
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Imagerie in vivo de tissu cérébral entièrement actif chez des larves et des juvéniles de poissons-zèbres éveillés par enlèvement du crâne et de la peau
Dans cet article
Résumé
Nous présentons ici une méthode pour imager le cerveau embryonnaire du poisson-zèbre in vivo jusqu’aux stades larvaire et juvénile. Cette procédure micro-invasive, adaptée des approches électrophysiologiques, donne accès aux détails cellulaires et subcellulaires des neurones matures et peut être combinée avec des études optogénétiques et neuropharmacologiques pour caractériser la fonction cérébrale et l’intervention médicamenteuse.
Résumé
Comprendre les changements éphémères qui se produisent au cours du développement et de la maturation du cerveau nécessite une imagerie détaillée à haute résolution dans l’espace et le temps à la résolution cellulaire et subcellulaire. Les progrès des technologies moléculaires et d’imagerie nous ont permis d’acquérir de nombreuses connaissances détaillées sur les mécanismes cellulaires et moléculaires du développement du cerveau dans l’embryon transparent de poisson-zèbre. Récemment, les processus de raffinement de la connectivité neuronale qui se produisent à des stades larvaires ultérieurs plusieurs semaines après la fécondation, qui sont par exemple le contrôle du comportement social, la prise de décision ou le comportement motivé par la motivation, sont passés au centre de la recherche. À ces stades, la pigmentation de la peau du poisson-zèbre interfère avec la pénétration de la lumière dans le tissu cérébral et les solutions pour les stades embryonnaires, par exemple l’inhibition pharmacologique de la pigmentation, ne sont plus réalisables.
Par conséquent, une solution chirurgicale mini-invasive pour l’accès microscopique au cerveau du poisson-zèbre éveillé est fournie qui est dérivée d’approches électrophysiologiques. Dans les téléostéens, la peau et le cartilage mou du crâne peuvent être soigneusement enlevés en micro-pelant ces couches, exposant les neurones sous-jacents et les voies axonales sans dommage. Cela permet d’enregistrer la morphologie neuronale, y compris les structures synaptiques et leur contenu moléculaire, et d’observer les changements physiologiques tels que les transitoires Ca2+ ou les événements de transport intracellulaire. En outre, l’interrogation de ces processus au moyen d’une inhibition pharmacologique ou d’une manipulation optogénétique est possible. Cette approche d’exposition cérébrale fournit des informations sur les changements structurels et physiologiques dans les neurones ainsi que sur la corrélation et l’interdépendance de ces événements dans les tissus cérébraux vivants de l’ordre de quelques minutes ou heures. La technique convient à l’imagerie cérébrale in vivo des larves de poisson zèbre jusqu’à 30 jours après la fécondation, le dernier stade de développement testé jusqu’à présent. Il donne ainsi accès à des questions aussi importantes que le raffinement et la mise à l’échelle synaptiques, le transport axonal et dendritique, le ciblage synaptique de la cargaison cytosquelettique ou l’expression dépendante de l’activité locale. Par conséquent, une large utilisation de cette approche de montage et d’imagerie peut être anticipée.
Introduction
Au cours des dernières décennies, le poisson-zèbre (Danio rerio) a évolué comme l’un des organismes modèles de vertébrés les plus populaires pour les études de développement embryonnaire et larvaire. La grande fécondité des femelles de poisson zèbre associée au développement rapide ex utero de l’embryon et à sa transparence au début du développement embryonnaire ne sont que quelques facteurs clés qui font du poisson-zèbre un organisme modèle puissant pour répondre aux questions de développement1. Les progrès des technologies de génétique moléculaire combinés à des études d’imagerie in vivo à haute résolution ont permis d’aborder les mécanismes biologiques cellulaires sous-jacents aux processus de développement2. En particulier, dans le domaine de la différenciation neuronale, de la physiologie, de la connectivité et de la fonction, le poisson-zèbre a mis en lumière l’interaction de la dynamique moléculaire, des fonctions cérébrales et du comportement de l’organisme avec des détails sans précédent.
Pourtant, la plupart de ces études sont limitées aux stades embryonnaires et larvaires précoces au cours de la première semaine de développement, car la transparence du tissu du système nerveux est progressivement perdue. À ces stades, le tissu cérébral est empêché d’y accéder par des approches de microscopie à haute résolution qui sont protégées par la différenciation et la pigmentation du crâne3.
Par conséquent, les questions clés de la différenciation neuronale, de la maturation et de la plasticité telles que le raffinement de la connectivité neuronale ou la mise à l’échelle synaptique sont difficiles à étudier. Ces processus cellulaires sont importants afin de définir les mécanismes cellulaires qui conduisent, par exemple, le comportement social, la prise de décision ou le comportement basé sur la motivation, domaines dans lesquels la recherche sur les larves de plusieurs semaines a récemment contribué à des résultats clés basés sur des études comportementales4.
Les approches pharmacologiques pour inhiber la pigmentation chez les larves de poisson zèbre pendant plusieurs semaines sont à peine réalisables ou peuvent même causer des effets néfastes5,6,7,8. Les souches doubles ou triples mutantes avec des défauts de pigmentation spécifiques, telles que le casper9 ou le cristal10,sont devenues des outils extrêmement précieux, mais sont laborieuses dans la reproduction, fournissent peu de progéniture et présentent le danger d’accumuler des malformations génétiques dues à une consanguinité excessive.
Ici, une procédure invasive minimale comme alternative est fournie qui est applicable à toute souche de poisson zèbre. Cette procédure a été adaptée à partir d’études électrophysiologiques pour enregistrer l’activité neuronale chez les larves de poisson zèbre vivantes et éveillées. Dans les téléostéens, la peau et le cartilage mou du crâne peuvent être soigneusement enlevés en micro-pelant ces couches, car elles ne sont pas étroitement imbriquées avec le système vasculaire cérébral. Cela permet d’exposer les tissus cérébraux contenant des neurones et des voies axonales sans dommage et d’enregistrer la morphologie neuronale, y compris les structures synaptiques et leur contenu moléculaire, qui à leur tour incluent l’observation de changements physiologiques tels que les transitoires Ca2+ ou les événements de transport intracellulaire pendant plusieurs heures. De plus, au-delà des caractérisations descriptives, l’accès direct au tissu cérébral permet d’interroger les fonctions neuronales matures au moyen de l’administration de substances neuropharmacologiques et d’approches optogénétiques. Par conséquent, de véritables relations structure-fonction peuvent être révélées dans le cerveau du poisson zèbre juvénile en utilisant cette stratégie d’exposition cérébrale.
Protocole
Tous les travaux d’animaux décrits ici sont conformes aux réglementations légales (directive européenne 2010/63). L’entretien et la manipulation du poisson ont été approuvés par les autorités locales et par le représentant du bien-être animal de la Technische Universität Braunschweig.
1. Préparation de liquide céphalo-rachidien artificiel (ACSF), d’agarose à faible fusion et d’aiguilles de verre pointues
- Préparer le FSA en dissolvant les produits chimiques énumérés aux concentrations suivantes dans l’eau distillée. 134 mM de NaCl (58,44 g/mol), 2,9 mM de KCl (74,55 g/mol), 2,1 mM de CaCl2 (110,99 g/mol), 1,2 mM de MgCl2 6xH2O (203,3 g/mol), 10 mM de HEPES (238,31 g/mol) et 10 mM de d-glucose (180,16 g/mol).
REMARQUE: Pour MgCl2, CaCl2et KCl, des solutions de 1 M sont préparées dans de l’eau stérile dessalée et stockées à 4 °C pour la préparation ultérieure de l’ACSF frais. Le glucose, l’HEPES et le NaCl sont dissous sous forme de composés solides dans la solution fraîche d’ACSF. Pour dissoudre les produits chimiques, suivez les instructions du fabricant. - Ajuster le pH de l’ACSF à 7,8 avec 10 M NaOH. La préparation du LCRS nécessite une mesure précise des produits chimiques et un ajustement fin du pH car il remplace le liquide céphalo-rachidien et maintient les conditions physiologiques requises pour que les neurones soient pleinement fonctionnels, sinon il pourrait causer un dysfonctionnement cérébral et la mort neuronale.
- Conserver le FSC fraîchement préparé à 4 °C pendant un maximum de 4 semaines. Pour les conditions de travail, aliquoter le volume requis d’ACSF pour la journée/l’expérience et pré-pré-température à 25-28 °C (et éventuellement l’oxygéner, étape 2.5)
REMARQUE: L’ASCF fraîchement préparée est bonne pour 1 jour. Si vous prévoyez de l’utiliser sur plusieurs jours, ACSF doit être filtré stérile. - Pour une anesthésie ultérieure des larves, préparer une solution mère de 50 mM de d-tubocurarine dans de l’eau distillée et conserver la solution à -20 °C sous forme d’aliquotes de 100 μL au congélateur jusqu’à ce que cela soit nécessaire.
- Pour incorporer le poisson, préparer 2,5 % d’agarose à faible fusion (LM) en dissolvant 1,25 g de LM-agarose (Table des matières) dans 50 mL ACSF et faire bouillir jusqu’à ce que l’agarose soit complètement dissoute.
REMARQUE: Alternativement, des concentrations plus élevées ou plus faibles de LM-agarose peuvent être utilisées en fonction de la configuration expérimentale. Cependant, si l’agarose est trop molle, elle ne pourra pas maintenir le poisson en position lors de l’ouverture du crâne. - Conservez l’agarose à 37 °C au bain-marie, pour éviter la solidification et parce que cette température ne nuira pas non plus aux larves lors de l’incorporation. Une fois l’agarose bouillie refroidie à 37 °C dans le bain-marie, ajoutez la quantité nécessaire de d-tubocurarine à l’agarose aliquote nécessaire pour que la journée atteigne une concentration de travail de 10 μM. Pour une utilisation future, conserver les restes d’agarose à 4 °C pour éviter toute contamination.
- Préparer des aiguilles de verre pointues et minces à partir de capillaires en verre(figure supplémentaire 1)à l’aide d’un tireur de micropipette avec les réglages suivants.
- Puller I, capillaire type 1: Chaleur 1: 65,8; Chaleur 2: 55,1; Traction en 2 étapes
Puller II, capillaire de type 2 : Chaleur = 700 ; Fil = 4; Vel = 55; Del = 130; Pul = 55; Traction en 1 étape.
REMARQUE: Les unités sont spécifiques pour chaque arracheur et capillaire en verre utilisés ici, respectivement (voir tableau des matériaux). D’autres capillaires et arracheurs peuvent également être utilisés pour préparer les aiguilles en verre. Mais les aiguilles en verre ne doivent pas être trop minces car elles pourraient se briser au contact du crâne. Capillaire: longueur: 100 mm (4 pouces); OD: 1,5 mm; ID: 0,84 mm; filament: Oui
- Puller I, capillaire type 1: Chaleur 1: 65,8; Chaleur 2: 55,1; Traction en 2 étapes
2. Anesthésie des larves et préparations pour l’incorporation
- Lorsque vous commencez l’expérience pour la journée, transférez les animaux nécessaires avec une pipette Pasteur en plastique dans une boîte de Petri de 90 mm de diamètre, qui est remplie soit de Danieau (pour les larves qui sont encore conservées dans une boîte de Petri avec Danieau), soit d’eau de l’installation de pêche (pour les larves âgées de plus de 7 dpf et conservées dans l’installation de pêche).
- Lorsque vous pipetez des poissons âgés de plus de 2 semaines, assurez-vous que l’ouverture de la pipette est suffisamment grande pour éviter de blesser les poissons lors de leur transfert. N’utilisez pas de filet car il endommagera physiquement en particulier les larves plus jeunes.
- Ajouter rotifera ou artemia nauplii adapté à la taille des larves conservées dans la boîte de Pétri, pour assurer le libre accès à la nourriture et l’état de santé maximal des larves et pour réduire le stress.
- Pour l’encastrement, transférer les larves sélectionnées dans une boîte de Petri de 35 mm de diamètre remplie de LCRS. Ajouter le volume nécessaire de d-tubocurarine pour atteindre une concentration de travail/dose efficace de 10 μM et attendre quelques minutes jusqu’à ce que les larves soient complètement immobilisées11.
REMARQUE: Lorsque le poisson vieillit ou si une anesthésie complète plus rapide est nécessaire (moins de 5 min), il est possible d’augmenter la concentration de d-tubocurarine (DL50 pour les souris est de 0,13 mg / kg par voie intraveineuse12). Il est également possible d’utiliser un anesthésique différent, tel que la α-bungarotoxine (concentration de travail: 1 mg / mL), qui a le même effet que le curare et maintient également le cerveau pleinement actif13. Si un cerveau pleinement actif n’est pas nécessaire pour le sujet d’intérêt, Tricaine à une dose non létale (0,02%) est également une option pour anesthésier complètement les larves. Cependant, la tricaïne bloque les canaux sodiques, altérant ainsi l’activité cérébrale14. - Préparez la chambre de montage en prenant le couvercle de la boîte de Petri de 35 mm de diamètre, retournez le couvercle à l’envers et placez un couvercle carré en verre (24 x 24 mm) sur le fond du couvercle. Reportez-vous à la figure 1 (partie supérieure) pour une description schématique de ces étapes. La surface plus lisse du verre empêche de glisser du bloc d’agarose, qui contient les larves pendant la procédure d’ouverture du crâne.
- Aliquotez la quantité d’ACSF nécessaire pour la journée dans un flacon approprié (par exemple, tube de 50 mL, bécher, bouteille Schott, etc.) et oxygénez-la avec du carbogène (5% CO2,95% O2). Si l’imagerie uniquement la morphologie (par exemple, les modèles de fluorescence), ACSF est toujours nécessaire pour assurer l’intégrité du cerveau et que les cellules ne sont pas influencées négativement par les effets d’osmolarité, mais l’oxygénation de l’ACSF n’est pas nécessaire. Cette étape ne doit être effectuée que lorsque l’activité cérébrale complète est nécessaire pour l’imagerie.
REMARQUE: Pour une saturation optimale en oxygène du milieu, ajoutez une pierre à air à l’extrémité du tube carbogène. Pour garantir un niveau d’oxygène suffisamment élevé, il est nécessaire d’échanger l’ACSF dans les chambres d’imagerie avec de l’ACSF fraîchement oxygéné toutes les 20 à 60 minutes, en fonction du nombre et de l’âge des larves intégrées dans la même chambre d’imagerie (par exemple, pour une seule larve intégrée, un échange ACSF toutes les heures est suffisant. Pour six larves de plus de 14 dpf intégrées en parallèle, l’échange d’ACSF toutes les 20 minutes est nécessaire) alors planifiez la quantité nécessaire d’ACSF saturé d’oxygène en fonction de l’expérience prévue.
3. Intégration des larves
- Transférer les larves entièrement anesthésisées avec une pipette Pasteur en plastique dans la chambre de montage préparée (à l’étape 2.4). Ensuite, retirez soigneusement l’excès de milieu pour éviter la dilution de la LM-agarose. Toutes les étapes suivantes doivent être effectuées sous un stéréomicroscope avec un grossissement suffisant.
REMARQUE: L’inclinaison de la chambre de montage peut aider à retirer complètement le support. - Passez immédiatement à l’étape suivante, en ajoutant une goutte de LM-agarose suffisamment importante sur le dessus des larves (environ 1 mL, selon la taille des larves) pour protéger les animaux du dessèchement et réduire le stress inutile.
- Orientez les larves en position avant que l’agarose ne se solidifie. Assurez-vous que la partie dorsale des larves est dirigée vers le haut. Assurez-vous également d’intégrer les larves aussi près que possible de la surface de l’agarose.
REMARQUE: Selon la taille et le nombre de larves prévues pour intégrer en même temps, il est possible d’ajuster la concentration d’agarose. Par exemple, pour 1 à 3 larves âgées de 30 dpf, une concentration de 1,8% à 2% de LM-agarose est recommandée. Pour 1 à 4 larves âgées de 7 dpf, il est plus pratique d’utiliser 2,5% de LM-agarose, tandis que, pour 5 à 8 larves, 2% est plus adapté. Si un cerveau pleinement actif est nécessaire, il est recommandé de n’intégrer que trois poissons en même temps pour réduire le temps nécessaire au fonctionnement des larves. En général, il est recommandé d’utiliser des concentrations plus faibles (1,8% à 2%) plus les larves vieillissent ou plus il est prévu d’incruster de larves en même temps. - Si les images sont enregistrées à l’aide d’un microscope inversé, coupez le bloc d’agarose contenant les larves en une petite forme cuboïde. Ceci est important pour transférer les larves dans la chambre d’imagerie plus tard. Si vous utilisez un microscope vertical, un tel rognage n’est pas nécessaire, car la chambre de montage peut également être utilisée comme chambre d’imagerie. Dans la figure 1 (partie supérieure), on peut trouver une description schématique de ces étapes.
4. Exposer le cerveau
REMARQUE: Toutes les étapes suivantes doivent être effectuées avec le plus grand soin pour ne pas blesser inutilement les larves. Si un cerveau pleinement actif est nécessaire pour l’expérience, gardez à l’esprit qu’à chaque seconde qui passe, alors que le poisson est encore entièrement monté dans l’agarose et a un crâne ouvert sans ACSF oxygéné, le cerveau souffrira d’un manque d’oxygène et se dessèchera également. Les effets de la carence en oxygène deviendront encore plus dramatiques, plus les larves incorporées sont âgées. Par conséquent, il est important d’effectuer la chirurgie non seulement dans les plus brefs délais, mais aussi avec une précision maximale pour ne pas évoquer de lésions cérébrales mécaniques avec l’aiguille. Une fois formés, les étapes 4.2 à 4.4 ne devraient pas prendre plus de 30 s par poisson.
- Commencez la chirurgie dès que l’agarose s’est solidifiée. Tout d’abord, coupez tout l’excès d’agarose au-dessus de la région du cerveau d’intérêt pour obtenir un accès libre à la tête et un espace de travail dégagé. Si la partie dorsale de la tête dépasse déjà de l’agarose, sautez cette étape.
- Selon la région d’intérêt, choisissez un endroit pour commencer la chirurgie. Prenez l’aiguille en verre et faites une petite incision à travers la peau, mais sans pénétrer trop profondément dans le tissu. Ce sera le point de départ pour décoller la peau superposée.
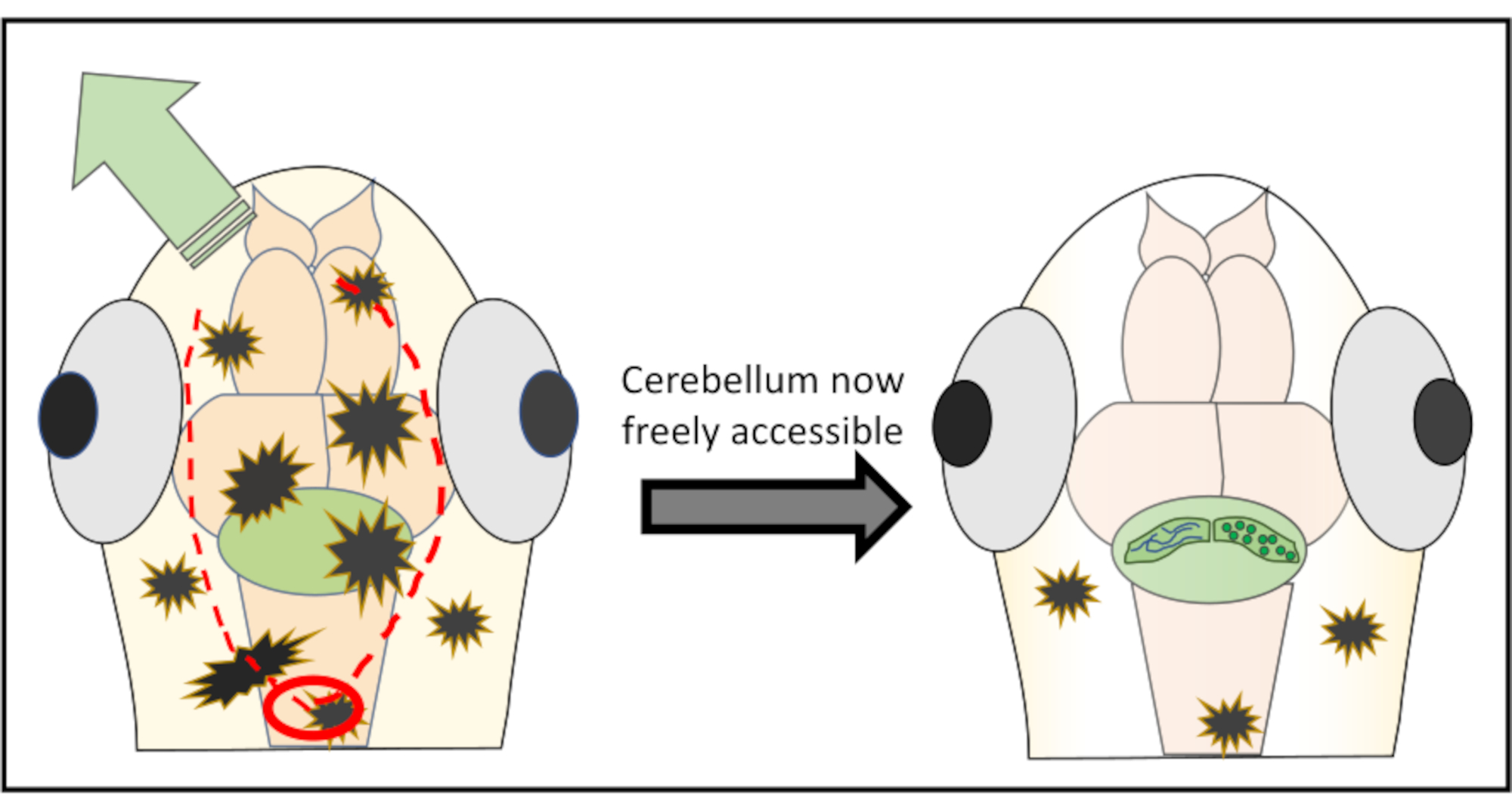
REMARQUE: Pour des résultats optimaux, ne commencez jamais directement au-dessus de la région d’intérêt pour réduire le risque d’endommager des structures importantes. Si nécessaire, il est possible de commencer même postérieurement au cerveau postérieur et de là travailler vers l’avant jusqu’à ce que la zone indésirable de la peau soit pelée. - Continuez avec de très petites coupures le long de la partie de la peau visant à enlever en déplaçant à peine l’aiguille juste sous la surface. La plupart du temps, il n’est pas nécessaire de se déplacer complètement autour du cerveau et de découper un morceau de peau et de crâne en forme de cercle, mais plutôt de faire deux incisions le long de la tête, puis de repousser la peau de l’un ou l’autre côté. La figure 2 montre une représentation schématique de la stratégie de coupe optimale pour obtenir un accès libre au cervelet.
REMARQUE: Cette micro-chirurgie est une procédure délicate et elle nécessitera probablement un peu d’entraînement pour enlever parfaitement la peau sans endommager le cerveau sous-jacent. Il est également recommandé de trouver la stratégie de coupe optimale pour la région du cerveau d’intérêt et de s’y tenir pendant la période de l’expérience. - Immédiatement après avoir retiré la peau de toutes les larves incrustées, versez du LCR (oxygéné) sur l’agarose pour inonder les particules de peau et le sang indésirables et pour garder le cerveau pleinement actif et le protéger du dessèchement.
REMARQUE: Si un cerveau sain est nécessaire pour l’expérience, il est recommandé d’aller pour un maximum de trois poissons à la fois. - Si vous utilisez un microscope vertical, commencez directement par l’imagerie.
- Lorsque vous utilisez un microscope inversé, faites glisser une petite spatule sous le bloc d’agarose cuboïde (étape 3.4).
- Ajouter une petite goutte de LM-agarose au fond de la chambre d’imagerie (par exemple, une boîte à fond de verre) et retourner immédiatement le bloc d’agarose contenant les larves avec la spatule à 180 ° et le pousser doucement au fond de la chambre d’imagerie, tandis que la goutte d’agarose liquide agit comme de la colle.
- Lorsque l’agarose s’est solidifiée, remplissez la chambre d’imagerie avec de l’ACSF (oxygéné), puis commencez l’imagerie. Voir la figure 1 (partie inférieure) pour une description schématique.
- Lorsque l’activité cérébrale complète est requise pour l’expérience, assurez-vous toujours que l’ACSF dans la chambre d’imagerie a un niveau d’oxygène suffisamment élevé. Pour ce faire, échangez soigneusement le milieu avec de l’ACSF fraîchement oxygéné lorsque cela est possible toutes les 20 à 60 minutes (en fonction du nombre et de la taille du poisson, de la taille et de la surface de la chambre d’imagerie et de la durée de l’imagerie).

Figure 1: Procédure schématique pour la préparation du poisson-zèbre à crâne ouvert pour l’imagerie in vivo de manière progressive. Les instructions de travail pour les différentes étapes peuvent être trouvées dans le graphique lui-même. Graphique conçu par Florian Hetsch et adapté par Paul Schramm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2: Représentation schématique détaillée de la micro-chirurgie réalisée pour enlever des morceaux de peau et de crâne au-dessus de la région cérébrale d’intérêt. Le cercle rouge marque l’endroit où la première coupe doit être faite. La ligne pointillée rouge délimite le chemin optimal pour couper avec l’aiguille afin d’obtenir un accès libre au cervelet sans l’endommager. La flèche verte marque la direction dans laquelle les morceaux excessifs de peau et de crâne peuvent facilement être repoussés. Assurez-vous de ne jamais pénétrer dans le tissu cérébral pendant toute la procédure. Après avoir réussi à décoller la peau, la région cérébrale d’intérêt (ici, le cervelet) sera librement accessible pour tout type d’imagerie in vivo à haute résolution. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Résultats
Graphique 3A,C montrent une larve de 14 dpf de la lignée transgénique Tg[-7.5Ca8:GFP]bz12[15] avec le crâne encore intact. Les cellules pigmentaires de la peau superposée sont réparties sur toute la tête et interfèrent avec le signal de fluorescence dans la région d’intérêt (ici, le cervelet). Avec la larve dans cette condition, il n’est pas possible d’obtenir des images haute rés...
Discussion
La méthode présentée fournit une approche alternative à l’isolement cérébral ou au traitement des larves de poisson zèbre avec des produits pharmaceutiques inhibant la pigmentation pour enregistrer des images à haute résolution des neurones dans leur environnement in vivo. La qualité des images enregistrées avec cette méthode est comparable aux images de cerveaux explantés, mais dans des conditions naturelles.
De plus, une perte d’intensité de fluorescence est évit?...
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous remercions tout particulièrement Timo Fritsch pour les excellents soins aux animaux et Hermann Döring, Mohamed Elsaey, Sol Pose-Méndez, Jakob von Trotha, Komali Valishetti et Barbara Winter pour leur soutien utile. Nous remercions également tous les autres membres du laboratoire Köster pour leurs commentaires. Le projet a été financé en partie par le projet 241961032 de la Fondation allemande pour la recherche (DFG, KO1949/7-2) (à RWK) et le Bundesministerium für Bildung und Forschung (BMBF; Era-Net NEURON II CIPRESS project 01EW1520 to JCM) est reconnu.
matériels
| Name | Company | Catalog Number | Comments |
| Calcium chloride | Roth | A119.1 | |
| Confocal Laser scanning microscope | Leica | TCS SP8 | |
| d-Glucose | Sigma | G8270-1KG | |
| d-Tubocurare | Sigma-Aldrich | T2379-100MG | |
| Glass Capillary type 1 | WPI | 1B150F-4 | |
| Glass Capillary type 2 | Harvard Apparatus | GC100F-10 | |
| Glass Coverslip | deltalab | D102424 | |
| HEPES | Roth | 9105.4 | |
| Hoechst 33342 | Invitrogen (Thermo Fischer) | H3570 | |
| Imaging chamber | Ibidi | 81156 | |
| Potassium chloride | Normapur | 26764298 | |
| LM-Agarose | Condalab | 8050.55 | |
| Magnesium chloride (Hexahydrate) | Roth | A537.4 | |
| Microscope Camera | Leica | DFC9000 GTC | |
| Needle-Puller type 1 | NARISHIGE | Model PC-10 | |
| Needle-Puller type 2 | Sutter Instruments | Model P-2000 | |
| Pasteur-Pipettes 3ml | A.Hartenstein | 20170718 | |
| Sodium chloride | Roth | P029.2 | |
| Sodium hydroxide | Normapur | 28244262 | |
| Tricain | Sigma-Aldrich | E10521-50G | |
| Waterbath | Phoenix Instrument | WB-12 | |
| 35 mm petri dish | Sarstedt | 833900 | |
| 90 mm petri dish | Sarstedt | 821473001 |
Références
- Embryology. Zebrafish Development Available from: https://embryology.med.unsw.edu.au/embryology/index.php/Zebrafish_Development (2020)
- Sassen, W. A., Köster, R. W. A molecular toolbox for genetic manipulation of zebrafish. Advances in Genomics and Genetics. Dove Medical Press. 2015 (5), 151-163 (2015).
- Singh, A. P., Nüsslein-Volhard, C. Zebrafish stripes as a model for vertebrate colour pattern formation. Current Biology. 25 (2), 81-92 (2015).
- Kalueff, A. V., et al. Time to recognize zebrafish 'affective' behavior. Brill: Behaviour. 149 (10-12), 1019-1036 (2012).
- Karlsson, J., von Hofsten, J., Olsson, P. -. E. Generating transparent zebrafish: a refined method to improve detection of gene expression during embryonic development. Marine Biotechnology. 3, 522-527 (2001).
- Bohnsack, B. L., Gallina, D., Kahana, A. Phenothiourea sensitizes zebrafish cranial neural crest and extraocular muscle development to changes in retinoic acid and IGF signaling. PloS One. 6, 22991 (2011).
- Elsalini, O. A., Rohr, K. B. Phenylthiourea disrupts thyroid function in developing zebrafish. Development Genes and Evolution. 212, 593-598 (2003).
- Baumann, L., Ros, A., Rehberger, K., Neuhauss, S. C. F., Segner, H. Thyroid disruption in zebrafish (Danio rerio) larvae: Different molecular response patterns lead to impaired eye development and visual functions. Aquatic Toxicology. 172, 44-55 (2016).
- White, R., et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell. 2, 183-189 (2008).
- Antinucci, P., Hindges, R. A crystal-clear zebrafish for in vivo imaging. Scientific Reports. 6, 29490 (2016).
- Burr, S. A., Leung, Y. L. Curare (d-Tubocurarine). Encyclopedia of Toxicology (3rd Edition). , 1088-1089 (2014).
- Gesler, H. M., Hoppe, J. 3,6-bis(3-diethylaminopropoxy) pyridazine bismethiodide, a long-acting neuromuscular blocking agent. The Journal of Pharmacology and Experimental Therapeutics. 118 (4), 395-406 (1956).
- Furman, B. . Alpha Bungarotxin. Reference Module in Biomedical Sciences. , (2018).
- Attili, S., Hughes, S. M. Anaesthetic tricaine acts preferentially on neural voltage-gated sodium channels and fails to block directly evoked muscle contraction. PLoS One. 9 (8), 103751 (2014).
- Namikawa, K., et al. Modeling neurodegenerative spinocerebellar ataxia type 13 in zebrafish using a Purkinje neuron specific tunable coexpression system. Journal of Neuroscience. 39 (20), 3948-3969 (2019).
- Hennig, M. Theoretical models of synaptic short term plasticity. Frontiers in Computational Neuroscience. 7 (45), (2013).
- Wang, Y., et al. Moesin1 and Ve-cadherin are required in endothelial cells during in vivo tubulogenesis. Development. 137, 3119-3128 (2010).
- Hobro, A., Smith, N. An evaluation of fixation methods: Spatial and compositional cellular changes observed by Raman imaging. Vibrational Spectroscopy. 91, 31-45 (2017).
- Knogler, L. D., Kist, A. M., Portugues, R. Motor context dominates output from purkinje cell functional regions during reflexive visuomotor behaviours. eLife. 8, 42138 (2019).
- Hsieh, J., Ulrich, B., Issa, F. A., Wan, J., Papazian, D. M. Rapid development of Purkinje cell excitability, functional cerebellar circuit, and afferent sensory input to cerebellum in zebrafish. Frontier in Neural Circuits. 8 (147), (2014).
- Scalise, K., Shimizu, T., Hibi, M., Sawtell, N. B. Responses of cerebellar Purkinje cells during fictive optomotor behavior in larval zebrafish. Journal of Neurophysiology. 116 (5), 2067-2080 (2016).
- Harmon, T. C., Magaram, U., McLean, D. L., Raman, I. M. Distinct responses of Purkinje neurons and roles of simple spikes during associative motor learning in larval zebrafish. eLife. 6, 22537 (2017).
- Zehendner, C. M., et al. Moderate hypoxia followed by reoxygenation results in blood-brain barrier breakdown via oxidative stress-dependent tight-junction protein disruption. PLoS One. 8 (12), 82823 (2013).
- Dhabhar, F. S. The short-term stress response - mother nature's mechanism for enhancing protection and performance under conditions of threat, challenge, and opportunity. Frontiers of Neuroendocrinology. 49, 175-192 (2018).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.