
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Essai de sédimentation basé sur la concanavaline A pour mesurer la liaison au substrat des phosphatases glucanes
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
Cette méthode décrit un essai de sédimentation in vitro à base de lectine pour quantifier l’affinité de liaison de la phosphatase glucane et de l’amylopectine. Ce test de co-sédimentation est fiable pour mesurer la liaison du substrat de la phosphatase glucane et peut être appliqué à divers substrats de glucane solubilisés.
Résumé
Les phosphatases glucanes appartiennent à la grande famille des phosphatases à double spécificité (DSP) qui déphosphorylent les substrats de glucanes, tels que le glycogène chez les animaux et l’amidon chez les plantes. Les structures cristallines de la phosphatase glucane avec des substrats de glucane modèles révèlent des interfaces distinctes de liaison au glucane constituées de domaines DSP et de liaison aux glucides. Cependant, les mesures quantitatives des interactions de la phosphatase glucane-glucane avec des substrats physiologiquement pertinents sont fondamentales pour la compréhension biologique de la famille des enzymes phosphatases glucane et la régulation du métabolisme énergétique. Ce manuscrit rapporte un essai de sédimentation in vitro basé sur la concanavaline A (ConA) conçu pour détecter l’affinité de liaison au substrat des phosphatases glucanes contre différents substrats de glucanes. Comme preuve de concept, la constante de dissociation (KD) de la phosphatase glucane Arabidopsis thaliana Starch Excess4 (SEX4) et de l’amylopectine a été déterminée. La caractérisation des mutants SEX4 et d’autres membres de la famille des enzymes phosphatases glucane démontre une fois de plus l’utilité de ce test pour évaluer la liaison différentielle des interactions protéine-glucides. Ces données démontrent la pertinence de ce test pour caractériser une large gamme de protéines interagissant avec l’amidon et le glycogène.
Introduction
Les phosphatases glucanes sont membres d’une sous-famille fonctionnellement diversifiée de phosphatases à double spécificité (DSP) au sein de la superfamille1 de la protéine tyrosine phosphatase (PTP). Ils ont été trouvés dans la plupart des formes de vie, y compris les organismes photosynthétiques très divergents, les humains, les vertébrés et certains invertébrés et protistes 2,3,4. Les plantes contiennent trois phosphatases glucanes connues : Starch Excess4 (SEX4), Like Sex Four1 (LSF1) et Like Sex Four2 (LSF2)5,6,7. Les plantes dépourvues de phosphatases glucanes présentent une diminution des taux de dégradation transitoire de l’amidon et d’accumulation d’amidon dans les feuilles 8,9. La laforine est le membre fondateur de la famille des phosphatases glucanes qui déphosphoryle le glycogène chez les vertébrés et les humains 3,10. Les mutations de la laforine entraînent la maladie neurodégénérative de Lafora, une forme autosomique récessive mortelle de l’épilepsie11. Les phosphatases glucanes sont nécessaires au métabolisme du glycogène et de l’amidon et sont apparues comme des enzymes importantes pour moduler la teneur en amidon des plantes et traiter la maladie neurodégénérative de Lafora12,13. Des études récentes de cristallographie aux rayons X sur des phosphatases de glucane avec des substrats de glucane modèles ont mis en lumière la liaison du substrat et le mécanisme catalytique de la déphosphorylation du glucane14,15,16,17. Cependant, la compréhension actuelle de la façon dont les phosphatases glucanes se lient à leurs substrats physiologiques est incomplète.
L’amidon est un polymère insoluble de glucose composé de 80% à 90% d’amylopectine et de 10% à 20% d’amylose18. Les substrats des phosphatases de glucane végétaux sont des molécules de glucides phosphorylés, telles que le glycogène et les granules d’amidon. Les résidus de glucosyle phosphorylés sont présents à un rapport phosphate:glucosyl de 1:600. Fait intéressant, les phosphates ne sont présents que sur les molécules d’amylopectine19. La plante principale glucane phosphatase SEX4 agit sur le granule d’amidon pour déphosphoryler les molécules d’amylopectine. La structure cristalline en rayons X de SEX4 combinée à des études de mutagénèse guidée par la structure a démontré les spécificités uniques du substrat de SEX4 pour différentes positions au sein d’une structure de glucane15. Nous avons récemment montré que l’activité biologiquement pertinente de SEX4 ne peut être observée que lorsqu’elle agit sur ses substrats d’amylopectine solubilisés20. Cependant, la compréhension des interactions glucane-SEX4 s’est avérée difficile en raison de la complexité structurelle du substrat, des spécificités de liaison plus larges et des faibles affinités de liaison entre la protéine et ses substrats. Ces problèmes ont entravé la capacité d’utiliser des méthodes couramment utilisées dans les interactions protéine-ligand, telles que la calorimétrie de titrage isotherme (ITC), la spectroscopie par résonance magnétique nucléaire (RMN) et les tests basés sur des tests immuno-enzymatiques (ELISA).
Fait intéressant, une grande partie de notre compréhension des interactions glucides-protéines provient de l’étude des lectines. La concanavaline A (ConA) est une famille de lectines de légumineuses extraites à l’origine du haricot jacquier. ConA lie les glucides avec une spécificité élevée, ce qui est avantageux pour son utilisation dans les applications de ciblage et d’administration de médicaments. La liaison de ConA à une variété de substrats contenant du α-D-mannosyl non réducteur et du α-D-glucosyl a été largement étudiée19,20. Les billes de sépharose liées à ConA disponibles dans le commerce sont couramment utilisées pour purifier les glycoprotéines et les glycolipides21. ConA se lie à ces glucanes via les groupes hydroxyles C3, C4 et C6 des résidus de glucose. Les billes ConA-Sepharose ont également été utilisées avec succès pour mesurer la liaison des interactions glycogène-protéine et amidon-protéine22,23. Dans cette étude, nous avons utilisé des billes ConA-Sepharose pour développer un test de liaison afin de mesurer les spécificités de liaison des interactions phosphatase-amylopectine glucane.
Auparavant, un test de sédimentation basé sur ConA a été utilisé pour évaluer la capacité de liaison du substrat de la phosphataseglucane 14,20,24. Dans cette étude, la même stratégie a été utilisée pour développer une nouvelle méthode permettant de déterminer l’affinité de liaison de la phosphatase glucane-glucane et des interactions glucidiques. Cette méthode présente également un avantage pour étudier diverses interactions glucides-protéines solubilisées.
Protocole
1. Préparation des billes de ConA-Sepharose
- Fabriquer 250 mL d’un tampon liant contenant 67 mM HEPES (pH 7,5), 10 mM MgCl 2 et 0,2 mM CaCl2. Ajuster le pH à l’aide d’une solution de NaOH 1 M.
- Pipeter 250 μL de suspension de billes ConA-Sepharose dans un tube microcentrifuge de 1,5 mL. Centrifuger le contenu à 10 000 x g pendant 30 s à 4 °C. Jetez le surnageant.
REMARQUE : 250 μL de billes de ConA-Sepharose dans un tube microcentrifuge de 1,5 mL sont nécessaires pour chaque concentration d’amylopectine utilisée pour le dosage. - Ajouter 750 μL du tampon de liaison à chaque tube contenant 250 μL de billes de ConA-Sepharose. Centrifuger les tubes à 10 000 x g pendant 1 min à 4 °C. Retirez le surnageant. Répétez cette étape 2x pour vous assurer que les perles sont correctement lavées et équilibrées avec le tampon de liaison.
2. Préparation de solutions d’amylopectine
- Préparer une solution mère d’amylopectine de pomme de terre à 10 mg/mL. L’amylopectine est insoluble dans l’eau et peut être solubilisée par la chaleur. Pour solubiliser, ajouter 0,1 g d’amylopectine de pomme de terre à 10 mL d’eau distillée. Chauffer la suspension au bain-marie à 80 °C pendant 1 h ou jusqu’à ce que la solution ne soit plus trouble.
- Laissez la solution revenir à température ambiante (RT), avec des vortex répétés pour éviter l’agglutination.
- Le traitement alcoolo-alcalin est une méthode alternative pour solubiliser les substrats d’amylopectine. Pour solubiliser à l’aide de cette méthode, suivez les étapes ci-dessous.
- Suspendre 0,5 g de substrat d’amylopectine dans 5 mL d’éthanol à 20 % et 5 mL de NaOH 2 M. Remuer vigoureusement le contenu pendant 15-20 min à RT.
- Ensuite, ajoutez 10 mL d’eau et ajustez le pH de la solution à 6,5 en ajoutant 2 M HCl. Augmenter le volume de la solution obtenue à 50 mL avec de l’eau distillée pour obtenir une solution d’amylopectine à 10 mg/mL.
- Diluer la solution d’amylopectine solubilisée à 10 mg/mL pour obtenir une série de 2 mL de solutions d’amylopectine diluées. Par exemple, effectuer des demi-dilutions de 10 mg/mL pour préparer une série de concentrations d’amylopectine (5 mg/mL, 2,5 mg/mL, 1,25 mg/mL, 0,625 mg/mL, 0,3125 mg/mL, 0,156 mg/mL, 0,078 mg/mL, 0,039 mg/mL, 0,019 mg/mL et 0 mg/mL).
3. Préparation de ConA-Sepharose: billes d’amylopectine
- Ajouter 250 μL de chaque solution d’amylopectine diluée à 1,5 mL de tubes microcentrifugés contenant 250 μL de billes de ConA-Sépharose prééquilibrées dans un tampon de liaison. Bien mélanger le contenu. Étiqueter les tubes avec la concentration d’amylopectine correspondante.
- Incuber le contenu sur une roue rotative à 4 °C pendant 30 min.
REMARQUE: Il n’y a aucun changement dans le complexe lié ConA-Sepharose:amylopectine au fil du temps après 20 min. Le temps d’incubation de 30 min a été choisi en variant les temps d’incubation de 10 min à 1 h pour assurer l’équilibre atteint. - Centrifuger les tubes à 10 000 x g pendant 1 min. Recueillir le surnageant dans un tube microcentrifuge de 1,5 mL nouvellement étiqueté. Conservez ces fractions surnageantes pour effectuer le test D-glucose12 (hydrolyse acide de l’amylopectine suivie de la détermination UV du glucose par dosage enzymatique). Cette étape est nécessaire pour s’assurer que toute l’amylopectine est liée aux billes.
- Ajouter 750 μL de tampon de liaison aux billes ConA-Sepharose:amylopectine. Centrifuger les tubes à 10 000 x g pendant 1 min. Jetez le surnageant pour éliminer toutes les molécules d’amylopectine non liées.
- Répétez l’étape 3.4 pour assurer un lavage suffisant. Chaque tube contient maintenant des billes de ConA-Sepharose liées à des quantités variables de substrats d’amylopectine.
4. Incubation de SEX4 avec ConA-Sepharose: billes d’amylopectine
- Mélanger 250 μL de billes d’amylopectine ConA-Sepharose:amylopectine avec 100 μL du tampon de liaison qui comprend 10 μg de protéine SEX4, 10 mM de dithiothréitol (DTT) et 10 μM de cocktail d’inhibiteurs de protéase (PIC). Notez que le volume total dans chaque tube est de 350 μL.
REMARQUE: Un cocktail d’inhibiteurs de protéase est ajouté par mesure de précaution pour éviter toute dégradation inutile de SEX4. Il s’agit d’une étape facultative. Dans ce test, la protéine recombinante Arabidopsis thaliana SEX4 (AtSEX4) est utilisée. La protéine purifiée contient un marqueur d’histidine N-terminal nécessaire à la détection de la protéine par chimiluminescence. Des informations détaillées sur les purifications de la phosphatase glucane sont décrites dans les publications précédentes14,20,24. - Incuber la suspension de billes de protéine et de ConA-Sepharose:amylopectine à 4 °C pendant 45 min en rotation douce.
REMARQUE: Le temps d’incubation de 45 minutes est choisi pour assurer l’équilibre du complexe. - Centrifuger les tubes à 10 000 x g pendant 1 min. Pipeter soigneusement 50 μL du surnageant à l’aide d’un embout de chargement de gel dans un nouveau tube microcentrifuge de 1,5 mL. Ajouter 20 μL de 4x colorant SDS-PAGE et 10 μL d’eau dans chaque tube contenant 50 μL des fractions surnageantes recueillies. Chauffer les échantillons à 95 °C pendant 10 min. Enregistrez ces exemples pour exécuter les gels SDS-PAGE. Assurez-vous que 10 nouveaux tubes étiquetés « surnageant (S) » ont les concentrations correspondantes dans le substrat.
- Ajouter 750 μL du tampon de liaison aux billes ConA-Sepharose:amylopectine: SEX4 pour éliminer toute protéine non liée des billes. Centrifuger les tubes à 10 000 x g pendant 1 min. Répétez cette étape une fois de plus pour assurer un lavage approprié. Jetez le surnageant.
- Ajouter 20 μL de 4x colorant SDS-PAGE et 80 μL d’eau distillée dans les tubes contenant des billes ConA-Sepharose:amylopectine:SEX4 lavées. Chauffer les échantillons à 95 °C pendant 10 min et centrifuger à 10 000 x g pendant 1 min.
- Jetez la pastille et conservez le surnageant pour faire fonctionner les gels SDS-PAGE. Pipeter 80 μL du surnageant dans de nouveaux tubes et les étiqueter comme « pastille (P) ».
5. Exécution des gels SDS-PAGE
- Chargez 40 μL des échantillons de protéines non liées (fabriqués à l’étape 2.3, étiquetés S) dans des puits de gel de polyacrylamide préfabriqué à 4 % à 12 %, de la concentration de substrat la plus faible à la plus élevée, mais gardez la première voie libre pour charger le marqueur de poids moléculaire de la protéine. Utilisez un deuxième gel pour charger 10 échantillons de protéines liées fabriqués à l’étape 2.5 (étiquetés comme P).
- Ajouter 1x tampon de fonctionnement SDS-PAGE fraîchement préparé aux deux chambres de l’appareil. Faites fonctionner le gel à 150 V pendant 35 minutes ou jusqu’à ce que le front du colorant atteigne le fond du gel.
- Retirez le gel de coulée de l’appareil et retirez les entretoises et les plaques de verre. Utilisez le gel séparé pour effectuer une analyse par transfert Western.
6. Western blot pour la détection par chimiluminescence14,15
REMARQUE : Cette méthode peut être facilement modifiée ou adaptée en fonction de l’équipement de transfert Western que les utilisateurs ont dans leurs laboratoires.
- Faire 1 L de tampon de transfert contenant 5,8 g de Tris base, 2,9 g de glycine, 0,37 g de SDS et 200 mL de méthanol.
- Transférer les protéines séparées par la taille du gel de polyacrylamide sur une membrane de nitrocellulose. Assembler brièvement les éponges, les papiers filtres, le gel et la membrane de nitrocellulose conformément au protocole de transfert occidental14,15. Fonctionnement à 70 V pendant 1 h.
- Pour éviter la liaison protéique non spécifique, incuber la membrane de nitrocellulose contenant la solution protéique de 1 % à 5 % d’albumine sérique bovine (BSA) ou de protéines de lait dans 50 mL de tampon TBST (20 mM Tris [pH 7,5], 150 mM NaCl, 0,1 % Tween 20) pendant 1 h. Lavez la membrane 3x à l’aide d’un tampon TBST pour éliminer toute solution bloquante non liée.
- Incuber la membrane avec un anticorps lié à la peroxydase de raifort (HRP) spécifique de la protéine marquée His pendant 1 h. Lavez la membrane 3x dans un tampon TBST pour éliminer tous les anticorps non liés. Utilisez une dilution de 1:2 000 des anticorps dirigés contre le TBST pour une reproductibilité et une sensibilité optimales.
- L’anticorps lié à l’enzyme HRP se lie spécifiquement au marqueur histidine de la protéine SEX4, qui produit une bande en présence de réactifs de chimiluminescence. Pour l’imagerie numérique, faire une solution de parties égales de solutions de substrat chimioluminescent (750 μL chacune) dans un tube de 1,5 mL. Incuber la membrane pendant au moins 5 minutes dans la solution.
- Placez la protéine membranaire vers le bas sur le scanner de transfert et exécutez le logiciel d’acquisition pour quantifier la protéine dans les fractions granulées et surnageantes.
7. Analyse des données
- Effectuez les mesures quantitatives du signal à l’aide du logiciel d’acquisition avec le scanner de transfert. Normaliser toutes les mesures quantitatives dans les fractions surnageantes et granulées à la protéine totale chargée.
NOTE: Le logiciel permet de quantifier l’intensité de chaque bande de protéines dans les fractions surnageant et granulé. - Dans l’expérience de liaison saturante, tracez le pourcentage de concentration liée aux protéines par rapport à l’amylopectine. Ajustez les données à Y = Bmax x X/(K D + X), en utilisant un logiciel d’analyse de données pour calculer KD.
NOTE: Bmax est la liaison spécifique maximale, l’axe des Y est le pourcentage de liaison protéique, l’axe X est la concentration d’amylopectine.

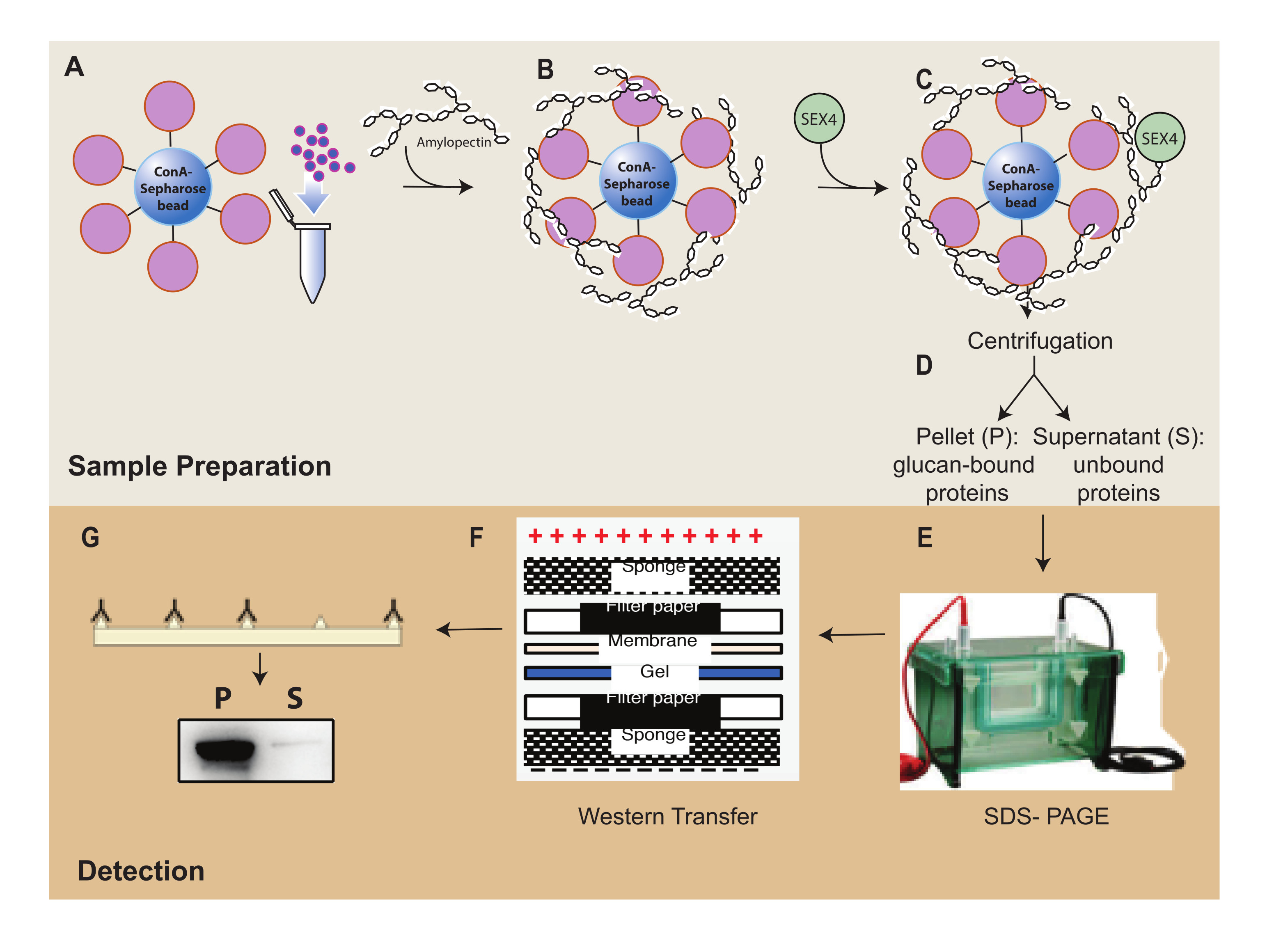
Figure 1 : Vue d’ensemble du flux de travail de l’essai de sédimentation ConA-Sepharose. (A) Préparation des billes ConA-Sepharose. (B) Incubation avec un substrat d’amylopectine. (C) Incubation avec la protéine SEX4. (D) Séparation des fractions protéiques liées et non liées par centrifugation. (E) Séparation des protéines par SDS-PAGE. (F) Analyse par transfert Western. (G) Détection par chimiluminescence de la protéine SEX4 marquée par His. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Résultats
L’une des principales caractéristiques de la famille des protéines de la phosphatase glucane est leur capacité à se lier aux substrats de glucanes. Tout d’abord, la capacité de liaison de SEX4 aux billes de ConA-Sepharose:amylopectine a été analysée à l’aide de SDS-PAGE (Figure 2A). L’albumine sérique bovine (BSA) a servi de témoin négatif pour détecter toute liaison non spécifique des protéines aux billes ConA-Sépharose:amylopectine. L’analyse SDS-PAGE des protéin...
Discussion
Cette étude démontre le développement réussi d’un nouveau test de sédimentation in vitro qui permet de déterminer l’affinité de liaison des interactions glucane-glucane phosphatase. La conception du dosage tire parti de la liaison spécifique de la lectine ConA aux glucanes via les résidus hydroxyles du glucose pour capturer indirectement les substrats glucidiques solubilisés sur les billes de sépharose. Cela permet la séparation des fractions protéiques liées et non liées par...
Déclarations de divulgation
Les auteurs ne déclarent aucun conflit d’intérêts.
Remerciements
Cette étude a été soutenue par le prix MCB-2012074 de la National Science Foundation. Les auteurs remercient le Dr Craig W. Vander Kooi du Département de biochimie et de biologie moléculaire de l’Université de Floride pour ses précieuses discussions et son soutien. Les auteurs remercient également le Dr Matthew S. Gentry du Département de biochimie et de biologie moléculaire de l’Université de Floride pour son soutien. Nous tenons à remercier la Dre Sara Lagalwar, présidente du programme de neurosciences du Collège Skidmore, de nous avoir permis d’utiliser le scanner par transfert C LICOR pour l’imagerie par transfert Western.
matériels
| Name | Company | Catalog Number | Comments |
| 6x-His Tag monoclonal antibody (HIS.H8), HRP | Therm Fisher Scientific | MA1-21315-HRP | |
| Biorad gel electrophoresis and Western blot kit | Biorad | 1703930 | |
| Calcium chloride | Sigma-Aldrich | 208291 | |
| C-Digit blot scanner | LICOR | 3600-00 | Blot scanner |
| Complete protease inhibitor cocktail | Sigma-Aldrich | 11836170001 | |
| Concanavalin A-sepharose beads | Sigma-Aldrich | C9017 | This product contains in 0.1 M acetate buffer, pH 6, containing 1 M NaCl, 1 mM CaCl2, 1 mM MnCl2, and 1 mM MgCl2 in 20% ethanol |
| Centrifuge | Eppendorf | 5425R | |
| Glycine | Fisher Scientific | BP381-5 | |
| GraphPad Prism 8.0 software | GraphPad | Version 8.0 | Data analysis software |
| HEPES | Sigma-Aldrich | H8651 | |
| Image Studio | LICOR | 3600-501 | Acquisition Software |
| Magnesium chloride | Sigma-Aldrich | M2670 | |
| Methanol | Fisher Scientific | A452SK-4 | |
| Sodium dodecyl sulfate | Fisher Scientific | PI28312 | |
| Potato amylopectin | Sigma-Aldrich | A8515 | |
| Precast SDSPAGE Gels | Genscript | M00653S | |
| Tris base | Fisher Scientific | BP154-1 | |
| Tween 20 | Fisher Scientific | MP1TWEEN201 | |
| Westernsure premium chemiluminescence substrate | LI-COR | 926-95000 |
Références
- Meekins, D. A., Vander Kooi, C. W., Gentry, M. S. Structural mechanisms of plant glucan phosphatases in starch metabolism. The FEBS Journal. 283 (13), 2427-2447 (2016).
- Gentry, M. S., et al. The phosphatase laforin crosses evolutionary boundaries and links carbohydrate metabolism to neuronal disease. The Journal of Cell Biology. 178 (3), 477-488 (2007).
- Worby, C. A., Gentry, M. S., Dixon, J. E. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. The Journal of Biological Chemistry. 281 (41), 30412-30418 (2006).
- Gentry, M. S., Pace, R. M. Conservation of the glucan phosphatase laforin is linked to rates of molecular evolution and the glucan metabolism of the organism. BMC Evolutionary Biology. 9, 138 (2009).
- Niittyla, T., et al. Similar protein phosphatases control starch metabolism in plants and glycogen metabolism in mammals. The Journal of Biological Chemistry. 281 (17), 11815-11818 (2006).
- Kotting, O., et al. STARCH-EXCESS4 is a laforin-like Phosphoglucan phosphatase required for starch degradation in Arabidopsis thaliana. The Plant Cell. 21 (1), 334-346 (2009).
- Comparot-Moss, S., et al. A putative phosphatase, LSF1, is required for normal starch turnover in Arabidopsis leaves. Plant Physiology. 152 (2), 685-697 (2010).
- Zeeman, S. C., Northrop, F., Smith, A. M., Rees, T. A starch-accumulating mutant of Arabidopsis thaliana deficient in a chloroplastic starch-hydrolysing enzyme. The Plant Journal: For Cell and Molecular Biology. 15 (3), 357-365 (1998).
- Kotting, O., et al. Identification of a novel enzyme required for starch metabolism in Arabidopsis leaves. The phosphoglucan, water dikinase. Plant Physiology. 137 (1), 242-252 (2005).
- Tagliabracci, V. S., et al. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proceedings of the National Academy of Sciences. 104 (49), 19262-19266 (2007).
- Gentry, M. S., Guinovart, J. J., Minassian, B. A., Roach, P. J., Serratosa, J. M. Lafora disease offers a unique window into neuronal glycogen metabolism. The Journal of Biological Chemistry. 293 (19), 7117-7125 (2018).
- Brewer, M. K., et al. Targeting pathogenic lafora bodies in lafora disease using an antibody-enzyme fusion. Cell Metabolism. 30 (4), 689-705 (2019).
- Santelia, D., Zeeman, S. C. Progress in Arabidopsis starch research and potential biotechnological applications. Current Opinion in Biotechnology. 22 (2), 271-280 (2011).
- Raththagala, M., et al. Structural mechanism of laforin function in glycogen dephosphorylation and lafora disease. Molecular Cell. 57 (2), 261-272 (2015).
- Meekins, D. A., et al. Phosphoglucan-bound structure of starch phosphatase Starch Excess4 reveals the mechanism for C6 specificity. Proceedings of the National Academy of Sciences. 111 (20), 7272-7277 (2014).
- Vander Kooi, C. W., et al. Structural basis for the glucan phosphatase activity of Starch Excess4. Proceedings of the National Academy of Sciences. 107 (35), 15379-15384 (2010).
- Meekins, D. A., et al. Structure of the Arabidopsis glucan phosphatase like sex four2 reveals a unique mechanism for starch dephosphorylation. The Plant Cell. 25 (6), 2302-2314 (2013).
- Smith, A. M., Zeeman, S. C. Starch: A flexible, adaptable carbon store coupled to plant growth. Annual Review of Plant Biology. 71, 217-245 (2020).
- Jane, J., Kasemuwan, T., Chen, J. F., Juliano, B. O. Phosphorus in rice and other starches. Cereal Foods World. 41 (11), 827-832 (1996).
- Mak, C. A., et al. Cooperative kinetics of the glucan phosphatase starch excess4. Biochemistry. 60 (31), 2425-2435 (2021).
- Campbell, K. P., MacLennan, D. H. Purification and characterization of the 53,000-dalton glycoprotein from the sarcoplasmic reticulum. The Journal of Biological Chemistry. 256 (9), 4626-4632 (1981).
- Campbell, K. P., MacLennan, D. H., Jorgensen, A. O., Mintzer, M. C. Purification and characterization of calsequestrin from canine cardiac sarcoplasmic reticulum and identification of the 53,000 dalton glycoprotein. The Journal of Biological Chemistry. 258 (2), 1197-1204 (1983).
- Davey, M. W., Sulkowski, E., Carter, W. A. Binding of human fibroblast interferon to concanavalin A-agarose. Involvement of carbohydrate recognition and hydrophobic interaction. Biochemistry. 15 (3), 704-713 (1976).
- Meekins, D. A., et al. Mechanistic insights into glucan phosphatase activity against polyglucan substrates. The Journal of Biological Chemistry. 290 (38), 23361-23370 (2015).
- Wilkens, C., et al. Plant α-glucan phosphatases SEX4 and LSF2 display different affinity for amylopectin and amylose. FEBS Letters. 590 (1), 118-128 (2016).
- Atanasova, M., Bagdonas, H., Agirre, J. Structural glycobiology in the age of electron cryo-microscopy. Current Opinion in Structural Biology. 62, 70-78 (2020).
- Doyle, M. L. Characterization of binding interactions by isothermal titration calorimetry. Current Opinion in Biotechnology. 8 (1), 31-35 (1997).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.