
Method Article
Microdissection laser industrialisée guidée par l’intelligence artificielle pour l’analyse protéomique à micro-échelle du microenvironnement tumoral
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
Ce protocole décrit un flux de travail à haut débit pour la segmentation basée sur l’intelligence artificielle des régions d’intérêt confirmées par la pathologie à partir d’images de coupes de tissus minces et colorées pour l’enrichissement des populations cellulaires résolues par histologie à l’aide de la microdissection laser. Cette stratégie comprend un nouvel algorithme permettant le transfert de démarcations désignant des populations cellulaires d’intérêt directement aux microscopes laser.
Résumé
Le microenvironnement tumoral (TME) représente un écosystème complexe composé de dizaines de types cellulaires distincts, y compris des populations de tumeurs, de stromas et de cellules immunitaires. Pour caractériser la variation au niveau du protéome et l’hétérogénéité tumorale à grande échelle, des méthodes à haut débit sont nécessaires pour isoler sélectivement les populations cellulaires discrètes dans les tumeurs malignes solides. Ce protocole décrit un flux de travail à haut débit, rendu possible par l’intelligence artificielle (IA), qui segmente les images de coupes de tissus minces colorées à l’hématoxyline et à l’éosine (H & E) dans des régions d’intérêt confirmées par la pathologie pour la récolte sélective de populations cellulaires résolues par histologie à l’aide de la microdissection laser (LMD). Cette stratégie comprend un nouvel algorithme permettant le transfert de régions indiquant des populations cellulaires d’intérêt, annotées à l’aide d’un logiciel d’image numérique, directement vers des microscopes laser, permettant ainsi des collections plus faciles. La mise en œuvre réussie de ce flux de travail a été réalisée, démontrant l’utilité de cette méthode harmonisée pour prélever sélectivement des populations de cellules tumorales à partir du TME pour une analyse protéomique quantitative et multiplexée par spectrométrie de masse à haute résolution. Cette stratégie s’intègre pleinement à l’examen de routine de l’histopathologie, tirant parti de l’analyse d’images numériques pour soutenir l’enrichissement des populations cellulaires d’intérêt et est entièrement généralisable, permettant des récoltes harmonisées de populations cellulaires à partir du TME pour des analyses multiomiques.
Introduction
Le TME représente un écosystème complexe peuplé d’un éventail très diversifié de types cellulaires, tels que les cellules tumorales, les cellules stromales, les cellules immunitaires, les cellules endothéliales, d’autres types de cellules mésenchymateuses et les adipocytes, ainsi qu’une matrice extracellulaire complexe1. Cet écosystème cellulaire varie à l’intérieur et entre les différents sites d’organes de la maladie, ce qui entraîne une hétérogénéité tumorale complexe 2,3. Des études récentes ont montré que les tumeurs hétérogènes et les tumeurs à faible cellularité tumorale (faible pureté) sont souvent en corrélation avec un mauvais pronostic de la maladie 2,3.
Pour comprendre l’interaction moléculaire entre les populations de cellules tumorales et non tumorales au sein du TME à grande échelle, des stratégies standardisées et à haut débit sont nécessaires pour récolter sélectivement des populations cellulaires distinctes d’intérêt pour l’analyse multiomique en aval. La protéomique quantitative représente une technique en évolution rapide et de plus en plus importante pour approfondir la compréhension de la biologie du cancer. À ce jour, la prépondérance des études utilisant la protéomique l’a fait avec des protéines extraites de préparations de tissus tumoraux entiers (par exemple, cryopulvérisées), ce qui a conduit à une insuffisance dans la compréhension de l’hétérogénéité au niveau du protéome dans le TME 4,5,6.
Le développement de stratégies de collecte d’échantillons qui s’intègrent de manière transparente aux flux de travail de pathologie clinique et les exploitent permettra une nouvelle génération de protéomiques résolues en histologie qui sont hautement complémentaires aux flux de travail de pathologie diagnostique de référence. La LMD permet la collecte directe et sélective de sous-populations cellulaires ou de régions d’intérêt (ROI) par inspection microscopique de fines coupes minces de tissus colorés histologiquement7. Les progrès majeurs récents en pathologie numérique et en analyse basée sur l’IA ont démontré la capacité d’identifier de manière automatisée des caractéristiques de composition et des retours sur investissement uniques au sein du TME, dont beaucoup sont en corrélation avec des altérations moléculaires et des caractéristiques cliniques de la maladie, telles que la résistance au traitement et le pronostic de la maladie8.
Le flux de travail décrit dans le protocole présenté ici exploite des solutions logicielles commerciales pour annoter sélectivement les ROI tumoraux dans les images histopathologiques numériques, et utilise des outils logiciels développés en interne pour transférer ces ROI tumoraux vers des microscopes laser pour la collecte automatisée de populations cellulaires discrètes d’intérêt qui s’intègre parfaitement aux flux de travail d’analyse multiomique en aval. Cette stratégie intégrée réduit considérablement le temps de l’opérateur LMD et minimise la durée pendant laquelle les tissus doivent être à température ambiante. L’intégration de la sélection automatisée des caractéristiques et de la récolte de LMD avec la protéomique quantitative à haut débit est démontrée par une analyse différentielle du TME de deux sous-types histologiques représentatifs du cancer épithélial de l’ovaire, le cancer de l’ovaire séreux de haut grade (HGSOC) et le carcinome à cellules claires de l’ovaire (OCCC).
Protocole
Tous les protocoles d’étude ont été approuvés pour une utilisation en vertu d’un protocole approuvé par la CISR de l’Ouest « An Integrated Molecular Analysis of Endometrial and Ovarian Cancer to Identify and Validate Clinically Informative Biomarkers » réputé exempté en vertu du règlement fédéral américain 45 CFR 46.102 (f). Tous les protocoles expérimentaux impliquant des données humaines dans cette étude étaient conformes à la Déclaration d’Helsinki. Le consentement éclairé a été obtenu de tous les sujets impliqués dans l’étude.
ATTENTION : Les réactifs suivants utilisés tout au long du protocole sont des cancérogènes connus ou soupçonnés et/ou contiennent des matières dangereuses : éthanol, eau DEPC, solution d’hématoxyline de Mayer, solution d’éosine Y, méthanol, acétonitrile et acide formique. Une manipulation appropriée, telle que décrite dans les fiches de données de sécurité (FDS) respectives, et l’utilisation d’équipements de protection individuelle (EPI) appropriés sont obligatoires.
1. Génération du fichier de données de liste de formes par défaut (.sld) contenant des fiducials calibrator
REMARQUE : Les étapes du protocole décrites dans cette section sont spécifiques à utiliser avec un microscope laser inversé et le logiciel associé (voir la Table des matériaux). La création d’un fichier .sld par défaut n’est nécessaire qu’une seule fois par microscope laser. Le fichier résultant peut être utilisé pour découper des fiducials dans toutes les diapositives PEN utilisées par la suite. Durée approximative : 5 min (une seule fois).
- Ouvrez le logiciel LMD et chargez la membrane en polyéthylène naphtalate (PEN) sur l’étage LMD face vers le bas, avec l’étiquette la plus proche de l’utilisateur. Décochez la case Fermer la ou les lignes sur le côté droit de la fenêtre du programme.
- Utilisez la fonction PtoP (point à point) sous fort grossissement (63x) pour dessiner trois flèches « V » servant de fiducials d’étalonnage. En partant d’un point externe sur le V, tracez une ligne jusqu’au point médian du V et cliquez un seul clic. Ensuite, tracez une deuxième ligne à partir du point central du V jusqu’à la fin du deuxième point V externe, puis double-cliquez pour créer une forme en V unique et non fermée à partir des deux lignes.
REMARQUE: Ces fiducials d’étalonnage doivent être placés dans trois coins de la diapositive: avant droite, arrière droit, arrière gauche. - Sélectionnez l’option AF (autofocus) avant la coupe . Coupez la diapositive en position 1, déplacez-la dans chacune des positions restantes de la diapositive et tracez avec précision les coupes d’étalonnage.
- Enregistrez le fichier .sld et sélectionnez l’option Enregistrer sans étalonnage dans la boîte de dialogue contextuelle pour éviter de couper les fiducials d’étalonnage dans la membrane.
REMARQUE : Un fichier .sld représentatif contenant des fiducials d’étalonnage standard pour quatre positions de diapositive est fourni dans le fichier supplémentaire 1.
2. Préparation des lames LMD
REMARQUE : Les étapes du protocole décrites dans cette section sont spécifiques à utiliser avec un microscope laser inversé et le logiciel associé (voir la Table des matériaux). Durée approximative : 5 min.
- Assurez-vous que la lame est complètement sèche avant de couper les fiducials d’étalonnage de référence. Ouvrez le logiciel LMD et ouvrez le fichier .sld d’étalonnage par défaut sous l’option Importer des formes .
- Sélectionnez l’option AF (autofocus) avant la coupe . Chargez la ou les lames avec le tissu tourné vers le bas et la glissière d’étiquette plus près de l’opérateur dans le porte-diapositives sur la scène LMD.
- À l’aide du microscope laser et du fichier .sld d’étalonnage par défaut, découpez les fiducials d’étalonnage dans la membrane PEN.
- FACULTATIF : Coupez les fiducials d’étalonnage dans la membrane PEN avant ou après que la ou les sections tissulaires soient/soient placées sur la lame. Si les fiducials d’étalonnage sont coupés avant la mise en place du tissu, assurez-vous que le tissu et/ou le fixateur ne se chevauchent pas avec les calibrateurs lorsque le tissu est placé sur la lame à l’étape 2.5. Si des fiducials d’étalonnage sont coupés après la mise en place de tissus, arrêtez-vous après la fin de l’étape 2.4 et passez à la section 3.
- Examinez tous les calibrateurs individuellement pour vous assurer que chaque coupe est complète et visible.
REMARQUE: Utilisez la fonction Déplacer et couper pour diriger manuellement le laser sur tous les fiducials d’étalonnage qui n’ont pas complètement coupé à travers la membrane PEN. - Placer la section de tissu congelée ou fixée au formol, incorporée à la paraffine (FFPE) sur la lame contenant les fiducials d’étalonnage.
3. Coloration tissulaire
REMARQUE: Durée approximative: 30 min.
- Fixer les lames de tissu LMD congelées dans de l’éthanol à 70% (EtOH) contenant des réactifs cocktails inhibiteurs de phosphatase pendant 5 min.
- Laver les lames dans de l’eau pyrocarbomate de diéthyle (DEPC) contenant des réactifs cocktails inhibiteurs de la phosphatase pendant 1 min.
- Lavez les lames à l’eau DEPC pendant 1 min.
- Incuber les lames dans la solution d’hématoxyline de Mayer pendant 3 min.
- Rincez les lames à l’eau DEPC pendant 3 min.
- Rincez les lames dans un échange frais d’eau DEPC pendant 1 min.
- Incuber les lames dans une solution aqueuse d’Eosin Y pendant 1 s.
- Rincez les lames 2 x 5 s dans 95% EtOH.
- Rincez les lames 3 x 10 s en 100% EtOH.
- Essuyez l’excès d’EtOH à l’arrière des glissières et laissez les lames sécher à l’air.
- Stockez les lames à -80 °C si la LMD ne doit pas être effectuée immédiatement.
4. Imagerie de diapositives
Remarque : Les étapes de protocole décrites dans cette section sont spécifiques aux diapositives numérisées (voir la table des matériaux) et aux images résultantes enregistrées en tant que fichiers .svs. Utilisez n’importe quel scanner et son logiciel associé qui génèrent des fichiers image dans un format que le logiciel d’analyse d’images (voir la Table des matériaux) peut ouvrir. Les types de fichiers utilisant des tiffs pyramidaux pris en charge incluent JPG, TIF, MRXS, QPTIFF, composant TIFF, SVS, AFI, SCN, LIF, DCM, OME. TIFF, ND2, VSI, NDPI, NDPIS, CZI, BIF, KFB et ISYNTAX. Durée approximative : 5 min.
- Allumez le scanner et ouvrez le logiciel du scanner de diapositives. Chargez la diapositive avec le tissu tourné vers le haut sur l’étage de diapositive unique dans le scanner. Assurez-vous que la lame est complètement sèche et placez doucement un couvercle sur le tissu. N’utilisez pas d’éthanol ou d’huile d’immersion sous le couvercle.
- Capturez l’image de la micrographie à l’aide de paramètres calibrés pour ajuster la membrane PEN au lieu de l’arrière-plan en verre et pour ignorer la coloration de la membrane d’arrière-plan, conformément aux instructions du fabricant.
- Ajustez la zone d’imagerie en faisant glisser et en redimensionnant le périmètre vert interne pour capturer toute la zone de la membrane PEN, selon les besoins. Ajoutez quatre points de mise au point sur le tissu en double-cliquant sur l’image d’aperçu de l’instantané et trois points de mise au point sur la membrane près des fiduciaires d’étalonnage (un point de mise au point pour chacun des trois fiducials d’étalonnage).
REMARQUE: Les quatre points de mise au point peuvent être placés presque n’importe où sur la section de tissu, bien que le placement sur un tissu trop taché et qui semble noir puisse entraîner l’échec de l’analyse. - Dans le menu Affichage , sélectionnez Moniteur vidéo. Ajustez manuellement la mise au point à l’aide du curseur de mise au point fine et/ou macro, selon les besoins, pour chaque point autour du tissu LMD. Capturez la numérisation de l’image sous un grossissement élevé (20x). Vérifiez que toutes les fiduciales d’étalonnage sont visibles et claires dans l’image enregistrée.
5. Sélection automatisée des caractéristiques à l’aide d’un logiciel d’analyse d’images
- Pour les collections de tumeurs entières (durée approximative: 5 min; en fonction du cas):
- Ouvrez le logiciel d’analyse d’images (voir la Table des matériaux). Sélectionnez Ouvrir les images et, dans la fenêtre contextuelle, sélectionnez le fichier image .svs généré par l’analyse de la diapositive sur le scanner AT2.
REMARQUE : Un fichier image .svs représentatif est fourni dans le fichier supplémentaire 2. - Accédez à l’onglet Annotations . Sélectionnez l’outil Plume dans la barre d’outils Annotation et dessinez une forme autour du tissu.
- Sélectionnez la forme et faites un clic droit sur l’image. Dans le menu déroulant Avancé, sélectionnez Partitionnement (en mosaïque). Définissez la taille et l’espace entre les vignettes sur 500 et 40, respectivement, puis sélectionnez OK pour générer les vignettes. Sélectionnez et supprimez la forme de périmètre utilisée pour générer les vignettes à l’étape 5.1.2.
- Sélectionnez le menu déroulant Actions de calque | Exportez pour enregistrer les annotations en mosaïque en tant que fichier .annotation.
REMARQUE: Un fichier d’annotation représentatif pour une collection complète de tissus tumoraux est fourni dans le fichier supplémentaire 3. - Créez un dossier pour la session ou le projet et enregistrez le fichier .annotation dans un sous-dossier étiqueté avec l’identificateur unique de la diapositive.
- Accédez à l’onglet Annotations . Sélectionnez le menu déroulant Actions de calque | Supprimer tous les calques pour supprimer toutes les annotations de l’image. Sélectionnez l’outil Plume et tracez une courte ligne à partir de la pointe interne de la pointe de flèche pour chaque fiducial d’étalonnage. Tracez les lignes à partir des marques dans l’ordre suivant : en haut à gauche, en haut à droite, en bas à droite.
- Sélectionnez le menu déroulant Actions de calque | Exportez pour enregistrer les annotations de ligne en tant que fichier .annotation. Ajoutez _calib au nom de fichier et placez le fichier dans le sous-dossier qui contient les coordonnées des formes en mosaïque.
REMARQUE : Un fichier _calib.annotation représentatif est fourni dans le fichier supplémentaire 4. - Copiez l’adresse du dossier principal du projet ou de la session. Ouvrez le script de génération d’importation XML, « Malleator » (disponible via https://github.com/GYNCOE/Mitchell.et.al.2022), à l’aide de l’environnement de développement intégré IDLE et collez l’adresse du dossier du projet entre les guillemets au bas du script.
- Sélectionnez le menu déroulant Exécuter | Exécutez Module pour exécuter le script.
REMARQUE : Le fichier d’importation LMD .xml sera généré dans le sous-dossier créé pour l’image/diapositive. Un dossier .xml représentatif est fourni dans le dossier supplémentaire 5.
- Ouvrez le logiciel d’analyse d’images (voir la Table des matériaux). Sélectionnez Ouvrir les images et, dans la fenêtre contextuelle, sélectionnez le fichier image .svs généré par l’analyse de la diapositive sur le scanner AT2.
- Pour les collections enrichies en LMD uniquement (durée approximative : 15 min ; en fonction du cas) :
- Ouvrez le logiciel d’analyse d’images (voir la Table des matériaux). Sélectionnez Ouvrir les images et, dans la fenêtre contextuelle, sélectionnez le fichier image .svs généré par l’analyse de la diapositive.
- Accédez à l’onglet Annotations . Sélectionnez et utilisez l’outil d’annotation Rectangle pour dessiner une zone autour du tissu.
- Sélectionnez l’annotation de la boîte et faites un clic droit sur l’image. Sélectionnez le menu déroulant Avancé | Option de partitionnement (mosaïque). Définissez la taille et l’espace entre les vignettes sur 500 et 40, respectivement, puis sélectionnez OK pour générer les vignettes. Sélectionnez et supprimez l’annotation de zone de périmètre utilisée pour générer les vignettes à l’étape 5.2.2.
- Sélectionnez le menu déroulant Actions de calque | Exportez pour enregistrer les annotations en mosaïque en tant que fichier .annotation.
- Placez une copie enregistrée de l’algorithme Python « Dapọ » (disponible via https://github.com/GYNCOE/Mitchell.et.al.2022), développé pour fusionner les couches d’annotation classées par l’IA, dans le même dossier que le fichier d’annotations en mosaïque.
- Copiez le nom du fichier d’annotation en mosaïque. Ouvrez le programme Python à l’aide de l’environnement de développement intégré IDLE et collez le nom du fichier d’annotation en mosaïque entre les guillemets au bas du programme.
- Sélectionnez le menu déroulant Exécuter | Exécutez Module. Attendez qu’un nouveau fichier soit généré qui aura toutes les annotations en mosaïque fusionnées sous un seul calque.
- Ouvrez le logiciel d’analyse d’images et accédez à l’onglet Annotations . Sélectionnez le menu déroulant Actions de calque | Supprimer tous les calques pour supprimer toutes les annotations de l’image.
- Sélectionnez le menu déroulant Actions de calque | Importer un fichier d’annotation local. Dans la fenêtre contextuelle, sélectionnez le fichier .annotation fusionné généré par le script. Assurez-vous que toutes les vignettes importées se trouvent sous le même calque d’annotation.
- Accédez à l’onglet Classificateur et suivez les instructions du fabricant pour générer des formes pour les rois. Avant d’exécuter le classificateur, sélectionnez la ou les couches d’annotation souhaitées (c’est-à-dire la ou les couches de tumeur ou de stroma) en cochant la ou les cases ROI de l’onglet Annotations , sous options avancées du classificateur. Utilisez l’option Calque d’annotation du menu Actions du classificateur pour exécuter le classificateur.
- Une fois l’analyse du classificateur terminée, accédez à l’onglet Annotations et sélectionnez la couche d’annotation générée à partir de l’analyse. Sélectionnez le menu déroulant Actions de calque | Supprimer tous les calques mais actuels pour supprimer tous les autres calques d’annotation de l’image.
- Sélectionnez le menu déroulant Actions de calque | Exportez pour enregistrer les annotations en tant que fichier .annotation. Créez un dossier pour la session ou le projet et enregistrez le fichier .annotation dans un sous-dossier étiqueté avec l’identificateur unique de la diapositive.
REMARQUE : Un fichier d’annotation représentatif pour une collection de tissus enrichis en LMD classifiée est fourni dans le fichier supplémentaire 6. - Accédez à l’onglet Annotations , sélectionnez la liste déroulante Actions de calque | Supprimer tous les calques pour supprimer toutes les annotations de l’image. Sélectionnez l’outil plume et tracez une courte ligne à partir de chaque fiducial d’étalonnage. Tracez des lignes à partir des marques dans l’ordre suivant : en haut à gauche, en haut à droite, en bas à droite.
- Sélectionnez le menu déroulant Actions de calque | Exportez pour enregistrer les annotations de ligne en tant que fichier .annotation. Ajoutez _calib au nom de fichier et placez le fichier dans le sous-dossier qui contient les coordonnées des formes en mosaïque.
- Copiez l’adresse du dossier principal du projet ou de la session. Ouvrez le script de génération d’importation XML, « Malleator » (disponible via https://github.com/GYNCOE/Mitchell.et.al.2022), à l’aide de l’environnement de développement intégré IDLE, puis collez l’adresse du dossier du projet entre les guillemets au bas du script.
- Sélectionnez le menu déroulant Exécuter | Exécutez Module pour exécuter le script.
REMARQUE : Le fichier d’importation LMD .xml sera généré à l’intérieur du sous-dossier créé pour l’image/diapositive.
6. Microdissection laser
REMARQUE : Les étapes du protocole décrites dans cette section sont spécifiques à utiliser avec un microscope laser inversé et le logiciel associé (voir la Table des matériaux). Temps approximatif: 2 h; selon le cas.
- Chargez la lame membranaire marquée (contenant des fiducials d’étalonnage) avec le tissu tourné vers le bas et le côté de l’étiquette plus près de l’opérateur dans le porte-lames sur l’étage du microscope laser.
- Sélectionnez Importer des formes dans le menu déroulant Fichier . Sélectionnez le fichier d’importation LMD .xml généré pour la diapositive. Sélectionnez Non dans la fenêtre contextuelle pour éviter de charger des points de référence à partir du fichier et Non dans la deuxième fenêtre contextuelle pour éviter d’utiliser des points de référence précédemment stockés pour l’étalonnage.
- Suivez les instructions de l’application LMD et alignez la croix d’étalonnage sur chacune des trois fiduciales d’étalonnage sur la diapositive. Recherchez les fiducials d’étalonnage qui apparaissent en haut à gauche, en haut à droite et en bas à droite de l’image de diapositive dans le logiciel d’analyse d’image qui correspondront aux points de référence dans les coins avant droit, arrière droit et arrière gauche, respectivement, de la lame LMD inversée sur l’étage du microscope. Basculez entre l’utilisation de l’objectif 5x pour localiser et l’objectif 63x pour aligner chaque fiducial d’étalonnage. Sélectionnez Non dans la fenêtre contextuelle pour éviter d’enregistrer les points de référence dans un fichier et OK dans la deuxième fenêtre contextuelle pour confirmer que la diapositive est insérée.
- Déplacez l’objectif 5x en position et sélectionnez Oui dans la fenêtre contextuelle pour utiliser le grossissement réel. Une fois que les formes importées apparaissent, concentrez l’appareil photo sur le tissu.
- Mettez en surbrillance et sélectionnez toutes les formes dans la fenêtre Liste des formes , faites-les glisser en place à l’aide d’une ou deux annotations dans le champ de vision en tant que références et alignez l’axe Z vertical pour la découpe avec le laser.
- Examinez les formes importées et affectez-les à la position de tube appropriée pour la collecte. Appuyez sur Démarrer la découpe pour démarrer le laser.
Remarque : Les formes importées dans le fichier .xml seront automatiquement affectées à la position « sans majuscule » dans la fenêtre Liste des formes . Pour prélever des tissus, les formes importées doivent être réaffectées à une position contenant un tube chargé.
7. Digestion des protéines par la technologie du cycle de pression (PCT)
REMARQUE: Temps approximatif: 4 h (3 h sans temps de séchage par centrifugeuse sous vide).
- Placer des tubes de 0,5 mL contenant les microtubes PCT coiffés contenant du tissu LMD récolté dans 20 μL de 100 mM teaB/10 % d’acétonitrile dans un thermocycleur et chauffer à 99 °C pendant 30 min, puis refroidir à 50 °C pendant 10 min.
- Faites tourner les tubes pendant 30 s à 4 000 × g , puis retirez les microtubes des tubes de 0,5 mL. À l’aide de l’outil MicroCap, retirez et jetez les MicroCaps des microTubes PCT. Ajouter la trypsine (voir le tableau des matériaux) à un rapport de 1 μg par tissu de 30 mm2 et insérer un MicroPestle dans le MicroTube à l’aide de l’outil MicroCap.
- Transférez les MicroTubes dans une cartouche de barocycle et assemblez la cartouche complète. Placez la cartouche dans la chambre de pression du barocycle et fixez le couvercle. Barocycle à 45 000 psi pendant 50 s et pression atmosphérique à 10 s à 50 °C pendant 60 cycles.
- Une fois le barocyclage terminé, transférer les microtubes dans un tube de microcentrifugation de 0,5 mL et centrifuger pendant 2 min à 4 000 × g.
- Retirez le microtube du tube de microcentrifugation de 0,5 mL à l’aide de l’outil de capuchon. Retirez soigneusement le pilon à l’aide de l’outil de capuchon et rincez la moitié inférieure du pilon avec 20 μL d’eau de qualité chromatographie de masse par chromatographie liquide (LC-MS) et collectez le lavage dans un tube de microcentrifugation propre de 0,5 mL.
- Tapotez doucement le MicroTube sur la paillasse pour déplacer le liquide vers le bas et transférez toute la solution du MicroTube dans le tube de microcentrifugation de 0,5 mL.
- Ajoutez 20 μL d’eau de qualité LC-MS au MicroTube et tapotez-le doucement sur la paillasse. Transférer la solution de lavage dans le tube de 0,5 mL et répéter cette étape de lavage une fois de plus.
- Centrifugeuse sous vide pour sécher les échantillons à ~2 μL et ajouter 100 μL de 100 mM TEAB, pH 8,0.
- Déterminer la concentration en peptides à l’aide d’un test colorimétrique (dosage de l’acide bicinchoninique (BCA); voir la table des matériaux) selon le protocole du fabricant.
8. Étiquetage de l’étiquette tandem-masse (TMT) et nettoyage EasyPep
REMARQUE: Temps approximatif: 7 h 20 min (2 h 20 min sans temps de séchage par centrifugeuse sous vide).
- Porter les réactifs d’étiquetage TMT isobares à température ambiante avant de les ouvrir. Ajouter 500 μL d’acétonitrile à 100 % à chaque flacon de TMT (5 mg). Incuber pendant 10 min avec des vortex occasionnels.
- Dissoudre 5 μg de l’échantillon peptidique dans 100 μL de 100 mM de TEAB, pH 8,0, et ajouter 10 μL d’un réactif TMT donné. Construire et inclure des pools de référence représentant chaque échantillon individuel de l’expérience dans chaque ensemble d’échantillons multiplex TMT afin de faciliter la quantification des échantillons sur plusieurs multiplexes TMT9. Incuber les réactions pendant 1 h à température ambiante avec agitation/tapotement occasionnel.
- Éteindre la réaction de marquage TMT en ajoutant 10 μL d’hydroxylamine à 5% et incuber pendant 30 min à température ambiante avec un taraudage occasionnel. Après trempe, mélanger les échantillons marqués TMT dans un seul tube et sécher à environ 200 μL.
- Ajouter 1 800 μL d’acide formique à 0,1 %. Vérifiez le pH avec du papier pH: si pH ~ 3, ajoutez 1 mL d’acide formique à 0,1%; si le pH >3, ajouter 10-20 μL d’acide formique à 5% jusqu’à pH ~3. Ajouter 0,1 % d’acide formique pour porter à un volume final de 3 mL.
- Retirez la languette au bas de la colonne de nettoyage des peptides, retirez le capuchon et placez-le dans un tube conique de 15 mL. Transférez l’échantillon étiqueté TMT dans la colonne et procédez au nettoyage conformément au protocole du fabricant.
- Centrifugeuse sous vide pour sécher les peptides élués à ~20 μL, transfert dans un flacon de LC à l’aide de 25 mM de bicarbonate d’ammonium pour un volume final de 80 μL et procéder au fractionnement hors ligne.
9. Fractionnement et mise en commun des échantillons multiplex TMT
REMARQUE: Temps approximatif: 3 h 30 min (1 h 30 min sans temps de séchage par centrifugeuse sous vide).
- Fractionner les multiplexes peptidiques marqués TMT par chromatographie de base en phase inversée en 96 fractions en développant un gradient linéaire croissant (0,69 % min-1) de la phase mobile B (acétonitrile) en phase mobile A (10 mM NH4HCO3, pH 8,0).
- Générez 36 fractions concaténées en regroupant des puits d’échantillonnage. Centrifuger sous vide pour sécher les fractions à ~2 μL et remettre en suspension dans 25 mM de bicarbonate d’ammonium (concentration finale 1,5 μg/10 μL), centrifuger à 15 000 × g pendant 10-15 min, et transférer dans des flacons LC pour analyse MS.
10. Chromatographie liquide spectrométrie de masse en tandem (LC-MS/MS)
REMARQUE: Temps approximatif: Méthode de l’instrument et conception expérimentale dépendants.
- Calibrez le spectromètre de masse selon les instructions/protocoles du fabricant.
- Préparer de nouvelles phases et normes mobiles et effectuer les préparations LC pré-série appropriées (y compris, mais sans s’y limiter, les solvants de purge, l’air de rinçage et les scripts de test d’étanchéité pour l’instrument référencé [voir le tableau des matériaux]). Équilibrez les colonnes pré- et analytiques et la boucle d’échantillonnage avant de commencer les analyses.
- Avant et entre les analyses multiplex TMT en série, validez que le système LC-MS répond aux mesures de performance précédemment étalonnées à l’aide de digestes peptidiques marqués TMT d’assurance qualité/ contrôle de la qualité (QA / QC) et (par exemple, MSPE (voir le tableau des matériaux), HeLa).
- Chargez les flacons d’échantillonneur automatique dans les positions appropriées dans l’échantillonneur automatique LC. Analysez les fractions individuelles avec une méthode de gradient/MS appropriée. Entrecouper un « lavage » avec un étalon peptidique (p. ex., étalonnage du temps de rétention peptidique [PRTC]) environ une fois par jour pour évaluer la performance chromatographique et spectrale de masse. Après l’analyse de chaque série d’échantillons de fractions multiplex TMT, exécutez les normes de référence QA/QC TMT pour évaluer les performances du système.
- Exécutez des routines d’évaluation du spectromètre de masse selon les normes de référence QA/QC TMT pour évaluer les performances post-échantillonnage, puis étalonnez le système comme à l’étape 10.1 pour le prochain ensemble d’échantillons.
11. Analyse des données bioinformatiques
REMARQUE: Temps approximatif: Dépend de la conception expérimentale.
- Transférez tous les exemples de données (par exemple, les fichiers .raw) vers un stockage réseau/lecteur d’ordinateur approprié.
- Recherchez toutes les fractions ensemble à l’aide de l’application d’analyse de données souhaitée (par exemple, Proteome Discover, Mascot) en utilisant les paramètres appropriés9 par rapport à une base de données de référence de protéines spécifiques à une espèce pour générer des correspondances spectrales peptidiques (PSM) et extraire les intensités du signal ionique rapporteur TMT. Filtrer les MSP en fonction des mesures de contrôle de la qualité appropriées et agréger les abondances normalisées, médianes du rapport ionique rapporteur TMT transformées log2 en abondances globales au niveau des protéines, comme décrit précédemment 3,9.
- Comparez les altérations protéiques dans des conditions d’intérêt à l’aide du logiciel d’analyse différentielle souhaité.
Résultats
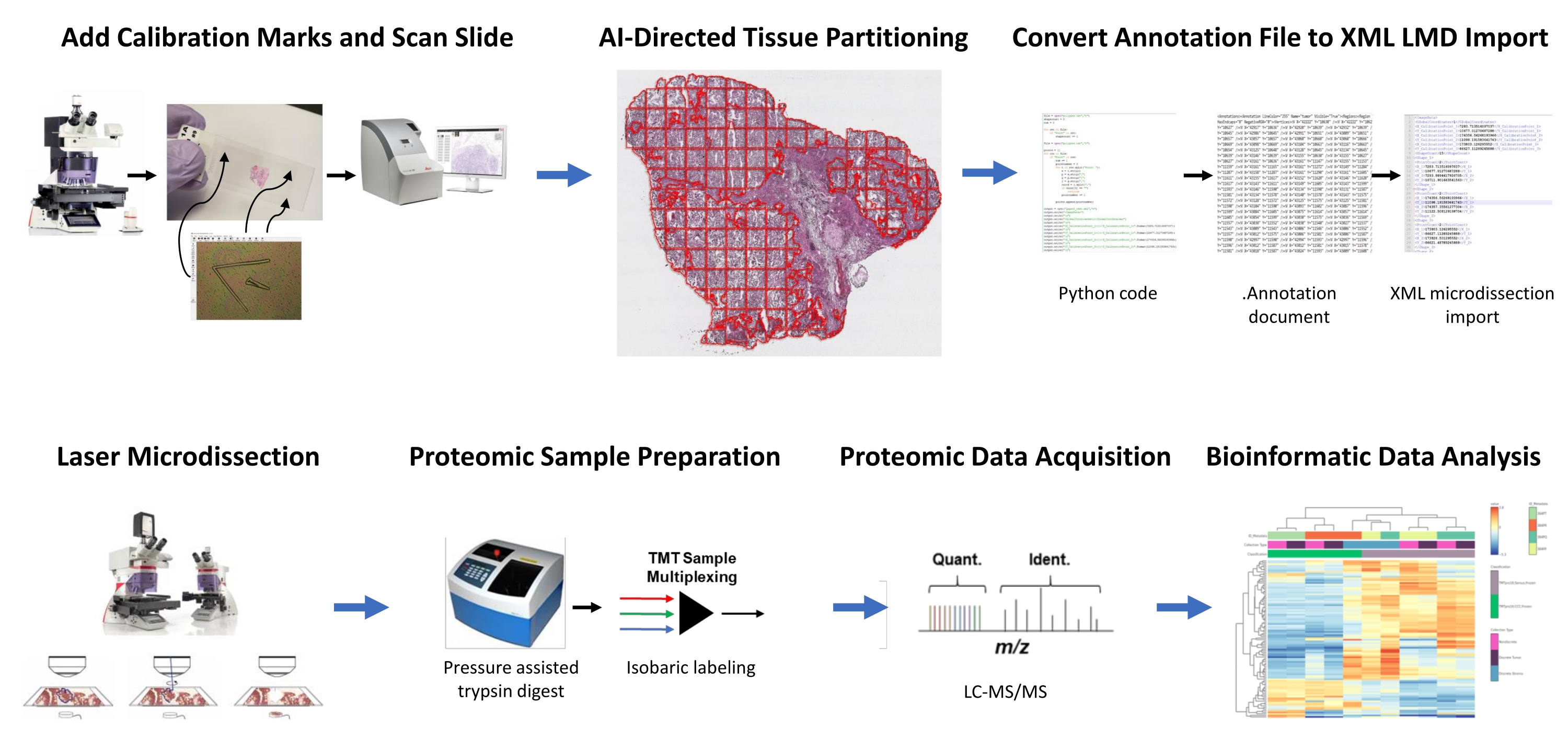
Des coupes minces de tissus fraîchement congelés de deux patients atteints de HGSOC et de deux patients OCCC ont été analysées à l’aide de ce flux de travail intégré d’identification, de segmentation, de LMD et d’analyse protéomique quantitative des tissus piloté par l’IA (Figure 1). Les sections représentatives de tissus colorés H & E pour chaque tumeur ont été examinées par un pathologiste certifié par le conseil d’administration; la cellularité tumorale variait de 70% à 99%. Les tissus ont été sectionnés finement sur des lames de membrane PEN (fichier supplémentaire 2) et prédécoupés avec des fiducials d’étalonnage (fichier supplémentaire 1), ce qui a permis d’intégrer les données d’orientation positionnelle des annotations générées dans le logiciel d’analyse d’images (voir la table des matériaux) avec l’orientation des coordonnées cartésiennes dans le logiciel LMD. Après la coloration H&E, des images haute résolution (20x) des lames PEN contenant le tissu et les calibrateurs ont été capturées.
Les populations de cellules tumorales et stromales dans les micrographies ont été segmentées à l’aide d’un logiciel d’analyse d’images (voir le tableau des matériaux) pour une récolte sélective par LMD, ainsi que des récoltes représentant l’ensemble de la section mince du tissu (par exemple, le tissu tumoral entier) (Figure 1). Des annotations non discriminantes pour les collections de tissus tumoraux entiers ont été générées en partitionnant toute la section tissulaire avec des carreaux de 500 μm2, laissant un espace de 40 μm entre les carreaux pour maintenir l’intégrité de la membrane PEN et empêcher la membrane de s’enrouler pendant la LMD. Sur les diapositives pour l’enrichissement en LMD résolu par histologie, le classificateur d’IA dans le logiciel d’analyse d’images (voir la table des matériaux) a été formé pour discriminer entre les cellules tumorales et stromales, ainsi que le fond de diapositive en verre vierge. Les régions représentatives de la tumeur, du stroma et du verre vierge ont été mises en évidence manuellement, et l’outil de classification a été utilisé pour segmenter ces ROI dans toute la section tissulaire. Les couches segmentées représentant le tissu entier, l’épithélium tumoral et le stroma ont été enregistrées séparément en tant que fichiers d’annotation individuels (fichier supplémentaire 3 et fichier supplémentaire 6). Dans une copie séparée du fichier image (sans les annotations ROI partitionnées), une courte ligne de l’extrémité la plus centrale de chacun des trois calibrateurs fiducial a été annotée et enregistrée en tant que fichier .annotation en utilisant le même nom de fichier que chacun des fichiers de couche d’annotation LMD, mais avec le suffixe « _calib » (fichier supplémentaire 4). Ces lignes ont été utilisées pour co-enregistrer la position des calibrateurs à membrane PEN avec les données de la liste de formes d’annotation dessinées dans le logiciel d’analyse d’images.
La présente étude fournit deux algorithmes, « Malleator » et « Dapọ » en Python pour prendre en charge ce flux de travail LMD piloté par l’IA, qui sont disponibles à https://github.com/GYNCOE/Mitchell.et.al.2022. L’algorithme Malleator extrait les coordonnées cartésiennes spécifiques pour toutes les annotations individuelles (ROI tissulaire et calibrateurs) des fichiers .annotation appariés et les fusionne en un seul fichier d’importation XML (Extensible Markup Language) (fichier supplémentaire 5). Plus précisément, l’algorithme Malleator utilise le nom de répertoire d’un dossier parent comme entrée pour rechercher tous les dossiers de sous-répertoires et génère des fichiers .xml pour tous les sous-dossiers qui n’ont pas encore de .xml fichier fusionné. L’algorithme Malleator fusionne toutes les couches d’annotation du logiciel d’analyse d’images (voir la table des matériaux) en une seule couche et convertit les données de liste de formes générées par l’IA, qui sont enregistrées en tant que type de fichier .annotation propriétaire, dans .xml format compatible avec le logiciel LMD. Après avoir fusionné les fichiers d’annotation et d’étalonnage, le fichier .xml généré par l’algorithme est enregistré et importé dans le logiciel LMD. De légers ajustements sont nécessaires pour ajuster manuellement l’alignement des annotations, ce qui sert également à enregistrer la position verticale (plan z) de l’étage de diapositive sur le microscope laser. L’algorithme Dapọ est utilisé spécifiquement pour les collections enrichies en LMD. Les tuiles partitionnées sont automatiquement affectées à des couches d’annotation individuelles par le logiciel d’analyse d’images. L’algorithme Dapọ fusionne toutes les vignettes partitionnées en une seule couche d’annotation avant l’utilisation de l’outil Classificateur, réduisant ainsi le temps d’exécution de l’analyse Classifier pour les collections enrichies LMD.
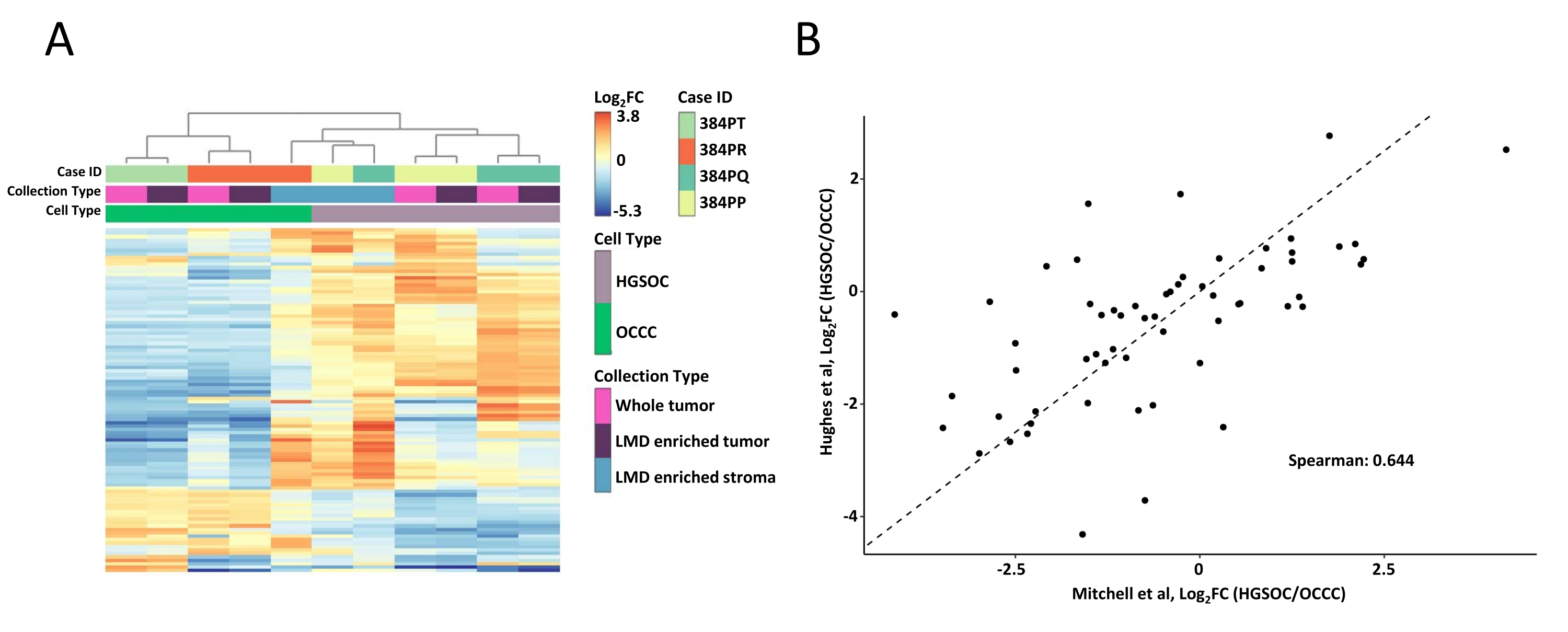
Les échantillons de tumeurs entières et de tissus enrichis en LMD ont été digérés, marqués avec des réactifs TMT, multiplexés, fractionnés hors ligne et analysés via une protéomique quantitative basée sur la SEP comme décrit précédemment9. Le rendement moyen en peptides (43-60 μg) et la récupération (0,46-0,59 μg/mm2) pour les échantillons prélevés à l’aide de ce flux de travail basé sur l’IA étaient comparables aux rapports précédents 9,10. Au total, 5 971 protéines ont été co-quantifiées dans tous les échantillons (tableau supplémentaire S1). Le regroupement hiérarchique non supervisé utilisant les 100 protéines les plus variables a entraîné une ségrégation des histotypes HGSOC et OCCC des échantillons de tumeurs entières enrichis en LMD (Figure 2A), similaire à celle décrite précédemment11. En revanche, les échantillons de stroma enrichis en LMD provenant du HGSOC et de l’OCCC se sont regroupés et indépendamment des échantillons de tumeur enrichis en LMD et de tumeurs entières. Parmi les 5 971 protéines quantifiées, 215 ont été significativement modifiées (LIMMA adj. p < 0,05) entre des collections tumorales entières provenant d’échantillons de HGSOC et d’OCCC (tableau supplémentaire S2). Ces protéines altérées ont été comparées à celles identifiées pour différencier le tissu tumoral HGSOC et OCCC par Hughes et al.11. Sur les 76 protéines de signature quantifiées par Hughes et coll., 57 ont été co-quantifiées dans cet ensemble de données et étaient fortement corrélées (Spearman Rho = 0,644, p < 0,001) (Figure 2B).

Figure 1 : Résumé du flux de travail intégré pour la sélection automatisée de la région tissulaire d’intérêt pour la microdissection laser pour la protéomique quantitative en aval. Les fiduciales d’étalonnage sont découpées sur des lames de membrane PEN pour co-enregistrer les données d’orientation positionnelle des segments dérivés de l’IA du retour sur investissement tissulaire dans le logiciel d’analyse d’images HALO, avec positionnement horizontal sur le microscope LMD. L’algorithme Malleator est utilisé pour fusionner les données de segmentation annotées sur toutes les couches d’annotation d’une diapositive avec le fichier de référence _calib et pour la convertir en un fichier .xml compatible avec le logiciel LMD. Les tissus récoltés par LMD pour l’analyse protéomique sont digérés et analysés par protéomique quantitative à haut débit, comme décrit précédemment9. Abréviations : LMD = microdissection laser ; ROI = région d’intérêt; TMT = étiquette de masse en tandem; Quant. = quantification; Ident. = identification; LC-MS/MS = chromatographie liquide-spectrométrie de masse en tandem. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2: Analyse des protéines dans des échantillons de tumeurs entières et enrichies en LMD. (A) Analyse hiérarchique non supervisée des 100 protéines les plus diversement abondantes dans des échantillons de tumeurs HGSOC et OCCC LMD enrichis et entiers. (B) Corrélation des abondances de protéines log2 fold-change entre les récoltes de tumeurs entières HGSOC et OCCC dans la présente étude (Mitchell et al., axe des x) et une étude similaire de Hughes et al. (axe des y)11. Abréviations : LMD = microdissection laser ; HGSOC = cancer de l’ovaire séreux de haut grade; OCCC = carcinome ovarien à cellules claires; log2FC = log2-transformée abondance protéomique. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Tableau supplémentaire S1: Abondances de 5 971 protéines co-quantifiées dans tous les échantillons de tumeurs entières enrichies en LMD provenant d’échantillons de tissus HGSOC et OCCC. Abréviations : LMD = microdissection laser ; HGSOC = cancer de l’ovaire séreux de haut grade; OCCC = carcinome ovarien à cellules claires. Veuillez cliquer ici pour télécharger ce tableau.
Tableau supplémentaire S2: Protéines exprimées différentiellement (215) dans des collections tumorales entières de HGSOC vs OCCC (LIMMA adj. p < 0,05). Abréviations : HGSOC = cancer de l’ovaire séreux de haut grade; OCCC = carcinome ovarien à cellules claires. Veuillez cliquer ici pour télécharger ce tableau.
Fichier supplémentaire 1 : Fichier de données de liste de formes représentatives (.sld) contenant des fiducials d’étalonnage standard pour quatre positions de diapositives. Le fichier peut être importé dans le logiciel LMD. Veuillez cliquer ici pour télécharger ce fichier.
Fichier supplémentaire 2 : Fichier image .svs représentatif d’une section de tissu haute résolution (20x) colorée par H&E. Le fichier peut être ouvert et visualisé à l’aide d’un logiciel d’analyse d’images ou d’un logiciel LMD. Abréviation : H&E = hématoxyline et éosine ; LMD = microdissection laser. Veuillez cliquer ici pour télécharger ce fichier.
Fichier supplémentaire 3: Fichier d’annotation représentatif de segments tumoraux entiers partitionnés. Le fichier peut être importé dans un logiciel d’analyse d’images. Veuillez cliquer ici pour télécharger ce fichier.
Fichier supplémentaire 4 : Fichier _calib.annotation représentatif des segments fiducial calibrateurs. Les informations de coordonnées représentent le positionnement oriental des courtes lignes d’étalonnage dessinées à partir de chaque fiducial à pointe de flèche. Le fichier peut être importé dans un logiciel d’analyse d’images. Veuillez cliquer ici pour télécharger ce fichier.
Fichier supplémentaire 5 : Fichier représentatif du langage de balisage extensible (.xml) généré par l’algorithme Malleator. Le fichier peut être importé dans le logiciel de microdissection laser. Veuillez cliquer ici pour télécharger ce fichier.
Fichier supplémentaire 6 : Fichier .annotation représentatif des segments partitionnés classés par IA pour les collections enrichies en LMD. Le fichier peut être importé dans un logiciel d’analyse d’images. Abréviations : IA = intelligence artificielle ; LMD = microdissection laser. Veuillez cliquer ici pour télécharger ce fichier.
Discussion
Bien qu’il y ait eu de multiples précédents d’étude visant à développer et / ou à améliorer les flux de travail pour l’enrichissement des sous-populations cellulaires cibles à partir de FFPE et / ou de tissus fraîchement congelés et des méthodologies pour maintenir la qualité des échantillons pendant le traitement 9,12,13,14,15, il existe un besoin important de développer des stratégies automatisées pour préparer des échantillons de tissus cliniques pour des analyses moléculaires afin de réduire la variabilité et augmenter la reproductibilité. Ce flux de travail décrit un protocole standardisé et semi-automatisé qui intègre les outils logiciels d’analyse d’images existants (voir la table des matériaux) pour la récolte résolue par histologie de populations de cellules discrètes par LMD à partir d’échantillons de tissus cliniques.
L’enrichissement en LMD à résolution spatiale des ROI capturant des populations cellulaires discrètes représente une étape de traitement tissulaire de nouvelle génération avant les analyses multiomiques afin d’améliorer la caractérisation moléculaire et l’identification et de faciliter la découverte de biomarqueurs sélectifs cellulaires. Ce protocole améliore les méthodologies existantes en réduisant l’exposition souvent longue des coupes de tissus à l’environnement ambiant qui est associée à la segmentation manuelle du retour sur investissement par un histologue (ce qui peut prendre >1 à 2 heures avant la collecte de LMD). Ce flux de travail permet plutôt au retour sur investissement d’être préidentifié par une classification et une segmentation guidées par l’IA. Limiter le temps de séjour des tissus réduira les variations fallacieuses dans les évaluations des cibles moléculaires hautement labiles, telles que les phosphopeptides et l’ARNm, ou pour les techniques analytiques à base d’anticorps qui reposent sur une protéine cible dans sa conformation native pour la détection.
La découpe de fiducials d’étalonnage soignés sur la lame de membrane PEN qui sont clairement visibles dans l’image de diapositive numérisée est l’un des éléments clés permettant l’intégration du logiciel d’analyse d’image (voir la table des matériaux) avec le flux de travail LMD. S’assurer que les calibrateurs ont un point précis (« propre ») au bas de la forme « V » permet de sélectionner un point précis dans le logiciel d’analyse d’image pour les lignes d’étalonnage à dessiner, comme décrit aux étapes 5.1.6 et 5.2.13. L’alignement de ces points lors de l’importation dans le logiciel LMD est essentiel pour superposer correctement les annotations (facilité par la génération d’un fichier .xml compatible à l’aide des algorithmes « Malleator » et / ou « Dapọ ») sur le retour sur investissement tissulaire pertinent sur la diapositive LMD physique. Il est nécessaire de mettre en évidence toutes les formes et collectivement de « glisser-déposer » en place même lorsque l’alignement est précis lors de l’importation dans le logiciel LMD pour enregistrer la position verticale (plan z) de l’étage de diapositive sur le microscope laser. Des ajustements mineurs au positionnement des annotations sur le roi des tissus peuvent également être effectués au cours de cette étape, si nécessaire.
Une limitation de la version actuelle de l’algorithme Malleator est qu’elle n’est pas compatible avec les outils de forme d’annotation prédéfinis fournis par le logiciel d’analyse d’images (voir la table des matériaux), bien que les futures mises à jour / versions de l’algorithme viseront à améliorer cette compatibilité. Le fichier .annotation pour les formes dessinées à l’aide de ces outils ne contient que deux ensembles de coordonnées x et y appariées pour chaque annotation, sans l’orientation spatiale complète autour de ces points. L’utilisation actuelle de ces outils entraîne la conversion des annotations en lignes droites définies par seulement deux points au cours du processus d’importation. La définition manuelle des segments de retour sur investissement des tissus est nécessaire pour une conversion réussie au format XML et à l’importation LMD. Cela peut être effectué en définissant manuellement chaque retour sur investissement avec des annotations polygonales individuelles à main levée spécifiques à la zone cible ou en appliquant une annotation circulaire ou rectangulaire approximative sur tous les segments de retour sur investissement des tissus, si vous le souhaitez, et sera compatible avec ce flux de travail.
Bien que le flux de travail présenté ici ait été démontré pour l’analyse protéomique d’échantillons de tissus cancéreux humains fraîchement congelés, ce flux de travail LMD piloté par l’IA peut être utilisé de manière équivalente avec les tissus FFPE, les types de tissus non cancéreux et ceux provenant de sources non humaines. Il peut également prendre en charge d’autres flux de travail de profilage moléculaire en aval, y compris les analyses transcriptomiques, génomiques ou phosphoprotéomiques. Ce flux de travail peut également tirer parti d’autres utilisations du logiciel d’analyse d’images (voir le tableau des matériaux), y compris avec des capacités associées au comptage de cellules ou à d’autres modules analytiques, y compris le module « Multiplex IHC » ou le « Tissue Microarray (TMA) Add-on ». Les applications futures de ce flux de travail pourraient également bénéficier de la prédéfinition du nombre de cellules par segment de retour sur investissement, garantissant ainsi des entrées cellulaires équivalentes dans plusieurs collections, ou en utilisant des méthodes alternatives pour définir les ROI cellulaires d’intérêt, telles que l’immunohistochimie ou la sociologie cellulaire.
Déclarations de divulgation
T.P.C. est membre de ThermoFisher Scientific, Inc SAB et reçoit un financement de recherche d’AbbVie.
Remerciements
Le financement de ce projet a été fourni en partie par le Defense Health Program (HU0001-16-2-0006 et HU0001-16-2-00014) à l’Uniformed Services University for the Gynecologic Cancer Center of Excellence. Les promoteurs n’ont joué aucun rôle dans la conception, l’exécution, l’interprétation ou la rédaction de l’étude. Démenti: Les opinions exprimées dans le présent document sont celles des auteurs et ne reflètent pas la politique officielle du département de l’Armée / Marine / Force aérienne, du ministère de la Défense ou du gouvernement des États-Unis.
matériels
| Name | Company | Catalog Number | Comments |
| 1260 Infinity II System | Agilent Technologies Inc | Offline LC system | |
| 96 MicroCaps (150uL) in bulk | Pressure Biosciences Inc | MC150-96 | |
| 96 MicroPestles in bulk | Pressure Biosciences Inc | MP-96 | |
| 96 MicroTubes in bulk (no caps) | Pressure Biosciences Inc | MT-96 | |
| 9mm MS Certified Clear Screw Thread Kits | Fisher Scientific | 03-060-058 | Sample vial for offline LC frationation and mass spectrometry |
| Acetonitrile, Optima LC/MS Grade | Fisher Chemical | A995-4 | Mobile phase solvent |
| Aperio AT2 | Leica Microsystems | 23AT2100 | Slide scanner |
| Axygen PCR Tubes with 0.5 mL Flat Cap | Fisher Scientific | 14-222-292 | Sample tubes; size fits PCT tubes and thermocycler |
| Barocycler 2320EXT | Pressure Biosciences Inc | 2320-EXT | Barocycler |
| BCA Protein Assay Kit | Fisher Scientific | P123225 | |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 11836170001 | |
| Easy-nLC 1200 | Thermo Fisher Scientific | Liquid Chromatography | |
| EasyPep Maxi Sample Prep Kit | Thermo Fisher Scientific | NCI5734 | Post-label sample clean up column |
| EASY-SPRAY C18 2UM 50CM X 75 | Fisher Scientific | ES903 | Analytical column |
| Eosin Y Solution Aqueous | Sigma Aldrich | HT110216 | |
| Formic Acid, 99+ % | Thermo Fisher Scientific | 28905 | Mobile phase additive |
| ggplot2 version 3.3.5 | CRAN | https://cran.r-project.org/web/packages/ggplot2/ | |
| HALO | Indica Labs | Image analysis software | |
| IDLE (Integrated Development and Learning Environment) | Python Software Foundation | ||
| iheatmapr version 0.5.1 | CRAN | https://cran.r-project.org/web/packages/iheatmapr/ | |
| iRT Kit | Biognosys | Ki-3002-1 | LC-MS QAQC Standard |
| limma version 3.42.2 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/limma.html | |
| LMD Scanning stage Ultra LMT350 | Leica Microsystems | 11888453 | LMD stage model outfitted with PCT tube holder |
| LMD7 (software version 8.2.3.7603) | Leica Microsystems | LMD apparatus (microscope, laser, camera, PC, tablet) | |
| Mascot Server | Matrix Science | Data analysis software | |
| Mass Spec-Compatible Human Protein Extract, Digest | Promega | V6951 | LC-MS QAQC Standard |
| Mayer’s Hematoxylin Solution | Sigma Aldrich | MHS32 | |
| PEN Membrane Glass Slides | Leica Microsystems | 11532918 | |
| Peptide Retention Time Calibration Mixture | Thermo Fisher Scientific | 88321 | LC-MS QAQC Standard |
| Phosphatase Inhibitor Cocktail 2 | Sigma Aldrich | P5726 | |
| Phosphatase Inhibitor Cocktail 3 | Sigma Aldrich | P0044 | |
| Pierce LTQ Velos ESI Positive Ion Calibration Solution | Thermo Fisher Scientific | 88323 | Instrument calibration solution |
| PM100 C18 3UM 75UMX20MM NV 2PK | Fisher Scientific | 164535 | Pre-column |
| Proteome Discoverer | Thermo Fisher Scientific | OPTON-31040 | Data analysis software |
| Python | Python Software Foundation | ||
| Q Exactive HF-X | Thermo Fisher Scientific | Mass spectrometer | |
| R version 3.6.0 | CRAN | https://cran-archive.r-project.org/bin/windows/base/old/2.6.2/ | |
| RColorBrewer version 1.1-2 | CRAN | https://cran.r-project.org/web/packages/RColorBrewer/ | |
| Soluble Smart Digest Kit | Thermo Fisher Scientific | 3251711 | Digestion reagent |
| TMTpro 16plex Label Reagent Set | Thermo Fisher Scientific | A44520 | isobaric TMT labeling reagents |
| Veriti 60 well thermal cycler | Applied Biosystems | 4384638 | Thermocycler |
| Water, Optima LC/MS Grade | Fisher Chemical | W6-4 | Mobile phase solvent |
| ZORBAX Extend 300 C18, 2.1 x 12.5 mm, 5 µm, guard cartridge (ZGC) | Agilent Technologies Inc | 821125-932 | Offline LC trap column |
| ZORBAX Extend 300 C18, 2.1 x 150 mm, 3.5 µm | Agilent Technologies Inc | 763750-902 | Offline LC analytical column |
Références
- Motohara, T., et al. An evolving story of the metastatic voyage of ovarian cancer cells: cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene. 38 (16), 2885-2898 (2019).
- Aran, D., Sirota, M., Butte, A. J. Systematic pan-cancer analysis of tumour purity. Nature Communications. 6, 8971 (2015).
- Hunt, A. L., et al. Extensive three-dimensional intratumor proteomic heterogeneity revealed by multiregion sampling in high-grade serous ovarian tumor specimens. iScience. 24 (7), 102757 (2021).
- Dou, Y., et al. Proteogenomic characterization of endometrial carcinoma. Cell. 180 (4), 729-748 (2020).
- Zhang, H., et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 166 (3), 755-765 (2016).
- Gillette, M. A., et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell. 182 (1), 200-225 (2020).
- Silvestri, A., et al. Protein pathway biomarker analysis of human cancer reveals requirement for upfront cellular-enrichment processing. Laboratory Investigation. 90 (5), 787-796 (2010).
- Echle, A., et al. Deep learning in cancer pathology: a new generation of clinical biomarkers. British Journal of Cancer. 124 (4), 686-696 (2021).
- Lee, S., et al. Molecular analysis of clinically defined subsets of high-grade serous ovarian cancer. Cell Reports. 31 (2), 107502 (2020).
- Xuan, Y., et al. Standardization and harmonization of distributed multi-center proteotype analysis supporting precision medicine studies. Nature Communications. 11 (1), 5248 (2020).
- Hughes, C. S., et al. Quantitative profiling of single formalin fixed tumour sections: proteomics for translational research. Scientific Reports. 6 (1), 34949 (2016).
- Espina, V., et al. A portrait of tissue phosphoprotein stability in the clinical tissue procurement process. Molecular & Cellular Proteomics. 7 (10), 1998-2018 (2008).
- Espina, V., Heiby, M., Pierobon, M., Liotta, L. A. Laser capture microdissection technology. Expert Review of Molecular Diagnostics. 7 (5), 647-657 (2007).
- Havnar, C. A., et al. Automated dissection protocol for tumor enrichment in low tumor content tissues. Journal of Visualized Experiments. (169), e62394 (2021).
- Mueller, C., et al. One-step preservation of phosphoproteins and tissue morphology at room temperature for diagnostic and research specimens. PLoS One. 6 (8), (2011).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.