
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Outils microfluidiques pour sonder les interactions fongiques-microbiennes au niveau cellulaire
Dans cet article
Résumé
En raison de l’opacité du sol, les interactions entre ses microbes constitutifs ne peuvent pas être facilement visualisées avec une résolution cellulaire. Ici, deux outils microfluidiques sont présentés, qui offrent de nouvelles opportunités pour étudier les interactions fongiques-microbiennes. Les appareils sont polyvalents et simples à utiliser, permettant un contrôle spatio-temporel élevé et une imagerie haute résolution au niveau cellulaire.
Résumé
Les champignons filamenteux sont des habitants prospères du sol et jouent un rôle majeur dans les écosystèmes du sol, tels que la décomposition de la matière organique et inorganique, ainsi que la régulation des niveaux de nutriments. Ils y trouvent également de nombreuses occasions d’interagir avec une variété d’autres microbes tels que des bactéries ou d’autres champignons. L’étude des interactions fongiques au niveau cellulaire, cependant, peut être difficile en raison de la nature du sol ressemblant à une boîte noire. De nouveaux outils microfluidiques sont en cours de développement pour l’étude des interactions fongiques; deux plateformes conçues pour étudier les interactions bactérien-fongique et fongique-fongique sont mises en évidence. Au sein de ces microcanaux, les interactions fongiques-microbiennes peuvent être surveillées dans des environnements physico-chimiques contrôlés à une résolution temporelle et spatiale plus élevée qu’auparavant. L’application de ces outils a donné lieu à de nombreuses nouvelles connaissances biologiques, telles que l’observation de l’attachement polaire bactérien aux hyphes ou la révélation d’antagonismes fongiques-fongiques non caractérisés. Une caractéristique clé de ces méthodologies concerne la facilité d’utilisation de cet outil par des non-experts, produisant des technologies hautement traduisibles pour une utilisation dans les laboratoires de microbiologie.
Introduction
Le sol est un environnement exceptionnellement diversifié contenant une abondance de micro-organismes qui jouent un rôle déterminant dans les cyclesdu carbone et du phosphore 1,2. Les champignons filamenteux sont une composante majeure de nombreux écosystèmes en tant que décomposeurs de matière organique et inorganique et peuvent améliorer la nutrition des plantes grâce à la formation de relations symbiotiques 3,4. Dans le sol, les champignons interagissent dynamiquement avec une multitude de microbes tels que d’autres champignons5, bactéries6, virus7 et nématodes8. Ces interactions ont des conséquences importantes sur la santé des sols et des plantes. Pourtant, en raison d’un manque de systèmes expérimentaux appropriés capables d’imager les micro-organismes en interaction avec une haute résolution, beaucoup restent indéfinis.
Les recherches concernant les interactions bactériens-fongiques (IFI) et les interactions fongiques-fongiques (IFF) ont des applications précieuses dans divers domaines, y compris les antimicrobiens en médecine et les agents de lutte biologique en agriculture. Par exemple, le champignon Coprinopsis cinerea produit le peptide copsine, dont il a été démontré qu’il présente une activité antibactérienne contre l’agent pathogène humain Listeria monocytogenes9. De même, le composé dérivé du champignon, la griséofulvine, est largement utilisé comme traitement des infections fongiques humaines et est en outre capable d’inhiber la croissance du champignon phytopathogène Alternaria solani10,11. Il a également été démontré que plusieurs souches de la bactérie bacillus subtilis sont des agents de lutte biologique efficaces de l’agent pathogène fongique des plantes Rhizoctonia solani12,13. Néanmoins, en raison des limites associées aux méthodologies traditionnelles, les IFI et les IFF sont mal compris au niveau des cellules individuelles.
Les études conventionnelles explorent généralement les IFI et les IFF à l’échelle macro en utilisant des plaques de gélose avec deux espèces ou plus en confrontation. Leur interaction est évaluée en mesurant les taux de croissance et la production de métabolites de l’espèce confrontante 14,15,16; cependant, cette méthodologie n’est résolue qu’au niveau de la colonie. Pour étudier les interactions au niveau cellulaire, les inoculants bactériens et fongiques peuvent être cultivés sur des lames de microscope en verre recouvertes d’agar qui sont ensuite imagées au microscope17. Néanmoins, il peut être difficile de suivre un seul hyphe à l’aide de lames de microscope en raison d’un manque de confinement, ce qui signifie que les images time-lapse sont plus difficiles à obtenir. En outre, la possibilité de confiner spatialement d’autres micro-organismes dans des régions définies du mycélium fongique ou de créer des environnements chimiques définis qui peuvent être perturbés, par exemple, n’est pas possible dans de telles configurations. La nature « boîte noire » du sol ajoute également à la complexité de l’étude des interactions fongiques-microbiennes au niveau des cellules individuelles18. En observant les espèces en interaction loin de l’incroyable diversité du microbiome du sol, la manière exacte dont les membres individuels interagissent peut être évaluée. Ainsi, il existe un besoin continu de plates-formes polyvalentes qui permettent l’imagerie unicellulaire à haute résolution des IFI et des IFF.
Les technologies microfluidiques, appelées systèmes de laboratoire sur puce, constituent une plate-forme idéale pour l’étude des BFI et des IFF au niveau des cellules individuelles. Le domaine de la microfluidique, issu des technologies développées pour l’analyse chimique et la microélectronique, a été adopté par les sciences biologiques19. Les technologies microfluidiques régulent de petits volumes de fluides au sein d’un réseau sur mesure de canaux miniaturisés, ayant au moins une dimension à l’échelle micrométrique, et leur utilisation dans la recherche biologique s’étend20. En particulier, des dispositifs microfluidiques ont été développés pour examiner la croissance des champignons filamenteux 21,22,23,24,25,26,27,28,29,30. L’un des avantages de l’utilisation de cette technologie est que le confinement des hyphes et la distribution des nutriments dans les microcanaux ressemblent davantage à la structure de l’environnement du sol que les méthodes conventionnelles d’agar31. Récemment, des plateformes microfluidiques ont été utilisées pour étudier les interactions entre les neutrophiles humains et les agents pathogènes fongiques32, les bactéries et les racines des plantes33, ainsi que les champignons et les nématodes34,35.
L’un des nombreux avantages de l’utilisation de la microfluidique pour étudier les interactions microbiennes comprend le contrôle spécifique de l’environnement des microcanaux. Par exemple, les régimes d’écoulement laminaire peuvent être exploités pour générer des gradients de concentration définis, ce qui est particulièrement utile lors de l’examen de la chimiotaxie bactérienne36. Un autre avantage est que la nature transparente du poly(diméthylsiloxane) (PDMS), un polymère élastomère biocompatible peu coûteux couramment utilisé dans la fabrication de dispositifs microfluidiques, facilite l’imagerie à haute résolution de cellules individuelles à l’aide de la microscopie à fond clair et à fluorescence37. De même, le confinement des microbes dans des microcanaux signifie que des expériences en accéléré de suivi de cellules individuelles peuvent être effectuées, permettant d’enregistrer et de quantifier les réponses cellulaires individuelles37. Enfin, comme les dispositifs microfluidiques peuvent être conçus pour être conviviaux, ils peuvent être facilement utilisés par des non-experts38.
Il est important de mieux connaître les interactions entre les micro-organismes vivant dans le sol pour améliorer les pratiques de gestion durable des écosystèmes qui maintiennent la biodiversité et atténuer l’impact des changements climatiques sur les environnements terrestres39. Ainsi, le développement de nouveaux outils microfluidiques est fondamental pour élargir la compréhension des champignons et de leurs interactions au niveau cellulaire. Le protocole ici se concentrera sur deux dispositifs microfluidiques produits pour l’étude des BFI40 et41 tels que représentés à la figure 1.

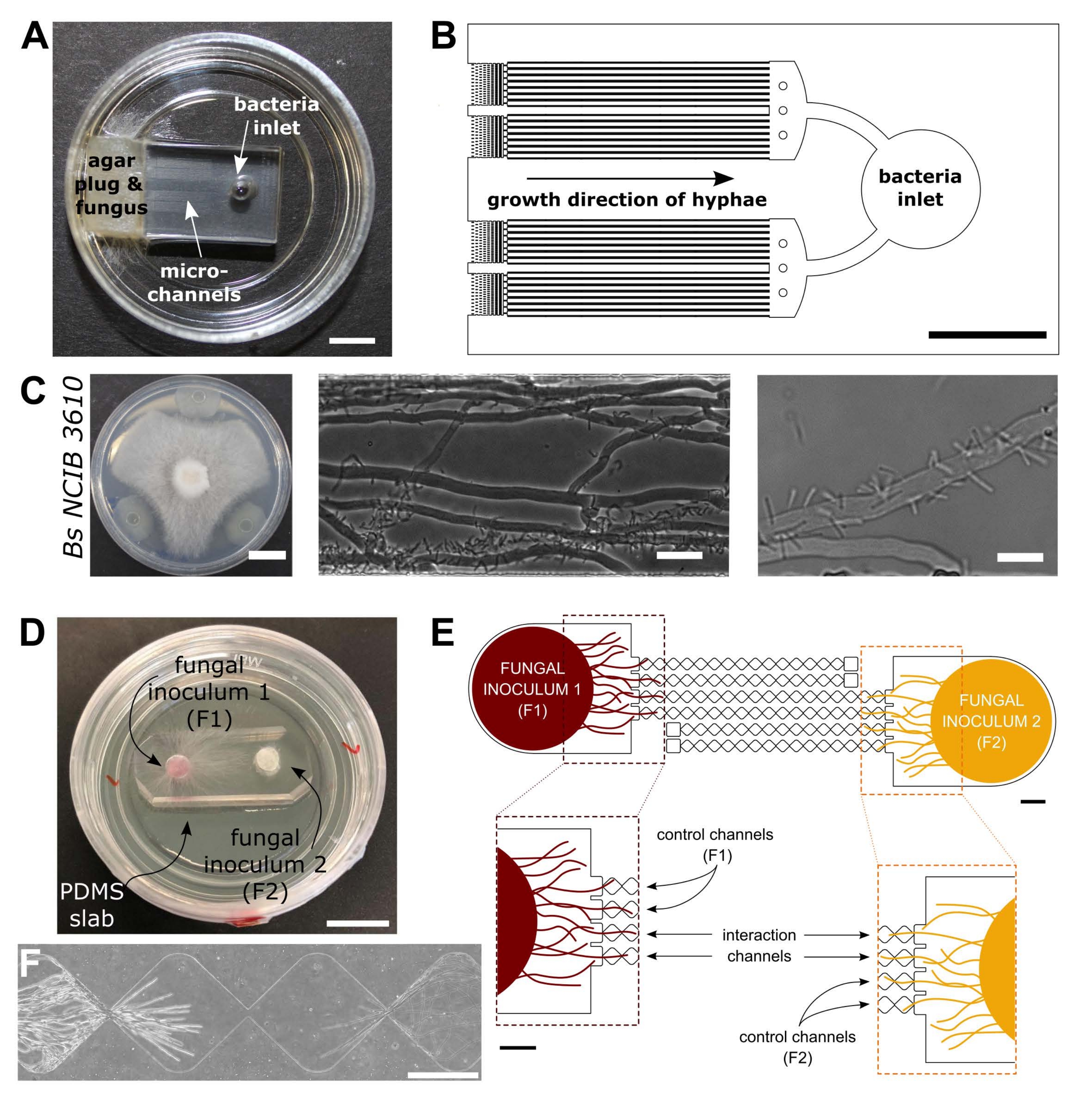
Figure 1 : Représentation visuelle et schématique des dispositifs d’interaction bactérienne-fongique (BFI) et d’interaction fongique (FFI). (A) Image du dispositif BFI. Un bouchon mycélien est placé à l’entrée d’une extrémité des microcanaux pour permettre la croissance hyphale dans l’appareil. L’entrée bactérienne se trouve à l’extrémité opposée. Barre d’échelle = 5 mm. (B) Vue d’ensemble schématique du dispositif BFI, décrivant le positionnement des entrées bactériennes et la direction de la croissance hyphale à travers les microcanaux d’interaction. Les canaux ont une profondeur de 10 μm, une largeur de 100 μm et une longueur de 7 mm, avec 28 canaux d’observation au total. (C) Essai de confrontation sur plaque de gélose entre Coprinopsis cinerea et Bacillus subtilis NCIB 3610, barre d’échelle = 20 mm (à gauche). Images microscopiques montrant l’interaction entre C. cinerea et B. subtilis NCIB 3610 dans le microcanal (milieu et droite), c’est-à-dire la fixation polaire des bactéries aux hyphes fongiques. Barre d’échelle = 25 μm (au milieu) et 10 μm (à droite). (D) Image du dispositif FFI collé à une boîte de Petri à fond de verre, double inoculée avec des bouchons mycéliens. Barre d’échelle = 1 cm. (E) Vue d’ensemble schématique du périphérique FFI. Deux bouchons d’inoculant fongiques sont introduits dans les entrées à chaque extrémité de l’appareil, permettant l’exploration hyphale des microcanaux. Les canaux de contrôle sont connectés à une seule entrée fongique et ont un canal sans issue, empêchant les interactions entre les champignons d’essai. Les canaux d’interaction relient les deux entrées fongiques et permettent des interactions hyphales entre les sujets testés au sein du microcanal. Chaque canal d’interaction se compose de 18 sections en forme de diamant, mesurant une longueur totale de 8,8 mm (490 x 430 μm par diamant), 10 μm de profondeur et ayant une région de connexion entre chaque diamant de 20 μm. Les types de canaux sont dupliqués, barres d’échelle = 1 mm. (F) Zone d’interaction entre deux fronts hyphales approchants, se développant à partir des extrémités opposées du canal d’interaction interconnecté. Image de microscopie à contraste de phase, barre d’échelle = 250 μm. Les panneaux de cette figure ont été modifiés à partir de Stanley et al., 2014 (A-C)40 et Gimeno et al., 2021 (D-F)41. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protocole
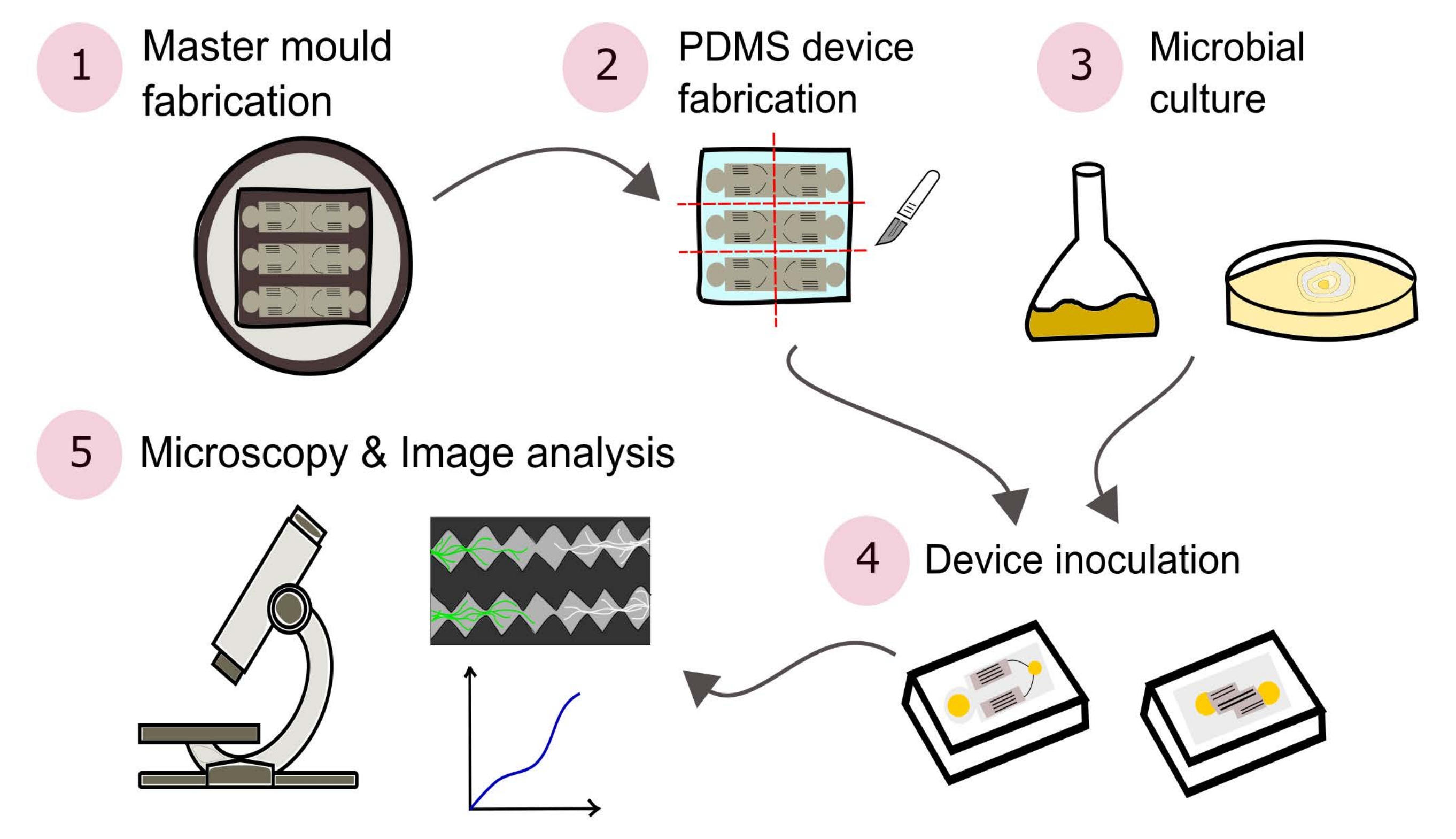
REMARQUE : Un résumé des procédures décrites dans ce protocole est illustré visuellement à la figure 2.

Figure 2 : Représentation schématique de la méthodologie présentée, composée de cinq grandes sections détaillées dans le présent protocole. Les conceptions d’appareils sont créées à l’aide d’un logiciel de conception assistée par ordinateur (CAO) et d’un moule maître fabriqué à l’aide de la photolithographie (1). Ceci est utilisé pour couler le poly(diméthylsiloxane) (PDMS), qui est ensuite coupé en dés en dalles et collé à des boîtes de Petri à fond de verre pour former les dispositifs microfluidiques (2). Les microbes à inclure dans l’étude sont cultivés (3) et utilisés pour inoculer les dispositifs (4). Les interactions sont étudiées à l’aide de la microscopie et quantifiées à l’aide de techniques d’analyse d’images (5). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
1. Fabrication de moules maîtres
- Production de photomasques
- Générez des conceptions de dispositifs microfluidiques à l’aide d’un logiciel de conception assistée par ordinateur (CAO). Les dimensions des dispositifs présentés sont données à la figure 1 et plus de détails sur les caractéristiques de conception spécifiques sont énumérés de manière exhaustive dans les publications respectives40,41.
- Exportez le fichier de conception CAO en utilisant un format approprié (par exemple, .dwg, .dxf). Imprimez un masque de photolithographie de film en envoyant le fichier de conception CAO exporté à un fournisseur commercial pour impression.
- Photolithographie
REMARQUE: Les étapes suivantes doivent être effectuées dans un environnement sans poussière et à lumière contrôlée, comme une hotte à flux laminaire ou une salle blanche. Les conditions expérimentales fournies ici sont données à titre indicatif et doivent être optimisées en interne. Les auteurs recommandent de rechercher une formation spécifique et de consulter les protocoles établis42.- Préparer une plaquette de silicium de 100 mm en la faisant cuire au four à 200 °C pendant 2 h. Spin-coat la plaquette de silicium avec la résine photosensible SU-8 2010, visant une épaisseur cible de 10 μm, en utilisant les conditions suivantes: 500 tr/ min pendant 10 s (accélération 100 tr / min / s) et 3 000 tr / min pour 45 s (accélération 300 tr / min / s).
ATTENTION: La résine photosensible SU-8 est dangereuse, faites attention lors de la manipulation et empêchez l’inhalation et le contact avec la peau. Il est inflammable, potentiellement cancérigène et toxique pour l’environnement. - Cuire la plaquette de silicium enrobée à 95 °C pendant 2,5 min (cuisson douce). Exposez la résine photosensible à la lumière ultraviolette (UV), à l’aide du masque de photolithographie sur film et d’une dose d’énergie de 140 mJ/cm2 à une longueur d’onde de 365 nm à l’aide d’un aligneur de masque.
- Cuire la plaquette de silicium enrobée à 95 °C pendant 3,5 min (cuisson après exposition). Immerger et agiter la plaquette de silicium dans une solution de développement pendant 3 min pour révéler les structures microfabriquées en retirant la résine photosensible non exposée.
ATTENTION: La solution de développement peut être inflammable, prenez les précautions appropriées lors de la manipulation et du stockage. - Rincer avec une solution de révélateur fraîche pendant 10 s. Rincer à l’alcool isopropylique pendant 10 s et sécher à l’air. Utilisez de l’air filtré et comprimé pour vous assurer que les structures sont bien sèches. Mesurez la hauteur des structures SU-8, par exemple, à l’aide d’un profilomètre.
- Silaniser chaque moule maître avec 50 μL de chlorotriméthylsilane en appliquant une pression de vide de 50 mbar pendant 2 h. Les auteurs notent que la re-silanisation du moule maître n’a pas été jugée nécessaire.
ATTENTION : Le chlorotriméthylsilane est une substance dangereuse. Portez un équipement de protection individuelle (EPI) approprié et manipulez-le avec soin. Évitez tout contact avec la peau et les yeux et empêchez l’inhalation. Tenir à l’écart des sources d’inflammation et utiliser dans un endroit bien ventilé.
- Préparer une plaquette de silicium de 100 mm en la faisant cuire au four à 200 °C pendant 2 h. Spin-coat la plaquette de silicium avec la résine photosensible SU-8 2010, visant une épaisseur cible de 10 μm, en utilisant les conditions suivantes: 500 tr/ min pendant 10 s (accélération 100 tr / min / s) et 3 000 tr / min pour 45 s (accélération 300 tr / min / s).
2. Fabrication de l’appareil
REMARQUE: Les étapes suivantes doivent être effectuées dans un environnement exempt de poussière, tel qu’une hotte à flux laminaire.
- Préparation de dalles de poly(diméthylsiloxane) (PDMS)
- Préparez environ 40 g de PDMS en mélangeant soigneusement la base et l’agent de durcissement dans un rapport de 10:1 à l’aide d’une spatule dans un gobelet en plastique propre. Dégazez le mélange pour éliminer toutes les bulles d’air en plaçant le gobelet en plastique contenant le PDMS dans une chambre à vide (pression de vide = 50 mbar) pendant 1 h.
- Fixez le moule principal dans un support en plastique à l’aide d’un ruban adhésif transparent. Nettoyez à l’aide d’air filtré comprimé pour éliminer les particules de poussière.
REMARQUE: Alternativement, la feuille d’aluminium peut être façonnée autour d’une boîte de Petri en verre, puis utilisée pour abriter le moule principal et contenir le PDMS43. - Versez le mélange PDMS sur le centre du moule maître, en vous assurant qu’il est sur une surface plane, et laissez-le se déposer.
REMARQUE: Le mélange PDMS doit être versé aussi près que possible de la surface du moule maître et un flux continu doit être maintenu pour minimiser l’introduction de bulles d’air. Les bulles d’air peuvent être éliminées en dirigeant l’air comprimé sur la bulle ou en les extrayant à l’aide d’une aiguille fine. - Couvrez le moule principal avec un couvercle en plastique pour empêcher les particules de poussière de se déposer à la surface du PDMS. Transférer le moule maître dans un four et durcir toute la nuit à 70 °C.
- Retirez le moule principal du four et laissez refroidir. Retirez le PDMS durci du moule maître et du cadre en plastique, en prenant soin d’éviter d’endommager le moule maître / PDMS.
- Placez du ruban adhésif transparent sur les microcanaux en relief dans le PDMS pour maintenir une surface exempte de poussière. Assurez-vous que la bande est retirée avant le collage.
- Coupez le PDMS en dalles (c’est-à-dire que si plusieurs dispositifs sont inclus dans la conception sur le moule maître, beaucoup peuvent être fabriqués à partir d’un seul moulage) comme indiqué par la conception à l’aide d’une guillotine montée ou d’une lame de rasoir. Lors de la coupe de l’ouverture latérale de la dalle BFI PDMS, assurez-vous que les microcanaux sont complètement ouverts (Figure 1A). Pour la dalle FFI PDMS, assurez-vous que chaque coin est coupé pour lui permettre de s’insérer dans la boîte de Petri à fond de verre illustrée à la figure 1D.
- Percez les trous d’entrée/sortie souhaités en fonction de la conception de l’appareil. Utilisez une fraise de précision pour percer des trous d’entrée de 3,18 mm et 4,75 mm pour les dispositifs BFI et FFI exemplaires, respectivement.
- Collage de dalles PDMS pour créer des périphériques
REMARQUE: Les étapes de lavage suivantes (2.2.1-2.2.2) utilisent un nettoyeur à ultrasons rempli d’eau purifiée (ddH2O) à 37 kHz. Le lavage des dalles PDMS permet d’améliorer le collageréussi 44 et de réduire le risque de contamination. Pour manipuler les dalles PDMS, utilisez des pinces propres et soulevez-les à l’aide des trous d’entrée pour éviter d’endommager les microcanaux ou la surface de l’appareil.- Immergez les dalles PDMS dans 0,5 M de NaOH et soniquez pendant 5 min. Rincer avec du ddH2O stérile. Transférer les dalles PDMS dans une solution d’éthanol à 70% et sonicer pendant 5 min. Rincer avec du ddHstérile 20.
- Immerger les dalles PDMS dans du ddH2O stérile et soniquer pendant 5 min. Retirez les dalles PDMS du ddH2O stérile, séchez-les à l’aide d’air comprimé filtré et placez-les dans une boîte de Petri carrée stérile.
- Placer la boîte de Petri carrée contenant les dalles PDMS dans un four à 70 °C pendant 1 h pour sécher. Retirer du four et laisser refroidir dans un environnement sans poussière. Éliminez toute poussière de la surface des dalles PDMS à l’aide de ruban adhésif et/ou d’air comprimé filtré.
- Activez les surfaces des dalles PDMS et des boîtes de Petri à fond de verre à coller à l’aide d’un nettoyeur plasma avec les réglages suivants: pression d’aspiration 0,75 mbar, puissance 50%, temps de revêtement 1 min. Placez les surfaces à activer (puis à coller) vers le haut dans le nettoyeur plasma.
- Retirez les dalles PDMS et les boîtes de Petri à fond de verre du nettoyeur plasma et liez en plaçant doucement les surfaces activées en contact les unes avec les autres. Collez les dalles BFI et FFI PDMS aux boîtes de Petri à fond de verre de 35 mm et 50 mm de diamètre (épaisseur de verre 0,17 mm), respectivement.
REMARQUE: Veillez à ne pas appliquer trop de pression lors du collage, car cela peut entraîner l’effondrement des microcanaux. - Vérifiez le collage réussi en essayant simplement de retirer la dalle PDMS de la boîte de Petri à fond de verre avec une pince à épiler. Visualisez les appareils à l’œil nu ou à l’aide de la microscopie générique pour éviter l’effondrement des entrées d’inoculation ou des microcanaux.
REMARQUE : Pour les conditions saturées (c.-à-d. saturées d’eau et/ou riches en nutriments), inclure l’étape 2.2.7 du protocole. Si des conditions insaturées dans l’eau sont requises, passez à l’étape 2.2.8. Les appareils peuvent être remplis d’eau ou de médias. - Remplissez les dispositifs immédiatement après le collage en pipetant 100 μL de la solution souhaitée pour le dispositif BFI (entrée bactérienne et ouverture latérale) ou 30 μL de fluide dans chaque entrée (60 μL au total) pour le dispositif FFI. Si des bulles d’air sont présentes, celles-ci se dissiperont environ 10 minutes après le remplissage car le PDMS est poreux.
- Ajouter ddH stérile2O (~100-200 μL) dans la boîte de Petri pour maintenir l’humidité.
3. Culture microbienne
REMARQUE : Les étapes suivantes fournissent une procédure microbiologique générale pour la culture fongique et bactérienne et doivent être effectuées dans des conditions stériles (c.-à-d. à l’aide d’une flamme ou d’une armoire de sécurité microbiologique) appropriées au niveau de confinement requis pour les microbes souhaités. Des exemples précis sont donnés à la fin de chaque section pour une espèce d’intérêt.
- Culture fongique
- Préparer le milieu de culture souhaité complété par de la gélose. Autoclaver le milieu à 121 °C pendant 15 min. Laisser refroidir le milieu à 50 °C et verser dans des boîtes de Petri de 9 cm de diamètre, en maintenant des conditions stériles.
- Utilisez un foreur de liège pour retirer un bouchon de gélose de 4 mm de diamètre contenant du mycélium d’une colonie de stock de réfrigérateur de la souche fongique souhaitée afin d’activer l’isolat. Ceci est effectué pour assurer une croissance standardisée et vigoureuse du champignon avant l’inoculation du dispositif.
REMARQUE: Les microbes peuvent également être activés à partir d’un stock de glycérol, c’est-à-dire des isolats fongiques stockés sur des bouchons de gélose dans une solution de glycérol à 50% à -70 ° C41. - Placez le côté du bouchon avec du mycélium en contact avec la surface de la gélose au centre de la boîte de Petri non ininoculée. Remplacez le couvercle sur le dessus de la boîte de Petri et scellez avant d’incuber à la température appropriée pour la souche souhaitée pendant la durée requise, généralement environ 3 à 4 jours.
NOTE: Exemples de conditions de culture pour Trichoderma rossicum: Gélose à l’extrait de malt, incubée à 25 °C dans l’obscurité pendant 48 h.
- Culture bactérienne
- Strier l’isolat bactérien désiré du stock source (p. ex., stock de glycérol ou colonie unique de la plaque d’agar) sur une plaque d’agar pour obtenir des colonies bactériennes uniques et assurer l’absence de contamination45. Scellez la plaque avec un film.
- Incuber à une température et à une durée spécifiques à l’isolat d’intérêt jusqu’à ce que des colonies individuelles soient observées.
- Préparez le bouillon de culture souhaité. Par exemple, ajouter 10 g de tryptone, 10 g de NaCl et 5 g d’extrait de levure pour 1 L de ddH2O pour préparer le milieu LB pour la culture de B. subtilis. Autoclaver le milieu à 121 °C pendant 15 min.
- Laissez le milieu refroidir à la température ambiante. Ajouter le milieu à une fiole de culture stérile à l’intérieur d’un environnement stérile.
- Touchez une seule colonie bactérienne de la plaque de gélose à l’aide d’une boucle d’inoculation stérile. Transférer la boucle inoculée dans le milieu de culture stérile en touchant brièvement le liquide avec la boucle.
- Scellez la fiole à l’aide d’un couvercle ou d’une feuille stérile et placez-la à l’intérieur d’un incubateur à agitation pendant la nuit en utilisant les paramètres appropriés à l’espèce sélectionnée.
NOTE: Exemples de conditions de culture pour B. subtilis: i) culture liquide - croissance aérobie à 37 °C à 200 tr/min en milieu LB et ii) culture sur plaque - température ambiante sur plaque de gélose LB. Reportez-vous aux documents FFI/BFI40,41 pour plus de détails sur la culture de différentes souches fongiques.
4. Inoculation de l’appareil
REMARQUE: Les étapes suivantes doivent avoir lieu à l’intérieur d’une hotte à flux laminaire à l’aide d’un équipement stérile.
- Inoculation fongique
- Utilisez un foreur de liège stérilisé (ø = 4 mm) pour retirer un bouchon d’agar de la colonie située à la périphérie d’une culture vieille de 3 jours (étape 3.1). Assurez-vous que le front hyphale en croissance reste intact.
- Introduisez le bouchon dans l’entrée fongique, côté mycélium vers le bas, avec la direction de croissance du front hyphale orientée vers les ouvertures des microcanaux pour encourager l’infiltration hyphale des canaux.
- Répétez les étapes 4.1.1 à 4.1.2 pour la deuxième espèce fongique (si vous utilisez le dispositif FFI), en introduisant la fiche dans l’entrée opposée. Si vous utilisez le périphérique BFI, omettez cette étape et passez à l’étape 4.1.4.
- Scellez la boîte de Petri avec un film transparent et incubez à 25-28 °C dans l’obscurité jusqu’à ce que l’imagerie commence. Déterminer le temps d’incubation avant l’imagerie en fonction de l’événement biologique prévu à observer, p. ex. confrontations fongiques-fongiques, et du taux de croissance des espèces incluses dans l’instrument.
- Inoculation bactérienne
- Diluer les bactéries d’une culture pendant la nuit (étape 3.2) dans un rapport de 1:25 en utilisant le même milieu de culture que celui détaillé à l’étape 3.2.3. Culture pendant 3 h à 37 °C.
- Laver les bactéries en granulant la culture à l’aide d’une centrifugeuse à 2000 x g pendant 10 min. Jeter le surnageant et remettre les cellules en suspension dans le volume désiré de 0,9 % p/v de solution de chlorure de sodium.
- Centrifuger à nouveau pour obtenir une pastille. Jeter le surnageant et remettre les cellules en suspension dans un milieu liquide (p. ex., C. cinerea minimal medium à un OD600 de 1). Optimiser la valeur OD600 pour la souche bactérienne en question.
- Retirez le dispositif BFI de l’incubateur et ouvrez-le dans un environnement stérile. Pipette 10 μL de suspension dans l’entrée bactérienne.
REMARQUE: Optimisez les temps exacts d’inoculation pour les interactions bactériennes-fongiques en question. Par exemple, introduire des bactéries dans le dispositif BFI 18 h après l’inoculation fongique si vous utilisez C. cinerea. - Scellez la boîte de Petri avec un film transparent et incubez à 25 °C dans l’obscurité jusqu’à ce que l’imagerie commence. Rangez l’appareil à la verticale.
5. Microscopie et analyse d’images
- Microscopie
REMARQUE: Le chercheur doit choisir la méthode d’imagerie appropriée en accord avec la nature de l’expérience à mener, par exemple, une épifluorescence à grand champ inversé ou une microscopie confocale. Un aperçu général a été fourni ici, car les détails spécifiques dépendront des attributs de la configuration microscopique choisie.- Allumez l’ordinateur du microscope, le corps principal du microscope (le cas échéant), la caméra, l’incubateur à température contrôlée et la ou les sources de lumière. Assurez-vous que le microscope a été correctement installé, par exemple, l’éclairage Köhler a été correctement appliqué pour un éclairage uniforme de l’échantillon. Lancez le progiciel d’imagerie.
REMARQUE: Lors de l’utilisation d’un incubateur à température contrôlée, il est important de permettre à la température du microscope de s’équilibrer pendant plusieurs heures avant de commencer une expérience. - Montez le dispositif microfluidique dans l’insert de scène. Assurez-vous que l’appareil est bien fixé, c’est-à-dire avec du ruban adhésif, pour éviter de déloger l’appareil pendant le mouvement actif de la scène.
- Acquérir des images des dispositifs inoculés, par exemple, des expériences en un seul point ou en accéléré. Des spécifications d’imagerie complètes pertinentes pour les expériences menées avec les dispositifs BFI et FFI sont fournies dans les publications respectivessusmentionnées 40,41.
REMARQUE: Les images en champ clair ont été acquises en utilisant la microscopie à contraste de phase pour visualiser la prolifération hyphale à travers les canaux de croissance à l’aide d’un logiciel de mise au point automatique et d’un grossissement de 10x, d’un grossissement de 0,30 NA (ouverture numérique) ou d’un grossissement de 20x, d’objectifs de 0,45 NA. L’excitation des protéines rapporteures fluorescentes a été réalisée à l’aide d’un moteur lumineux à diode électroluminescente de haute puissance avec des longueurs d’onde spécifiques au fluorophore. - Exportez les images dans un format approprié pour le traitement ultérieur de l’image. Par exemple, .tiff.
- Allumez l’ordinateur du microscope, le corps principal du microscope (le cas échéant), la caméra, l’incubateur à température contrôlée et la ou les sources de lumière. Assurez-vous que le microscope a été correctement installé, par exemple, l’éclairage Köhler a été correctement appliqué pour un éclairage uniforme de l’échantillon. Lancez le progiciel d’imagerie.
- Analyse d’images
NOTE: Les auteurs recommandent Fidji46 comme outil d’analyse d’images, mais d’autres progiciels sont disponibles. Voici des exemples d’analyses d’images effectuées à l’aide de Fidji à partir des publications présentées sur les appareils BFI et FFI. Ces étapes sont spécifiques à un Mac et peuvent différer légèrement si vous utilisez un PC.- Mesures du taux de croissance hyphale
NOTE: Cette méthode a été utilisée dans le manuscrit BFI40 pour mesurer les taux de croissance des hyphes individuels.- Téléchargez, installez et lancez Fiji. Importez la séquence d’images à partir d’une expérience time-lapse en sélectionnant Fichier > Importer > séquence d’images. Recherchez le dossier dans lequel les données sont stockées et sélectionnez Ouvrir. Dans la fenêtre Options de séquence , sélectionnez Préférences, puis OK.
- Sélectionnez l’icône Ligne droite dans la barre d’outils principale. Placez le début de la ligne droite à l’extrémité de l’extrémité hyphale en croissance en cliquant puis en faisant glisser le curseur vers un autre point de la fenêtre. Une ligne jaune apparaîtra avec trois cases indiquant le début, le milieu et la fin de la ligne.
- Passez à l’image suivante de la séquence d’images en maintenant la touche Ctrl enfoncée et >. Placez l’extrémité de la ligne droite à l’extrémité de l’hyphe en croissance en sélectionnant et en faisant glisser la case carrée vers la bonne position.
- Maintenez les touches Ctrl et M enfoncées pour mesurer la longueur de la ligne en pixels. Une fenêtre Résultats apparaîtra avec les données mesurées. Définissez les données affichées dans la fenêtre Résultats comme suit : cliquez sur la fenêtre Résultats , puis sélectionnez Résultats > Définir les mesures.
- Passez à l’image suivante de la séquence d’images en maintenant la touche Ctrl enfoncée, puis >. Placez le début de la ligne droite à l’extrémité de l’hyphe en croissance en sélectionnant et en faisant glisser la case carrée à la bonne position.
- Maintenez la touche Ctrl enfoncée , puis sur M pour mesurer la longueur de la ligne en pixels. Une fenêtre Résultats apparaîtra avec les données mesurées.
- Répétez les étapes 5.2.1.5-5.2.1.6 jusqu’à ce que vous ayez fini de mesurer la croissance de l’hyphe en pixels.
- Sélectionnez toutes les données dans la fenêtre Résultats . Copiez et collez dans un autre logiciel, par exemple une feuille de calcul, pour traiter les données. Tracez la croissance hyphale (en pixels ou en micromètres) en fonction du temps et calculez les taux de croissance moyens. Effectuer au moins trois répliques biologiques par expérience.
- Mesures de l’intensité de fluorescence
NOTE: Cette méthode a été utilisée dans la publicationFFI 41 pour évaluer le changement d’intensité de fluorescence dans les hyphes de Fusarium graminearum 8/1-wt-GFP au contact de Clonostachys rosea 016 en fonction du temps.- Téléchargez, installez et lancez Fiji. Importez une séquence d’images à partir d’une expérience time-lapse en sélectionnant Fichier > Importer > Séquence d’images. Recherchez le dossier dans lequel les données sont stockées et sélectionnez Ouvrir. Dans la fenêtre Options de séquence, sélectionnez Préférences , puis OK.
- Spécifiez une région d’intérêt (ROI) pour mesurer l’intensité de fluorescence absolue d’un hyphe à l’aide de l’outil rectangulaire, situé dans la barre d’outils principale. La taille du carré peut être définie exactement comme suit : Modifier > Sélection > Spécifier ; le retour sur investissement peut également être enregistré pour référence ultérieure dans le gestionnaire de retour sur investissement en sélectionnant Modifier > sélection > Ajouter au gestionnaire.
- Mesurez l’intensité de fluorescence absolue (valeur grise moyenne) dans le retour sur investissement défini pour chaque image de l’ensemble de la séquence ou de la pile d’images comme suit : Pile d’images > > Pile de mesures. La fenêtre Résultats s’ouvrira automatiquement une fois que toutes les images de la pile auront été traitées.
REMARQUE: Les données affichées dans la fenêtre Résultats peuvent être définies comme suit: cliquez sur la fenêtre Résultats , puis sélectionnez Résultats > Définir les mesures. Assurez-vous que la valeur grise moyenne a été sélectionnée. - Sélectionnez toutes les données dans la fenêtre Résultats . Copiez et collez dans un autre logiciel, par exemple une feuille de calcul, pour tracer les intensités de fluorescence absolues du retour sur investissement spécifié en fonction du temps.
- Répétez les étapes 5.2.2.2-5.2.2.4 pour recueillir des mesures d’intensité de fluorescence absolue pour chaque roi, c’est-à-dire sur l’hyphe d’intérêt, à côté de l’hyphe d’intérêt ou dans le canal de contrôle correspondant.
- Calculer les intensités de fluorescence relatives appropriées en unités arbitraires (UA), par exemple en divisant l’intensité de fluorescence absolue du ROI [hyphe d’intérêt] par l’intensité de fluorescence absolue du ROI [canal de contrôle]. Consultez la publication41 de la FFI pour plus de détails.
- Effectuer au moins trois répliques biologiques par expérience et tracer les intensités de fluorescence relatives en fonction du temps.
- Mesures du taux de croissance hyphale
Access restricted. Please log in or start a trial to view this content.
Résultats
Des résultats représentatifs sont présentés à partir des dispositifs exemplaires BFI40 et FFI41. Les mesures du taux de croissance de l’hyphalie peuvent facilement être obtenues à l’aide de ces dispositifs en combinaison avec des techniques de microscopie de base. La figure 3A-B illustre les interactions bactériens-fongiques entre C. cinerea hyphae et B. subtilis NCIB 3610. La pré...
Access restricted. Please log in or start a trial to view this content.
Discussion
Cet article présente un protocole pour l’étude des interactions fongiques-microbiennes à l’aide de la microfluidique des canaux. Les auteurs visent à démontrer la polyvalence de ces dispositifs et à encourager l’adaptation en fonction des intérêts du chercheur. En utilisant les dispositifs BFI et FFI exemplaires, les interactions fongiques-microbiennes peuvent être étudiées plus en détail qu’auparavant. En supprimant la complexité et l’hétérogénéité du fond du sol, en modérant la croissance d...
Access restricted. Please log in or start a trial to view this content.
Déclarations de divulgation
Les auteurs ne déclarent aucun intérêt concurrent.
Remerciements
Nous reconnaissons le soutien financier du département de bio-ingénierie de l’Imperial College de Londres et du Leverhulme Trust (référence de subvention de recherche: RPG-2020-352).
Access restricted. Please log in or start a trial to view this content.
matériels
| Name | Company | Catalog Number | Comments |
| Agar | Difco Laboratories | 214010 | Used to solidify culture medium for bacterial and fungal cultivation within Petri dishes |
| Aluminum foil | Fisher Scientific Ltd | 11759408 | |
| AutoCAD 2021 | Autodesk, USA | ||
| Autoclave (VX-75) | Systec | ||
| Centrifuge (5810R) | Eppendorf | ||
| Chlorotrimethysilane | Merck Life Sciences | 386529 | CAUTION: Chlorotrimethylsilane is a hazardous substance. Wear appropriate PPE and handle with care. Avoid contact with skin and eyes and prevent inhalation. Keep away from sources of ignition and use in a well-ventilated area. |
| Cork borer | SLS | COR1000 | |
| Developer solution (mr-Dev 600) | Microresist Technologies | CAUTION: mr-Dev 600 developer solution is flammable | |
| Erlenmeyer flasks | VWR | 214-1108 | e.g. 200 mL; choose size to suit your exact needs |
| Ethanol (70% v/v) | Fisher Scientific Ltd | E/0650DF/15 | Diluted from 99.8% (Analytical Reagent Grade) |
| Fiji | ImageJ | Exemplar software package for imaging processing | |
| Filtered, compressed air | Available as standard in most labs. Altervatively, an oil-free compressor with air regulator can be used. | ||
| Flat-headed wafer tweezers | SLS | INS5026 | |
| Forceps | Fisher Scientific Ltd | 10008051 | Bent, sharp |
| Glass bottom petri dish | World Precision Instruments | FD35-100 | 35 mm |
| Glass bottom petri dish | World Precision Instruments | FD5040-100 | 50 mm |
| Glass crystallisation dishes | VWR | 216-1865 | Used for washing of PDMS slabs |
| Glass crystallisation dishes | VWR | 216-1866 | Used in the development of master moulds |
| Glass media bottles | Fisher Scientific Ltd | 15456113 | e.g. 250 mL; choose size to suit your exact needs |
| Glass syringe (Hamilton) | Fisher Scientific Ltd | 10625251 | Used for dispensing chlorotrimethylsilane |
| Hot plate (HP 160 III BM) | SAWATEC | ||
| Inoculation loop | VWR | COPA175CS01 | |
| Isopropyl alcohol | Sigma-Aldrich | W292907 | |
| Laminar flow hood | Air Science (PCR) | Exemplar laminar flow hood used for device fabrication | |
| LB medium | Fisher Scientific Ltd | BP9723-500 | Exemplar nutrient broth for bacterial overnight culture |
| Light emitting diode light engine (LedHUB) | Omicron-Laserage Laserprodukte GmbH | Exemplar light source that can be used for imaging fungal-microbial interactions (fluorescence) | |
| MA6 Ultraviolet mask aligner | Suss Microtec | ||
| Malt extract | VWR | 84618 | Used to make exemplar fungal culture medium (Malt extract agar) |
| Mask Writer | Applied Materials | 4700DP | Example of a mask writer which can be used to print photo-mask for photolithography |
| Master mould plastic mount | 3D-printed bespoke holder manufactured in-house | ||
| Microbiological safety cabinet (BioMat2) | Contained Air Solutions | Exemplar MSC used for microbial culture and device inoculation | |
| Milli-Q purified water | Available as standard in biology labs. | ||
| NaOH | Fisher Scientific Ltd | BP359-500 | |
| NIS-Elements Advanced Research imaging software | Nikon | Exemplar software package for image acquisition | |
| NIS-Elements Free Viewer | Nikon | Exemplar software package for viewing acquired images | |
| Oven (Binder BD115) | Fisher Scientific Ltd | 15602126 | Used for curing poly(dimethylsiloxane)(PDMS) |
| Oven (CLO-2AH-S) | KOYO | Used for preparing silicon wafers | |
| Parafilm | Bemis | HS234526B | transparent film |
| Petri dishes, square sterile | Fisher Scientific Ltd | 11708573 | 120.5 mm |
| Petri dishes, sterile | Fisher Scientific Ltd | 15370366 | 90 mm |
| Photolithography mask | Micro Lithography Services Ltd. UK | ||
| Plasma cleaner (Zepto) | Diener Electronic | 100012601 | |
| Plastic cup | Semadeni | 8323 | |
| Plastic spatula | Semadeni | 3340 | |
| Portable precision balance (OHAUS Scout) | Fisher Scientific Ltd | 15519631 | Used for weighing PDMS, media components etc. |
| Precision cutter | Syneo | HS1251135P1183 | Cutting edge diameter: 3.18 mm |
| Precision cutter | Syneo | HS1871730P1183S | Cutting edge diameter: 4.75 mm |
| Profilometer | Bruker | Dektak XT-stylus | |
| Razor blades | Häberle Labortechnik | 9156110 | |
| Refridgerator | Haden | 4-6 °C | |
| Retiga R1 CCD camera | Qimaging | Exemplar camera that can be used for imaging fungal-microbial interactions | |
| Scotch magic tape | Office Depot | 3969954 | 19 mm invisible tape; clear tape |
| Shaking incubator (Cole-Parmer SI500) | Fisher Scientific Ltd | 10257954 | |
| Silicon wafer | Inseto | 100 mm | |
| Soda lime glass plate | Inseto | 125 mm x 125 mm x 2 mm. Used to hold photolithography mask in mask aligner | |
| Sodium chloride | Sigma-Aldrich | S7653 | |
| Spincoater | SAWATEC | SM-180-BM | |
| SU-8 2010 photoresist | MicroChem | CAUTION: SU-8 photoresist is hazardous, take care when handling and prevent inhalation and contact with skin. Flammable, potentially carcinogenic and toxic to the environment. | |
| Sylgard 184 elastomer kit | VWR | 634165S | Used for the preparation of poly(dimethylsiloxane)(PDMS) devices |
| Temperature controlled incubator | Okolab | Exemplar incubator that can be used for imaging fungal-microbial interactions | |
| Ti2-E inverted epifluorescence microscope | Nikon | MEA54000 | Exemplar microscope that can be used for imaging fungal-microbial interactions |
| Ultrasonic cleaner S-Line | Fisher Scientific Ltd | FB15050 | |
| Vacuum desiccator | Fisher Scientific Ltd | 10528861 | Silianisation and PDMS degassing should be conducted in separate desiccators |
| x10/0.3 NA CFI Plan Fluor DL objective lens | Nikon | MRH20105 | Exemplar objective lens that can be used for imaging fungal-microbial interactions |
| x20/0.45 NA CFI Plan Fluor DL objective lens | Nikon | MRH48230 | Exemplar objective lens that can be used for imaging fungal-microbial interactions |
Références
- Zhu, Y. -G., Miller, R. M. Carbon cycling by arbuscular mycorrhizal fungi in soil-plant systems. Trends in Plant Science. 8 (9), 407-409 (2003).
- Dai, Z., et al. Long-term nutrient inputs shift soil microbial functional profiles of phosphorus cycling in diverse agroecosystems. The ISME Journal. 14 (3), 757-770 (2020).
- Op De Beeck, M., et al. Regulation of fungal decomposition at single-cell level. The ISME Journal. 14 (4), 896-905 (2020).
- Bender, S. F., et al. Symbiotic relationships between soil fungi and plants reduce N2O emissions from soil. The ISME Journal. 8 (6), 1336-1345 (2014).
- Dullah, S., et al. Melanin production and laccase mediated oxidative stress alleviation during fungal-fungal interaction among basidiomycete fungi. IMA Fungus. 12 (1), 33(2021).
- Deveau, A., et al. Bacterial-fungal interactions: ecology, mechanisms and challenges. FEMS Microbiology Reviews. 42 (3), 335-352 (2018).
- Bian, R., et al. Facilitative and synergistic interactions between fungal and plant viruses. Proceedings of the National Academy of Sciences of the United States of America. 117 (7), 3779-3788 (2020).
- Jiang, X., Xiang, M., Liu, X. Nematode-trapping fungi. Microbiology Spectrum. 5 (1), (2017).
- Essig, A., et al. a novel peptide-based fungal antibiotic interfering with the peptidoglycan synthesis. Journal of Biological Chemistry. 289 (50), 34953-34964 (2014).
- Tang, H. -Y., Zhang, Q., Li, H., Gao, J. -M. Antimicrobial and allelopathic metabolites produced by Penicillium brasilianum. Natural Product Research. 29 (4), 345-348 (2015).
- Bai, Y. -B., et al. Antifungal activity of griseofulvin derivatives against phytopathogenic fungi In vitro and In vivo and three-dimensional quantitative structure-activity relationship analysis. Journal of Agricultural and Food Chemistry. 67 (22), 6125-6132 (2019).
- Solanki, M. K., et al. Characterization of antagonistic-potential of two Bacillus strains and their biocontrol activity against Rhizoctonia solani in tomato. Journal of Basic Microbiology. 55 (1), 82-90 (2015).
- Jamali, H., Sharma, A., Srivastava, A. K. Biocontrol potential of Bacillus subtilis RH5 against sheath blight of rice caused by Rhizoctonia solani. Journal of Basic Microbiology. 60 (3), 268-280 (2020).
- Válková, H., Novotný, Č, Malachová, K., Šlosarčíková, P., Fojtík, J. Effect of bacteria on the degradation ability of Pleurotus ostreatus. Science of The Total Environment. 584-585, 1114-1120 (2017).
- Leyva-Rojas, J. A., Coy-Barrera, E., Hampp, R. Interaction with soil bacteria affects the growth and amino acid content of Piriformospora indica. Molecules. 25 (3), Basel, Switzerland. 572(2020).
- Dullah, S., et al. Fungal interactions induce changes in hyphal morphology and enzyme production. Mycology. 12 (4), 279-295 (2021).
- Marfetán, J. A., Romero, A. I., Folgarait, P. J. Pathogenic interaction between Escovopsis weberi and Leucoagaricus sp.: mechanisms involved and virulence levels. Fungal Ecology. 17, 52-61 (2015).
- Cortois, R., De Deyn, G. B. The curse of the black box. Plant and Soil. 350 (1), 27-33 (2012).
- Whitesides, G. M. The origins and the future of microfluidics. Nature. 442 (7101), 368-373 (2006).
- Sackmann, E. K., Fulton, A. L., Beebe, D. J. The present and future role of microfluidics in biomedical research. Nature. 507 (7491), 181-189 (2014).
- Hanson, K. L., et al. Fungi use efficient algorithms for the exploration of microfluidic networks. Small. 2 (10), 1212-1220 (2006).
- Held, M., Edwards, C., Nicolau, D. V. Probing the growth dynamics of Neurospora crassa with microfluidic structures. Fungal Biology. 115 (6), 493-505 (2011).
- Thomson, D. D., et al. Contact-induced apical asymmetry drives the thigmotropic responses of Candida albicans hyphae. Cellular Microbiology. 17 (3), 342-354 (2015).
- Lee, K. K., Labiscsak, L., Ahn, C. H., Hong, C. I. Spiral-based microfluidic device for long-term time course imaging of Neurospora crassa with single nucleus resolution. Fungal Genetics and Biology. 94, 11-14 (2016).
- Asenova, E., Lin, H. Y., Fu, E., Nicolau, D. V., Nicolau, D. V. Optimal fungal space searching algorithms. IEEE Transactions on NanoBioscience. 15 (7), 613-618 (2016).
- Soufan, R., et al. Pore-scale monitoring of the effect of microarchitecture on fungal growth in a two-dimensional soil-like micromodel. Frontiers in Environmental Science. 6, (2018).
- Uehling, J. K., et al. Microfluidics and metabolomics reveal symbiotic bacterial-fungal interactions between Mortierella elongata and Burkholderia include metabolite exchange. Frontiers in Microbiology. 10, 2163(2019).
- Millet, L. J., et al. Increasing access to microfluidics for studying fungi and other branched biological structures. Fungal Biology and Biotechnology. 6 (8), 1-14 (2019).
- Baranger, C., Fayeulle, A., Le Goff, A. Microfluidic monitoring of the growth of individual hyphae in confined environments. Royal Society Open Science. 7 (8), 191535(2020).
- Aleklett, K., Ohlsson, P., Bengtsson, M., Hammer, E. C. Fungal foraging behaviour and hyphal space exploration in micro-structured Soil Chips. The ISME Journal. 15 (6), 1782-1793 (2021).
- Aleklett, K., et al. Build your own soil: exploring microfluidics to create microbial habitat structures. The ISME Journal. 12 (2), 312-319 (2018).
- Ellett, F., Jorgensen, J., Frydman, G. H., Jones, C. N., Irimia, D. Neutrophil interactions stimulate evasive hyphal branching by Aspergillus fumigatus. PLOS Pathogens. 13 (1), 1006154(2017).
- Massalha, H., Korenblum, E., Malitsky, S., Shapiro, O. H., Aharoni, A. Live imaging of root-bacteria interactions in a microfluidics setup. Proceedings of the National Academy of Sciences of the United States of America. 114 (17), 4549-4554 (2017).
- Schmieder, S. S., et al. Bidirectional propagation of signals and nutrients in fungal networks via specialized hyphae. Current Biology. 29 (2), 217-228 (2019).
- Tayyrov, A., Stanley, C. E., Azevedo, S., Künzler, M. Combining microfluidics and RNA-sequencing to assess the inducible defensome of a mushroom against nematodes. BMC Genomics. 20 (1), 243(2019).
- Stanley, C. E., Grossmann, G., Casadevall i Solvas, X., deMello, A. J. Soil-on-a-Chip: microfluidic platforms for environmental organismal studies. Lab on a Chip. 16 (2), 228-241 (2016).
- Stanley, C. E., vander Heijden, M. G. A. Microbiome-on-a-Chip: new frontiers in plant-microbiota research. Trends in Microbiology. 25 (8), 610-613 (2017).
- Ortseifen, V., Viefhues, M., Wobbe, L., Grünberger, A. Microfluidics for biotechnology: bridging gaps to foster microfluidic applications. Frontiers in Bioengineering & Biotechnology. 8, 589074(2020).
- Jansson, J. K., Hofmockel, K. S. The soil microbiome-from metagenomics to metaphenomics. Current Opinion in Microbiology. 43, 162-168 (2018).
- Stanley, C. E., et al. Probing bacterial-fungal interactions at the single cell level. Integrative Biology (Camb). 6 (10), 935-945 (2014).
- Gimeno, A., et al. A versatile microfluidic platform measures hyphal interactions between Fusarium graminearum and Clonostachys rosea in real-time. Communications Biology. 4 (1), 262(2021).
- Duffy, D. C., McDonald, J. C., Schueller, O. J. A., Whitesides, G. M. Rapid prototyping of microfluidic systems in poly(dimethylsiloxane). Analytical Chemistry. 70 (23), 4974-4984 (1998).
- Stanley, C. E., et al. Fabrication and use of the dual-flow-RootChip for the imaging of Arabidopsis roots in asymmetric microenvironments. Bio-protocol. 8 (18), 3010(2018).
- Choi, C. -H., Lee, H., Weitz, D. A. Rapid patterning of PDMS microfluidic device wettability using syringe-vacuum-induced segmented flow in nonplanar geometry. ACS Applied Materials & Interfaces. 10 (4), 3170-3174 (2018).
- Sanders, E. R. Aseptic laboratory techniques: plating methods. Journal of Visualized Experiments. (63), e3064(2012).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Harting, R., et al. Pseudomonas strains induce transcriptional and morphological changes and reduce root colonization of Verticillium spp. Frontiers in Microbiology. 12, 652468(2021).
- Boenisch, M. J. Structural and molecular characterisation of the penetration process of Fusarium graminearum during Fusarium head blight infection. , Staats-und Universitätsbibliothek Hamburg Carl von Ossietzky. (2013).
- Eynck, C., Koopmann, B., Grunewaldt-Stoecker, G., Karlovsky, P., von Tiedemann, A. Differential interactions of Verticillium longisporum and V. dahliae with Brassica napus detected with molecular and histological techniques. European Journal of Plant Pathology. 118 (3), 259-274 (2007).
- Ghanem, N., Stanley, C. E., Harms, H., Chatzinotas, A., Wick, L. Y. Mycelial effects on phage retention during transport in a microfluidic platform. Environmental Science & Technology. 53 (20), 11755-11763 (2019).
- Alrifaiy, A., Lindahl, O. A., Ramser, K. Polymer-based microfluidic devices for pharmacy, biology and tissue engineering. Polymers. 4 (3), 1349-1398 (2012).
- Duncombe, T. A., Tentori, A. M., Herr, A. E. Microfluidics: reframing biological enquiry. Nature Reviews Molecular Cell Biology. 16 (9), 554-567 (2015).
- Hoelzle, D., et al. Microfluidic device design, fabrication, and testing protocols. Protocol Exchange. , (2015).
Access restricted. Please log in or start a trial to view this content.
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.