
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Imagerie à long terme de populations neuronales identifiées à l’aide de microprismes chez des animaux se déplaçant librement et avec la tête fixe
Dans cet article
Résumé
Lorsqu’elle est intégrée à une plaque de tête et à une conception optique compatible avec les microscopes à un ou deux photons, la lentille à microprisme présente un avantage significatif pour mesurer les réponses neuronales dans une colonne verticale dans diverses conditions, y compris des expériences bien contrôlées dans des états de tête fixe ou des tâches comportementales naturelles chez des animaux en mouvement libre.
Résumé
Avec les progrès de la microscopie multiphotonique et des technologies moléculaires, l’imagerie par fluorescence se développe rapidement pour devenir une approche puissante pour étudier la structure, la fonction et la plasticité des tissus cérébraux vivants. Par rapport à l’électrophysiologie conventionnelle, la microscopie à fluorescence peut capturer l’activité neuronale ainsi que la morphologie des cellules, permettant des enregistrements à long terme des populations de neurones identifiées à une résolution unicellulaire ou subcellulaire. Cependant, l’imagerie haute résolution nécessite généralement une configuration stable et fixe à la tête qui limite le mouvement de l’animal, et la préparation d’une surface plane en verre transparent permet de visualiser les neurones à un ou plusieurs plans horizontaux, mais est limitée dans l’étude des processus verticaux à différentes profondeurs. Ici, nous décrivons une procédure pour combiner une fixation de plaque de tête et un microprisme qui donne une imagerie multicouche et multimodale. Cette préparation chirurgicale donne non seulement accès à toute la colonne du cortex visuel de la souris, mais permet une imagerie à deux photons en position fixe de la tête et une imagerie à un photon dans un paradigme en mouvement libre. En utilisant cette approche, on peut échantillonner des populations cellulaires identifiées à travers différentes couches corticales, enregistrer leurs réponses sous des états fixes et mobiles et suivre les changements à long terme sur des mois. Ainsi, cette méthode fournit un test complet des microcircuits, permettant une comparaison directe des activités neuronales évoquées par des stimuli bien contrôlés et sous un paradigme comportemental naturel.
Introduction
L’avènement de l’imagerie fluorescente à deux photons in vivo 1,2, combinant les nouvelles technologies des systèmes optiques et des indicateurs de fluorescence génétiquement modifiés, est devenu une technique puissante en neurosciences pour étudier la structure, la fonction et la plasticité complexes du cerveau vivant 3,4. En particulier, cette modalité d’imagerie offre un avantage inégalé par rapport à l’électrophysiologie traditionnelle en capturant à la fois la morphologie et les activités dynamiques des neurones, facilitant ainsi le suivi à long terme des neurones identifiés 5,6,7,8.
Malgré ses atouts notables, l’application de l’imagerie par fluorescence à haute résolution nécessite souvent une configuration statique et fixe à la tête qui limite la mobilité de l’animal 9,10,11. De plus, l’utilisation d’une surface en verre transparent pour visualiser les neurones restreint les observations à un ou plusieurs plans horizontaux, limitant ainsi l’exploration de la dynamique des processus verticaux qui s’étendent sur différentes profondeurs corticales12.
En remédiant à ces limitations, la présente étude décrit une procédure chirurgicale innovante qui intègre la fixation de la plaque de tête, le microprisme et le miniscope pour créer une modalité d’imagerie avec des capacités multicouches et multimodales. Le microprisme permet d’observer le traitement vertical le long de la colonne corticale 13,14,15,16, ce qui est essentiel pour comprendre comment l’information est traitée et transformée lorsqu’elle se déplace à travers différentes couches du cortex et comment le traitement vertical est modifié lors des changements plastiques. De plus, il permet d’imager les mêmes populations neuronales dans un paradigme de tête fixe et dans un cadre en mouvement libre, englobant les cadres expérimentaux polyvalents 17,18,19 : par exemple, la fixation de la tête est souvent nécessaire pour des paradigmes bien contrôlés comme l’évaluation de la perception sensorielle et les enregistrements stables sous le paradigme à 2 photons, tandis que le mouvement libre offre un environnement plus naturel et plus flexible pour les études comportementales. Par conséquent, la capacité d’effectuer une comparaison directe dans les deux modes est cruciale pour approfondir notre compréhension des microcircuits qui permettent des réponses flexibles et fonctionnelles.
Essentiellement, l’intégration de la fixation de la plaque frontale, du microprisme et du miniscope dans l’imagerie de fluorescence offre une plate-forme prometteuse pour sonder les subtilités de la structure et de la fonctionnalité du cerveau. Les chercheurs peuvent échantillonner des populations cellulaires identifiées à différentes profondeurs couvrant toutes les couches corticales, comparer directement leurs réponses dans des paradigmes bien contrôlés et naturels, et surveiller leurs altérations à long terme sur des mois20. Cette approche offre des informations précieuses sur la façon dont ces populations neuronales interagissent et changent au fil du temps dans différentes conditions expérimentales, offrant une fenêtre sur la nature dynamique des circuits neuronaux.
Protocole
Toutes les expériences ont été menées conformément à la loi britannique de 1986 sur les animaux (procédures scientifiques) en vertu de licences personnelles et de projets approuvées et délivrées par le ministère de l’Intérieur britannique après un examen éthique approprié. Lignées transgéniques adultes CaMKII-TTA ; GCaMP6S-TRE21 ont été élevés et leur progéniture a été utilisée dans l’expérience. Pour la sécurité des expérimentateurs et le maintien de conditions stériles, toutes les procédures ont été effectuées dans des conditions aseptiques et avec un équipement de protection individuelle complet.
1. Préparation préopératoire
- Pour minimiser l’œdème, administrer la dexaméthasone (0,2 mg/kg) par voie sous-cutanée, 12 à 24 heures avant la chirurgie.
- Stériliser tous les outils chirurgicaux dans un autoclave et stériliser la zone chirurgicale avec de l’acide hypochloreux stabilisé avec de l’eau distillée et de l’éthanol à 70 % avant la chirurgie. Assurez-vous que tout l’équipement chirurgical est allumé.
- Anesthésier l’animal (24 semaines, mâle pesant 31 g) à l’aide d’isoflurane avec une dose d’induction de 5 %, qui est réduite à 1 % à 2 % une fois que la souris est sur le cadre stéréotaxique, avec O2 maintenu entre 1 et 2 L/min. Injecter des AINS (carprofène, 2,5 mg/kg) par voie sous-cutanée.
- Vérifiez l’absence de réflexe de pincement des orteils pour évaluer la profondeur de l’anesthésie (augmenter la concentration d’isoflurane par incréments de 0,5 % si réflexe observé).
- Rasez la tête de l’animal, à l’aide d’une tondeuse, de derrière les oreilles jusqu’à légèrement au-dessus des yeux. Nettoyez cette zone avec une lingette imbibée d’alcool et une solution de povidone iodée, en veillant à éviter tout contact avec les yeux de l’animal.
- Montez l’animal sur le coussin chauffant homéotherme et le cadre stéréotaxique équipé de barres d’oreille et de dents et fixez la tête. Assurez-vous que la tête est stable, car cela est crucial pour que la procédure suivante soit réussie (Figure 1).
- Appliquez une pommade ophtalmique sur les yeux de l’animal pour éviter qu’ils ne sèchent pendant la chirurgie et couvrez-les d’une feuille d’aluminium pour les protéger de la lumière. Couvrir l’animal d’une couverture chirurgicale stérile.

Figure 1 : Préparation préopératoire. La souris est placée sur le cadre stéréotaxique, fixé par un nez et des barres d’oreille. La souris est placée sur un tapis chauffant à température régulée. Les yeux sont recouverts d’une pommade ophtalmique et d’une feuille d’aluminium. La tête est rasée et le crâne est exposé. Une couverture stérile est placée sur l’animal. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
2. Craniotomie
- À l’aide de ciseaux chirurgicaux, incisez la peau le long de la ligne médiane de la zone rasée de la tête pour exposer le crâne.
- Nettoyez le crâne à l’aide d’un coton-tige stérile et de peroxyde d’hydrogène dilué (3 % p/v de 35 % H2O2 dans 97 % dH2O) pendant 1 à 3 s pour éliminer tout tissu conjonctif (Figure 2A). Séchez le crâne avec une goutte d’éthanol à 70 % et un nouveau coton-tige stérile.
- Alignez le crâne antérieurement/postérieurement (AP) et médialement/latéralement (ML) pour assurer un site d’implantation précis. Pour ce faire, mesurez la profondeur dorso-ventrale (DV) du crâne à la fois au niveau du bregma et du lambda et assurez-vous que la différence entre les deux est de <0,03 mm. Pour l’alignement médiolatéral, mesurez les points équidistants sur les deux os pariétaux à partir de la ligne médiane et assurez-vous à nouveau que la différence de DV est de <0,03 mm.
- En utilisant le bregma comme origine, trouvez et marquez la zone corticale souhaitée ; ici, il s’agit du cortex visuel primaire monoculaire (V1), AP : -3,5 mm, ML : -2,5 mm.
- Utilisez un foret à trépan (1,8 mm de diamètre) et un foret dentaire (vitesse de 10 000 tr/min) pour exposer le cortex, en veillant à ce que la marque de la zone corticale souhaitée (monoculaire V1) soit située dans le tiers inférieur de la fenêtre du foret.
- Assurez-vous que l’angle du foret est perpendiculaire à la courbure du crâne. Cela assurera une craniotomie uniforme et évitera d’endommager la dure-mère ou le cortex.
- Percez jusqu’à ce qu’il y ait une diminution de la résistance, puis arrêtez-vous (Figure 2B). Retirez délicatement le fragment d’os détaché à l’aide d’une pointe d’aiguille 23G (Figure 2C).
- Nettoyez le cortex exposé avec de la mousse chirurgicale saturée de liquide céphalo-rachidien artificiel froid (ACSF) pour éliminer tous les débris et arrêter tout saignement qui pourrait survenir.
- Gardez toujours le cortex exposé hydraté, en utilisant de l’ACSF froid, tout au long de la chirurgie.

Figure 2 : Craniotomie. (A) L’incision cutanée entre le bregma et le lambda est montrée. Le tissu conjonctif a été retiré de la surface exposée. (B) Craniotomie par tréphine avant le retrait du fragment d’os. (C) Craniotomie après ablation d’un fragment d’os, montrant une dure-mère et un cortex intacts (la barre d’échelle représente 0,5 mm). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
3. Incision prédécoupée
REMARQUE : Pour être pris en compte lors de l’incision prédécoupée, l’incision et l’implantation du microprisme devront être antérieures à la région d’intérêt (ROI) d’imagerie. Ceci afin de permettre un champ de vision complet et précis. Dans le cadre de ce protocole, l’incision sera réalisée le long de l’axe médiolatéral, et le microprisme est orienté face à la postérieure (Figure 3B).
- Pour faciliter l’insertion et soulager la pression dans le cortex lors de l’insertion du microprisme, faites une incision.
- Fixez le couteau chirurgical au support de bras stéréotaxique et orientez la lame ou le bras stéréotaxique de manière à ce qu’il coupe le long de l’axe ML.
- Déplacez le couteau à la coordonnée AP souhaitée (AP : -3,4 mm) ; le prisme doit être devant le ROI, alors faites l’incision 100 μm en avant de la coordonnée ROI AP d’imagerie (-3,5 mm).
- Maintenant, déplacez le couteau vers le bord médial de la craniotomie, où il rencontre le crâne, abaissez lentement le couteau jusqu’à ce qu’il atteigne l’os, puis arrêtez-vous. Comme l’épaisseur de l’os est de 200 μm, intégrez cette valeur dans la profondeur d’insertion totale (voir calcul de l’étape 5.1).
- L’imagerie optimale est au centre du prisme, c’est-à-dire 500 μm ; par conséquent, assurez-vous que cette profondeur s’aligne sur la profondeur de la colonne corticale (ROI DV : - 0,35 mm).
- En incorporant l’épaisseur du crâne dans le calcul de la profondeur, utilisez l’équation ci-dessous, qui détermine la profondeur de l’incision prédécoupée par rapport à la surface du crâne. Pour ce protocole, la profondeur d’implantation est calculée comme suit :
Épaisseur osseuse (200 μm) + retour sur investissement de l’imagerie (par exemple, 350 μm) + profondeur restante du microprisme (500 μm) = 1 050 μm
- En incorporant l’épaisseur du crâne dans le calcul de la profondeur, utilisez l’équation ci-dessous, qui détermine la profondeur de l’incision prédécoupée par rapport à la surface du crâne. Pour ce protocole, la profondeur d’implantation est calculée comme suit :
- Assurez-vous que la longueur de l’incision est supérieure à 1 mm, mais pas excessive ; par conséquent, une distance de 1,2 mm est idéale, la coordonnée ML monoculaire étant au milieu de cette distance.
- Lorsque vous êtes prêt à effectuer l’incision, retirez l’excès d’ACSF afin que la vision ne soit pas obscurcie (Figure 3A).
- Déplacez le couteau du bord médial de la craniotomie à la coordonnée médiale de départ de l’incision. Lentement (10 μm/s) descendez le couteau dans le cortex.
- Une fois la dure-mère percée et le couteau pénétré dans le cortex, appliquez une goutte d’ACSF froid sur le cortex pour garder le tissu lubrifié et hydraté pendant l’incision.
- Une fois la profondeur finale atteinte, commencez à déplacer le couteau le long de l’axe ML (à une vitesse de 10 μm/s).
- Continuez à observer les tissus environnants pendant que l’incision est pratiquée. Si le tissu traîne avec le couteau, déplacez le couteau de haut en bas plusieurs fois pour vous assurer que le tissu est coupé, puis continuez latéralement, en n’oubliant pas de remettre le couteau à sa profondeur finale.
- Une fois terminé, soulevez lentement le couteau. Si du sang émerge pendant l’incision, utilisez ce temps pour nettoyer le site de l’incision avec de la mousse chirurgicale imbibée d’ACSF pour diluer le sang et expulser le sang dans l’incision. N’oubliez pas de laisser une mousse chirurgicale fraîche et saturée sur le cortex exposé jusqu’au moment d’insérer le microprisme.

Figure 3 : Implantation de microprismes. (A) Incision prédécoupée. (B) Schéma de la lentille à microprisme intégrée démontrant sa position dans le cortex (C) Lentille à microprisme intégrée dans la bonne orientation pour prédécouper l’incision avant l’insertion dans le cortex (la barre d’échelle représente 0,5 mm). (D) Exemple d’accumulation de ciment autour de la lentille intégrée pour sécuriser sa fixation au crâne. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
4. Insertion de microprisme et implantation de plaque de tête
- La lentille à microprisme est composée d’une lentille à gradient d’indice fixée à un prisme à l’extrémité distale de la lentille, qui est intégrée dans une plaque de base. Fixez le microprisme au kit d’implant.
- Assurez-vous que le côté imagerie du prisme est opposé à la vis de la plaque de base. Pour faciliter l’insertion et la mise en place du microprisme, fixez-le au cadre stéréotaxique et orientez le prisme de manière à ce qu’il s’aligne avec l’incision (Figure 3C).
- Abaissez lentement le microprisme dans le site d’incision (10 μm/s). N’oubliez pas de retirer l’ACSF lors de l’insertion initiale du prisme, mais une fois dans le cortex, rincez avec de l’ACSF froid pour lubrifier l’insertion.
- Le cortex doit rester stable pendant que le prisme est abaissé dans l’incision ; sinon, ajoutez de l’ACSF supplémentaire et agitez le cortex en déplaçant le prisme de haut en bas pour détacher le cortex du prisme.
- Une fois la profondeur finale atteinte, séchez la surface corticale exposée à l’aide d’un tissu stérile, en prenant soin de ne pas toucher le prisme.
- Couvrez la zone corticale exposée entourant le prisme, ainsi que la lentille, avec une couche protectrice d’adhésif en silicone, minimisant l’excès d’adhésif sur le crâne environnant et la lunette factice.
- Une fois durci (5 à 10 min), fixez la plaque frontale au crâne pour stabiliser la tête pendant l’imagerie de la tête.
- Assurez-vous que la plaque de tête est suffisamment postérieure pour ne pas interférer avec la mise en place de l’implant et pour permettre une application adéquate du ciment pour bien fixer l’implant.
- Assurez-vous que la ligne médiane de la plaque frontale se trouve légèrement à droite de la lentille implantée pour vous assurer que les deux côtés de la plaque frontale peuvent être fixés à la scène de tête lors de la réalisation d’expériences fixées sur la tête
- Appliquez du ciment adhésif sur la plaque de tête et le crâne.
- Préparez le ciment dentaire adhésif en mélangeant 1 cuillère de poudre de ciment opaque avec 4 gouttes de milieu de mélange et en appliquant une goutte de catalyseur.
- Placez du ciment sur la plaque frontale et le crâne et maintenez la plaque frontale en place jusqu’à ce qu’elle soit durcie, en vous assurant qu’elle est parallèle aux barres auriculaires (par un examen visuel, inspectez à la fois par le dessus et derrière la tête de l’animal).
- Appliquez du ciment adhésif pour couvrir le reste du crâne et des tissus exposés, en incorporant le microprisme (jusqu’à la base de la plaque de base) et la plaque frontale.
- Ne mettez pas de ciment sur la plaque de base, le microscope factice ou l’un de ses composants. Continuez à appliquer le ciment adhésif jusqu’à ce que le microprisme et la plaque de tête soient recouverts et stables (Figure 3D).
- Lorsque le ciment a durci, détachez le microscope factice en déplaçant lentement le bras stéréotaxique vers le haut tout en stabilisant le microprisme avec une pince (ils sont reliés par des aimants, donc une certaine résistance pendant la séparation peut être ressentie).
- Insérez le couvercle de protection sur l’objectif et serrez la vis pour le fixer en place.
- Retirer l’animal du cadre stéréotaxique, le laisser récupérer dans une boîte de récupération chaude et administrer une solution saline stérile réchauffée à 0,9 % par voie sous-cutanée (3 % du poids corporel).
- Une fois que l’animal est réveillé et en mouvement, replacez-le dans une cage propre à logement unique. Surveillez l’animal et administrez une analgésie post-opératoire supplémentaire conformément à la politique de l’établissement local en matière d’analgésie.
- Attendez 4 semaines après la chirurgie, l’animal devrait être prêt pour l’imagerie.
5. Imagerie calcique à un photon des couches corticales chez des souris se déplaçant librement
REMARQUE : Il est essentiel d’utiliser les images capturées à chaque fois lors de la session d’imagerie d’origine pour garantir une acquisition précise du plan d’imagerie prévu. Ces repères identifiés, ainsi que les neurones, jouent un rôle essentiel dans le processus d’alignement décrit en détail à l’étape 9 du protocole. Lors de l’acquisition de données à un photon, le miniscope est à la fois le système d’imagerie et la source laser. L’excitation utilise des LED d’une plage de puissance de 0 à 2 mW/mm2 sur la surface avant de l’objectif. Le laser utilise une longueur d’onde d’excitation de 455 ± 8 nm (lumière bleue) pour la signalisation GCaMP. Le curseur de mise au point de l’objectif peut être utilisé pour régler la mise au point (axe Z), qui est représentée sur l’interface par 0-1000, où 0 représente une distance de travail de 0 μm et 1000 représente la distance de travail maximale de 300 μm.
- Avant d’acquérir des données, laissez l’animal s’acclimater à la pièce et à l’arène ouverte pendant 1 h avant la session d’enregistrement.
- Avant l’imagerie, désinfectez et nettoyez le tout avec des désinfectants appropriés (par exemple, de l’acide hypochloreux stabilisé avec de l’eau distillée et de l’éthanol à 70 %).
- Configurez le boîtier DAQ en le connectant à un ordinateur et en lançant le logiciel d’acquisition de données. Établissez une connexion directe via un câble Ethernet pour minimiser les pertes d’images ; cependant, le mode de connexion sans fil peut suffire en fonction de la puissance de la connexion sans fil.
- Fixez le miniscope à la plaque de base de l’animal sous une légère peau.
- Tout d’abord, retirez le couvercle de protection de la plaque de base en dévissant la vis de réglage. Tenez le couvercle par son ouverture avec une pince. Ensuite, fixez le miniscope à la plaque de base, où se trouve le couvercle.
- Vérifiez l’orientation du miniscope par rapport à la plaque de base avant de l’installer de manière à ce que le côté avec le marquage de la vis, fasse face à la vis.
- Une fois le miniscope fixé, serrez la vis de réglage pour le stabiliser. N’avancez la vis de réglage que jusqu’à ce qu’une certaine résistance se fasse sentir. Un serrage excessif de la vis de réglage pourrait endommager le miniscope et doit donc être évité.
- Connectez le miniscope au boîtier DAQ et préparez le logiciel pour l’enregistrement.
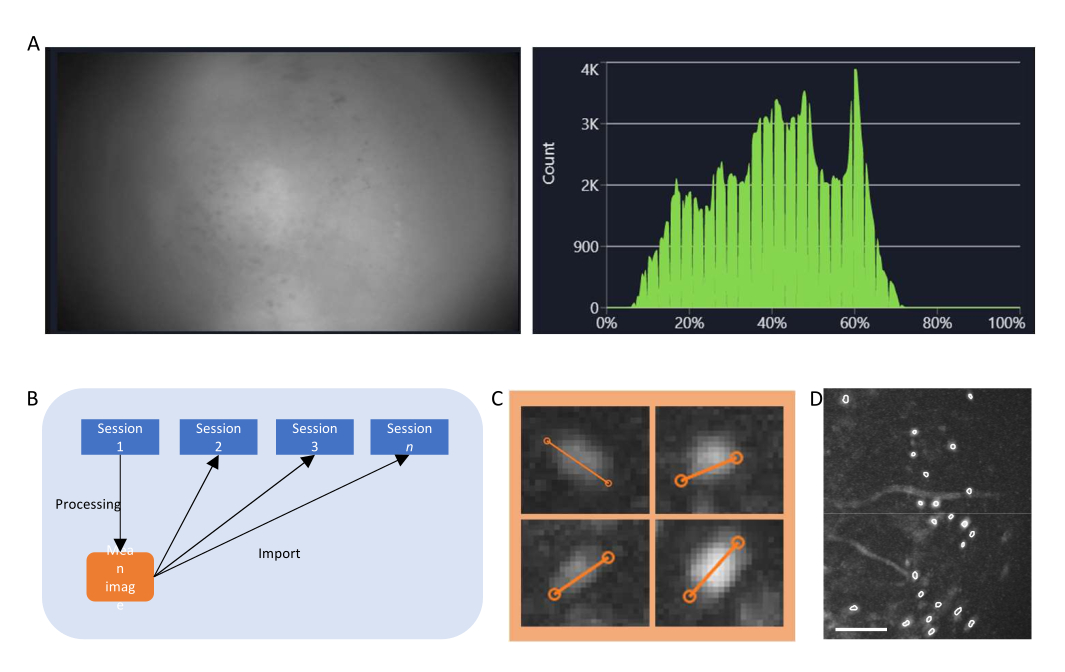
- Dans le logiciel d’acquisition de données, activez le flux pour le miniscope et ajustez les paramètres d’enregistrement (fréquence d’images de l’image, gain, puissance du voyant, valeur efocus) pour obtenir un champ de vision clair (Figure 4A).
- Activez la fenêtre de l’histogramme et ajustez le gain et la puissance de la LED de manière à ce que l’intensité enregistrée se situe entre 35 % et 70 %.
- Si vous effectuez une étude longitudinale, reportez-vous aux enregistrements ou aux instantanés pris lors d’une session précédente et ajustez la valeur efocus afin que le même plan d’imagerie soit clairement visible.
- Démarrez l’expérience et l’acquisition des données.
- Une fois l’expérience terminée, retirez l’animal de l’appareil comportemental.
- Sous une légère peau, desserrez la vis de réglage et retirez le miniscope de la plaque de base de l’animal.
- Remettez le couvercle de protection sur la plaque de base et stabilisez-le avec la vis de réglage.
- Remettre l’animal dans sa cage d’origine (passer à l’étape 7 si vous souhaitez traiter des données à un photon).

Figure 4 : Acquisition et traitement des données avec un logiciel. (A) Une image montrant le flux en temps réel du miniscope. Il est recommandé d’ajuster la valeur de mise au point de l’objectif, afin qu’une vue claire soit visible dans la fenêtre de streaming, ainsi que le gain et la puissance du laser d’imagerie (B) Graphique schématique illustrant le flux de travail d’alignement recommandé pour les sessions enregistrées à différents moments. Il est recommandé de générer une image moyenne à partir de la première session, en suivant les instructions du logiciel de traitement des données. Cette image doit être utilisée comme image de référence lors de la correction de mouvement pour les sessions suivantes. (C) Exemples de quatre cellules de la même image ΔF/F projetée au maximum. Une ligne orange est tracée sur chaque cellule pour mesurer son diamètre en pixels, dont la moyenne est prise comme argument d’entrée pour l’algorithme d’identification des cellules (en haut à gauche : 13, en haut à droite : 11, en bas à gauche : 12, en bas à droite : 13). (D) Sortie de l’algorithme d’identification des cellules après curation manuelle (image recadrée). Les contours blancs représentent les cellules identifiées (la barre d’échelle représente 100 μm). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
6. Imagerie calcique à deux photons des couches corticales chez des souris à tête fixe
REMARQUE : Pour la microscopie à balayage laser à deux photons, la source lumineuse est un laser ultrarapide accordable avec une longueur d’onde d’excitation de 920 nm. La puissance d’excitation, mesurée à l’objectif, était généralement comprise entre 100 et 150 mW et ajustée à chaque session pour atteindre des niveaux de fluorescence similaires. La lumière d’émission a été filtrée par un filtre d’émission (525/70 nm) et mesurée par un tube photomultiplicateur indépendant (PMT), appelé canal vert. Les images ont été acquises avec un objectif à immersion aérienne 20x (NA = 0,45, distance de travail de 6,9 à 8,2 mm).
- Avant d’acquérir des données, habituez l’animal à l’appareil dans les jours précédents (pour que l’animal se familiarise avec la configuration comportementale). Laissez l’animal passer 15 à 30 minutes par jour, pendant 2 à 3 jours, à explorer l’installation ou jusqu’à ce qu’il affiche un comportement naturaliste avant de commencer l’acquisition des données.
- Allumez le système d’imagerie à deux photons, lancez le logiciel d’acquisition et allumez le laser. Assurez-vous que les lasers sont fermés et que les PMT sont éteints avant de continuer.
- Nettoyez l’appareil d’imagerie à deux photons avec de l’acide hypochloreux stabilisé avec de l’eau distillée et de l’éthanol à 70 %.
- Assurez-vous d’ajuster l’appareil à la taille de l’animal. Fixez délicatement la souris et sa plaque frontale à la configuration Head-Stage et vissez-la en place pour stabiliser la tête de la souris.
- Une fois fixé, retirez le couvercle de l’objectif (reportez-vous à l’étape 5.4.1) et alignez l’objectif de manière à ce qu’il soit sur la plaque de base.
- Utilisez les commandes d’épifluorescence et de stade XYZ pour mettre au point le tissu cortical.
- Une fois que les couches corticales sont visibles, changez le microscope pour permettre l’imagerie à deux photons (remplacez le miroir par le miroir dichroïque, fermez l’obturateur fluorescent et éteignez le laser d’épifluorescence et le moniteur). Assurez-vous d’éteindre les lumières principales afin de protéger les PMT lors de l’imagerie.
- Configurez des paramètres pour optimiser les fichiers d’image acquis.
- Utilisez le mode d’acquisition par résonance pour l’imagerie calcique car il peut capturer le déclenchement rapide des signaux GCaMP.
- Ajustez la puissance laser, le gain PMT, le zoom et les tables de conversion (LUT) pour obtenir une image optimale et reportez-vous à l’image à un photon pour vous assurer que le plan focal correct est imagé.
- Commencez l’imagerie des couches corticales avec le système à deux photons avec surveillance comportementale synchronisée et entrées de stimulus (le cas échéant).
- Assurez-vous de conserver ces paramètres d’acquisition et les distances XYZ si vous souhaitez reproduire des images identiques dans le temps.
- Pour capturer une pile z des couches corticales, suivez les étapes décrites ci-dessous.
- Trouvez le plan à partir duquel démarrer la pile z, ajustez les paramètres d’acquisition pour optimiser l’image et marquez-le comme point de départ sur le logiciel.
- Ensuite, à l’aide de la commande Z, descendez dans la pile, en ajustant uniquement la puissance laser pour maintenir une luminance constante de la pile, et marquez la fin de la pile sur le logiciel.
- Étape critique : à l’aide de l’option Gradient exponentiel relatif sous l’onglet Gradient de puissance laser , permettez au logiciel de calculer l’augmentation de la puissance du laser lorsqu’il se déplace dans la pile z. Assurez-vous de noter les valeurs de puissance laser du point final dans le tableau fourni par le logiciel pour lui permettre de calculer le gradient.
- Une fois les paramètres de la pile z définis, ajustez la taille du pas (μm).
REMARQUE : La taille du pas déterminera le temps nécessaire, le nombre de tranches et la qualité des détails de la pile. Des pas plus petits se traduiront par un temps d’acquisition plus long, une augmentation du nombre de tranches et de meilleurs détails par rapport à des pas plus grands. Les piles Z sont utilisées pour faciliter le recalage des images à un et deux photons, car elles mettent en évidence les points de repère ou les caractéristiques anatomiques.
- Pour acquérir une série chronologique (série T) de changements calciques dans les neurones, trouvez un plan focal optimal à l’aide des commandes de la platine XYZ et ajustez la puissance laser, le gain PMT, le zoom et les LUT.
- Sous l’onglet Série T du logiciel d’acquisition, définissez les paramètres de fréquence d’acquisition pour qu’ils correspondent aux données acquises à l’aide du système d’imagerie à un photon.
REMARQUE : La correspondance de la fréquence rendra les données 1P et 2P comparables, et elle est mieux adaptée à l’algorithme d’identification des cellules utilisé dans les étapes de traitement des données. Plusieurs déclencheurs et d’autres modes d’acquisition peuvent être incorporés dans l’acquisition de la série T.
- Sous l’onglet Série T du logiciel d’acquisition, définissez les paramètres de fréquence d’acquisition pour qu’ils correspondent aux données acquises à l’aide du système d’imagerie à un photon.
- Début de l’acquisition de la série T.
7. Traitement des données d’imagerie calcique à un photon
- Pour l’enregistrement de vidéos à un photon, utilisez le logiciel de traitement des données fourni avec le système miniscope.
- Tout d’abord, pré-traitez le film par sous-échantillonnage spatial et temporel. En général, un sous-échantillonnage spatial du film d’un facteur deux réduirait considérablement le temps de traitement sans compromettre gravement la précision de l’identification des cellules.
- Définissez le facteur de sous-échantillonnage temporel de manière à ce que la fréquence d’images du film soit ramenée à environ 10 Hz, ce qui convient mieux à l’algorithme d’identification des cellules utilisé dans les étapes suivantes.
- Si vous acquérez plusieurs films le même jour d’imagerie, combinez-les en une seule série chronologique avant l’étape de prétraitement pour les traiter ensemble.
- Facultatif : Appliquez un filtre passe-bande spatial à la vidéo pour supprimer les fréquences spatiales inférieures et supérieures, ce qui permet d’obtenir une vidéo plus fluide avec un contraste plus élevé.
- Enregistrez le film à l’aide de la fonctionnalité de correction de mouvement du logiciel. Cela enregistre le film et corrige les artefacts de mouvement causés par le mouvement du miniscope par rapport à la surface d’image.
- Étape critique : Si vous effectuez une étude longitudinale, enregistrez les films dans le même champ de vision, par exemple, l’image moyenne du film prise le premier jour d’imagerie (Figure 4B).
- Calculez le ΔF/F du film à l’aide de la tabulation correspondante et projetez le film pour générer une image de projection maximale du film ΔF/F. Cette image montrera des régions qui présentent des changements dans les niveaux de fluorescence, potentiellement des neurones individuels, et pourrait être utilisée pour mesurer le diamètre moyen des neurones (Figure 4C).
- Vous pouvez également mesurer le diamètre des cellules sur le film corrigé du mouvement, où les neurones présentent une fluorescence claire.
- Identifiez les cellules à l’aide des algorithmes du logiciel.
- Bien que deux options (PCA-ICA et CNMF-E) soient disponibles à cette étape, utilisez CNMF-E pour cette étude. Entrez le diamètre moyen des cellules en pixels et exécutez l’algorithme pour générer un ensemble de cellules contenant des régions d’intérêt (ROI) qui démontrent des activités de type cellulaire.
- Sélectionnez manuellement les zones d’intérêt qui sont des cellules (ont une morphologie semblable à celle d’une cellule et une activité22,23, et se trouvent dans le champ de vision) parmi celles qui ne le sont pas, et validez l’ensemble de cellules organisées (Figure 4D).
- Exportez les traces de calcium de chaque retour sur investissement pour une analyse plus approfondie.
8. Traitement des données d’imagerie calcique à deux photons
- Pour les films d’enregistrement à deux photons, utilisez un package python conçu pour traiter les données d’analyse du calcium à deux photons.
- Tout d’abord, combinez les images prises dans une série T dans une pile .tiff comme décrit ci-dessous.
- Dans l’interface Options d’exécution , ajustez les paramètres, y compris la valeur tau et la fréquence d’images, afin qu’ils correspondent au GCaMP utilisé et à la fréquence d’images de l’enregistrement.
- Facultatif : Réglez le paramètre do_registration sur 1 pour enregistrer le film. Cela équivaut à l’étape de correction de mouvement décrite ci-dessus.
- Facultatif : Réglez le paramètre anatomical_only sur 1 pour détecter les zones d’intérêt à l’aide des caractéristiques anatomiques en plus de la dynamique de fluorescence. Cela nécessite la saisie du diamètre de la cellule, alors prenez des mesures à l’aide du logiciel de traitement d’image. Ceci est généralement recommandé, car cela génère des retours sur investissement avec des formes plus naturelles (Figure 5A-C).
- Une fois tous les paramètres définis, exécutez l’algorithme pour qu’il effectue tous les calculs ensemble. Reportez-vous à l’interface utilisateur graphique (GUI) pour vérifier la progression.
- Une fois que c’est fait, revenez à l’interface de sélection des cellules pour une conservation manuelle des résultats d’identification des cellules.
- Enregistrez une image de la cellule organisée au-dessus de la projection maximale du film. Celle-ci sera utilisée ultérieurement comme image de référence pour l’enregistrement des données d’enregistrement d’un photon.
- L’algorithme enregistre ensuite automatiquement les résultats. npy, accessible ultérieurement avec Python. Vous pouvez également enregistrer les résultats dans d’autres fichiers formatés pour une analyse plus approfondie dans d’autres logiciels.

Figure 5 : Identification des cellules à l’aide d’un logiciel de traitement à deux photons. (A) Image représentative de l’identification des cellules prise à partir du logiciel de traitement à deux photons. En définissant Anatomical_only paramètre sur 0 mais en gardant tous les autres paramètres identiques, plusieurs non-cellules sont présentes dans la zone entre les lignes pointillées qui interfèrent avec la curation manuelle des cellules réelles. (B) Exemples de mesures du diamètre des cellules prises à partir de (A), à l’aide d’un logiciel de traitement d’images (en haut à gauche ; 7,5 pixels, en haut à droite ; 9, en bas à gauche ; 6,5, en bas à droite ; 7,5). (C) Image représentative de l’identification des cellules. Lorsque vous réglez Anatomical_only paramètre sur 1 et que vous saisissez le diamètre moyen de la cellule pris à partir de (B) dans l’algorithme de diamètre de cellule, aucune cellule n’est présente dans la zone entre les lignes pointillées (les barres d’échelle représentent 200 μm). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
9. Enregistrement d’ensembles de cellules identifiés dans toutes les modalités d’imagerie
- Effectuer l’enregistrement des cellules identifiées à partir d’enregistrements à un et deux photons avec l’algorithme d’enregistrement et d’analyse d’images multimodales (MIRA), disponible via l’interface Python du logiciel d’imagerie à un photon.
- Cet algorithme aligne les données à un photon et à deux photons via un recalage non rigide. Voir l’ensemble des cahiers de démonstration en ligne trouvés sur le site Web et utilisés pour cette étude.
REMARQUE : Les ordinateurs portables ont été écrits de manière à ce que tout le traitement soit effectué dans le logiciel 1P et ne sont donc pas compatibles avec le logiciel de traitement 2P. Par conséquent, ne suivez que certaines des étapes des carnets de notes pour cette étude.
- Cet algorithme aligne les données à un photon et à deux photons via un recalage non rigide. Voir l’ensemble des cahiers de démonstration en ligne trouvés sur le site Web et utilisés pour cette étude.
- Suivez les étapes décrites dans les carnets de démonstration, qui impliquent la génération d’une image structurelle pour chaque modalité d’imagerie. Par défaut, cela implique de générer une projection maximale de la pile z à deux photons et une image moyenne de l’enregistrement à un photon. Vous pouvez également utiliser une image moyenne de l’enregistrement à deux photons.
- Lorsque vous y êtes invité, filtrez les images par bande passante spatiale pour mieux visualiser les points de repère et les réorienter pour qu’ils correspondent.
- Sélectionnez des points de repère correspondants sur les deux images (Figure 6A).
- Utilisez-les pour calculer la déformation nécessaire pour aligner les deux images. En général, 3 à 5 points de repère devraient suffire.
- L’algorithme calcule la déformation en fonction d’une combinaison de points de repère et de similitude d’images. Optimiser le poids relatif accordé aux deux facteurs jusqu’à ce que des résultats satisfaisants soient obtenus.
- Déformer la carte cellulaire acquise lors d’une session à un photon pour générer une nouvelle carte cellulaire alignée sur les données à deux photons.
- Importez ensuite cette carte de cellule déformée dans le logiciel de traitement 1P pour générer une image avec l’image de projection maximale du film à deux photons en arrière-plan.
- Exportez cette image à des fins d’enregistrement.
- Dans un logiciel de programmation, alignez les deux images générées jusqu’à présent (carte de cellule à deux photons sur l’image de projection maximale à deux photons) et déformez la carte de cellule à un photon sur l’image de projection maximale à deux photons (Figure 6B, C).
- Pour ce faire, pour cette étude, utilisez une application d’estimation d’alignement qui permet à l’utilisateur de comparer les résultats de différentes techniques d’enregistrement. Étant donné que les deux images ont le même arrière-plan, la technique de corrélation de phase, avec un recalage rigide, était suffisante.
- Une fois l’enregistrement terminé, scannez l’image maintenant enregistrée pour les ROI qui se chevauchent. Il s’agit de ROI qui sont actifs dans les deux sessions d’enregistrement, qui pourraient être utilisés dans une analyse plus approfondie.

Figure 6 : Enregistrement intermodal des cellules à l’aide du flux de travail MIRA. (A) Image représentative du flux de travail d’alignement des cellules. L’image moyenne des données à un photon est affichée à gauche, et celle des données à deux photons est affichée à droite. Les points de repère correspondants des deux images sont sélectionnés et étiquetés dans le logiciel par un schéma de couleurs aléatoires (cercles rouges). (B) Des images alignées sur des exemples montrant les deux ensembles de cellules identifiés, un photon (violet) et deux photons (vert), sont superposées à l’image moyenne des données à deux photons. (C) Image de la région marquée par la case blanche en (B), les cellules alignées sont représentées ici sous forme de contours verts et violets superposés. Dans tous les panneaux, la barre d’échelle représente 200 μm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Résultats
La méthode d’imagerie calcique chronique multicouche in vivo de la même population neuronale sur une période de plusieurs semaines, en utilisant des modalités d’imagerie à un et deux photons, dans des conditions de mouvement libre et de fixation de la tête, a été montrée. Ici, la capacité d’identifier des populations neuronales correspondantes sous imagerie à un photon pendant que l’animal explorait une arène ouverte dans l’obscurité a été démontrée (Figure 7A
Discussion
Ici, nous avons montré la capacité d’observer et de comparer directement les neurones dans des conditions de tête fixe et de mouvement libre dans les mêmes populations neuronales. Bien que nous ayons démontré l’application dans le cortex visuel, ce protocole peut être adapté à une multitude d’autres zones du cerveau, à la fois les zones corticales et les noyaux profonds 24,25,26,27,28, ainsi que d’autres configurations d’acquisition de données et comportementales
Déclarations de divulgation
Les auteurs ne déclarent aucun intérêt financier concurrent ou conflit d’intérêts.
Remerciements
Nous remercions Mme Charu Reddy et le professeur Matteo Carandini (Cortex Lab) pour leurs conseils sur le protocole chirurgical et le partage de souches de souris transgéniques. Nous remercions le Dr Norbert Hogrefe (Inscopix) pour ses conseils et son aide tout au long du développement de la chirurgie. Nous remercions Mme Andreea Aldea (Sun Lab) pour son aide dans la configuration chirurgicale et le traitement des données. Ce travail a été soutenu par la Moorfields Eye Charity.
matériels
| Name | Company | Catalog Number | Comments |
| 0.9% Sodium Chloride solution for infusion (Vetivex 11) 250ml | Dechra | 20091607 | Saline for hydration and drug reconsitution |
| 18004-1 Trephine 1.8mm diameter bur | FST | 18004-18 | Drill bit |
| 1ml syringe | Terumo | MDSS01SE | 1ml syringe |
| 23G x 5/8 inch 6% LUER needle | Terumo | NN-2316R | 23G needle |
| 71000 Automated stereotaxic apparatus w/ built-in software | RWD | - | RWD |
| Absorbable Haemostatic Gelatin Sponge (10x10x10mm) | Surgispon | SSP-101010 | gel-foam |
| Alcohol pads 70% isopropyl alcohol | Braun | 9160612 | Alcohol pads |
| Aluminium foil | Any retailer | - | Foil to cover eyes during surgery |
| Articifical Cerebrospinal Fluid | Tocris Bioscience a Bio-Techne Brand | 3525/25ML | ACSF |
| Automated microinjection pump | WPI | 8091 | |
| Betadine solution (10% iodinated Povidone) 500ml | Videne/Ecolab | 3030440 | Betadine |
| Bruker Ultime 2Pplus (customised) | Bruker | - | Two-photon imaging system |
| Cardiff Aldasorber | Vet-Tech | AN006 | Anaesthesia absorber |
| CFI S Plan Fluor ELWD ADM 20XC | Nikon | MRH48230 | 20x objective lens |
| Compact Anaesthesia system - single gas - isoflurane K/F, with oxygen concentrator model: ZY-5AC and scavenging unit | Vet-Tech | AN001 | Compact anaesthesia system |
| Contec Prochlor | Aston Pharma | AP2111L1 | Disinfectant (hypochlorous acid) |
| Dexamethasone Sodium Phosphate Injection, USP, 4mg/ml, NDC: 0641-6145-25 | Hikma | Covetrus:70789 | Dexamethasone |
| Dissecting Knife, cutting edge 4mm, thickness 0.5mm, stainless steel | Fine Science Tools | 10055-12 | Knife for incisino of cortex |
| Dual-Sided, Non-Puncture Mouse & Neonatal Rat Ear Bars | Stoelting | 51649 | Ear bar |
| Dummy microscope | Inscopix | Dummy microscope | To help with implantation |
| Ethanol (100%) | VWR | 40-1712-25 | Used to make 70% ethanol |
| Fisherbrand Nitrile Indigo Disposable Gloves PPE Cat III | FischerScientific | 17182182 | Gloves |
| Homeothermic Monitor 50-7222-F | Harvard Apparatus | 50-7222-F | Homeothermic monitoring system/heating pad |
| Image processing software | ImageJ | - | Image processing software |
| Inscopix Data Processing Software (IDPS) | Inscopix | - | One-photon calcium imaging processing software |
| Insight Duals-232, S/N 2043 | InSight | Insight Spectra X3 | Two-photon imaging laser |
| IsoFlo 250ml 100% w/w inhalation | Zoetis | WM 42058/4195 | Isoflurane |
| Kwik-Sil Low Toxicity Silicone Adhesive | World Precision Intruments (WPI) | KWIK-SIL | Silicone adhesive |
| MICROMOT mains adapter NG 2/S, w/ Drill unit 60/E | PROXXON | NO 28 515 | Handheld drill |
| nVoke Integrated Imaging and Optogenetics System package | Inscopix | - | One-photon Imaging system and software |
| ProView Implant Kit | Inscopix | ProView Implant Kit | Dummy microscope, stereotaxic arm and attachment |
| ProView Prism Probe | Inscopix | 1050-002203 | Microprism lens |
| Rimadyl (50mg/ml) | Zoetis | VM 42058/4123 | Carprofen |
| Stereotaxis Microscope on Articulated arm with table clamp | WPI | PZMTIII-AAC | Microscope |
| Super-Bond Universal kit, SUN Medical | Prestige-Dental | K058E | Adhesive cement |
| Two-photon calcium image software | Suite2P | - | Two-photon calcium imaging processing software |
| Vapouriser | Vet-Tech | - | Isoflurane vapouriser |
| Xailin Lubricating Eye Ointment 5g | Xailin-Night | MLG/28/1551 | Ophthalmic ointment |
Références
- Denk, W., Strickler, J. H., Webb, W. W. Two-photon laser scanning fluorescence microscopy. Science. 248 (4951), 73-76 (1990).
- Svoboda, K., Yasuda, R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 50 (6), 823-839 (2006).
- Dombeck, D. A., Khabbaz, A. N., Collman, F., Adelman, T. L., Tank, D. W. Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron. 56 (1), 43-57 (2007).
- Vaziri, A., Emiliani, V. Reshaping the optical dimension in optogenetics. Curr Opin Neurobiol. 22 (1), 128-137 (2012).
- Holtmaat, A., et al. high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 4 (8), 1128-1144 (2009).
- Sun, Y. J., Sebastian Espinosa, J., Hoseini, M. S., Stryker, M. P. Experience-dependent structural plasticity at pre- and postsynaptic sites of layer 2/3 cells in developing visual cortex. Proc Natl Acad Sci U S A. 116 (43), 21812-21820 (2019).
- Andermann, M. L., Kerlin, A. M., Reid, R. C. Chronic cellular imaging of mouse visual cortex during operant behavior and passive viewing. Front Cell Neurosci. 4, 3 (2010).
- Sofroniew, N. J., Flickinger, D., King, J., Svoboda, K. A large field of view two-photon mesoscope with subcellular resolution for in vivo imaging. Elife. 5, 14472 (2016).
- Puścian, A., Benisty, H., Higley, M. J. NMDAR-dependent emergence of behavioral representation in primary visual cortex. Cell Rep. 32 (4), 107970 (2020).
- Trachtenberg, J. T., et al. Long-term in vivo. imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 420 (6917), 788-794 (2002).
- Seaton, G., et al. Dual-component structural plasticity mediated by αCaMKII autophosphorylation on basal dendrites of cortical layer 2/3 neurones. J Neurosci. 40 (11), 2228-2245 (2020).
- Helmchen, F., Denk, W. Deep tissue two-photon microscopy. Nat Methods. 2 (12), 932-940 (2005).
- Andermann, M. L., et al. Chronic cellular imaging of entire cortical columns in awake mice using microprisms. Neuron. 80 (4), 900-913 (2013).
- Chia, T. H., Levene, M. J. Microprisms for in vivo multilayer cortical imaging. J Neurophysiol. 102 (2), 1310-1314 (2009).
- Low, R. J., Gu, Y., Tank, D. W. Cellular resolution optical access to brain regions in fissures: Imaging medial prefrontal cortex and grid cells in entorhinal cortex. Proc Natl Acad Sci U S A. 111 (52), 18739-18744 (2014).
- Buxhoeveden, D. P., Casanova, M. F. The minicolumn hypothesis in neuroscience. Brain. 125, 935-951 (2002).
- Chen, S., et al. Miniature fluorescence microscopy for imaging brain activity in freely-behaving animals. Neurosci Bull. 36 (10), 1182-1190 (2020).
- Gulati, S., Cao, V. Y., Otte, S. Multi-layer cortical Ca2+ imaging in freely moving mice with prism probes and miniaturized fluorescence microscopy. J Vis Exp. (124), e55579 (2017).
- Resendez, S. L., et al. Visualization of cortical, subcortical, and deep brain neural circuit dynamics during naturalistic mammalian behavior with head-mounted microscopes and chronically implanted lenses. Nat Protoc. 11 (3), 566-597 (2016).
- Guo, Z. V., et al. Procedures for behavioral experiments in head-fixed mice. PLoS One. 9 (2), 88678 (2014).
- Wekselblatt, J. B., Flister, E. D., Piscopo, D. M., Niell, C. M. Large-scale imaging of cortical dynamics during sensory perception and behavior. J Neurophysiol. 115 (6), 2852-2866 (2016).
- Pnevmatikakis, E. A., et al. Simultaneous denoising, deconvolution, and demixing of calcium imaging data. Neuron. 89 (2), 285-299 (2016).
- Zhou, P., et al. Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. Elife. 7, 28728 (2018).
- Beckmann, L., et al. Longitudinal deep-brain imaging in mouse using visible-light optical coherence tomography through chronic microprism cranial window. Biomed Opt Express. 10 (10), 5235-5250 (2019).
- Wenzel, M., Hamm, J. P., Peterka, D. S., Yuste, R. Reliable and elastic propagation of cortical seizures in. Cell Rep. 19 (13), 2681-2693 (2017).
- Heys, J. G., Rangarajan, K. V., Dombeck, D. A. The functional micro-organization of grid cells revealed by cellular-resolution imaging. Neuron. 84 (5), 1079-1090 (2014).
- Barson, D., Hamodi, A. S. Simultaneous mesoscopic and two-photon imaging of neuronal activity in cortical circuits. Nat Methods. 17 (1), 107-113 (2020).
- Paquelet, G. E., et al. Single-cell activity and network properties of dorsal raphe nucleus serotonin neurons during emotionally salient behaviors. Neuron. 110 (16), 2664-2679 (2022).
- Yang, Q., et al. Transparent microelectrode arrays integrated with microprisms for electrophysiology and simultaneous two-photon imaging across cortical layers. bioRxiv. , (2022).
- Priestley, J. B., Bowler, J. C., Rolotti, S. V., Fusi, S., Losonczy, A. Signatures of rapid plasticity in hippocampal CA1 representations during novel experiences. Neuron. 110 (12), 1978-1992 (2022).
- Zong, W., et al. Miniature two-photon microscopy for enlarged field-of-view, multi-plane and long-term brain imaging. Nat Methods. 18 (1), 46-49 (2021).
- Engelbrecht, C. J., et al. Ultra-compact fiber-optic two-photon microscope for functional fluorescence imaging in vivo. Opt Express. 16 (8), 5556-5564 (2008).
- Suzuki, M., Aru, J., Larkum, M. E. Double-μ Periscope, a tool for multilayer optical recordings, optogenetic stimulations or both. Elife. 10, 72894 (2021).
- Stibůrek, M., et al. 110 μm thin endo-microscope for deep-brain in vivo observations of neuronal connectivity, activity and blood flow dynamics. Nat Commun. 14 (1), 1897 (2023).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.