 s ou jusqu’à ~70 °C et verser dans un appareil à gel. Préparer un colorant de charge de formamide (dénaturant) contenant 95% de formamide + 1 mM d’EDTA et de bleu de bromophénol. Mélanger le colorant de chargement avec chaque échantillon et charger le mélange sur le gel de polyacrylamide.
REMARQUE: Utilisez l’équipement de protection individuelle approprié lors de la manipulation du formamide car il est cancérigène. Le tableau 11 contient un exemple de tableau de chargement de gel. - Faites couler le gel à 270 V pendant 35 min, ou jusqu’à ce que le front de teinture migre vers la fin. Placez le gel dans une boîte à gel et colorez avec du colorant cyanine pour les acides nucléiques pendant 15 minutes à température ambiante avant l’imagerie.
REMARQUE: Utilisez l’équipement de protection individuelle approprié lorsque vous manipulez un colorant cyaniné car il est combustible.
11. Conjugaison de l’oligonucléotide à l’analyse SNAP T7 RNAP et PAGE
- Préparer les réactifs pour le couplage à l’échelle analytique de l’oligonucléotide BG à l’ARNP SNAP T7 : effectuer 9 dilutions d’oligo d’ADN simple brin (ADNSS) avec ddH2O pour créer des rapports oligo:RNAP allant de 5:1 à 1:5. Diluer le stock de protéines à 50 μM.
NOTE: Des exemples de ratios peuvent être trouvés dans le tableau 12; ces rapports sont calculés à l’aide d’une concentration rNAP de 50 μM. - Pour chaque dilution d’oligo d’ADNSs, faire 10 μL du mélange réactionnel contenant 2 μL de tampon SNAP, 4 μL d’oligonucléotide BG et 4 μL de SNAP T7 RNAP.
REMARQUE : Le tableau 13 contient des formules de réaction pour la réaction d’étiquetage de la balise SNAP.- Préparer deux autres échantillons témoins : 1) un contrôle RNAP en remplaçant BG-oligonucléotide par ddH2O ; 2) un contrôle de l’ADN en remplaçant SNAP T7 RNAP par ddH2O (pour la plus faible concentration d’oligonucléotides de SNAP T7 RNAP). Incuber tous les échantillons à température ambiante pendant 1 h et conserver sur la glace jusqu’à ce que nécessaire.
- Mettre en place onze réactions de 10 μL en ajoutant 2 μL de chaque échantillon à 4 μL de tampon SNAP et 2 μL de colorant à charge protéique, et chauffer à 70 °C pendant 10 min. Chargez 2 μL de chaque échantillon sur le gel protéique Bis-Tris à 4-12 % et effectuez l’électrophorèse sur gel sur glace à 200 V pendant 35 min.
NOTE: Le tableau 14 contient des formules de réaction pour les échantillons de chargement de gel.- Lavez les FDS via 3x échange d’eau sur un agitateur, chaque lavage dure 10 minutes chacun. Coloration avec un colorant cyanine pour les acides nucléiques pendant 15 min avant l’imagerie. Tacher à nouveau le gel en utilisant 20 mL de teinture bleue Coomassie pendant 1 h. Décolorer avec ddH2O pendant 1 h (ou pendant la nuit) avant l’imagerie.
REMARQUE: Dans le gel, l’une des réactions produira la polymérase la plus attachée avec le moins d’excès d’oligonucléotide BG libre; c’est le ratio optimal.
- Lavez les FDS via 3x échange d’eau sur un agitateur, chaque lavage dure 10 minutes chacun. Coloration avec un colorant cyanine pour les acides nucléiques pendant 15 min avant l’imagerie. Tacher à nouveau le gel en utilisant 20 mL de teinture bleue Coomassie pendant 1 h. Décolorer avec ddH2O pendant 1 h (ou pendant la nuit) avant l’imagerie.
- Préparer des réactifs pour le couplage à l’échelle préparative BG-oligonucléotide à SNAP T7 RNAP. Effectuer la réaction de couplage avec le rapport optimal trouvé dans l’échelle analytique.
REMARQUE: Minimiser l’exposition aux protéines à la température ambiante en plaçant la protéine sur la glace lorsqu’elle n’est pas utilisée.
12. Purification du SNAP-T7 attaché aux oligonucléotides à l’aide de colonnes d’échange d’ions
- Suivez les instructions du fabricant pour la configuration du tube s’il s’écarte des instructions répertoriées ici. Préparez un tampon de purification avec un pH supérieur au point isoélectrique de la protéine.
NOTE: Pour l’exemple de protéine dans ce protocole, un tampon de purification de 10 mM de tampon phosphate de sodium (pH 7) a été utilisé.- Préparer 1 000 μL de tampon d’élution contenant des concentrations finales de 50 mM de Tris et de 0,5 M de NaCl. Mélanger 50 μL de 1 M Tris, 100 μL de 5 M NaCl et 850 μL de ddH2O.
NOTE: Le tableau 15 contient la formule de réaction pour le tampon d’élution.
- Préparer 1 000 μL de tampon d’élution contenant des concentrations finales de 50 mM de Tris et de 0,5 M de NaCl. Mélanger 50 μL de 1 M Tris, 100 μL de 5 M NaCl et 850 μL de ddH2O.
- Placer une colonne dans un tube de centrifugeuse de 2 mL et laver avec un tampon de purification à 2 000 × g pendant 15 min, ou jusqu’à ce que tout le tampon ait été élué. Jetez le tampon élué.
- Diluer chaque échantillon avec un tampon de purification à un rapport tampon de purification:échantillon de 3:1 et charger l’échantillon dans la colonne 400 μL à la fois. Tourner à 2 000 × g pendant 10 min, ou jusqu’à ce que tout le tampon ait été élué. Collectez le flux traversant et étiquetez-le comme flow-through.
- Ajouter 400 μL de tampon de purification au centre de la colonne. Tourner à 2 000 × g pendant 15 min, ou jusqu’à ce que tout le tampon ait été élué. Collectez le flux et étiquetez-le comme lavage 1. Répétez deux fois de plus pour le lavage 2 et le lavage 3.
- Ajouter 50 μL de tampon d’élution au centre de la colonne. Tourner à 2 000 × g pendant 5 min, ou jusqu’à ce que tout le tampon ait été élué. Collectez le flux et étiquetez-le comme éluant 1. Répétez deux fois de plus pour éluer 2 et éluer 3.
- La piscine élue 1, 2 et 3 (étiquetez cette éluat totale),laissant une petite fraction de chaque éluat pour le gel, et mesurent l’absorbance à 260 nm (A260) et 280 nm (A280). Après la mesure, ajouter le glycérol à un rapport de 1:1 et conserver à -20 °C jusqu’à une utilisation ultérieure.
- Utilisez une unité de filtre centrifuge (0,5 mL; 30 kDa) pour échanger un tampon total élué avec 2x tampon de stockage (~ 1:100) (étiquetez ce produit). Mesure A260/280 à nouveau. Ajouter le glycérol à un rapport de 1:1 et conserver à -20 °C jusqu’à nouvel usage.
- Chargez chaque éluat : écoulement, lavage 1-3, éluat total et produit dans un gel Bis-Tris SDS-PAGE à 4-12%, accompagné d’une échelle à protéines. Exécuter à 200 V pendant 35 min, ou jusqu’à ce que le front de teinture migre vers la fin.
13. Démonstration du contrôle à la demande de l’activité de l’ARN polymérase attachée
- Préparer 5x tampon de recuit contenant 25 mM de Tris, 5 mM d’EDTA et 25 mM de chlorure de magnésium (MgCl2). Mélanger 2,4 μL de chaque gabarit (1 μM) avec 5 μL de tampon de recuit et 14,2 μL de ddH2O pour former 25 μL de cage d’ADNds de 1 μM. Incuber cette solution à 75 °C pendant 2 min. De même, recuit les brins de sens et d’antisens du modèle d’ADN d’aptamère vert promoteur et malachite. Préparer une solution de 1mM d’oxalate vert de malachite.
REMARQUE: Le tableau 16 contient la formule de réaction pour 5x tampon de recuit, le tableau 17 contient la formule de réaction pour le recuit de deux modèles d’ADNSs. - Incuber le SNAP T7 RNAP attaché avec la cage dsDNA dans un rapport molaire de 1:5 à température ambiante pendant 15 min jusqu’à une concentration finale de 500 nM RNAP. Garder sur la glace jusqu’à ce que nécessaire.
- Préchauffez le lecteur de plaques à 37 °C. Mettre en place trois réactions IVT de 25 μL sur la glace
- Mettre en place une réaction contenant le SNAP T7RNAP en cage avec des facteurs de transcription d’acides nucléiques. Mélanger 2,5 μL de 10x tampon IVT, 1 μL de mélange rNTP de 25 mM, 1 μL de vert malachite 1 mM, 2,5 μL du mélange RNAP-cage, 2,5 μL chacun des brins d’oligonucléotides du facteur de transcription A et B de 1 μM, et 3 μL de 1 mM de gabarit d’aptamère vert malachite dans 10 μL de ddH2O.
- Mettre en place une réaction contenant le SNAP T7RNAP en cage sans facteurs de transcription des acides nucléiques. Mélanger 2,5 μL de 10x tampon IVT, 1 μL de mélange rNTP de 25 mM, 1 μL de vert malachite 1 mM, 2,5 μL du mélange RNAP-cage et 3 μL de gabarit d’aptamère vert malachite de 1 mM dans 15 μL de ddH2O.
- Configurez une réaction contenant uniquement un tampon. Mélanger 2,5 μL de 10x tampon IVT, 1 μL de mélange rNTP de 25 mM, 1 μL de vert malachite 1 mM et 3 μL de 1 mM de gabarit d’aptamère vert malachite dans 17,5 μL de ddH2O.
NOTE : Le tableau 18 contient une référence générale pour les réactions de transcription in vitro.
- Transférer chaque réaction sur une plaque de 384 puits. Surveiller la transcription de l’aptamère vert malachite sur un lecteur de plaque de fluorescence pendant 2 h à 37 °C et avec une excitation de 610 nm et une émission de 655 nm. Une fois terminé, gardez la plaque sur la glace jusqu’à ce que nécessaire.
- Tampon micro-ondes 0,5x TBE pendant 2 min 30 s ou jusqu’à ~70 °C. Faites fonctionner les produits d’ARN de chaque puits dans un gel de polyacrylamide TBE-urée dénaturant à 12% dans le tampon chauffé 0,5x TBE à 280 V pendant 20 min, ou jusqu’à ce que le front de teinture atteigne la fin. Tacher le gel avec une coloration d’acide nucléique de colorant cyanine pendant 10 minutes sur un agitateur orbital avant l’imagerie.
NOTE: Le tableau 19 contient la formule de réaction pour un gel dénaturant 12% TBE-Urea PAGE.
Résultats

Figure 5: Analyse SDS-PAGE de l’expression snap T7 RNAP et du test de transcription in vitro. (A) Analyse de purification de la protéine SNAP T7 RNAP, poids moléculaire SNAP T7 RNAP: 119.4kDa. FT = écoulement à partir de la colonne, W1 = fractions d’élution du tampon de lavage contenant des impuretés, E1-3 = fractions d’élution c...
Discussion
Cette étude démontre une approche inspirée de la nanotechnologie de l’ADN pour contrôler l’activité de l’ARN polymérase T7 en couplant de manière covalente un RNAP T7 recombinant marqué N-terminal SNAP avec un oligonucléotide fonctionnalisé BG, qui a ensuite été utilisé pour programmer les réactions TMDSD. De par sa conception, la balise SNAP a été positionnée à la terminaison N de la polymérase, car la terminaison C de l’ARNR T7 de type sauvage est enfouie dans le noyau de la structure protéi...
Déclarations de divulgation
Il n’y a pas d’intérêts financiers concurrents à déclarer par l’un des auteurs.
Remerciements
L.Y.T.C reconnaît le généreux soutien du Fonds-exploration Nouvelles frontières en recherche (NFRF-E), de la subvention de découverte du Conseil de recherches en sciences naturelles et en génie du Canada (CRSNG) et de l’initiative Medicine by Design de l’Université de Toronto, qui reçoit un financement du Fonds d’excellence en recherche Canada First (CFREF).
matériels
| Name | Company | Catalog Number | Comments |
| 0.5% polysorbate 20 (TWEEN 20) | BioShop | TWN510.5 | |
| 0.5M ethylenediaminetetraacetic acid (EDTA) | Bio Basic | SD8135 | |
| 10 mM sodium phosphate buffer (pH 7) | Bio Basic | PD0435 | Tablets used to make 10 mM buffer |
| 10% ammonium persulfate (APS) | Sigma Aldrich | A3678-100G | |
| 100 kDa Amicon Ultra-15 Centrifugal Filter Unit | Fisher Scientific | UFC910008 | |
| 100% acetone | Fisher Chemical | A18P4 | |
| 100% ethanol (EtOH) | House Brand | 39752-P016-EAAN | |
| 10x in vitro transcription (IVT) buffer | New England Biolabs | B9012 | |
| 10x Tris-Borate-EDTA (TBE) buffer | Bio Basic | A0026 | |
| 1M Isopropyl β- d-1-thiogalactopyranoside (IPTG) | Sigma Aldrich | I5502-1G | |
| 1M sodium bicarbonate buffer | Sigma Aldrich | S6014-500G | |
| 1M Tris(hydroxymethyl)aminomethane (Tris) | Sigma Aldrich | 648311-1KG | |
| 1X Tris-EDTA (TE) buffer | ThermoFisher | 12090015 | |
| 2M imidazole | Sigma Aldrich | 56750-100G | |
| 2-mercaptoethanol (BME) | Sigma Aldrich | M3148 | |
| 3M sodium acetate | Bio Basic | SRB1611 | |
| 40% acrylamide (19:1) | Bio Basic | A00062 | |
| 4x LDS protein sample loading buffer | Fisher Scientific | NP0007 | |
| 5M sodium chloride (NaCl) | Bio Basic | DB0483 | |
| 5mM dithiothreitol (DTT) | Sigma Aldrich | 43815-1G | |
| 6x gel loading dye | New England Biolabs | B7024S | |
| agarose B powder | Bio Basic | AB0014 | |
| BG-GLA-NHS | New England Biolabs | S9151S | |
| BL21 competent E. coli | Addgene | C2530H | |
| BLUeye prestained protein ladder | FroggaBio | PM007-0500 | |
| bromophenol blue | Bio Basic | BDB0001 | |
| coomassie blue (SimplyBlue SafeStain) | ThermoFisher | LC6060 | |
| cyanine dye (SYBR Gold nucleic acid gel stain) | Fisher Scientific | S11494 | |
| cyanine dye (SYBR Safe nucleic acid gel stain) | Fisher Scientific | S33102 | |
| dry dimethyl sulfoxide (DMSO) | Fisher Scientific | D12345 | |
| formamide | Sigma Aldrich | F9037-100ML | |
| glycerol | Bio Basic | GB0232 | |
| kanamycin sulfate | BioShop | KAN201.5 | |
| lysogeny broth | Sigma Aldrich | L2542-500ML | |
| malachite green oxalate | Sigma Aldrich | 2437-29-8 | |
| N,N,N'N'-Tetramethylethane-1,2-diamine (TEMED) | Sigma Aldrich | T9281-25ML | |
| NuPAGE MES SDS running buffer (20x) | Fisher Scientific | LSNP0002 | |
| NuPAGE Novex 4-12% Bis-Tris gel 1.0 mm 12-well | Life Technologies | NP0322BOX | |
| oligonucleotide (cage antisense) | IDT | N/A | TATAGTGAGTCGTATTAATTTG |
| oligonucleotide (cage sense) | IDT | N/A | TCAGTCACCTATCTGTTTCAAA TTAATACGACTCACTATA |
| oligonucleotide (malachite green aptamer antisense) | IDT | N/A | GGATCCATTCGTTACCTGGCT CTCGCCAGTCGGGATCCTATA GTGAGTCGTATTACAGTTCCAT TATCGCCGTAGTTGGTGTACT |
| oligonucleotide (malachite green aptamer sense) | IDT | N/A | TAATACGACTCACTATAGGATC CCGACTGGCGAGAGCCAGGT AACGAATGGATCC |
| oligonucleotide (Transcription Factor A) | IDT | N/A | AGTACACCAACTACGAGTGAG |
| oligonucleotide (Transcription Factor B) | IDT | N/A | TCAGTCACCTATCTGGCGATAA TGGAACTG |
| oligonucleotide with 3’ Amine modification (tether) | IDT | N/A | GCTACTCACTCAGATAGGTGAC TGA/3AmMO/ |
| Pierce strong ion exchange spin columns | Fisher Scientific | 90008 | |
| plasmid encoding SNAP T7 RNAP and kanamycin resistance genes | Genscript | N/A | custom gene insert |
| protein purification column (HisPur Ni-NTA spin column) | Fisher Scientific | 88226 | |
| rNTP mix | New England Biolabs | N0466S | |
| Roche mini quick DNA spin column | Sigma Aldrich | 11814419001 | |
| Triton X-100 | Sigma Aldrich | T8787-100ML | |
| Ultra Low Range DNA ladder | Fisher Scientific | 10597012 | |
| urea | BioShop | URE001.1 |
Références
- Cherry, K. M., Qian, L. Scaling up molecular pattern recognition with DNA-based winner-take-all neural networks. Nature. 559 (7714), 370-376 (2018).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475 (7356), 368-372 (2011).
- Chen, Y. -. J., et al. Programmable chemical controllers made from DNA. Nature Nanotechnology. 8 (10), 755-762 (2013).
- di Bernardo, D., Marucci, L., Menolascina, F., Siciliano, V. Predicting synthetic gene networks. Synthetic Gene Networks: Methods and Protocols. 813, 57-81 (2012).
- Xiang, Y., Dalchau, N., Wang, B. Scaling up genetic circuit design for cellular computing: advances and prospects. Natural Computing. 17 (4), 833-853 (2018).
- Gould, N., Hendy, O., Papamichail, D. Computational tools and algorithms for designing customized synthetic genes. Frontiers in Bioengineering and Biotechnology. 2, (2014).
- MacDonald, J. T., Siciliano, V. Computational sequence design with R2oDNA Designer. Mammalian Synthetic Promoters. 1651, 249-262 (2017).
- Cervantes-Salido, V. M., Jaime, O., Brizuela, C. A., Martínez-Pérez, I. M. Improving the design of sequences for DNA computing: A multiobjective evolutionary approach. Applied Soft Computing. 13 (12), 4594-4607 (2013).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. Journal of Computational Chemistry. 32 (1), 170-173 (2011).
- Fornace, M. E., Porubsky, N. J., Pierce, N. A. A unified dynamic programming framework for the analysis of interacting nucleic acid strands: enhanced models, scalability, and speed. ACS Synthetic Biology. 9 (10), 2665-2678 (2020).
- Wetterstrand, K. DNA sequencing costs: Data. Genome.gov. , (2020).
- Lopez, R., Wang, R., Seelig, G. A molecular multi-gene classifier for disease diagnostics. Nature Chemistry. 10 (7), 746-754 (2018).
- Pardee, K., et al. low-cost detection of Zika virus using programmable biomolecular components. Cell. 165 (5), 1255-1266 (2016).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406 (6796), 605-608 (2000).
- Lin, K. N., Volkel, K., Tuck, J. M., Keung, A. J. Dynamic and scalable DNA-based information storage. Nature Communications. 11 (1), 2981 (2020).
- Yurke, B., Mills, A. P. Using DNA to power nanostructures. Genetic Programming and Evolvable Machines. 4 (2), 111-122 (2003).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318 (5853), 1121-1125 (2007).
- Wang, B., Thachuk, C., Ellington, A. D., Winfree, E., Soloveichik, D. Effective design principles for leakless strand displacement systems. Proceedings of the National Academy of Sciences. 115 (52), 12182-12191 (2018).
- Machinek, R. R. F., Ouldridge, T. E., Haley, N. E. C., Bath, J., Turberfield, A. J. Programmable energy landscapes for kinetic control of DNA strand displacement. Nature Communications. 5 (1), 5324 (2014).
- Cabello-Garcia, J., Bae, W., Stan, G. -. B. V., Ouldridge, T. E. Handhold-mediated strand displacement: a nucleic acid-based mechanism for generating far-from-equilibrium assemblies through templated reactions. bioRxiv. , (2020).
- Brophy, J. A. N., Voigt, C. A. Principles of genetic circuit design. Nature Methods. 11 (5), 508-520 (2014).
- Khalil, A. S., et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 150 (3), 647-658 (2012).
- Swank, Z., Laohakunakorn, N., Maerkl, S. J. Cell-free gene-regulatory network engineering with synthetic transcription factors. Proceedings of the National Academy of Sciences. 116 (13), 5892-5901 (2019).
- Howland, S. W., Tsuji, T., Gnjatic, S., Ritter, G., Old, L. J., Wittrup, K. D. Inducing efficient cross-priming using antigen-coated yeast particles. Journal of immunotherapy. 31 (7), 607 (2008).
- Abil, Z., Ellefson, J. W., Gollihar, J. D., Watkins, E., Ellington, A. D. Compartmentalized partnered replication for the directed evolution of genetic parts and circuits. Nature Protocols. 12 (12), 2493-2512 (2017).
- Baugh, C., Grate, D., Wilson, C., Doudna, J. A. 2.8 Å crystal structure of the malachite green aptamer11. Journal of Molecular Biology. 301 (1), 117-128 (2000).
- Chou, L. Y. T., Shih, W. M. In vitro transcriptional regulation via nucleic acid-based transcription factors. ACS Synthetic Biology. 8 (11), 2558-2565 (2019).
- Lykke-Andersen, J., Christiansen, J. The C-terminal carboxy group of T7 RNA polymerase ensures efficient magnesium ion-dependent catalysis. Nucleic Acids Research. 26 (24), 5630-5635 (1998).
- Pu, J., Disare, M., Dickinson, B. C. Evolution of C-terminal modification tolerance in full-length and split T7 RNA Polymerase biosensors. Chembiochem. 20 (12), 1547-1553 (2019).
- Gardner, L. P., Mookhtiar, K. A., Coleman, J. E. Initiation, elongation, and processivity of carboxyl-terminal mutants of T7 RNA polymerase. Biochemistry. 36 (10), 2908-2918 (1997).
- Yin, J., Lin, A. J., Golan, D. E., Walsh, C. T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nature Protocols. 1 (1), 280-285 (2006).
- Warden-Rothman, R., Caturegli, I., Popik, V., Tsourkas, A. Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Analytical Chemistry. 85 (22), 11090-11097 (2013).
- Zhang, W. -. B., Sun, F., Tirrell, D. A., Arnold, F. H. Controlling macromolecular topology with genetically encoded SpyTag-SpyCatcher chemistry. Journal of the American Chemical Society. 135 (37), 13988-13997 (2013).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.