
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Bio-impression 3D d’hydrogels photoaccordables pour étudier l’activation des fibroblastes
Dans cet article
Résumé
Cet article décrit comment bio-imprimer en 3D des hydrogels photoaccordables pour étudier le raidissement de la matrice extracellulaire et l’activation des fibroblastes.
Résumé
Les hydrogels photoaccordables peuvent se transformer spatialement et temporellement en réponse à l’exposition à la lumière. L’incorporation de ces types de biomatériaux dans des plateformes de culture cellulaire et le déclenchement dynamique de changements, tels que l’augmentation de la rigidité microenvironnementale, permettent aux chercheurs de modéliser les changements dans la matrice extracellulaire (MEC) qui se produisent au cours de la progression de la maladie fibrotique. Dans cet article, une méthode de bio-impression 3D d’un biomatériau hydrogel photoaccordable capable de deux réactions de polymérisation séquentielles dans un bain de support de gélatine est présentée. La technique de bio-impression FRESH (Freeform Reversible Embedding of Suspended Hydrogels) a été adaptée en ajustant le pH du bain de support pour faciliter une réaction d’addition de Michael. Tout d’abord, la bio-encre contenant du méthacrylate de poly(éthylène glycol)-alpha (PEGαMA) a réagi hors stœchiométrie avec un agent de réticulation dégradable par cellule pour former des hydrogels mous. Ces hydrogels mous ont ensuite été exposés à un photo-initateur et à la lumière pour induire l’homopolymérisation des groupes qui n’ont pas réagi et rigidifier l’hydrogel. Ce protocole couvre la synthèse d’hydrogel, la bio-impression 3D, le phototiffening et la caractérisation des paramètres pour évaluer l’activation des fibroblastes dans les structures 3D. La méthode présentée ici permet aux chercheurs de bio-imprimer en 3D une variété de matériaux qui subissent des réactions de polymérisation catalysées par le pH et pourraient être mis en œuvre pour concevoir divers modèles d’homéostasie, de maladie et de réparation des tissus.
Introduction
La bio-impression 3D est une technologie transformatrice qui permet aux chercheurs de déposer avec précision des cellules et des biomatériaux dans des volumes 3D et de recréer la structure hiérarchique complexe des tissus biologiques. Au cours de la dernière décennie, les progrès de la bio-impression 3D ont permis de créer des tissus cardiaques humains battants1, des modèles fonctionnels des tissus rénaux2, des modèles d’échange gazeux dans les poumons3 et des modèles tumoraux pour la recherche sur le cancer4. L’invention des techniques de bio-impression 3D embarquée, telles que la bio-impression FRESH (Freeform Reversible Embedding of Suspended Hydrogel), a permis de reproduire des structures complexes de tissus mous telles que les vaisseaux sanguins pulmonaires5 et même le cœur humain6 en 3D. La bio-impression 3D FRESH facilite l’impression couche par couche de bio-encres souples et à faible viscosité par extrusion dans un bain de support de cisaillement. Le bain de support est constitué d’un matériau tel que des microparticules de gélatine compactes qui agissent comme un plastique Bingham et maintiennent la forme et la structure prévues de la bio-encre après l’impression. Une fois que la construction imprimée s’est solidifiée, le bain de support peut être dissous en augmentant la température à 37 °C7.
Un article de synthèse récent a résumé les matériaux qui ont été bio-imprimés en 3D dans diverses publications à l’aide de la technique FRESH. Ces matériaux d’origine naturelle vont du collagène de type I à l’acide hyaluronique méthacrylé et représentent plusieurs mécanismes de gélification différents7. La plupart des études de recherche réalisées à l’aide de cette technique de bio-impression 3D utilisent des biomatériaux statiques qui ne changent pas en réponse à des stimuli externes. Des biomatériaux hydrogels photoaccordables dynamiques ont été utilisés par notre laboratoire et d’autres 8,9,10,11,12 pour modéliser une variété de maladies fibrotiques. Contrairement aux biomatériaux statiques, les bioencres photoaccordables permettent de créer un modèle ramolli avec une valeur de module d’élasticité plus faible, puis de le rigidifier pour explorer les réponses cellulaires à l’augmentation du raidissement microenvironnemental.
Les maladies fibrotiques sont caractérisées par une augmentation de la production de matrice extracellulaire qui peut provoquer des cicatrices et des raidissements13. Le raidissement des tissus peut entraîner d’autres blessures et la destruction des tissus touchés, causant des dommages permanents aux organes et même la mort ; Les troubles fibrotiques sont responsables d’un tiers de la mortalité dans le monde. Les fibroblastes produisent une matrice extracellulaire excessive et aberrante dans cet état pathologique14,15. L’augmentation de la prolifération des fibroblastes et le dépôt de matrice extracellulaire rigidifient davantage le tissu et activent une boucle de rétroaction positive profibrotique16,17,18,19. L’étude de l’activation des fibroblastes est essentielle à la compréhension des maladies fibrotiques. Nous présentons ici l’hypertension artérielle pulmonaire humaine (HTAP) comme un exemple d’un trouble fibrotique dans lequel il est important d’imiter la géométrie 3D du vaisseau sanguin à l’aide de la bio-impression 3D et d’introduire les capacités de raidissement dynamique des hydrogels photoaccordables. L’HTAP est une affection dans laquelle la pression dans les artères pulmonaires principales dépasse les niveaux normaux et exerce une pression sur le cœur, augmentant l’activation des fibroblastes adventiaux de l’artère pulmonaire humaine (HPAAF) et rigidifiant les tissus des vaisseaux sanguins16,17,18,19. Une formulation de bio-encre à base de poly(éthylène glycol)-alpha méthacrylate (PEGαMA) photoréglable permet un raidissement temporel des constructions et aide à modéliser à la fois les tissus sains et la progression de la maladie 5,8,9,10. L’exploitation de cette caractéristique unique permet de quantifier l’activation et la prolifération de HPAAF en réponse au raidissement microenvironnemental en 3D et peut fournir des informations précieuses sur les mécanismes cellulaires impliqués dans cette maladie. Le protocole décrit ici permettra aux chercheurs de créer des modèles 3D qui récapitulent les changements dans le microenvironnement extracellulaire au cours de la progression de la maladie ou de la réparation tissulaire et d’étudier l’activation des fibroblastes.
Protocole
1. Synthèse et caractérisation de PEGαMA
REMARQUE : La synthèse du poly(éthylène glycol)-alpha méthacrylate (PEGαMA) a été adaptée de Hewawasam et al. et réalisée dans des conditions sans humidité9.
- Peser les réactifs.
REMARQUE : Par exemple, peser 5 g de PEG-hydroxyle à 8 bras (PEG-OH) de 10 kg/mol et 0,38 g d’hydrure de sodium (NaH) (voir le tableau des matériaux). - Ajouter une barre d’agitation dans une fiole Schlenk de 250 ml et purger avec de l’argon.
- Dissoudre le PEG-OH dans le plus faible volume de tétrahydrofurane anhydre (THF) nécessaire à la dissolution dans le flacon de Schlenk.
REMARQUE : Environ 80 mL de THF dissoudront 5 g de PEG-OH. Ajoutez la quantité minimale de THF nécessaire pour dissoudre le PEG-OH. - Ajouter 3 fois l’excès molaire de NaH au mélange réactionnel et remuer à température ambiante (RT) pendant 30 min.
- Ajouter 6 fois l’excès molaire d’éthyle 2-(bromométhyl)acrylate (EBrMA, voir tableau des matériaux) goutte à goutte dans la fiole de Schlenk et couvrir le récipient de réaction avec du papier d’aluminium pour le protéger de la lumière. Remuez la réaction à température ambiante pendant environ 48 h.
REMARQUE : Pour 5 g de PEG-OH et 0,38 g de NaH, utilisez 3,68 mL d’EBrMA pour cette réaction. - Ajoutez quelques gouttes d’acide acétique 1 N pour éteindre la réaction. Filtrez la solution sous vide à l’aide d’un adjuvant de filtration.
REMARQUE : L’ajout d’acide acétique produira des bulles de gaz. Arrêtez d’ajouter des gouttes d’acide acétique lorsque les bulles cessent de se former, car cela indique que le mélange a été trempé avec succès. - Concentrer le filtrat sur un évaporateur rotatif et précipiter dans de l’éther diéthylique à 4 °C. Laisser le précipité à l’abri de la lumière à 4 °C pendant 12 à 18 h.
- Ajoutez un papier filtre Whatman dans un entonnoir Buchner. Versez lentement le mélange réactionnel sur le papier filtre et utilisez l’aspiration sous vide pour séparer le précipité de l’éther diéthylique. Recueillir le précipité dans une fiole de filtration sèche et propre.
- Séchez le produit sous vide pendant au moins 5 h ou toute la nuit à température ambiante et dissolvez-le dans le volume minimum d’eau déminéralisée nécessaire. Transvaser le produit dissous dans une tubulure de dialyse (voir le tableau des matériaux) et dialyser contre 3,5 L d’eau déminéralisée pendant au moins quatre jours. Changez l’eau de dialyse toutes les 12 h.
REMARQUE : Le produit apparaîtra comme une poudre solide blanche pure complètement sèche après le séchage sous vide. - Congelez le produit et lyophilisez-le pendant environ 72 h ou jusqu’à ce qu’il soit complètement sec.
- Dissoudre le produit dans le chloroforme D (CDCl 3). Exécutez l’échantillon à l’aide d’une RMN de 1H avec un protocole qui effectue 248 balayages avec un temps de relaxation de 2,5 s.
- Vérifier la fonctionnalisation et la pureté du produit en étalonnant le pic de solvantCDCl 3 à 7,26 PPM. Intégrez le pic pour les protons du squelette PEG (d3.71) et calibrez l’intégration à 114.
- Intégrer les crêtes restantes : RMN PEGαMA 1 H (300 MHz, CDCl 3) : d (ppm) 1,36(t, 3H, CH 3-),3,71 (s, 114H, PEG CH 2-CH 2), 4,29 (t, s, 4H, -CH 2-C(O)-O-O, -O-CH 2-C(=CH 2)-), 5,93 (q, 1H, -C=CH 2), 6,34 (q, 1H, -C=CH 2) et comparer l’intégration pour les pics du groupe d’extrémités des alcènes αMA à la valeur attendue (1H) sur la base de l’étalonnage du squelette PEG (Graphique 1).
REMARQUE : Additionnez les deux pics étiquetés « a » (Figure 1) et multipliez-les par 100 pour obtenir le pourcentage moyen de fonctionnalisation du PEGαMA.

Figure 1 : La RMN des protons a confirmé la réussite de la fonctionnalisation de PEGαMA. L’analyse RMN a été réalisée dans le chloroforme-D (CDCl3) et a montré une fonctionnalisation de 96,5%. RMN PEGαMA 1 H (300 MHz, CDCl3) : d (ppm) 1,36(t, 3H, CH 3-),3,71 (s, 114H, PEG CH 2-CH 2), 4,29 (t, s, 4H, -CH 2-C(O)-O-O, -O-CH2-C(=CH 2)-), 5,93 (q, 1H, -C=CH 2), 6,34 (q, 1H, -C=CH 2). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
2. Conception du modèle et configuration de la bio-imprimante 3D
REMARQUE : Une imprimante 3D disponible dans le commerce (voir le tableau des matériaux) a été modifiée en remplaçant l’extrudeuse thermoplastique par une extrudeuse à pousse-seringue sur mesure et adaptée de Hinton et al.20. Des designs open-source sont disponibles en ligne : https://3d.nih.gov/users/awfeinberg.
- Ouvrez le logiciel Fusion 360 (voir Tableau des matériaux) et créez une conception de cylindre creux 3D assistée par ordinateur.
REMARQUE : Un fichier téléchargeable qui peut être utilisé pour cette étape et qui imite la géométrie des vaisseaux sanguins se trouve dans le fichier supplémentaire 1. - Enregistrez le fichier et ouvrez-le dans le logiciel Slic3r (voir la table des matériaux). Vérifiez que tous les paramètres sont comme vous le souhaitez, puis appuyez sur le bouton d’exportation du G-code. Enregistrez le G-code sur l’ordinateur.
- Ouvrez le logiciel Pronterface (voir la table des matériaux) et téléchargez le fichier G-code.
REMARQUE : Le logiciel Pronterface s’interface avec la bio-imprimante et fournit un contrôle suffisant de l’entrée matérielle. Un fichier G-code utilisable se trouve dans le fichier supplémentaire 2. - Transférez la bio-imprimante et toutes les pièces associées dans une enceinte de sécurité biologique (BSC) à l’aide de techniques aseptiques.
- Assemblez une pointe d’aiguille émoussée de 30 g de 0,5 po de longueur (voir le tableau des matériaux) à la seringue en verre d’impression et mettez-la de côté.
- Branchez le cordon d’alimentation de la bio-imprimante dans une prise. Appuyez sur le bouton d’alimentation rouge situé à l’avant de la bio-imprimante pour l’allumer. Connectez le cordon USB (Universal Serial Bus) entre l’ordinateur et la bio-imprimante et assurez-vous que toutes les connexions filaires sont établies et branchées.
3. Préparation du bain de support et des réactifs
REMARQUE : Effectuez toutes les étapes dans une enceinte de sécurité biologique en utilisant des techniques aseptiques.
- Préparer un milieu de culture cellulaire composé de substrat basal SmBM (CC-3181) et de suppléments SmGM-2 SingleQuots (CC-4149), à l’exclusion du sérum de veau fœtal (FBS), conformément aux instructions du fabricant. Conserver à 4 °C jusqu’à utilisation.
- Aliquote 50 mL du milieu de culture cellulaire et ajouter 1 % v/v de FBS (CC-4149) (voir le tableau des matériaux) pour obtenir un milieu à faible teneur en sérum. Conserver à 4 °C jusqu’à utilisation.
- Remettre en suspension la poudre de boue de gélatine conformément aux instructions du fabricant en utilisant un milieu de culture cellulaire stérile sans FBS comme solvant (voir le tableau des matériaux). Immédiatement avant l’utilisation, ajustez le pH final de la boue de gélatine à un pH de 9 à l’aide de 2 M d’hydroxyde de potassium (KOH) et/ou de 2 M d’acide chlorhydrique (HCl) pour ajuster le pH de la solution au besoin à l’aide d’un pH-mètre.
- Remplir le nombre désiré de puits d’une plaque de 24 puits, chacun rempli à moitié environ, en utilisant 1 mL de boue de gélatine par puits à l’aide d’une seringue sans aiguille.
REMARQUE : Remplissez uniformément le centre des puits et confirmez qu’il n’y a pas de poches d’air. Tapotez la plaque pour aider à répartir uniformément la boue de microparticules. Ajustez la hauteur et le volume de la boue selon vos besoins pour vous adapter à la taille et à la forme de chaque bio-impression. Les utilisateurs peuvent créer une seringue maison pour transférer la bouillie de gélatine dans chaque puits. Pour ce faire, il suffit d’ajouter un piston de seringue de taille correcte dans un tube à essai de 50 ml contenant déjà la bouillie de gélatine compactée au fond. Lors de l’insertion du piston, insérez un petit fil guide le long du tube à essai pour aider l’air à s’échapper, puis retirez-le lorsque le piston est en contact avec la boue de gélatine. Immédiatement avant l’utilisation, coupez l’extrémité du tube à essai avec une lame de rasoir pour créer un trou d’où la bouillie de gélatine peut sortir et appuyez sur le piston. - Placez la plaque remplie à 24 puits au centre de la platine de la bio-imprimante et fixez-la à la platine.
REMARQUE : La figure 2 montre une configuration de bio-imprimante générique. Placez un élastique autour de la platine d’impression pour fixer une plaque de 24 puits à la plate-forme et empêcher tout mouvement.

Figure 2 : Configuration de base de la bio-impression 3D. La bio-imprimante a été installée dans un environnement stérile tel qu’une enceinte de sécurité biologique, et la tête d’impression a été assemblée de manière à ce que la seringue et l’aiguille en verre soient abaissées verticalement dans la zone d’impression du bain de support située en dessous. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
4. Culture cellulaire
REMARQUE : Effectuez toutes les étapes dans une enceinte de sécurité biologique en utilisant des techniques aseptiques.
- Décongeler les cellules HPAAF (obtenues dans le commerce, voir le tableau des matériaux) et les étendre dans des flacons en plastique traités pour la culture tissulaire T-75 contenant du milieu de base SmBM (CC-3181) et tous les suppléments SmGM-2 SingleQuots (CC-4149) conformément aux instructions du fabricant (voir le tableau des matériaux).
REMARQUE : Des protocoles standard de culture cellulaire pour les cellules adhérentes doivent être utilisés, en maintenant les cellules à 37 °C et 5 % de CO2 et en réapprovisionnant le milieu tous les quelques jours. - Une fois que les HPAAF ont atteint ~80-90% de confluence, aspirez le milieu et rincez les cellules une fois avec une solution saline tamponnée au phosphate (PBS).
- Ajouter environ 4 mL de trypsine-EDTA préchauffée à 0,05 % dans chaque fiole T-75. Inclinez le flacon pour vous assurer que toute la surface de culture cellulaire est recouverte d’une solution de trypsine-EDTA à 0,05 %. Incuber les flacons pendant 3 à 5 minutes à 37 °C et vérifier le décollement des cellules.
- Une fois que les cellules flottent, ajoutez au moins 6 mL de milieu Eagle modifié (DMEM) de Dulbecco dans chaque fiole et transférez les cellules dans un tube conique de 50 mL.
- Centrifuger la suspension cellulaire à 300 x g pendant 5 min à température ambiante pour granuler les cellules. Aspirer le surnageant de la pastille cellulaire et remettre les cellules en suspension dans un milieu de 1 à 3 ml avec FBS à l’aide d’une pipette de 1000 μL, en assurant une suspension unicellulaire.
- Transférez 10 μL de la suspension cellulaire dans un tube de microcentrifugation. Ajouter 10 μL de solution de Trypan Blue et bien mélanger. Utilisez 10 μL de ce mélange pour compter les cellules dans un hémocytomètre à l’aide d’un microscope à lumière inversée.
REMARQUE : Pour atteindre une concentration finale de bio-encre de 4 x 106 cellules/mL, 800 000 fibroblastes ont été mis de côté pour chaque 200 μL de bio-encre.
5. Préparation de l’encre bio hydrogel
REMARQUE : La préparation de l’encre biologique a été adaptée de Davis-Hall et al.5. Les étapes 5.1 à 5.2 peuvent être effectuées en parallèle avec les étapes 4.1 à 4.3 afin de minimiser le temps entre le prélèvement des cellules et la remise en suspension dans la bio-encre. Effectuer des étapes dans une enceinte de sécurité biologique à l’aide d’une technique aseptique.
- Préparer une solution de tris(2-carboxyéthyl) phosphine (TCEP, voir le tableau des matériaux) de pH 7 de 20 mM et un filtre stérile à l’aide d’un filtre à seringue de 0,2 μm. Immédiatement avant l’utilisation, ajouter 2 M de KOH et/ou 2 M de HCl pour ajuster le pH de la solution au besoin. Mesurez avec un pH-mètre et ajustez en conséquence.
REMARQUE : Le TCEP réduit les liaisons disulfure. - Préparer une solution mère de PEGαMA remise en suspension à 0,25 mg/mL dans un milieu de culture cellulaire stérile sans FBS, des solutions mères à 250 mM de 1,4-dithiothréitol (DTT), de réticulant dégradable MMP2 et de peptide CGRGDS (RGD) (voir le tableau des matériaux), en remettant en suspension le tout dans du TCEP stérile de 20 mM, et une solution mère à 15 % en poids de poly(oxyde d’éthylène) (PEO) dans de l’eau distillée (DI) à l’aide de pipettes au besoin.
- En suivant le tableau 1 à titre indicatif, combinez les quantités respectives nécessaires de PEGαMA, de DTT, de réticulant dégradable MMP2, de PEO, de CGRGDS et de milieux de culture cellulaire à faible teneur en sérum avec les fibroblastes dans un tube conique de 50 ml.
REMARQUE : Il est recommandé de vérifier le pH avec des bandelettes de pH après avoir ajouté tous les milieux de culture cellulaire sauf le milieu de culture cellulaire, car cette combinaison devrait donner un pH très proche de 6,2. Si d’autres ajustements du pH sont nécessaires, gardez une trace du volume supplémentaire nécessaire pour ajuster le pH de la solution de précurseur. Porter le volume total à 200 μL en ajoutant le volume restant du milieu de culture cellulaire moins tout volume ajouté lors de l’ajustement final du pH. - Mélangez la bio-encre à l’aide d’une pipette à déplacement positif pour vous assurer que les cellules sont des cellules uniques et confirmez que la solution de précurseur finale a un pH de 6,2 pour éviter la polymérisation catalysée par les bases pendant la bio-impression 3D.
- Chargez la bio-encre dans la seringue en verre en retirant le piston et en utilisant une seringue séparée avec une pointe d’aiguille émoussée de calibre 15 de 1,5 po de longueur (voir le tableau des matériaux) attachée pour transférer la bio-encre du tube de centrifugation à la seringue, en prenant soin d’éviter la formation de bulles d’air dans la solution.
- Placez la seringue en verre à l’intérieur de la tête d’impression et fixez les composants de la tête d’impression afin que tout soit fermement assemblé et prêt pour l’impression.
REMARQUE : À ce stade, la seringue en verre à l’intérieur de la tête d’impression doit être munie d’une pointe d’aiguille émoussée de calibre 30 de 0,5 po de longueur pour l’impression.
| Composant | Concentration de la solution mère | Montant à ajouter |
| PEGαMA | 0,25 mg/ml | 140 μL |
| TNT | 250 mètres d’épaisseur | 12,24 μL |
| Réticulant dégradable MMP2 | 250 mètres d’épaisseur | 5,25 μL |
| RGD | 250 mètres d’épaisseur | 1,6 μL |
| Entreprise d’externalisation des ressources | 15 % en poids | 33,33 μL |
| Milieux d’activation et/ou réactifs d’ajustement du pH | - | 7,58 μL |
| Fibroblastes | - | 800000 cellules |
Tableau 1 : Exemples de volumes nécessaires pour préparer 200 μL de bio-encre (solution de précurseur d’hydrogel et cellules de fibroblastes).
6.3D bio-impression
REMARQUE : Effectuez toutes les étapes dans une enceinte de sécurité biologique en utilisant des techniques aseptiques.
- À l’aide des flèches directionnelles du logiciel Pronterface, ajustez manuellement la position de l’aiguille d’extrusion au centre d’un puits et à l’intérieur de la boue du bain de support. Laissez au moins 1 mm de boue de bain de support sous la pointe de l’aiguille.
REMARQUE : Le logiciel n’a pas la capacité de dire où se trouve l’aiguille dans l’espace. C’est à l’utilisateur de déplacer l’aiguille en cliquant manuellement sur les flèches dans le logiciel (par exemple, en cliquant sur la flèche vers le haut, l’aiguille sera déplacée vers le haut ou loin de la plate-forme d’impression, etc.). Manœuvrez l’aiguille avec précaution pour vous assurer qu’elle ne heurtera aucune limite du puits. - Une fois que la pointe de l’aiguille est située au centre de la boue dans le puits, appuyez sur le bouton de démarrage de la face avant et attendez que l’impression soit terminée pour réaliser des constructions, comme le montre la figure 3A.
REMARQUE : Pour bioimprimer une construction à l’aide du fichier fourni (fichier supplémentaire 1), il faudra environ 3 minutes. Il faut environ 5 minutes pour orienter et déplacer l’aiguille, puis imprimer complètement une construction du début à la fin. - Répétez les étapes 6.1 à 6.2 jusqu’à ce que le nombre de constructions bio-imprimées souhaité soit atteint.
REMARQUE : Il est recommandé de créer plus de constructions que nécessaire pour tenir compte des impressions infructueuses. En cas de défaillance, passez au puits suivant, réinitialisez tout et répétez à nouveau les étapes 6.1 à 6.2. - Laissez la plaque de puits à température ambiante et couvrez-la dans le BSC pendant 1 h après la fin de l’impression pour permettre la polymérisation catalysée par la base de l’hydrogel photoaccordable.
- Placez la plaque de puits contenant les constructions bio-imprimées en 3D dans un incubateur stérile à 37 °C et laissez-les pendant 12 à 18 h pour faire fondre la boue du bain de support.

Figure 3 : Schéma expérimental. Ce protocole a été décrit en trois étapes principales : (A) Bio-impression 3D de tubes creux PEGαMA avec des cellules intégrées pour imiter la vascularisation pulmonaire. (B) Photo-initiation de la réaction d’homopolymérisation pour rigidifier le microenvironnement cellulaire. (C) Évaluation des marqueurs cellulaires de prolifération et d’activation. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
7.3D bio-imprimé construction culture et phototiffening
REMARQUE : Toutes les étapes doivent être effectuées dans une enceinte de sécurité biologique en utilisant des techniques aseptiques.
- Préparer une solution mère de phényl-2,4,6-triméthylbenzoylphosphinate (LAP) de phényl-2,6-triméthylbenzoylphosphinate (LAP) de 2,2 mM (voir le tableau des matériaux) dans du PBS et un filtre stérile à l’aide d’un filtre seringue de 0,2 μm. Conservez la solution LAP à l’abri de la lumière.
- Après 12-18 h, changez le support entourant les constructions bio-imprimées. Retirez manuellement le support et le bain de support de gélatine fondue dans les puits et veillez à ne pas perturber les constructions bio-imprimées.
REMARQUE : Il est utile de retirer lentement le support tout en tenant la plaque à un angle de 45° afin que les constructions émergent dans le puits et puissent être vues. Un cylindre d’hydrogel transparent doit être identifiable dans chaque puits avec une impression réussie (figure 4A). - Ajouter un volume approprié de milieu à faible teneur en sérum dans chaque puits.
REMARQUE : Pour une plaque à 24 puits, un support de 700 μL par puits doit recouvrir complètement les constructions bioimprimées. Ajustez au besoin. - Remettez la plaque dans l’incubateur et changez le milieu sur les échantillons tous les 3 jours ou conformément au plan expérimental.
- Vingt-quatre heures avant le point de temps de rigidification souhaité, retirez le milieu des échantillons et remplacez-le par un milieu à faible teneur en sérum complété par du LAP stérile de 2,2 mM.
REMARQUE : Pour marquer la structure par fluorescence, gonflez les constructions bioimprimées en 3D dans du PBS complété par 10 μM de méthacryloxyéthyl thiocarbamoyl rhodamine B (voir T able des matériaux) pendant la nuit, puis rigidifiez comme décrit àl’étape 7.6 pour marquer la structure par fluorescence. Transférer dans du PBS pendant 2 jours à 4 °C pour éliminer l’excès de rhodamine et imager à l’aide d’un filtre TRITC (Figure 4B,C). - Au moment souhaité du temps de raidissement, retirez la moitié du média des puits à rigidifier et placez la plaque avec le couvercle retiré sous la lumière UV. Allumez la lumière UV et rigidifiez ces constructions en appliquant une lumière de 10 mW/cm2 365 nm pendant 5 min à l’aide d’Omnicure (voir le tableau des matériaux) et d’un filtre passe-bande de 365 nm (Figure 3B).
REMARQUE : Utilisez le radiomètre/photomètre pour confirmer que l’intensité lumineuse est correcte avant d’exposer les cellules à la lumière UV. - Retirez le milieu restant de ces puits et ajoutez un nouveau milieu à faible teneur en sérum dans chaque puits. Remettez la plaque dans l’incubateur.
- Sortez la plaque de l’incubateur et effectuez l’étude d’activation des fibroblastes au moment souhaité en suivant l’étape 9.

Figure 4 : Les structures d’hydrogel bio-imprimées en 3D ont soutenu la viabilité cellulaire au fil du temps. (A) Photographie d’une structure d’hydrogel imprimée en 3D dans une plaque à 24 puits. (B) Projection d’intensité maximale d’un hydrogel imprimé en 3D PEGαMA marqué par fluorescence. Barre d’échelle = 1 mm. La microscopie à fort grossissement a montré des pores dans la structure de l’hydrogel induits par les microparticules de gélatine dans le bain de support de bio-impression FRESH. (C) Un tube PEGαMA imprimé en 3D avec des régions raidies marquées par fluorescence et imagé au microscope confocal (pile z de 100 μm affichée sous forme de projection d’intensité maximale) a montré un contrôle spatial du raidissement en 3D. Barre d’échelle = 500 μm. (D) Viabilité HPAAF dans les constructions bio-imprimées en 3D mesurée par des tests Live/Dead. Les constructions d’une épaisseur de 300 μm et d’une épaisseur de 4 × 106 cellules/ml ont surpassé toutes les autres conditions à chaque instant. La viabilité a atteint son apogée le jour 7. Cette condition et ce point temporel ont été sélectionnés pour de futures expériences. Les colonnes indiquent la ± moyenne MEB, n = 3. *, p < 0,05, ANOVA, Tukey HSD. (E) Images confocales représentatives de cellules dans des constructions 3D colorées avec un réactif vivant/mort au jour 7, le point de temps avec la plus grande viabilité globale. Les cellules vivantes marquées par la calcéine AM en vert et les cellules mortes marquées par l’iodure de propidium en rouge. La colonne la plus à droite montre que la condition la plus performante avait une distribution cellulaire uniforme et un pourcentage élevé de cellules vivantes. Barre d’échelle = 500 μm. Reproduit avec la permission de Davis-Hall et al.5. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
8. L’évaluation de la viabilité des fibroblastes
- Aux moments de viabilité souhaités, colorer à l’aide de calcéine AM et d’iodure de propidium (voir le tableau des matériaux).
REMARQUE : Pour résumer, les milieux doivent être retirés de chaque puits et les constructions doivent être rincées avec du PBS stérile. Incuber les constructions dans une solution de coloration vivante/morte pendant 40 min à 37°C sur une bascule. La solution de coloration doit contenir de la calcéine AM (dilution 1 :1000) et de l’iodure de propidium (dilution 1 :1000) pour identifier les cellules vivantes ou mortes lors de l’imagerie. - Transférez les constructions dans un PBS stérile et imagez-les immédiatement sur un microscope à fluorescence confocale. Acquérir trois images différentes de 100 μm par échantillon et par point temporel et exprimer la viabilité sous la forme du pourcentage moyen de cellules vivantes (Figure 4D,E).
9. L’évaluation de l’activation des fibroblastes
- Préparer de l’albumine sérique bovine (BSA) à 3 % p/v et de l’albumine sérique bovine à 0,1 % v/v Tween 20 dans du PBS. Cette solution sera appelée solution d’immunofluorescence (FI).
- Aux moments souhaités, retirez les milieux des puits d’échantillonnage et rincez les constructions avec du PBS. Remplacer le PBS par du paraformaldéhyde (PFA) à 4 % et conserver ces échantillons à 37 °C pendant 30 min sur une bascule. Ensuite, remplacez le PFA à 4 % par 100 mM de glycine dans du PBS et laissez ces échantillons sur une bascule à température ambiante (RT) pendant encore 15 minutes.
- Ensuite, transférez ces échantillons dans des cryomoules Tissue-Tek, recouvrez complètement l’échantillon d’une solution de température de coupe optimale (OCT) (voir le tableau des matériaux) et laissez l’OCT se diffuser dans les échantillons pendant 12 à 18 h à 4 °C.
- Congeler les échantillons imbibés d’OCT dans du 2-méthylbutane à l’aide d’azote liquide. Remplissez une boîte en polystyrène ou un autre récipient approprié avec de l’azote liquide, puis placez un deuxième récipient rempli de 2-méthylbutane dans l’azote liquide de manière à ce qu’il soit au moins à moitié immergé. À l’aide d’une pince, maintenez chaque cryomoule contenant un échantillon recouvert d’OCT dans du 2-méthylbutane refroidi à l’azote liquide jusqu’à ce qu’il soit visiblement congelé. Ces échantillons peuvent être conservés à -80 °C jusqu’à ce qu’ils soient prêts pour la cryosection.
ATTENTION : L’équipement de protection individuelle (EPI) tel que les gants de protection contre le froid, un tablier de protection contre le froid et un écran facial fourni dans la trousse de sécurité cryogénique (voir le tableau des matériaux) doit être utilisé lors de la manipulation de l’azote liquide. - Cryosectionner les échantillons OCT congelés à -22 °C et fixer des tranches de 10 μm d’épaisseur sur des lames de microscope en verre chargées positivement. Préparez trois lames de microscope avec au moins 3 à 5 cryocoupes par lame pour chaque échantillon d’hydrogel 3D.
REMARQUE : Les lames de microscope peuvent être stockées à ce stade à -80 °C si nécessaire comme point d’arrêt. - Fixez les cryosections dans de l’acétone glacée pendant 15 min pour aider les cryosections à adhérer aux lames. Rincez délicatement les cryosections avec de l’eau RT pour éliminer tout COT restant. Laissez sécher ces échantillons et tracez les contours des cryocoupes à l’aide d’un stylo hydrophobe (voir le tableau des matériaux).
- Perméabiliser les échantillons à température ambiante avec 0,2 % v/v Triton X-100 dans du PBS pendant 10 min, puis bloquer les coupes avec 5 % de BSA w/v dans du PBS pendant 1 h à RT.
- Ajouter l’anticorps primaire anti-actine alpha du muscle lisse humain (αSMA) de souris (dilution 1 :250) (voir le tableau des matériaux) à la solution FI. Conservez ces échantillons sectionnés avec l’anticorps primaire pendant la nuit à 4 °C. Rincez les échantillons 3 fois avec une solution IF.
- Incuber les coupes dans une solution IF contenant l’anticorps secondaire anti-souris Alexa Fluor 555 de chèvre (dilution 1 :250) et deux gouttes d’ActinGreen 488 ReadyProbe (voir tableau des matériaux) par millilitre de solution IF. Couvrir les échantillons pour toutes les étapes suivantes avec du papier d’aluminium pour les protéger de la lumière et laisser la solution d’anticorps secondaire rester sur les échantillons pendant 1 h à RT.
- Rincez les sections 3 fois avec une solution IF. Incubez-les dans 300 nM 4',6-diamidino-2-phylindole (DAPI) dans de l’eau DI pendant 15 min à RT. Effectuez un rinçage final des sections 3x avec de l’eau DI.
- À l’aide de 10 μL d’un réactif antidécoloration disponible dans le commerce (voir le tableau des matériaux), feuilletez les sections en utilisant des méthodes standard.
REMARQUE : Les lames montées peuvent être stockées à l’abri de la lumière dans un congélateur à -80 °C jusqu’à ce qu’elles soient nécessaires pour l’imagerie. - Imagez les cryocoupes à l’aide d’un microscope à fluorescence (Figure 3C et Figure 5B). Imaginez trois sections aléatoires par diapositive à l’aide d’un objectif 10x.
REMARQUE : Les images doivent être prises dans les canaux DAPI, FITC et TRITC. - Téléchargez les images dans ImageJ (NIH). Quantifier le pourcentage de cellules αSMA positives comme mesure de l’activation des fibroblastes (Figure 5A) en divisant le nombre total de cellules αSMA-positives par le nombre total de noyaux cellulaires pour chaque champ de vision.

Figure 5 : Activation des fibroblastes dans des modèles bio-imprimés en 3D de l’adventite artérielle pulmonaire. (A) Activation fibrotique dans des hydrogels 3D mous et rigidifiés mesurée par l’expression de l’αSMA. Les HPAAF dans les constructions rigides étaient significativement plus positifs pour l’αSMA que les cellules dans les constructions molles. Les colonnes représentent la moyenne ± SEM, n = 3. *, p < 0,05, test U de Mann-Whitney. (B) Images confocales représentatives de l’immunomarquage de l’αSMA, de l’actine et du DAPI dans des hydrogels 3D mous et rigidifiés. Les HPAAF dans les constructions rigides ont montré une immunofluorescence αSMA plus répandue que les cellules dans les constructions molles. Barre d’échelle = 250 μm. (C) Prolifération des fibroblastes dans les constructions bio-imprimées en 3D souples et rigides mesurées par la positivité EdU. Les HPAAF dans les constructions rigides étaient significativement plus positifs pour l’EdU que les cellules dans les constructions molles. Les colonnes représentent la moyenne ± SEM, n = 3. *, p < 0,05, test U de Mann-Whitney. (D) Images confocales représentatives de l’immunomarquage pour les colorants EdU et Hoechst dans des hydrogels 3D mous et rigidifiés. Les HPAAF dans les constructions rigides ont montré une immunofluorescence EdU plus répandue que les cellules dans les constructions molles. Barre d’échelle = 300 μm. Reproduit avec la permission de Davis-Hall et al.5. Veuillez cliquer ici pour voir une version agrandie de cette figure.
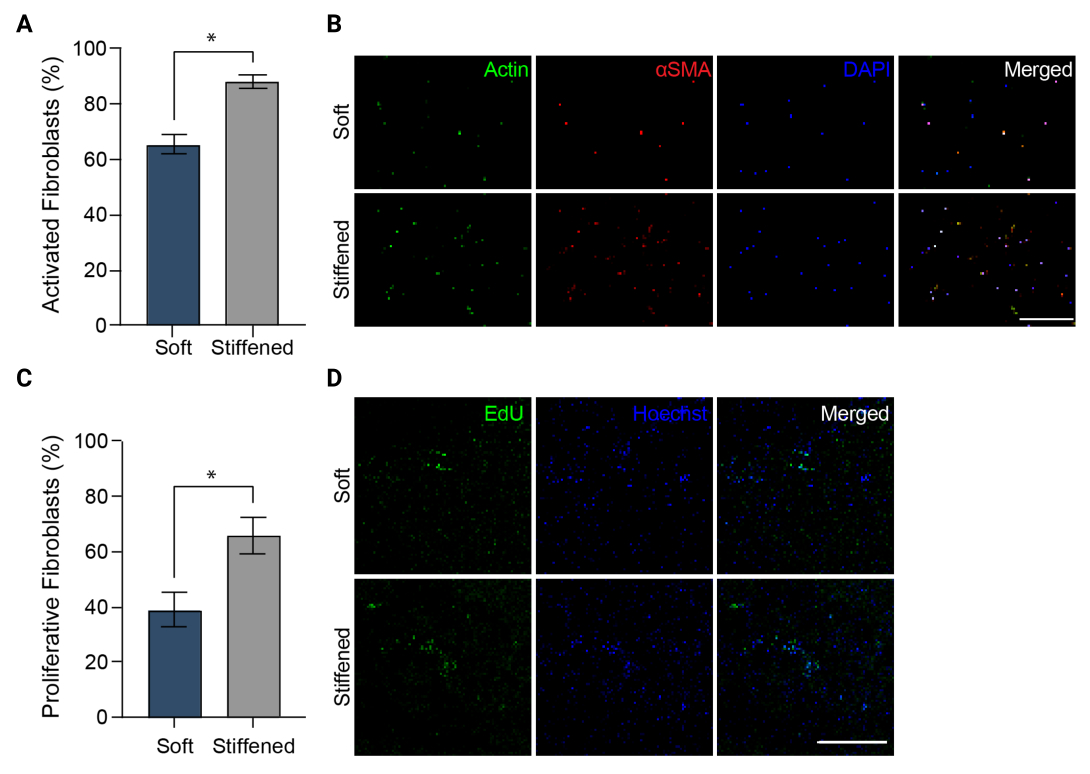{kind=link}
10. L’évaluation de la prolifération des fibroblastes
- Vingt-quatre heures avant le moment de prolifération souhaité, retirer le milieu de culture cellulaire de chaque puits et le remplacer par un milieu à faible teneur en sérum complété par une solution d’EdU à 10 μM provenant de la trousse de prolifération cellulaire disponible dans le commerce (voir le tableau des matériaux). Remettez les échantillons dans l’incubateur pour une incubation pendant la nuit.
- Au moment souhaité de prolifération, fixer les échantillons incubés avec de l’EdU en utilisant 4% de PFA à 37 °C pendant 30 min sur une bascule. Remplacer la solution de PFA à 4 % par 100 mM de glycine dans du PBS et incuber les échantillons à 37 °C pendant au moins 15 minutes. Ajoutez le Hoechst à une concentration appropriée pendant 30 minutes, puis rincez les constructions 2x avec du PBS.
REMARQUE : Les échantillons peuvent être conservés à l’abri de la lumière à 4 °C jusqu’à l’imagerie. - Imagez tous les échantillons d’EdU fixés et stockés à l’aide d’un microscope à fluorescence et des paramètres et filtres suggérés par le fabricant de la trousse de prolifération cellulaire (Figure 3C et Figure 5D). Acquérez trois images z-stack différentes de 100 μm par échantillon et créez des projections maximales à partir de chacune de ces z-stacks. Mesurer la prolifération de l’HPAAF en comptant le nombre de cellules EdU positives et en divisant par le nombre total de cellules identifiées par la contre-coloration de Hoechst dans les images de projection maximales (Figure 5C).
Résultats
Ce protocole décrit comment bio-imprimer en 3D des hydrogels photoaccordables dans un bain de support pour créer des constructions capables de raidissement dynamique et temporel pour étudier l’activation des fibroblastes dans des géométries qui imitent les tissus humains. Tout d’abord, le protocole expliquait comment synthétiser le PEGαMA, l’épine dorsale de ce système polymère photoaccordable. Les mesures de spectroscopie par résonance magnétique nucléaire (RMN) ont montré une fonctionnalisation réu...
Discussion
Les réactions de polymérisation en deux étapes en réponse à une exposition contrôlée à la lumière peuvent rigidifier les biomatériaux avec un contrôle spatial et temporel. Plusieurs études ont exploité cette technique pour évaluer les interactions cellule-matrice dans diverses plateformes 5,8,9,10,11,21,22,23.
Déclarations de divulgation
Les auteurs n’ont aucun conflit d’intérêts à divulguer. Des parties de ce manuscrit sont reproduites avec la permission d’IOP © Publishing https://doi.org/10.1088/1758-5090/aca8cf. 5 Tous droits réservés.
Remerciements
Les auteurs tiennent à remercier le Dr Adam Feinberg (Université Carnegie Mellon) et ceux qui ont organisé l’atelier 3D Bio-Printing Open-Source. Ces personnes ont permis d’apprendre les techniques de bio-impression FRESH et de construire la bio-imprimante 3D utilisée pour ces études. De plus, les auteurs tiennent à remercier Biorender.com, qui a été utilisé pour produire des figures dans ce manuscrit. Ce travail a été soutenu par de multiples groupes ou sources de financement, notamment la Rose Community Foundation (DDH et CMM), une bourse de recherche sur les maladies vasculaires pulmonaires du Colorado (DDH et CMM), la National Science Foundation sous le prix 1941401 (CMM), le ministère de l’Armée sous le prix W81XWH-20-1-0037 (CMM), le National Cancer Institute du NIH sous le prix R21 CA252172 (CMM), le Ludeman Family Center for Women’s Health Research du campus médical Anschutz de l’Université du Colorado (DDH et CMM), l’Institut national du cœur, des poumons et du sang des National Institutes of Health dans le cadre des prix R01 HL080396 (CMM), R01 HL153096 (CMM), F31 HL151122 (DDH) et T32 HL072738 (DDH et AT).
matériels
| Name | Company | Catalog Number | Comments |
| AccuMax Radiometer/Photometer Kit | Spectronics Corporation | XPR-3000 | To measure light intensity, used for photostiffening |
| Acetic Acid | Fisher Scientific | BP2401-500 | Used during PEGaMA synthesis |
| Acetone | Fisher Scientific | A184 | Used with the cryosections |
| ActinGreen 488 ReadyProbes | Fisher Scientific | R37110 | Used for staining |
| Aluminum Foil | Reynolds | F28028 | |
| Anhydrous Tetrahydrofuran (THF) | Sigma-Aldrich | 401757-1L | Used during PEGaMA synthesis |
| Argon Compressed Gas | Airgas | AR R300 | Used during PEGaMA synthesis |
| 8 Arm Poly(ethylene glycol)-hydroxyl (PEG-OH) | JenKem Technology | 8ARM-PEG-10K | Used during PEGaMA synthesis |
| 365 nm Bandpass Filter | Edmund Optics | 65-191 | Used for photostiffening |
| Bovine Serum Albumin (BSA) | Fisher Scientific | BP9700-100 | Used during staining process |
| Buchner Funnel | Quark Glass | QFN-8-14 | Used during PEGaMA synthesis |
| Calcein AM | Invitrogen | 65-0853-39 | Used during staining process |
| Celite 545 (Filtration Aid) | EMD Millipore | CX0574-1 | Used during PEGaMA synthesis |
| Charged Microscope Slides | Globe Scientific | 1358W | |
| Chloroform-d | Sigma-Aldrich | 151823-10X0.75ML | Used to characterize PEGaMA |
| Click-iT Plus EdU Cell Proliferation Kit | Invitrogen | C10637 | Used for staining |
| 50 mL Conical Tubes | CELLTREAT | 667050B | |
| Cryogenic Safety Kit | Cole-Parmer | EW-25000-85 | |
| Cryostat | Leica | CM 1850-3-1 | |
| Dialysis Tubing | Repligen | 132105 | |
| 4’,6-Diamidino-2-Phylindole (DAPI) | Sigma-Aldrich | D9542-1MG | Used for staining |
| Diethyl Ether | Fisher Scientific | E1384 | Used during PEGaMA synthesis |
| 1,4-Dithiothreitol (DTT) | Sigma-Aldrich | 10197777001 | Bioink component |
| Dulbecco's Modified Eagle's Medium (DMEM) | Cytiva | SH30271.FS | |
| Filter Paper | Whatman | 1001-090 | Used during PEGaMA synthesis |
| Freezone 2.5L Freeze Dry System | Labconco | LA-2.5LR | Lyophilizer |
| Fusion 360 | Autodesk | N/A | Software download |
| 2.5 mL Gastight Syringe | Hamilton | 81420 | Used for bioprinting |
| 15 Gauge 1.5" IT Series Tip | Jensen Global | JG15-1.5X | Used for bioprinting |
| 30 Gauge 0.5" HP Series Tip | Jensen Global | JG30-0.5HPX | Used for bioprinting |
| Goat Anti-Mouse Alexa Fluor 555 Antibody | Fisher Scientific | A21422 | Used for staining |
| Glycine | Fisher Scientific | C2H5NO2 | Used during staining process |
| Hemocytometer | Fisher Scientific | 1461 | |
| Hoechst | Thermo Scientific | 62249 | Used during staining process |
| Human Pulmonary Artery Adventitial Fibroblasts (HPAAFs) | AcceGen | ABC-TC3773 | From a 2-year-old male patient |
| Hydrochloric Acid (HCl) | Fisher Scientific | A144-500 | Used to pH adjust solutions |
| ImageJ | National Institutes of Health (NIH) | N/A | Free software download |
| ImmEdge® Pen | Vector Laboratories | H-4000 | Used during staining process |
| Incubator | VWR | VWR51014991 | |
| LifeSupport Gelatin Microparticle Slurry (Gelatin Slurry) | Advanced Biomatrix | 5244-10GM | Used for bioprinting |
| Light Microscope | Olympus | CKX53 | Inverted light microscope |
| Lithium Phenyl-2,4,6-Trimethylbenzoylphosphinate (LAP) | Sigma-Aldrich | 900889-5G | Photoinitiator used for photostiffening |
| Liquid Nitrogen | N/A | N/A | |
| LulzBot Mini 2 | LulzBot | N/A | Bioprinter adapted |
| Methacryloxyethyl Thiocarbamoyl Rhodamine B | Polysciences Inc. | 669775-30-8 | |
| 2-Methylbutane | Sigma-Aldrich | M32631-4L | |
| Microman Capillary Pistons CP1000 | VWR | 76178-166 | Positive displacement pipette tips |
| MMP2 Degradable Crosslinker (KCGGPQGIWGQGCK) | GL Biochem | N/A | Bioink component |
| Mouse Anti-Human αSMA Monoclonal Antibody | Fisher Scientific | MA5-11547 | Used for staining |
| OmniCure Series 2000 | Lumen Dynamics | S2000-XLA | UV light source used for photostiffening |
| Paraformaldehyde (PFA) | Electron Microscopy Sciences | 15710 | Used to fix samples |
| pH Meter | Mettler Toledo | FP20 | |
| pH Strips | Cytiva | 10362010 | |
| Phosphate Buffered Saline (PBS) | Hyclone Laboratories, Inc. | Cytiva SH30256.FS | |
| Pipette Set | Fisher Scientific | 14-388-100 | |
| 10 µL Pipette Tips | USA Scientific | 1120-3710 | |
| 20 µL Pipette Tips | USA Scientific | 1183-1510 | |
| 200 µL Pipette Tips | USA Scientific | 1111-0700 | |
| 1000 µL Pipette Tips | USA Scientific | 1111-2721 | |
| Poly(Ethylene Glycol)-Alpha Methacrylate (PEGαMA) | N/A | N/A | Refer to manuscript for synthesis steps |
| Poly(Ethylene Oxide) (PEO) | Sigma-Aldrich | 372773-250G | Bioink component |
| Positive Displacement Pipette | Fisher Scientific | FD10004G | 100-1000 µL |
| Potassium Hydroxide (KOH) | Sigma-Aldrich | 221473-500G | Used to pH adjust solutions |
| ProLong Gold Antifade Reagent | Invitrogen | P36930 | Used during staining process |
| Pronterface | All3DP | N/A | Software download |
| Propidium Iodide | Sigma-Aldrich | P4864-10ML | Used for staining |
| RGD Peptide (CGRGDS) | GL Biochem | N/A | Bioink component |
| Rocker | VWR | 10127-876 | |
| Rotary Evaporator | Thomas Scientific | 11100V2022 | Used during PEGaMA synthesis |
| Rubber Band | Staples | 808659 | |
| Schlenk Flask | Kemtech America | F902450 | Used during PEGaMA synthesis |
| Slic3r | Slic3r | N/A | Software download |
| Smooth Muscle Cell Growth Medium-2 (SmGM-2) BulletKit | Lonza | CC-3182 | Kit contains CC-3181 and CC-4149 components |
| Sodium Hydride | Sigma-Aldrich | 223441-50G | Used during PEGaMA synthesis |
| Sorvall ST 40R Centrifuge | Fisher Scientific | 75-004-525 | |
| Stir Bar | VWR | 58948-091 | |
| Syringe Filter | VWR | 28145-483 | Used to sterile filter solutions |
| T-75 Tissue-Cultured Treated Flask | VWR | 82050-856 | Used for cell culture work |
| Tissue-Tek Cyromold | Sakura | 4557 | |
| Tissue-Tek O.C.T Compound (OCT) | Sakura | 4583 | |
| Tris(2-Carboxyethyl) Phosphine (TCEP) | Sigma-Aldrich | C4706-2G | |
| Triton X-100 | Fisher Bioreagents | C34H622O11 | Used during staining process |
| Trypan Blue | Sigma-Aldrich | T8154-20ML | Used for cell culture work |
| 0.05% Trypsin-EDTA | Gibco | 25-300-062 | Used for cell culture work |
| Tween 20 | Fisher Bioreagents | C58H114O26 | Used during staining process |
| Upright Microscope | Olympus | BX63F | Fluorescent microscope capabilities |
| Water Bath | PolyScience | WBE20A11B | |
| 24-Well Tissue Culture Plates | Corning | 3527 |
Références
- Ahrens, J. H., et al. Programming cellular alignment in engineered cardiac tissue via bioprinting anisotropic organ building blocks. Advanced Materials. 34 (26), e2200217 (2022).
- Lin, N. Y. C., et al. Renal reabsorption in 3D vascularized proximal tubule models. Proceedings of the National Academy of Sciences of the United States of America. 116 (12), 5399-5404 (2019).
- Grigoryan, B., et al. Multivascular networks and functional intravascular topologies within biocompatible hydrogels. Science. 364 (6439), 458-464 (2019).
- Kang, Y., Datta, P., Shanmughapriya, S., Ozbolat, I. T. 3D bioprinting of tumor models for cancer research. ACS Applied Biomaterials. 3 (9), 5552-5573 (2020).
- Davis-Hall, D., Thomas, E., Pena, B., Magin, C. M. 3D-bioprinted, phototunable hydrogel models for studying adventitial fibroblast activation in pulmonary arterial hypertension. Biofabrication. 15 (1), (2022).
- Mirdamadi, E., Tashman, J. W., Shiwarski, D. J., Palchesko, R. N., Feinberg, A. W. FRESH 3D bioprinting of a full-size model of the human heart. ACS Biomaterials Science & Engineering. 6 (11), 6453-6459 (2020).
- Shiwarski, D. J., Hudson, A. R., Tashman, J. W., Feinberg, A. W. Emergence of FRESH 3D printing as a platform for advanced tissue biofabrication. APL Bioengineering. 5 (1), 010904 (2021).
- Petrou, C. L., et al. Clickable decellularized extracellular matrix as a new tool for building hybrid hydrogels to model chronic fibrotic diseases in vitro. Journal of Materials Chemistry B. 8 (31), 6814-6826 (2020).
- Hewawasam, R. S., Blomberg, R., Serbedzija, P., Magin, C. M. Chemical modification of human decellularized extracellular matrix for incorporation into phototunable hybrid hydrogel models of tissue fibrosis. ACS Applied Materials & Interfaces. 15 (12), 15071-15083 (2023).
- Saleh, K. S., et al. Engineering hybrid hydrogels comprised healthy or diseased decellularized extracellular matrix to study pulmonary fibrosis. Cellular and Molecular Bioengineering. 15 (5), 505-519 (2022).
- Guvendiren, M., Burdick, J. A. Stiffening hydrogels to probe short- and long-term cellular responses to dynamic mechanics. Nature Communications. 3, 792 (2012).
- Rosales, A. M., Vega, S. L., DelRio, F. W., Burdick, J. A., Anseth, K. S. Hydrogels with reversible mechanics to probe dynamic cell microenvironments. Angewandte Chemie International Edition English. 56 (40), 12132-12136 (2017).
- Wynn, T. A., Ramalingam, T. R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nature Medicine. 18 (7), 1028-1040 (2012).
- Huertas, A., Tu, L., Humbert, M., Guignabert, C. Chronic inflammation within the vascular wall in pulmonary arterial hypertension: more than a spectator. Cardiovascular Research. 116 (5), 885-893 (2020).
- Kendall, R. T., Feghali-Bostwick, C. A. Fibroblasts in fibrosis: novel roles and mediators. Frontiers in Pharmacology. 5, 123 (2014).
- Parker, M. W., et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. The Journal of Clinical Investigation. 124 (4), 1622-1635 (2014).
- Habiel, D. M., Hogaboam, C. Heterogeneity in fibroblast proliferation and survival in idiopathic pulmonary fibrosis. Frontiers in Pharmacology. 5, 2 (2014).
- Hu, C. J., Zhang, H., Laux, A., Pullamsetti, S. S., Stenmark, K. R. Mechanisms contributing to persistently activated cell phenotypes in pulmonary hypertension. The Journal of Physiology. 597 (4), 1103-1119 (2019).
- Li, M., et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. The Journal of Immunology. 187 (5), 2711-2722 (2011).
- Hinton, T. J., et al. Three-dimensional printing of complex biological structures by freeform-reversible embedding of suspended hydrogels. Science Advances. 1 (9), e1500758 (2015).
- Brown, T. E., et al. Secondary photocrosslinking of click hydrogels to probe myoblast mechanotransduction in three dimensions. Journal of the American Chemical Society. 140 (37), 11585-11588 (2018).
- Ondeck, M. G., et al. Dynamically stiffened matrix promotes malignant transformation of mammary epithelial cells via collective mechanical signaling. Proceedings of the National Academy of Sciences of the United States of America. 116 (9), 3502-3507 (2019).
- Caliari, S. R., et al. Stiffening hydrogels for investigating the dynamics of hepatic stellate cell mechanotransduction during myofibroblast activation. Scientific Reports. 6, 21387 (2016).
- Liu, F., et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. Journal of Cell Biology. 190 (4), 693-706 (2010).
- Tschumperlin, D. J., Ligresti, G., Hilscher, M. B., Shah, V. H. Mechanosensing and fibrosis. The Journal of Clinical Investigation. 128 (1), 74-84 (2018).
- Chelladurai, P., Seeger, W., Pullamsetti, S. S. Matrix metalloproteinases and their inhibitors in pulmonary hypertension. European Respiratory Journal. 40 (3), 766-782 (2012).
- Caracena, T., et al. Alveolar epithelial cells and microenvironmental stiffness synergistically drive fibroblast activation in three-dimensional hydrogel lung models. Biomaterials Science. 10 (24), 7133-7148 (2022).
- Ruskowitz, E. R., DeForest, C. A. Proteome-wide analysis of cellular response to ultraviolet light for biomaterial synthesis and modification. ACS Biomaterials Science & Engineering. 5 (5), 2111-2116 (2019).
- Kruse, C. R., et al. The effect of pH on cell viability, cell migration, cell proliferation, wound closure, and wound reepithelialization: In vitro and in vivo study. Wound Repair and Regeneration. 25 (2), 260-269 (2017).
- Filippi, M., et al. Perfusable biohybrid designs for bioprinted skeletal muscle tissue. Advanced Healthcare Materials. , e1500758 (2023).
- Matthiesen, I., et al. Astrocyte 3D culture and bioprinting using peptide-functionalized hyaluronan hydrogels. Science and Technology of Advanced Materials. 24 (1), 2165871 (2023).
- Xu, L., et al. Bioprinting a skin patch with dual-crosslinked gelatin (GelMA) and silk fibroin (SilMA): An approach to accelerating cutaneous wound healing. Materials Today Bio. 18, 100550 (2023).
- Bliley, J. M., Shiwarski, D. J., Feinberg, A. W. 3D-bioprinted human tissue and the path toward clinical translation. Science Translational Medicine. 14 (666), eabo7047 (2022).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.