
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Méthodes in vitro ascendantes pour tester l’organisation ultrastructurale, le remodelage de la membrane et le comportement de sensibilité à la courbure des septines
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
Les septines sont des protéines cytosquelettiques. Ils interagissent avec les membranes lipidiques et peuvent détecter mais aussi générer une courbure de membrane à l’échelle du micron. Nous décrivons dans ce protocole des méthodologies in vitro ascendantes pour l’analyse des déformations membranaires, la liaison de la septine sensible à la courbure et l’ultrastructure du filament de septine.
Résumé
Le remodelage membranaire se produit constamment au niveau de la membrane plasmique et dans les organites cellulaires. Pour décortiquer complètement le rôle de l’environnement (conditions ioniques, compositions protéiques et lipidiques, courbure membranaire) et des différents partenaires associés à des processus spécifiques de remodelage membranaire, nous entreprenons des approches ascendantes in vitro . Ces dernières années, on a manifesté un vif intérêt pour révéler le rôle des protéines septines associées aux principales maladies. Les septines sont des protéines cytosquelettiques essentielles et omniprésentes qui interagissent avec la membrane plasmique. Ils sont impliqués dans la division cellulaire, la motilité cellulaire, la neuro-morphogenèse et la spermiogenèse, entre autres fonctions. Il est donc important de comprendre comment les septines interagissent et s’organisent au niveau des membranes pour induire ensuite des déformations membranaires et comment elles peuvent être sensibles à des courbures membranaires spécifiques. Cet article vise à déchiffrer l’interaction entre l’ultra-structure des septines au niveau moléculaire et le remodelage membranaire se produisant à l’échelle du micron. À cette fin, des complexes de levure bourgeonnante et de septine de mammifères ont été exprimés et purifiés de manière recombinante. Une combinaison de tests in vitro a ensuite été utilisée pour analyser l’auto-assemblage des septines au niveau de la membrane. Des bicouches lipidiques soutenues (SLB), des vésicules unilamellaires géantes (GUV), de grandes vésicules unilamellaires (LUV) et des substrats ondulés ont été utilisés pour étudier l’interaction entre l’auto-assemblage de la septine, le remodelage de la membrane et la courbure de la membrane.
Introduction
Les septines sont des protéines formant des filaments cytosquelettiques qui interagissent avec les membranes lipidiques. Les septines sont omniprésentes chez les eucaryotes et essentielles à de nombreuses fonctions cellulaires. Ils ont été identifiés comme les principaux régulateurs de la division cellulaire chez les levures bourgeonnantes et les mammifères 1,2. Ils sont impliqués dans les événements de remodelage membranaire, la ciliogenèse3 et la spermiogenèse4. Dans les cellules de mammifères, les septines peuvent également interagir avec l’actine et les microtubules 5,6,7 dans un liant de Rho GTPases (BORG) de manière 8. Dans divers tissus (neurones9, cils3, spermatozoïdes10), les septines ont été identifiées comme régulateurs des barrières de diffusion pour les composants liés à la membrane11. Il a également été démontré que les septines régulent le blebbing de la membrane et la formation de saillies12. Les septines, étant des protéines multitâches, sont impliquées dans l’émergence de diverses maladies répandues13. Leur mauvaise régulation est associée à l’émergence de cancers14 et de maladies neurodégénératives15.
Selon l’organisme, plusieurs sous-unités septines (deux chez Caenorhabditis elegans à 13 chez l’homme) s’assemblent pour former des complexes dont l’organisation varie de façon tissulaire16. Le bloc de construction septine de base rassemble deux à quatre sous-unités, présentes en deux exemplaires et auto-assemblées de manière palindromique en forme de tige. Dans la levure bourgeonnante, les septines sont octamériques17,18. In situ, les septines sont souvent localisées sur des sites à courbure micrométrique; On les trouve aux sites de constriction de division, à la base des cils et des dendrites, et à l’anneau des spermatozoïdes19,20. Au niveau de la membrane, le rôle des septines semble être double : elles sont impliquées dans le remodelage de la bicouche lipidique et dans le maintien de l’intégrité de la membrane21. Par conséquent, l’étude des propriétés biophysiques des protéines formant des filaments de septine et / ou des sous-unités à la membrane est cruciale pour comprendre leur rôle. Pour disséquer les propriétés spécifiques des septines dans un environnement bien contrôlé, des approches in vitro ascendantes sont appropriées. Jusqu’à présent, seuls quelques groupes ont décrit les propriétés biophysiques des septines in vitro20,22,23. Par conséquent, par rapport à d’autres filaments cytosquelettiques, les connaissances actuelles sur le comportement des septines in vitro restent limitées.
Ce protocole décrit comment l’organisation des filaments de septine, le remodelage de la membrane et la sensibilité à la courbure peuvent être analysés19. À cette fin, une combinaison de méthodes de microscopie optique et électronique (microscopie à fluorescence, cryo-microscopie électronique [cryo-EM] et microscopie électronique à balayage [MEB]) a été utilisée. Le remodelage membranaire de vésicules unilamellaires géantes (GUV) de taille micrométrique est visualisé à l’aide de la microscopie optique à fluorescence. L’analyse de l’arrangement et de l’ultrastructure des filaments de septine liés aux vésicules lipidiques est réalisée à l’aide de cryo-EM. L’analyse de la sensibilité à la courbure de la septine est réalisée à l’aide de MEB, en étudiant le comportement des filaments de septine liés aux bicouches lipidiques soutenues par des solides déposés sur des substrats ondulés de courbures variables, ce qui permet d’analyser la sensibilité à la courbure pour les courbures positives et négatives. Par rapport à l’analyse précédente20,24, nous proposons ici d’utiliser une combinaison de méthodes pour analyser en profondeur comment les septines peuvent s’auto-assembler, déformer la membrane de manière synergique et être sensibles à la courbure. Ce protocole est considéré comme utile et adaptable à toute protéine filamenteuse qui présente une affinité pour les membranes.
Protocole
1. Détermination du remodelage de la membrane à l’aide de vésicules unilamellaires géantes (GUV)
REMARQUE: Dans cette section, les GUV sont générés pour imiter les déformations membranaires éventuellement induites par les septines dans un contexte cellulaire. En effet, dans les cellules, les septines se retrouvent fréquemment sur des sites à courbures micrométriques. Les GUV ont des tailles allant de quelques à quelques dizaines de micromètres et peuvent être déformés. Ils sont donc appropriés pour doser toute déformation induite par la septine à l’échelle micrométrique. Les lipides fluorescents, ainsi que les septines marquées par fluorescence (utilisant la protéine fluorescente verte [GFP]), sont utilisés pour suivre le comportement des lipides et des protéines par microscopie à fluorescence.
- Préparation des tampons et des solutions
- Préparer le tampon de croissance GUV (50 mM NaCl, 50 mM saccharose et 10 mM Tris [pH = 7,8]) et le tampon d’observation (75 mM NaCl et 10 mM Tris [pH = 7,8]).
- Mesurer (à l’aide d’un osmomètre commercial) et ajuster l’osmolarité des tampons d’observation et de croissance (170 mOsmol· L-1, en théorie) en ajoutant de petites quantités de NaCl jusqu’à ce que leurs osmolarités respectives soient égales. Filtrez les tampons à l’aide d’un filtre de 0,2 μm. Aliquote du tampon de croissance et le conserver à -20 °C pour une utilisation ultérieure. Stocker le tampon d’observation à 4 °C.
REMARQUE : La différence d’osmolarité entre les deux tampons ne doit pas dépasser 5 %. - Préparer une solution de β-caséine à 5 mg·mL-1 dans le tampon d’observation. Assurer la dissolution complète (après plusieurs heures à 4 °C sous agitation magnétique). Filtrer la solution avec un filtre de 0,2 μm, aliquote, et conserver à -20 °C.
- Préparer les mélanges lipidiques à une concentration lipidique totale de 3 mg·mL-1 dans le chloroforme. Utiliser une composition (mol%) de 56,8% de L-α-phosphatidylcholine (EggPC), 15% de cholestérol, 10% de 1,2-dioléoyl-sn-glycéro-3-phosphoéthanolamine (DOPE), 10% de 1,2-dioléoyl-sn-glycéro-3-phospho-L-sérine (DOPS), 8% de cerveau L-α-phosphatidylinositol-4,5-bisphosphate (PI(4,5)P 2) et0,2% de Bodipy-TR-céramide pour améliorer les interactions protéine-lipide et favoriser l’incorporation de PI(4,5)P225.
REMARQUE : Manipulez le chloroforme sous une hotte à l’aide de gants en nitrile et de lunettes de sécurité. Pipeter les solutions de chloroforme avec des seringues en verre et éviter le plastique car le chloroforme dissout le plastique. Avant et après utilisation, rincer les seringues en pipetant du chloroforme 5x-10x. Utilisez des seringues séparées pour pipeter des lipides fluorescents spécifiques afin de prévenir la contamination croisée. Les lipides peuvent être conservés dans du chloroforme à -20 °C dans un flacon en verre ambré recouvert de téflon. Les flacons doivent être remplis d’argon avant la fermeture et scellés avec du parafilm pour éviter toute oxydation lipidique. - Électroformation de GUV à l’aide d’une configuration de fils de platine
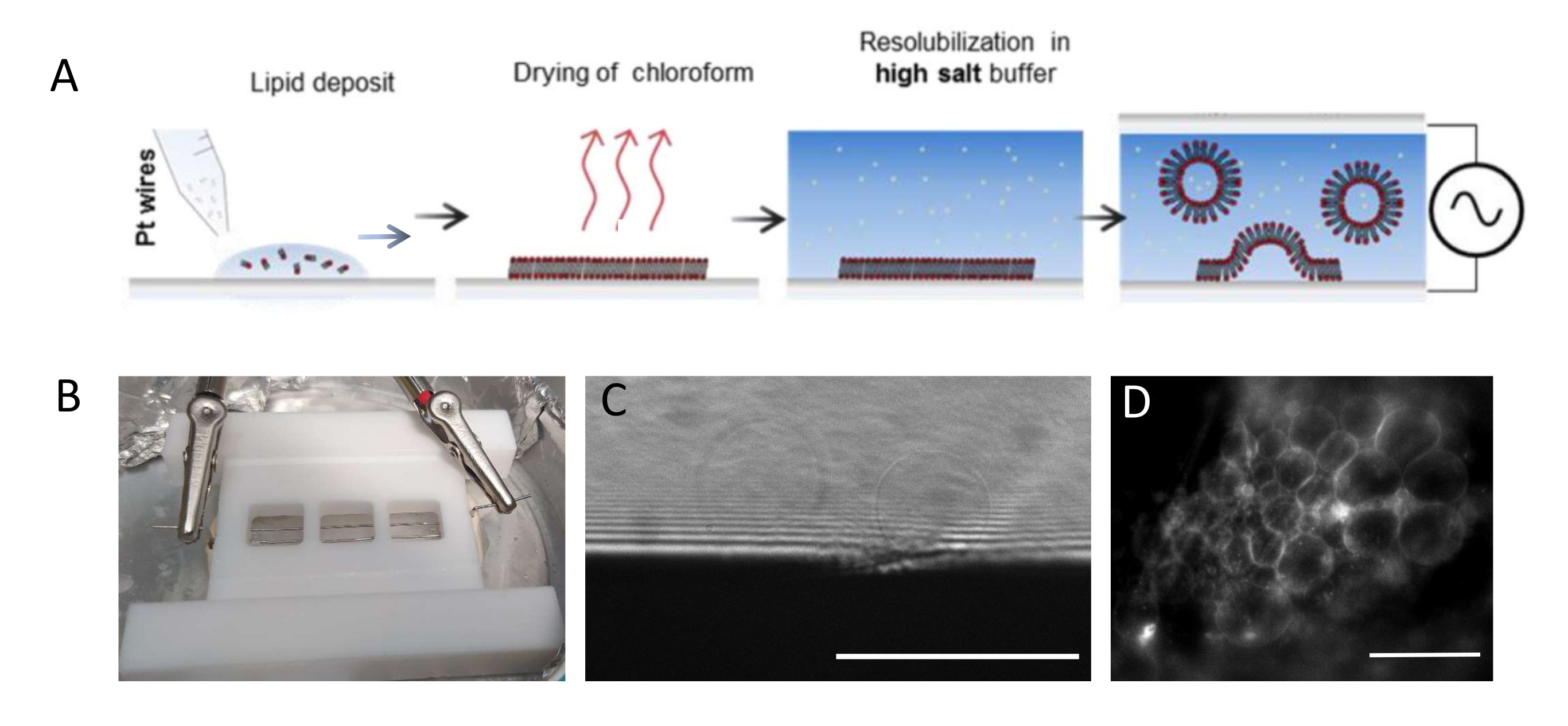
NOTE: La figure 1 présente le schéma des étapes expérimentales et une image de la chambre.- Nettoyez soigneusement la chambre et les fils de platine comme suit pour éliminer les résidus lipidiques.
- Plonger les fils et la chambre dans l’acétone et soniquer pendant 10 min. Essuyez-les soigneusement avec un mouchoir en papier à l’aide d’acétone.
- Assemblez la chambre en insérant les fils, plongez à nouveau dans l’acétone et sonicez pendant 10 min. Essuyez une fois de plus avec de l’acétone, en vous assurant que les fils sont complètement nettoyés. Plonger la chambre dans l’éthanol, sonifier pendant 10 minutes et essuyer avec de l’éthanol.
- Enfin, plongez la chambre dans de l’eau désionisée, soniquez pendant 10 min et séchez avec un jet d’azote ou d’air.
NOTE: La chambre en téflon (Figure 1B) a été fabriquée sur mesure dans l’atelier interne. Il accueille trois compartiments qui peuvent être scellés des deux côtés à l’aide de lamelles de couverture en verre. Les fils de platine peuvent être insérés dans la chambre à travers des trous de 1,3 mm de diamètre. - Après avoir nettoyé la chambre, déposer 3-4 gouttes par compartiment (chaque goutte est d’environ 0,1 μL)) du mélange lipidique 3 mg·mL-1 sur chaque fil de platine. Tournez les fils de 180° et déposez 3-4 gouttelettes lipidiques par compartiment du côté opposé de chaque fil de platine. Assurez-vous que les gouttes ne sont pas en contact les unes avec les autres. Environ 5 μL du mélange lipidique sont nécessaires par chambre entière.
- Placez la chambre de croissance dans une chambre à vide pendant 30 minutes pour éliminer toute trace de chloroforme.
REMARQUE: Le vide profond (0,1 mbar) est préférable. Lorsqu’ils sont séchés, les lipides sont vulnérables à l’oxydation et, par conséquent, ne doivent pas être laissés dans l’air pendant plus de quelques minutes. - Déposer de la graisse sous vide poussé au fond de la chambre (le côté le plus proche des fils) le long de la périphérie des trois compartiments à l’aide d’une seringue et appuyer une glissière propre (22 mm x 40 mm) contre la graisse pour assurer une étanchéité parfaite. Sceller les deux extrémités de la chambre (c.-à-d. aux sites d’entrée et de sortie des fils) à l’aide de pâte à sceller (plaques de cire). De même, appliquez de la graisse sous vide de l’autre côté de la chambre.
- Remplir les compartiments avec un tampon de croissance (~1 mL par chambre) à l’aide d’une pipette. Ne pas agiter la solution trop rapidement ou trop fortement pour éviter tout détachement du film lipidique des fils. Scellez hermétiquement le haut de la chambre à l’aide d’une glissière de couverture de 22 mm x 40 mm en la pressant contre la graisse. Pour éviter la formation de bulles d’air, appuyez doucement sur le couvercle en verre du centre vers les bords.
- Placez la chambre dans un réfrigérateur à 4 °C et connectez les fils à un générateur de fonction d’onde (fonction sinusoïdale à 500 Hz). Fixer une tension effective de 350 mV pour une période de croissance plus courte (c.-à-d. 6 h) ou de 250 mV pour une période de croissance plus longue (c.-à-d. 12-16 h), comme déjà présenté et optimisé dans une étude de Beber et al.25.
- Retirez les lamelles de couverture, essuyez le scellant et la graisse et retirez les fils. Lavez et frottez la chambre avec un mouchoir en papier en utilisant de l’eau et de l’éthanol (≥70%) en alternance.
REMARQUE: La tension optimale et l’échelle de temps pour la croissance des GUV dépendent de nombreux paramètres, de la concentration en sel tampon à la géométrie de la chambre (c’est-à-dire la distance entre les fils et la taille de la chambre). Utilisez la même chambre chaque fois que l’expérience est répétée pour assurer la reproductibilité. Les fils sont à proximité du fond de la chambre afin que les lipides puissent être imagés par microscopie fluorescente. Imagez les lipides à chaque étape (voir Figure 1C,D) pour vous assurer que le processus d’électroformation est réussi.
- Incubation de septine avec les vésicules
- Collectez les GUV des fils à l’aide d’embouts de pipettes prédécoupés (ouverture de ~ 1 mm) qui sont amenés à proximité des fils. Ensuite, pipeter la solution tout le long du fil. Cette procédure empêche la génération de fortes coulées lamellaires qui pourraient perturber les GUV. Après cette étape, il n’est plus nécessaire de couper les pointes des pipettes. En effet, les coulées lamellaires n’endommagent pas les GUVs dans la solution.
NOTE: En raison de la présence de PI(4,5)P 2 dans le mélange lipidique, les GUVs qui ont été collectés ne doivent pas être stockés plus de2-3 heures avant l’expérience. En effet, PI(4,5)P2 se solubilise rapidement et les septines ne se lient plus aux membranes quelques heures après leur formation. Cependant, une fois que les septines sont liées à la membrane, elles restent liées pendant quelques jours. - Diluer la solution mère de septines dans Tris 10 mM (pH 8) exclusivement pour atteindre une osmolarité égale à celle du tampon de croissance ; Si nécessaire, diluer davantage la solution de septine dans le tampon d’observation. Ajouter le volume prévu de GUVs collectés (50-100 μL pour un volume total de 200 μL). Effectuer l’incubation directement dans la chambre d’observation après passivation avec de la caséine β (voir ci-dessous). Un temps d’attente de 20 à 30 minutes est nécessaire pour atteindre l’équilibre.
NOTE: L’expression et la purification des complexes octamères septiniques (levures humaines ou bourgeonnantes) sont décrites en détail dans d’autres articles17. En bref, les septines ont été exprimées dans Escherichia coli, purifiées en laboratoire en utilisant des étapes d’affinité, d’exclusion de taille et de chromatographie par échange d’ions, et stockées à -80 °C dans une solution aqueuse de 50 mM de Tris-HCl (pH 8), 300 mM KCl et 5 mM MgCl2 à ~1 mg·mL-1 (3 μM) concentration. Une concentration élevée en sel est utilisée pour éviter l’agrégation de septines. Les complexes septiniques ne doivent pas être concentrés à travers un dispositif de centrifugation par filtre, ce qui induit l’agrégation et réduit ainsi le rendement en protéines.
- Collectez les GUV des fils à l’aide d’embouts de pipettes prédécoupés (ouverture de ~ 1 mm) qui sont amenés à proximité des fils. Ensuite, pipeter la solution tout le long du fil. Cette procédure empêche la génération de fortes coulées lamellaires qui pourraient perturber les GUV. Après cette étape, il n’est plus nécessaire de couper les pointes des pipettes. En effet, les coulées lamellaires n’endommagent pas les GUVs dans la solution.
- Imagerie avec microscope confocal et/ou à disque rotatif
- Pour éviter que les GUV ne collent à la surface et/ou n’explosent, passivez la chambre d’observation en l’incubant avec une solution de caséine β 5 mg·mL-1 pendant 30 min.
- Retirer la solution de caséine β et transférer la solution septine-GUV (étape 1.4.2.) dans la chambre d’observation à l’aide d’une pipette. Laissez les GUVs sédimenter au fond de la chambre pendant 10-15 min.
REMARQUE: La différence de composition entre l’intérieur du GUV et le tampon externe crée à la fois une discordance de densité et d’indice de réfraction. En raison de l’inadéquation de l’indice de réfraction, les GUV sont visibles avec la microscopie optique à lumière de transmission. - À l’aide de la microscopie confocale, visualisez le signal fluorescent des lipides pour vérifier la qualité des GUV et l’état de lamellarité de la membrane. Évaluer la densité des septines liées aux GUV en enregistrant le signal fluorescent des septines après avoir effectué un étalonnage approprié25. Effectuer des acquisitions Z-stack à un intervalle spatial de 0,4 μm pour analyser et visualiser les déformations 3D des vésicules induites par l’interaction entre les septines et la membrane.
REMARQUE : Des objectifs d’immersion dans l’huile avec des grossissements de 60 ou 100 fois ont été utilisés. Des microscopes confocaux ou à disque rotatif standard (Table of Materials) avec des tailles de pixels de 250 nm et 110 nm, respectivement, ont été utilisés. Il faut adapter les conditions d’imagerie à un équipement donné. Aucun agent anti-photoblanchiment spécifique n’a été ajouté à la solution.

Figure 1: Électroformation des GUV. (A) Représentation schématique du procédé d’électroformation à l’aide de fils de platine. (B) Image du dispositif artisanal en téflon assemblé avec des fils de platine utilisés pour générer des GUV par électroformation. Les fils ont un diamètre de 0,5 mm et sont espacés de 3 mm. (C) GUVs (objets sphériques) observés par microscopie optique à transmission au cours du processus de croissance. La zone opaque au bas de l’image est le fil de platine. (D) GUVs (objets fluorescents ronds) observés par microscopie fluorescente pendant la croissance sur le fil de platine. Barres d’échelle = 100 μm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
2. Analyse de l’organisation ultrastructurale des filaments de septine par cryo-microscopie électronique
REMARQUE: Les vésicules ne conviennent pas à l’imagerie avec les méthodes standard de microscopie électronique. En effet, les échantillons sont séchés à l’aide de méthodes standard de coloration négative. Lors de la déshydratation, les vésicules sont susceptibles de subir des déformations non spécifiques, entraînant fréquemment des protrusions lipidiques. La cryo-microscopie électronique est donc une bien meilleure stratégie pour observer des déformations spécifiques des vésicules. En utilisant cryo-EM, les échantillons sont incorporés dans une fine couche (~100-200 nm) de glace vitrifiée, ce qui préserve les échantillons près de l’état natif. Les GUV sont cependant trop grands (plusieurs dizaines de micromètres) pour être encastrés dans de la glace mince et donc imagés par microscopie électronique à transmission. Par conséquent, de grandes vésicules unilamellaires (LUV), dont le diamètre varie de ~50-500 nm, sont générées pour déterminer comment les septines peuvent déformer les vésicules et comment elles s’arrangent sur les vésicules.
- Génération de grandes vésicules unilamellaires (LUV)
- Préparer 50 μg de mélange lipidique solubilisé dans du chloroforme avec une composition molaire de 57% EggPC, 15% de cholestérol, 10% DOPE, 10% DOPS et 8% cerveau PI(4,5)P2), qui est optimisé pour améliorer l’interaction de la septine avec la membrane dans un flacon en verre.
- Sécher la solution sous flux d’argon pour générer un film lipidique séché dans le flacon. Mettez le flacon sous vide pendant 30 minutes pour sécher complètement le lipide.
- Résoudre le film lipidique dans 50 μL de solution aqueuse (50 mM Tris-HCl [pH 8]; 50 mM KCl; 2 mM MgCl2) pour obtenir une concentration finale de 1 mg·mL-1, vortex pendant 10 s, et transférer la solution dans un tube.
NOTE: Les LUV doivent être utilisés immédiatement pour l’incubation avec des septines. Sinon, l’interaction protéine-lipide sera faible en raison de la solubilisation PI(4,5)P2. Ce processus brut de resolubilisation génère une population hétérogène de vésicules de diamètres allant de 50 nm à 500 nm. Par conséquent, toute une gamme de diamètres et donc de courbures sont analysés simultanément.
- Incuber les septines avec les lipides solubilisés à des concentrations finales de lipides et de septines de 0,1 mg·mL-1 (environ 300 nM) et 20 nM, respectivement, dans un tampon riche en sel (50 mM Tris-HCl [pH 8], 300 mM KCl, 2 mM MgCl2). Incuber l’échantillon pendant 1 h à température ambiante.
- Congélation pour vitrifier l’échantillon
- Grilles de carbone à décharge luminescente (300 mailles) côté carbone pendant 30 s à 5 mA à l’aide d’un générateur de plasma et insérer la grille dans une machine de congélation plongeante dans un environnement humide.
- Adsorber 4 μL de l’échantillon (étape 2.2.) du côté du carbone évacué par lueur de la grille. Immédiatement avant l’adsorption de l’échantillon, ajouter des billes d’or de 5 à 10 nm dans la solution, au cas où les échantillons seraient utilisés pour générer des séries inclinées par cryotomographie.
NOTE: La densité des perles d’or doit être criblée et ajustée empiriquement et dépend du fournisseur. La densité optimisée est de 10-15 perles d’or dans le champ de vision. - Sécher les échantillons du côté nu pour aspirer la goutte d’échantillon du côté opposé.
REMARQUE : Le temps de buvard est généralement de 4 s, et la position du papier filtre et la force de buvard sont ajustées empiriquement en testant et en criblant d’autres positions du papier filtre afin d’optimiser la qualité de la glace (épaisseur) et la densité du matériau. - Transférez les grilles dans un microscope ou stockez-les dans un récipient d’azote liquide.
REMARQUE: L’utilisation de grilles de carbone trouées est essentielle pour tenir compte de la polydispersité de la taille des vésicules. L’éponge l’échantillon du côté opposé favorise une meilleure adsorption du matériel biologique sur la grille.
- Imagerie par cryo-microscopie électronique
- Insérez les grilles dans un microscope électronique (EM) équipé pour l’observation cryo-EM. Examinez toute la grille en générant une carte de l’échantillon entier à faible grossissement (généralement à un grossissement de 120x) pour sélectionner des zones affichant une meilleure glace (c.-à-d. mince et bien vitrifiée).
- Pour la collecte de données cryo-EM 2D, collectez des images à une taille de pixel d’environ 2 Å par pixel pour vérifier la qualité de l’échantillon. Assurez-vous que les filaments de septine et la bicouche lipidique sont visibles.
- Pour la collecte de données de cryotomographie électronique, sélectionnez les zones d’intérêt affichant des vésicules déformées. Assurez-vous qu’il y a suffisamment de perles d’or (au moins 10) dans le champ de vision.
- Selon le logiciel utilisé pour la collecte de données en série inclinée, sélectionnez les positions de mise au point et de suivi pour être suffisamment éloignées de la zone d’intérêt. Collectez les séries inclinées en faisant varier l’angle d’inclinaison de -60° à +60°, en collectant une image tous les 2°-3° degrés.
NOTE: La dose totale doit être d’environ ou moins de 100 électrons/Å2. La taille des pixels varie de 1,3 Å à 2,1 Å, selon le microscope utilisé pour la collecte des données. Idéalement, un schéma symétrique angulaire pour la collecte de données est préféré comme suit: 0°, -3°, +3°, -6°, +6°, -9°, +9° [...] -60°, +60°. Cependant, les goniomètres des microscopes à entrée latérale n’offrent pas une stabilité mécanique suffisante pour réaliser ces schémas symétriques. Alternativement, l’acquisition peut être commencée à 0° à 34°, suivie d’une deuxième séquence angulaire de -2° à -60°, et terminée par une séquence finale de 36° à 60°. Le but est de collecter les premières images (avec les dommages de rayonnement les plus faibles) aux angles les plus bas. De plus, pour visualiser l’ultrastructure des vésicules et des filaments de septine liés, un microscope cryo-EM standard peut être utilisé (200 kV, filament d’hexaborure de lanthane (LaB6) et entrée latérale de l’échantillon). Cependant, si l’on souhaite poursuivre le traitement de l’image (moyenne sous-tomogramme, par exemple), il est préférable d’utiliser des microscopes à canon à émission de champ (FEG) de dernière génération équipés de détecteurs directs. Dans ce protocole, nous limitons notre description à l’acquisition de reconstructions 3D et laissons de côté la moyenne des sous-tomogrammes.
- Reconstruction 3D à partir de la cryotomographie et de la segmentation
- Utilisez la suite logicielle IMOD (Table of Materials) pour l’alignement d’images en série inclinée et la reconstruction 3D26,27. Dans IMOD, effectuez un alignement de série d’inclinaison basé sur le positionnement fiduciaire (perles d’or). En outre, si nécessaire, effectuer la détermination et la correction de la fonction de transfert de contraste (CTF) dans IMOD26. Enfin, réalisez une reconstruction 3D avec IMOD, en suivant chaque étape avec rigueur.
- Segmentez manuellement les bicouches lipidiques et les filaments de septine à l’aide de 3Dmod27 de la suite logicielle IMOD, pour l’affichage.
3. Analyse de la sensibilité à la courbure de la septine à l’aide du MEB
REMARQUE: Pour comprendre comment les septines peuvent être sensibles aux courbures micrométriques, une approche in vitro a été utilisée pour incuber des complexes de filaments de septine avec des bicouches lipidiques solides déposées sur des motifs ondulés à l’échelle micrométrique.
- Conception d’une réplique ondulée de NOA (Norland optical adhesive) à partir de motifs ondulés en polydiméthylsiloxane (PDMS)
- Utiliser des motifs ondulés PDMS d’amplitude de 250 nm et de périodicité latérale de 2 μm ou d’autres dimensions pour tester les courbures appropriées pour la protéine d’intérêt.
REMARQUE : Les modèles ondulés PDMS sont conçus et générés comme décrit dans Nania et al.28,29. - Dans un environnement de salle blanche, déposer 5 μL de NOA liquide sur une lamelle circulaire en verre de 1 cm de diamètre et placer le gabarit PDMS sur la goutte. Traiter à la lumière UV (320 nm) pendant 5 min pour photopolymériser le NOA liquide en un mince film polymère. Ensuite, retirez délicatement le gabarit PDMS de la feuille de couverture avec le NOA fraîchement polymérisé.
REMARQUE: NOA est une colle optiquement transparente commune adaptée à l’imagerie par microscopie optique. De plus, le NOA est résistant aux processus de fixation chimique et de coloration effectués avant l’imagerie SEM. Les résines NOA 71 et NOA 81 peuvent être utilisées avec des résultats similaires. Le modèle PDMS initial pouvait être utilisé plusieurs fois pour produire des répliques NOA. Les répliques de NOA obtenues peuvent être conservées dans une boîte à température ambiante pendant des mois.
- Utiliser des motifs ondulés PDMS d’amplitude de 250 nm et de périodicité latérale de 2 μm ou d’autres dimensions pour tester les courbures appropriées pour la protéine d’intérêt.
- Génération d’une bicouche lipidique soutenue et incubation de protéines
- Traiter les films NOA à l’aide d’un purificateur plasma d’air pendant 5 minutes pour rendre la surface hydrophile.
- Préparer une solution de petites vésicules unilamellaires (VUS) avec une composition molaire de 57% d’EggPC, 15% de cholestérol, 10% de DOPE, 10% de DOPS et 8% de PI(4,5)P2 dans le cerveau à une concentration lipidique totale de 1 mg·mL-1. Préparer les VUS en remettant en suspension un film lipidique séché dans le tampon d’observation, comme décrit à l’étape 2.1.3. Sonifier doucement la solution à l’aide d’un sonicateur de bain pendant 5-10 minutes jusqu’à ce que la solution soit transparente.
REMARQUE: La solution des SUV peut être conservée congelée pendant plusieurs semaines à -20 °C. - Insérez les lamelles de couverture soutenant les modèles d’avis d’anomalie dans les puits des boîtes de culture cellulaire. Déposer 100 μL de solution SUV à 1 mg·mL-1 sur les modèles de NOA fraîchement déchargés (étape 3.2.1.) et incuber pendant 30 min à température ambiante. Cette étape induit la fusion des VUS avec la surface du diagramme NOA pour générer une bicouche lipidique supportée.
- Rincez abondamment les lames 6x avec le tampon septine (50 mM Tris-HCl [pH 8], 50 mM KCl, 2 mM MgCl2) pour éliminer le SUV non fusionné. Après chaque rinçage, ne laissez jamais l’échantillon sécher complètement.
- Diluer la solution mère de septine octamère dans un tampon de septine à haute teneur en sel (50 mM Tris-HCl [pH 8], 300 mM KCl, 2 mM MgCl2) en utilisant un tampon septine (50 mM Tris-HCl [pH 8], 50 mM KCl, 2 mM MgCl2) à des concentrations finales allant de 10 nM à 100 nM et des volumes de 1 mL. Incuber la solution protéique sur les lames pendant ~1 h à température ambiante.
REMARQUE: Le volume est suffisamment grand pour que l’évaporation ne soit pas un problème.
- Préparation des échantillons pour l’analyse SEM
NOTE: Divers protocoles ont été développés en microscopie électronique pour analyser les structures protéiques et leur assemblage. Le protocole actuel préserve l’organisation des filaments de septine, en utilisant un protocole de fixation dérivé de Svitkina et al.30 C’est facile à mettre en œuvre. En outre, ce protocole optimise les observations SEM haute résolution.- Préparation des réactifs et des solutions mères
REMARQUE: Ce protocole nécessite plusieurs réactifs et solutions qui peuvent être préparés à l’avance ou juste avant l’incubation. Suivez les instructions fournies pour éviter tout artefact ou manque de réactivité chimique.- Solution mère de cacodylate de sodium 0,2 M : Pour préparer cette solution à double résistance, dissoudre 2,14 g de poudre de cacodylate de sodium dans ~ 40 mL d’eau distillée sous agitation magnétique. Après dissolution complète, ajuster le pH à 7,4 en ajoutant doucement 0,1 M HCl (~1 mL pour 50 mL de solution) et compléter le volume final avec de l’eau distillée. Cette solution peut être conservée pendant 24-48 h à 4°C.
- 2% de glutaraldéhyde (GA) dans du cacodylate de sodium 0,1 M (solution fixatrice): Préparer cette solution juste avant utilisation en diluant du GA de qualité EM très pure avec une solution de cacodylate de sodium 0,2 M (voir ci-dessus). Utilisez une solution GA commerciale de 25 % à 50 % en raison de sa capacité de stockage optimisée à 4 °C. Pour préparer 10 mL de solution fixatrice, diluer 0,8 mL de solution commerciale de GA à 25 % avec 4,2 mL d’eau distillée et 5 mL de cacodylate de sodium 0,2 M.
- Tétroxyde d’osmium à 1 % (OsO 4) dans du cacodylate de sodium 0,1 M : Préparer cette deuxième solution fixatrice juste avant utilisation en diluant la solution mère commerciale à 4 % d’OsO4 avec du cacodylate de sodium 0,2 M (voir ci-dessus). Pour 4 mL de solution fixatrice d’OsO 4, diluer 1 mL de solution commerciale d’OsO 4 à4 % avec 1 mL d’eau distillée et 2 mL de cacodylate de sodium 0,2 M.
REMARQUE: Utilisez une solution mère OsO 4 commerciale à4 % dans les ampoules en verre décolleables en raison de leurs propriétés de stockage et de manipulation. Notez qu’OsO4 est très réactif. Assurez-vous que sa couleur est légèrement jaune et non foncée. - 1% d’acide tannique (TA) dans l’eau: Préparer cette solution juste avant utilisation. Préparer la solution de TA pour obtenir une concentration finale de 1% TA dans de l’eau distillée à température ambiante. Dissoudre 10 mg dans 1 mL d’eau distillée et vortex pendant quelques minutes. La solution de TA ne peut pas être conservée et doit être filtrée avec un filtre de 0,2 μm avant utilisation.
- Solution d’acétate d’uranyle (UA) à 1 % dans l’eau Préparer la solution d’AU pour obtenir une concentration finale de 1 % d’UA dans l’eau distillée. Dissoudre 10 mg dans 1 mL d’eau distillée et vortex pendant au moins 30 minutes à 1 h en remuant ou en agitant à température ambiante. Cette solution peut être conservée pendant 1 mois à 4 °C mais peut facilement précipiter et doit donc être filtrée avec un filtre de 0,2 μm avant utilisation.
- Séries graduées de solutions d’éthanol Préparer des solutions d’éthanol à 50 %, 70 %, 95 % et 100 % dans de l’eau. Préparer le bain final à partir de bouteilles d’éthanol 100 % ou d’éthanol à 100 % nouvellement ouvertes qui ont été déshydratées pendant au moins 24 h avec des tamis moléculaires (diamètre nominal des pores = 4 Å) pour éliminer des échantillons toute trace d’eau qui pourrait interférer avec le séchage et le revêtement. Veillez à ne pas secouer cette solution car elle pourrait provoquer la remise en suspension des particules de silicate.
NOTE: Soyez prudent lorsque vous manipulez les réactifs chimiques utilisés dans ce protocole. Le glutaraldéhyde, le tétroxyde d’osmium, le cacodylate de sodium et l’acétate d’uranyle sont tous hautement toxiques, l’acétate d’uranyle étant également radioactif. Toute manipulation des réactifs et de leurs déchets doit être effectuée à l’aide d’une protection individuelle (gants, sarraus, lunettes de sécurité) et collective (hottes et écrans en plexiglas), selon les procédures spécifiques au laboratoire.
- Fixation de l’échantillon
- Lavez les échantillons (c.-à-d. lames d’ANO avec des bicouches lipidiques soutenues fusionnées et des protéines incubées) avec du PBS. Remplacer le PBS par la solution fixatrice GA préchauffée à 37 °C et laisser la réaction se dérouler pendant 15 min. Les échantillons peuvent ensuite être conservés à 4 °C.
- Retirer la solution fixatrice et laver les échantillons fixés 3x avec 0,1 M de cacodylate de sodium (5 min par lavage) en secouant doucement.
- Incuber les échantillons dans la solution fixatrice OsO4 pendant 10 min avec une protection maximale contre la lumière pour permettre la fixation des structures membraneuses et augmenter la conductivité électrique des échantillons. Retirer la solution fixatrice et laver les échantillons 3x dans de l’eau distillée (5 min par lavage) en agitant doucement.
- Incuber les échantillons lavés dans la solution fixatrice TA filtrée pendant 10 minutes maximum. Retirer la solution de TA et laver les échantillons 3x dans de l’eau distillée (5 min par lavage) en secouant doucement.
- Incuber les échantillons lavés dans une solution fixatrice UA fraîchement filtrée pendant 10 min avec une protection maximale contre la lumière. Retirer la solution d’UA et laver les échantillons 3x dans de l’eau distillée (5 min par lavage) en agitant doucement.
- Déshydratation des échantillons et séchage au point critique
NOTE: Le séchage à l’air n’est pas autorisé pour éviter d’endommager les échantillons par des tensions superficielles indésirables lors du passage de l’état liquide à l’état gazeux. Deux méthodes ont été établies dans la ME: la méthode physique permettant d’atteindre l’état supercritique de CO2 et de contourner son point critique (31 °C, 74 bar), ou la méthode chimique d’évaporation de l’hexaméthyldisilazane (HMDS), un agent desséchant à tension superficielle réduite. Le CO2 et le HMDS sont peu miscibles avec l’eau. Par conséquent, toutes les traces d’eau doivent être remplacées par un solvant de transition (éthanol) pour éviter tout dommage ultérieur au cours des processus de séchage.- Incuber les échantillons pendant 2-3 min dans chaque solution d’éthanol, en commençant par un bain d’éthanol (anhydre) à 50% à 100%.
REMARQUE: Comme l’évaporation de l’éthanol entre les bains est rapide, la manipulation de l’échantillon doit également être rapide pour éviter tout séchage à l’air. - Transférez les lames de verre à l’intérieur du sécheur à point critique prérempli d’éthanol et suivez les instructions du fabricant.
REMARQUE: Dans ce protocole, un appareil automatique a été utilisé, mais n’importe quel système peut être utilisé. Les protocoles peuvent différer pour chaque appareil mais doivent être optimisés pour éliminer complètement l’éthanol (25 bains dans ce protocole) et réduire les vitesses pour les différents échanges de solvants (entrées de CO 2 et sorties éthanol/CO2) et la dépressurisation finale (près de 1 h dans ce protocole). - À la fin du séchage du point critique, conserver immédiatement les échantillons dans un dessiccateur jusqu’à ce qu’ils soient montés et revêtements. Comme les échantillons séchés sont très hygroscopiques, enduisez-les (voir ci-dessous) dès que possible.
REMARQUE: Alternativement, HMDS peut offrir une méthode moins chère et plus rapide. Bien que HDMS n’ait jamais été testé sur nos échantillons, cette approche a produit de bons résultats pour l’observation des protéines à la face interne des membranes cellulaires31,32.
- Incuber les échantillons pendant 2-3 min dans chaque solution d’éthanol, en commençant par un bain d’éthanol (anhydre) à 50% à 100%.
- Montage et revêtement d’échantillons.
REMARQUE: Étant donné que les échantillons biologiques présentent de mauvaises propriétés de conductivité électrique, ils doivent être recouverts d’un film métallique conducteur avant l’observation SEM. La pulvérisation cathodique plasma-magnétron est donc utilisée.- Attachez le bordereau de couverture aux talons qui seront utilisés pour les observations futures du SEM. Utilisez de la peinture argentée en raison de sa conductivité électrique améliorée, par rapport aux disques de carbone. Ajoutez une bande de peinture argentée sur la face supérieure du bordereau de couverture et assurez-vous que la connexion avec le talon est satisfaisante. Évitez tout agglomérat de peinture.
REMARQUE : La bande de peinture argentée doit être mince pour éviter tout contact entre l’échantillon et la lentille d’objectif du MEB lorsque vous travaillez à haute résolution (c.-à-d. à faible distance de travail). - Attendez que le solvant s’évapore complètement.
REMARQUE: La durée de cette étape peut varier en fonction de la quantité et de l’épaisseur du dépôt de peinture argentée. Cette étape peut être raccourcie en utilisant un clocher ou l’enrobage et un aspirateur primaire pendant 10-30 min. - Utiliser un appareil équipé d’une tête de pulvérisation magnétron plasma et d’un étage rotatif-planétaire, et suivre les protocoles standard fournis par le fabricant. Ici, l’appareil a été évacué à 2,5 x 10-5 mbar, purgé 1x avec de l’argon de haute qualité, puis ajusté à 8,0 x 10-3 mbar.
- Effectuer une pré-pulvérisation cathodique (120 mA pendant 60 s) pour enlever la couche d’oxyde à la surface. Ensuite, déposez 1,5 nm de tungstène (90 mA, distance de travail = 50 mm) à l’aide d’un moniteur d’épaisseur de film.
NOTE: L’enduit doit être parfaitement nettoyé pour assurer la reproductibilité de l’évaporation du film. Le revêtement doit être arrêté une fois que l’épaisseur visée a été atteinte. L’épaisseur finale du film est calculée et corrigée par la suite. Les films d’environ 1,5 nm ont, en moyenne, une post-correction de 0,7 nm à l’aide de notre appareil. Comme ces valeurs de correction sont proches de la cible, une série de revêtements est effectuée pour évaluer puis soustraire cette correction de la valeur cible. - Conservez les échantillons sous vide pour les protéger de l’air ambiant jusqu’à et pendant toutes les analyses SEM.
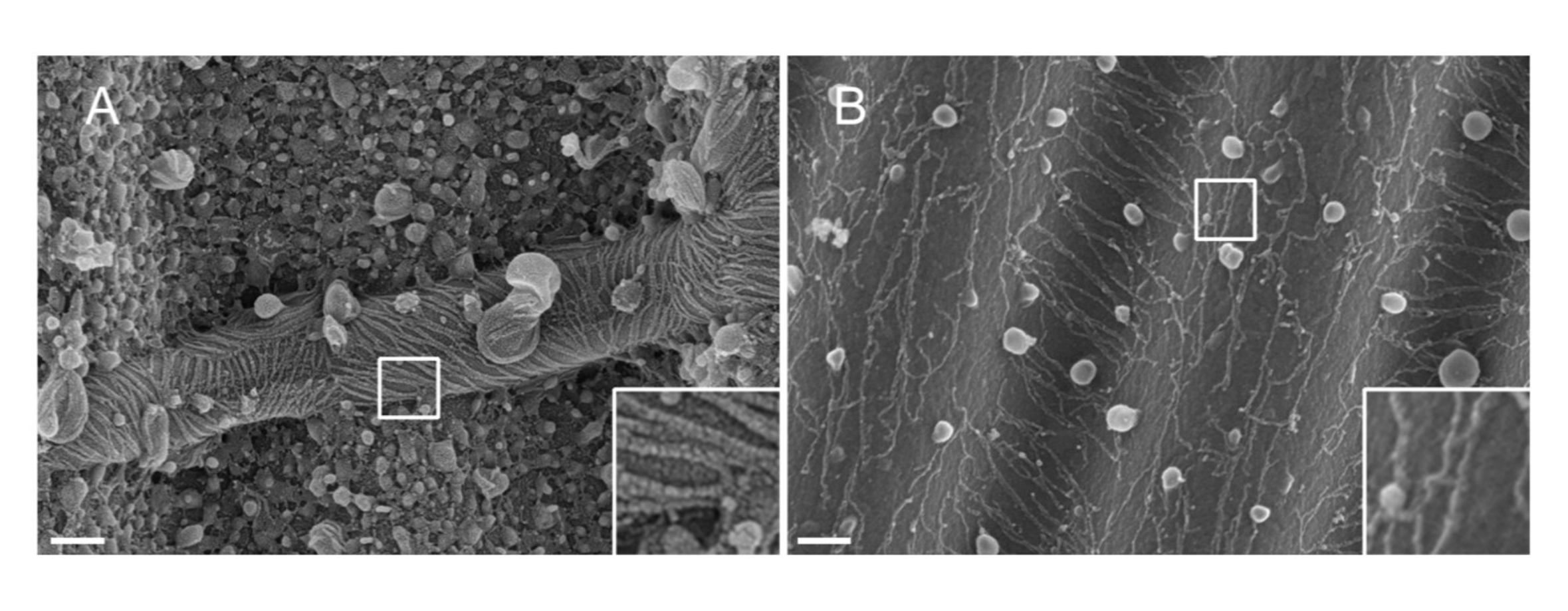
NOTE: La nature du métal utilisé pour pulvériser des matières. Le Pt, bien qu’il s’agisse d’un matériau courant, donne un revêtement de mauvaise qualité avec une épaisseur de film Pt adaptée aux filaments de septine (1,5 nm). À haute résolution, un film Pt de 1,5 nm manque de cohésivité; la taille des grappes de Pt et des filaments de septine devient ainsi similaire, ce qui entraîne des interprétations erronées au cours du processus de segmentation des filaments33 (voir figure 2A, encadré). Le tungstène est une bonne alternative au Pt car il présente une granulométrie plus petite, à peine visible au MEB haute résolution (voir Figure 2B, encadré). Néanmoins, le tungstène pur est facilement oxydé, ce qui conduit à de forts artefacts d’effet de charge lors de l’observation SEM si la procédure détaillée à l’étape 3.3.4. n’est pas strictement suivi.
- Attachez le bordereau de couverture aux talons qui seront utilisés pour les observations futures du SEM. Utilisez de la peinture argentée en raison de sa conductivité électrique améliorée, par rapport aux disques de carbone. Ajoutez une bande de peinture argentée sur la face supérieure du bordereau de couverture et assurez-vous que la connexion avec le talon est satisfaisante. Évitez tout agglomérat de peinture.
- Préparation des réactifs et des solutions mères
- Acquérir les images à l’aide d’un microscope MEB à émission de champ (FESEM).
REMARQUE : Les technologies MEB ont récemment été mises à niveau pour améliorer la résolution, et les technologies dépendent du fabricant (p. ex. , optique électronique, décélération du faisceau, lentille magnétique). Ce gain de résolution (accessible auprès de plusieurs constructeurs), notamment à une faible tension d’accélération (proche du nanomètre à 1 kV), est nécessaire pour résoudre des structures nanométriques similaires aux réseaux de septines.- Obtenez une imagerie haute résolution grâce à la détection d’électrons secondaires primaires (SE1) avec le détecteur « dans la lentille » en utilisant les paramètres suivants.
- Réglez la tension d’accélération sur 3 kV. Fixez le courant du faisceau avec l’ouverture de 20 μm (pour les colonnes Zeiss Gemini I, ou équivalent à 34 pA) ou avec l’ouverture de 15 μm (pour les colonnes Zeiss Gemini I, ou équivalent à 18,5 pA), si la suppression des effets de charge est nécessaire.
- Pour les observations, utilisez des résolutions allant de 21,25 nm/pixel à 1,224 nm/pixel, et pour l’analyse des données, utilisez une résolution de ~5,58 nm/pixel (grossissement de 20 000x selon la référence Polaroid 545).
- Réglez la distance de travail entre 1 mm et 2 mm pour une observation à haute résolution, et environ 3 mm si une profondeur de champ accrue est nécessaire. Ajustez la vitesse de numérisation et l’intégration de la ligne en continu pour garantir un rapport signal sur bruit constant avec un temps d’acquisition d’environ 30 à 45 s par image.

Figure 2 : Effet du matériau déposé sur les filaments de septine sur les motifs PDMS ondulés. MEB de filaments de septine recouverts par pulvérisation cathodique soit de (A) 1,5 nm de platine, présentant le motif de « sol fissuré aride » typique d’un manque de cohésion entre les grappes de noyaux de platine, soit (B) de 1,5 nm de tungstène recouvert d’une couche lisse et cohésive. Barre d’échelle = 200 nm. Les cases carrées blanches représentent les vues agrandies en bas à droite. Les globules sphériques sont de petites vésicules lipidiques interagissant avec les septines. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Résultats
Déformations des GUV
Des images confocales typiques de fluorescence de GUV remodelés après avoir été incubés avec des septines sont présentées à la figure 3, dans des conditions où les septines polymérisent. Les GUV nus (Figure 3A) étaient parfaitement sphériques. Lors de l’incubation avec plus de 50 nM de filaments de septine de levure bourgeonnante, les vésicules sont apparues déformées. Jusqu’à une concentration de 100...
Discussion
Comme indiqué ci-dessus, un mélange lipidique a été utilisé qui améliore l’incorporation de PI(4,5)P2 dans la bicouche lipidique et facilite ainsi les interactions septine-membrane. En effet, nous avons montré ailleurs25 que les septines de levure bourgeonnantes interagissent avec les vésicules de manière spécifique à PI(4,5)P2. Cette composition lipidique a été ajustée empiriquement à partir du criblage de plusieurs compositions et est maintenant largement u...
Déclarations de divulgation
Les auteurs n’ont aucun conflit d’intérêts.
Remerciements
Nous remercions Patricia Bassereau et Daniel Lévy pour leurs précieux conseils et discussions. Ces travaux ont bénéficié du soutien de l’ANR (Agence Nationale de la Recherche) pour le financement du projet « SEPTIME », ANR-13-JSV8-0002-01, de l’ANR septimorf ANR-17-CE13-0014, et du projet « SEPTSCORT », ANR-20-CE11-0014-01. B. Chauvin est financé par l’Ecole Doctorale « ED564: Physique en Ile de France » et la Fondation pour lea Recherche Médicale. K. Nakazawa a été soutenu par Sorbonne Université (AAP Emergence). G.H. Koenderink a été soutenu par la Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO/OCW) par le biais du « BaSyC-Building a Synthetic Cell ». Subvention de gravitation (024.003.019). Nous remercions le Labex Cell(n)Scale (ANR-11-LABX0038) et Paris Sciences et Lettres (ANR-10-IDEX-0001-02). Nous remercions l’Imagerie Cellulaire et Tissulaire (PICT-IBiSA), Institut Curie, membre de l’Infrastructure Nationale de Recherche Français France-BioImagerie (ANR10-INBS-04).
matériels
| Name | Company | Catalog Number | Comments |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine | Avanti Polar Lipids | 850725 | |
| 1,2-dioleoyl-sn-glycero-3-phospho-L-serine | Avanti Polar Lipids | 840035 | |
| Bath sonicator | Elma | Elmasonic S10H | |
| Bodipy-TR-Ceramide | invitrogen, Thermo Fischer scientific | 11504726 | |
| Chemicals: NaCl, Tris-HCl, sucrose, KCl, MgCl2, B-casein, chloroform, sodium cacodylate, tannic acid, ethanol | Sigma Aldrich | ||
| Confocal microscope | nikon | spinning disk or confocal | |
| Critical point dryer | Leica microsystems | CPD300 | |
| Deionized water generator | MilliQ | F1CA38083B | MilliQ integral 3 |
| Egg L-α-phosphatidylcholine | Avanti Polar Lipids | 840051 | |
| Field Emission Gun SEM (FESEM) | Carl Zeiss | Gemini SEM500 | |
| Glutaraldehyde 25 %, aqueous solution | Thermo Fischer scientific | 50-262-19 | |
| High vacuum grease, Dow corning | VWR | ||
| IMOD software | https://bio3d.colorado.edu/imod/ | software suite for tilted series image alignment and 3D reconstruction | |
| Lacey Formvar/carbon electron microscopy grids | Eloise | 01883-F | |
| Lipids | Avanti Polar Lipids | ||
| L-α-phosphatidylinositol-4,5-bisphosphate | Avanti Polar Lipids | 840046 | |
| Metal evaporator | Leica microsystems | EM ACE600 | |
| NOA (Norland Optical Adhesives), NOA 71 and NOA 81 | Norland Products | NOA71, NOA81 | |
| Osmium tetraoxyde 4% | delta microscopies | 19170 | |
| Osmometer | Löser | 15 M | |
| Plasma cleaner | Alcatel | pascal 2005 SD | |
| Plasma generator | Electron Microscopy Science | ||
| Plunge freezing equipment | leica microsystems | EMGP | |
| Transmission electron microscope | Thermofischer | Tecnai G2 200 kV, LaB6 | |
| Uranyl acetate | Electron Microscopy Science | 22451 | this product is not available for purchase any longer |
| Wax plates, Vitrex | VWR |
Références
- Finger, F. P. Reining in cytokinesis with a septin corral. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology. 27 (1), 5-8 (2005).
- Barral, Y., Kinoshita, M. Structural insights shed light onto septin assemblies and function. Current Opinion in Cell Biology. 20 (1), 12-18 (2008).
- Hu, Q., et al. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 329 (5990), 436-439 (2010).
- Lin, Y. -. H., Kuo, Y. -. C., Chiang, H. -. S., Kuo, P. -. L. The role of the septin family in spermiogenesis. Spermatogenesis. 1 (4), 298-302 (2011).
- Addi, C., Bai, J., Echard, A. Actin, microtubule, septin and ESCRT filament remodeling during late steps of cytokinesis. Current Opinion in Cell Biology. 50, 27-34 (2018).
- Spiliotis, E. T., Kesisova, I. A. Spatial regulation of microtubule-dependent transport by septin GTPases. Trends in Cell Biology. 31 (12), 979-993 (2021).
- Spiliotis, E. T., Nakos, K. Cellular functions of actin- and microtubule-associated septins. Current Biology: CB. 31 (10), 651-666 (2021).
- Salameh, J., Cantaloube, I., Benoit, B., Poüs, C., Baillet, A. Cdc42 and its BORG2 and BORG3 effectors control the subcellular localization of septins between actin stress fibers and microtubules. Current Biology: CB. 31 (18), 4088-4103 (2021).
- Ewers, H., Tada, T., Petersen, J. D., Racz, B., Sheng, M., Choquet, D. A septin-dependent diffusion barrier at dendritic spine necks. PloS One. 9 (12), 113916 (2014).
- Myles, D. G., Primakoff, P., Koppel, D. E. A localized surface protein of guinea pig sperm exhibits free diffusion in its domain. The Journal of Cell Biology. 98 (5), 1905-1909 (1984).
- Luedeke, C., Frei, S. B., Sbalzarini, I., Schwarz, H., Spang, A., Barral, Y. Septin-dependent compartmentalization of the endoplasmic reticulum during yeast polarized growth. The Journal of Cell Biology. 169 (6), 897-908 (2005).
- Gilden, J. K., Peck, S., Chen, Y. -. C. M., Krummel, M. F. The septin cytoskeleton facilitates membrane retraction during motility and blebbing. The Journal of Cell Biology. 196 (1), 103-114 (2012).
- Dolat, L., Hu, Q., Spiliotis, E. T. Septin functions in organ system physiology and pathology. Biological Chemistry. 395 (2), 123-141 (2014).
- Angelis, D., Spiliotis, E. T. Septin mutations in human cancers. Frontiers in Cell and Developmental Biology. 4, 122 (2016).
- Takehashi, M., et al. Septin 3 gene polymorphism in Alzheimer's disease. Gene Expression. 11 (5-6), 263-270 (2004).
- Shuman, B., Momany, M. Septins from protists to people. Frontiers in Cell and Developmental Biology. 9, 824850 (2022).
- Bertin, A., et al. Saccharomyces cerevisiae septins: supramolecular organization of heterooligomers and the mechanism of filament assembly. Proceedings of the National Academy of Sciences of the United States of America. 105 (24), 8274-8279 (2008).
- Iv, F., et al. Insights into animal septins using recombinant human septin octamers with distinct SEPT9 isoforms. Journal of cell science. 134 (15), (2021).
- Beber, A., et al. Membrane reshaping by micrometric curvature sensitive septin filaments. Nature communications. 10 (1), 420 (2019).
- Bridges, A. A., Jentzsch, M. S., Oakes, P. W., Occhipinti, P., Gladfelter, A. S. Micron-scale plasma membrane curvature is recognized by the septin cytoskeleton. The Journal of Cell Biology. 213 (1), 23-32 (2016).
- Patzig, J., et al. Septin/anillin filaments scaffold central nervous system myelin to accelerate nerve conduction. eLife. 5, 17119 (2016).
- Szuba, A., et al. Membrane binding controls ordered self-assembly of animal septins. eLife. 10, 63349 (2021).
- Tanaka-Takiguchi, Y., Kinoshita, M., Takiguchi, K. Septin-mediated uniform bracing of phospholipid membranes. Current Biology: CB. 19 (2), 140-145 (2009).
- Bertin, A., et al. Phosphatidylinositol-4,5-bisphosphate promotes budding yeast septin filament assembly and organization. Journal of Molecular Biology. 404 (4), 711-731 (2010).
- Beber, A., et al. Septin-based readout of PI(4,5)P2 incorporation into membranes of giant unilamellar vesicles. Cytoskeleton. 76 (4,5), 92-103 (2019).
- Mastronarde, D. N., Held, S. R. Automated tilt series alignment and tomographic reconstruction in IMOD. Journal of Structural Biology. 197 (2), 102-113 (2017).
- Kremer, J. R., Mastronarde, D. N., McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. Journal of Structural Biology. 116 (1), 71-76 (1996).
- Nania, M., Foglia, F., Matar, O. K., Cabral, J. T. Sub-100 nm wrinkling of polydimethylsiloxane by double frontal oxidation. Nanoscale. 9 (5), 2030-2037 (2017).
- Nania, M., Matar, O. K., Cabral, J. T. Frontal vitrification of PDMS using air plasma and consequences for surface wrinkling. Soft Matter. 11 (15), 3067-3075 (2015).
- Svitkina, T. M., Borisy, G. G. Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods in Enzymology. 298, 570-592 (1998).
- Franck, A., et al. Clathrin plaques and associated actin anchor intermediate filaments in skeletal muscle. Molecular Biology of the Cell. 30 (5), 579-590 (2019).
- Elkhatib, N., et al. Tubular clathrin/AP-2 lattices pinch collagen fibers to support 3D cell migration. Science. 356 (6343), (2017).
- Stokroos, I., Kalicharan, D., Van Der Want, J. J., Jongebloed, W. L. A comparative study of thin coatings of Au/Pd, Pt and Cr produced by magnetron sputtering for FE-SEM. Journal of Microscopy. 189, 79-89 (1998).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.