Visualizzazione dei focolai di DNA a singolo filamento nella fase G1 del ciclo cellulare
In questo articolo
Riepilogo
Il seguente protocollo presenta la rilevazione di focolai di DNA a singolo filamento nella fase G1 del ciclo cellulare utilizzando la sincronizzazione del ciclo cellulare seguita dalla colorazione immunofluorescente RPA2.
Abstract
Il DNA ha vie di riparazione cellulare dedicate in grado di far fronte a lesioni che potrebbero derivare da fonti sia endogene che esogene. La riparazione del DNA richiede la collaborazione tra numerose proteine, responsabili della copertura di un'ampia gamma di compiti, dal riconoscimento e segnalazione della presenza di una lesione del DNA alla sua riparazione fisica. Durante questo processo, vengono spesso create tracce di DNA a singolo filamento (ssDNA), che alla fine vengono riempite da DNA polimerasi. La natura di queste tracce di ssDNA (sia in termini di lunghezza che di numero), insieme alla polimerasi reclutata per colmare queste lacune, sono specifiche del percorso di riparazione. La visualizzazione di queste tracce di ssDNA può aiutarci a comprendere le complicate dinamiche dei meccanismi di riparazione del DNA.
Questo protocollo fornisce un metodo dettagliato per la preparazione di cellule sincronizzate G1 per misurare la formazione di focolai di ssDNA in seguito a stress genotossico. Utilizzando un approccio di immunofluorescenza facile da utilizzare, visualizziamo l'ssDNA colorando per RPA2, un componente del complesso A della proteina di replicazione eterotrimerica (RPA). RPA2 si lega e stabilizza gli intermedi di ssDNA che si verificano in seguito a stress o replicazione genotossici per controllare la riparazione del DNA e l'attivazione del checkpoint del danno al DNA. La colorazione con 5-etinil-2'-desossiuridina (EdU) viene utilizzata per visualizzare la replicazione del DNA per escludere eventuali cellule della fase S. Questo protocollo fornisce un approccio alternativo ai saggi convenzionali non denaturanti a base di 5-bromo-2'-desossiuridina (BrdU) ed è più adatto per la rilevazione di focolai di ssDNA al di fuori della fase S.
Introduzione
Per sostenere la vita, le cellule esaminano e riparano costantemente il DNA per mantenere la loro integrità genomica. Le cellule possono accumulare vari tipi di danni al DNA a causa di fonti di stress del DNA sia endogene (ad esempio, ossidazione, alchilazione, deaminazione, errori di replicazione) che esogene (ad esempio, UV, radiazioni ionizzanti). La mancata riparazione di queste lesioni provoca apoptosi, arresto del ciclo cellulare o senescenza e può portare a malattie1. Le lesioni del DNA possono essere affrontate da una delle seguenti principali vie di riparazione del DNA: DR (direct reversal repair), che ripara principalmente le basi alchilate2; BER (riparazione per escissione di basi), che prende di mira gli errori di basi del DNA non ingombranti e le rotture del DNA a singolo filamento (SSB)3; NER (riparazione per escissione nucleotidica) che corregge le lesioni del DNA voluminose e distorcenti l'elica4; MMR (riparazione del mismatch) che prende di mira principalmente i mismatch del DNA, i loop di inserzione/delezione (IDL) e alcuni danni alla base5; NHEJ (giunzione delle estremità non omologhe) e HRR (riparazione della ricombinazione omologa) che sono entrambi attivi nelle rotture del DNA a doppio filamento (DSB)6; e TLS (sintesi translesionale), che è un meccanismo di bypass della lesione del DNA7. Sebbene questi percorsi abbiano specificità distinte del substrato, ci sono alcune sovrapposizioni tra di loro per garantire la ridondanza per una riparazione efficiente. Comprendere l'azione delle diverse vie di riparazione del DNA nelle varie fasi del ciclo cellulare è fondamentale in quanto questi fattori di riparazione del DNA potrebbero fungere da bersagli essenziali per gli approcci terapeutici per il trattamento del cancro, dell'invecchiamento e dei disturbi neurologici 8,9.
Il DNA a singolo filamento (ssDNA) viene generato durante tutto il ciclo cellulare a causa della riparazione delle lesioni del DNA generate da agenti dannosi per il DNA sia endogeni che esogeni. In seguito a stress genotossico, l'ssDNA viene generato abbondantemente nelle fasi S e G2 in cui HRR e MMR hanno la loro massima attività e quando il meccanismo di replicazione si blocca o collassa quando si incontrano lesioni del DNA 6,10,11. Anche altre vie di riparazione del DNA (ad esempio, NHEJ/giunzione delle estremità mediata dalla microomologia (MMEJ)/ricottura a singolo filamento [SSA]) generano ssDNA durante la riparazioneDSB 12. Queste tracce di ssDNA di solito derivano dalla resezione del DNA, effettuata da esonucleasi come EXO1, DNA2 e CtIP durante HR e MMR, endonucleasi come XPF e XPG durante NER, o attraverso l'azione combinata di POLB e FEN1 durante BER 4,13,14,15,16,17,18,19 . A causa del lavoro del meccanismo di replicazione, le tracce di ssDNA vengono generate anche quando le elicasi del DNA svolgono il DNA davanti alle polimerasi replicative legate al PCNA20. Al contrario, nella fase G1, la mancanza di HRR e di replicazione del DNA e l'attività limitata di MMR riducono l'estensione delle tracce di ssDNA generate e sono quindi più difficili da rilevare 10,11,21.
Le tracce cellulari di ssDNA sono strutture altamente sensibili che devono essere protette per evitare la formazione di DSB. Ciò si ottiene rivestendo le tracce di ssDNA con RPA. L'RPA è un abbondante complesso proteico eterotrimerico composto da più subunità (RPA1, RPA2 e RPA3, note anche come RPA70, RPA32 e RPA14, rispettivamente), che sono ubiquitariamente espresse durante tutto il ciclo cellulare22. Ogni subunità RPA contiene un dominio di legame al DNA (DBD), in grado di interagire con 4-6 nucleotidi, e le subunità combinate formano un nucleo di trimerizzazione stabile. Complessivamente, l'RPA si lega a circa 20-30 nucleotidi con affinità sub-nanomolare23,24.
I metodi convenzionali utilizzano la microscopia a immunofluorescenza (IF) per visualizzare i focolai di ssDNA marcando la 5-bromo-2'-deossiuridina (BrdU) incorporata nel DNA genomico utilizzando gli anticorpi BrdU25. Questo approccio si basa sul fatto che gli anticorpi BrdU possono rilevare BrdU solo nell'ssDNA25 esposto. Sebbene questo approccio sia semplice, presenta anche alcune limitazioni. Ad esempio, le cellule vengono pretrattate per incorporare BrdU prima dell'inizio dell'esperimento, il che richiede molto tempo e può interferire con gli effettori a valle. Pertanto, il rilevamento dell'ssDNA basato su BrdU è limitato alle cellule in replicazione e non può essere utilizzato per le cellule quiescenti. Ciò esclude l'applicazione di questo metodo per studiare la riparazione del DNA in cellule non replicanti, nonostante la sua importanza in diverse malattie come il cancro e la neurodegenerazione 5,26. Inoltre, poiché le strutture di BrdU e EdU sono molto simili, la maggior parte degli anticorpi BrdU mostra una crossreattività nei confronti di EdU, che deve essere presa in considerazione quando si mirano a esperimenti di doppia marcatura27. La colorazione RPA è stata precedentemente utilizzata per mostrare i focolai di ssDNA principalmente nelle cellule in fase S; tuttavia, alcuni articoli lo hanno utilizzato con successo anche al di fuori della fase S 28,29,30,31,32,33,34,35. Il seguente protocollo utilizza in modo efficiente le proprietà dell'RPA, consentendo la visualizzazione dei focolai di ssDNA a seguito di danni al DNA nella fase G1 del ciclo cellulare (sebbene possa essere utilizzato in tutte le fasi del ciclo cellulare).
Protocollo
1. Mantenimento delle cellule epiteliali pigmentate retiniche immortalizzate hTERT (RPE1)
- Mantenere linee cellulari RPE1 nel terreno di coltura modificato (DMEM) di Dulbecco integrato con il 10% di siero fetale bovino inattivato al calore (Hi-FBS) e 100 μg/mL di penicillina-streptomicina (d'ora in poi denominata terreno di coltura) in un incubatore umidificato con il 5% di CO2 a 37 °C. Per la coltura di routine, far crescere le cellule RPE1 in un piatto trattato con coltura tissutale da 15 cm e dividerle quando si raggiunge l'80-90% di confluenza (~16-18 × 106 cellule per piatto da 15 cm).
- Durante la scissione, rimuovere il terreno e sciacquare le cellule con 10 mL di soluzione salina tamponata con fosfato (PBS).
- Aggiungere 3 ml di tripsina-EDTA allo 0,05% fino a coprire l'intera superficie del piatto. Mantenere le cellule a 37 °C con la tripsina fino a quando non si staccano.
- Dopo la tripsinizzazione, risospendere le cellule con il terreno di coltura e farle girare a 150 × g per 5 minuti a temperatura ambiente (RT, 22-25 °C). Rimuovere il surnatante e risospendere delicatamente le cellule in 10 mL di terreno di coltura.
- Seminare 1,6-1,8 × 10-6 cellule in un nuovo piatto da 15 cm (~1 mL di sospensione cellulare).
NOTA: Tutte le colture tissutali devono essere eseguite secondo i livelli di sicurezza BSL-2. Il tempo di incubazione per la tripsinizzazione dipende dalla confluenza cellulare. Di solito, il processo richiede 2-3 minuti per essere completato per una piastra confluente al 90%. Le cellule devono essere sottoposte a screening per la contaminazione da micoplasmi su base regolare con kit disponibili in commercio (vedere esempi nella tabella dei materiali).
2. Knocking del gene di interesse da parte dei siRNA (GOI)
- Seminare 1,0 × 106 cellule RPE1 in una piastra di 10 cm trattata con coltura tissutale con 10 ml di terreno di coltura il giorno prima della trasfezione.
- Il giorno della trasfezione, complessare il siRNA. Per una piastra da 10 cm, utilizzare una concentrazione finale di 20 nM siRPA2 e 12 μL di reagente di trasfezione a base lipidica in 500 μL di terreno di trasfezione a basso contenuto di siero. Mescolare delicatamente tutti i componenti agitando la provetta e incubare a RT (22-25 °C) per 5 min.
- Aggiungere la miscela di siRNA complessata alle cellule goccia a goccia e incubare le cellule con il siRNA per 48 ore.
3. Sincronizzazione delle celle RPE1 in fase G0
- Tripsinizzare le cellule RPE1 dal passaggio 2.3 come descritto nel paragrafo 1 (~2 × 106 cellule).
- Trasferire la sospensione cellulare in provette da centrifuga da 15 mL e centrifugarla a 150 × g, RT (22-25 °C) per 5 min.
- Rimuovere il surnatante e risospendere le cellule in 12 mL di PBS. Centrifugare le cellule a 150 × g a RT (22-25 °C) per 5 min. Ripetere due volte la rimozione del surnatante e la centrifugazione.
- Risospendere le cellule in 10 mL di DMEM esente da siero integrato con 100 μg/mL di Penicillina-Streptomicina, 1 mM di piruvato di sodio, 15 mM di HEPES e piastrellarle su un piatto di coltura tissutale di 10 cm.
NOTA: Se le cellule tendono ad aggregarsi, risospenderle in solo 1 mL di DMEM senza siero e pipettarle su e giù 5 volte utilizzando un puntale P1000 per rimuovere i grumi prima di diluire la sospensione fino a un volume finale di 10 mL. - Dopo 24 ore di fame sierica, introdurre il secondo ciclo di silenziamento utilizzando la stessa procedura descritta nel paragrafo 2 aggiungendo il siRNA complessato alle cellule affamate di siero.
- Conservare le cellule RPE1 in DMEM privo di siero per 72 ore prima di procedere al rilascio di G1.
4. Rivestimento del vetrino coprioggetto e rilascio delle cellule in fase G1
- Sterilizzare le pinzette con etanolo al 70% e posizionare un singolo vetrino coprioggetti (12 mm di diametro e spessore #1,5 [0,17 mm]) in un pozzetto di una piastra da 24 pozzetti.
- Diluire la matrice di rivestimento di vitronectina con PBS per ottenere una concentrazione finale di 10 μg/mL. Aggiungere 500 μL della soluzione di vitronectina in ciascun pozzetto contenente i vetrini coprioggetti e incubare per 1 ora a RT.
- Rimuovere la soluzione di rivestimento e lavare i vetrini coprioggetti con 1 mL di PBS.
- Staccare le cellule RPE1 affamate di siero dalla piastra trattata con coltura tissutale di 10 cm utilizzando 1 mL di tripsina allo 0,05% dopo un lavaggio PBS per 1 minuto a 37 °C.
NOTA: Le cellule si staccano molto più velocemente dopo la fame di siero. Prestare attenzione quando si lavano le cellule con PBS e utilizzare tempi di tripsinizzazione brevi. - Per inattivare la tripsina, risospendere le cellule RPE1 in un totale di 6 mL di terreno di coltura. Rimuovere la tripsina inattivata facendo girare le cellule con 150 × g a RT (22-25 °C) per 5 minuti.
- Risospendere le cellule in 1 mL di terreno di coltura e misurare il numero di cellule.
- Seminare 4 × 104 cellule RPE1 sul vetrino coprioggetto rivestito per un totale di 500 μL di terreno di coltura.
NOTA: Assicurarsi che la vitalità cellulare sia superiore al 90% prima di procedere alle fasi a valle. La vitalità cellulare potrebbe essere valutata rapidamente mediante colorazione blu tripano durante la fase di conteggio delle cellule. - Dopo 6 ore di placcatura delle cellule nel terreno di coltura, le cellule rilasciate da G0 saranno nella fase iniziale di G1. Eseguire esperimenti in G1 in questa finestra di 6-12 ore prima che le cellule inizino ad entrare nella fase S.
- Prima di introdurre danni al DNA, pulsare le cellule con 10 μM di 5-etinil-2'-desossiuridina (EdU) per 30 minuti a 37 °C, diluito in terreno di coltura.
- Rimuovere il terreno contenente EdU e inseguire le cellule con 10 μM di timidina per 10 minuti a 37 °C per evitare l'incorporazione residua di EdU durante l'induzione del danno al DNA.
- Rimuovere il terreno con timidina e trattare le cellule con 250 μM H2O2 per 1 ora, diluite in terreno di coltura.
5. Colorazione in immunofluorescenza dell'ssDNA
- Lavare le cellule una volta con 1 mL di PBS RT (22-25 °C) per rimuovere il terreno e i componenti sierici.
NOTA: Essere delicati durante il lavaggio delle celle per evitare il distacco e l'asciugatura. Non elaborare più pozzi contemporaneamente. - Pre-estrazione: incubare le cellule lavate in 1 mL di tampone di estrazione CSK (Tabella 1) per 5 minuti a RT (22-25 °C).
NOTA: La preestrazione CSK rimuove tutte le proteine non legate alla cromatina, inclusa l'RPA2 solubile.
ATTENZIONE: Triton X-100 è dannoso se ingerito e può causare irritazioni cutanee e danni agli occhi. - Rimuovere il tampone CSK dalle cellule e fissarle direttamente aggiungendo 0,5 mL di soluzione di paraformaldeide al 3,6% (in PBS) contenente Triton X-100 allo 0,05% per 10 minuti a RT (22-25 °C).
ATTENZIONE: È importante preparare il 3.6% di PFA dal 32% di PFA appena stoccato. La paraformaldeide può causare gravi danni agli occhi, irritazione della pelle e irritazione delle vie respiratorie. - Lavare le cellule una volta con 1 mL di PBS contenente lo 0,05% di Triton X-100 per rimuovere il PFA.
- Permeabilizzare ulteriormente le cellule utilizzando 1 mL di PBS contenente lo 0,5% di Triton X-100 per 15 minuti a RT (22-25 °C).
- Reazione click-IT EdU per visualizzare le cellule in replicazione (fase S)
- Rimuovere la soluzione di permeabilizzazione e lavare le cellule 2 volte utilizzando 1 mL di tampone bloccante (Tabella 1).
ATTENZIONE: L'albumina sierica bovina (BSA) può causare irritazione delle vie respiratorie. - Aggiungere 1 mL di tampone bloccante (Tabella 1) e far oscillare delicatamente la piastra contenente vetrino coprioggetti per 10 minuti a RT (22-25 °C).
- Rimuovere il tampone bloccante e aggiungere 500 μL di cocktail di reazione a scatto contenente picolil azide 647 (Tabella 1). Incubare i vetrini coprioggetti per 30 minuti a RT (22-25 °C) con un leggero dondolio ed eseguire incubazioni a valle al buio.
NOTA: Quando si utilizzano gli anticorpi BrdU, utilizzare il doppio della quantità (1 mL) e del tempo (60 min) per la reazione a scatto come raccomandato dal produttore per garantire che la reazione sia satura e che l'EdU incorporato sia etichettato. Ciò limita la cross-reattività degli anticorpi BrdU27.
- Rimuovere la soluzione di permeabilizzazione e lavare le cellule 2 volte utilizzando 1 mL di tampone bloccante (Tabella 1).
- Rimuovere la miscela di reazione a scatto e lavare le celle 2 volte con PBS con Triton X-100 allo 0,05% per 10 minuti a RT (22-25 °C) (Figura 1 e Figura 2).
- Aggiungere 1 mL di tampone bloccante e incubare a RT (22-25 °C) per 30 min. In alternativa, mantenere le cellule in tampone bloccante a 4 °C per una notte.
- Applicare l'anticorpo primario (anti-RPA2 ratto, diluizione 1:1.000) per 2 ore a RT (22-25 °C) in 250-500 μL di tampone bloccante con leggera oscillazione.
- Lavare le cellule 2 volte con PBS contenente lo 0,05% di Triton X-100 per rimuovere rapidamente la maggior parte della soluzione anticorpale.
- Continuare a lavare le celle per 3 x 10 min con tampone bloccante a RT (22-25 °C).
- Applicare l'anticorpo secondario (anti-ratto Alexa-488, diluizione 1:1.000) in 250-500 μL di tampone bloccante a RT (22-25 °C) per 2 ore con un'oscillazione delicata.
- Lavare le cellule con tampone bloccante 2 volte per rimuovere rapidamente la maggior parte dell'anticorpo secondario. Continuare a lavare le celle per 3 x 10 minuti con PBS contenente lo 0,05% di Triton X-100 a RT (22-25 °C).
- Per contrastare i nuclei, lavare le cellule una volta con PBS contenente lo 0,05% di Triton X-100 e 1 μg/mL 4',6-diamidino-2-fenilindolo (DAPI) per 10 minuti a RT (22-25 °C). Lavare le celle una volta con PBS per 5 minuti a RT (22-25 °C).
- Montare il vetro di copertura sui vetrini del microscopio utilizzando 10 μL di mezzo di montaggio/vetrino coprioggetto. Immergere i vetrini coprioggetti in acqua distillata prima di montarli per eliminare eventuali cristalli di sale. Eseguire l'immagine dei vetrini il giorno successivo e conservarli a 4 °C per settimane (Figura 3).
6. Acquisizione e quantificazione delle immagini
- Per acquisire immagini, utilizzare qualsiasi microscopio a epifluorescenza disponibile dotato di set di filtri di routine per visualizzare i canali DAPI, FITC e Cy5 con un ingrandimento di almeno 60-63x, un'apertura numerica elevata e obiettivi a olio per visualizzare i fuochi nucleari.
NOTA: L'eccitazione DAPI ottimale è ~359 nm; L'eccitazione di Alexa 488 è ~488 nm; mentre l'eccitazione di Alexa 647 è di ~647 nm. - Per l'analisi delle immagini, aprire i file di immagine in Fiji/ImageJ.
- Realizzare maschere nucleari utilizzando la colorazione DAPI (Figura 4A-F e Video supplementare S1).
- Aprire l'immagine DAPI.
- Selezionare Processo | Migliora contrasto e imposta i pixel saturi su 0,35.
- Fare clic su Elabora | Binario | Converti in maschera. Scegli Binario | Riempi fori e fai clic su Analizza | Analizza le particelle. Impostare la dimensione su 10-Infinity.
- In Gestione ROI, fai clic su Mostra tutto.
- Individuazione dei focolai RPA2 nel nucleo (Figura 4G,H e video supplementare S1)
- Aprire l'immagine RPA2.
- Scegli Processo | Trova Maxima. Impostare la prominenza su un valore che evidenzi i fuochi RPA2 (tra 500 e 750), separandoli dallo sfondo.
- Infine, fai clic sul pulsante Misura in Gestione ROI.
- Calcola il numero totale di focolai di ssDNA nucleare dividendo il valore nella colonna RawinDen per 255 (il valore massimo dell'intensità dei pixel in ogni focola).
- Eseguire analisi statistiche utilizzando lo strumento software statistico preferito.
NOTA: Escludere dall'analisi tutte le cellule EdU-positive e le maschere DAPI segmentate in modo errato.
- Realizzare maschere nucleari utilizzando la colorazione DAPI (Figura 4A-F e Video supplementare S1).
Risultati Rappresentativi
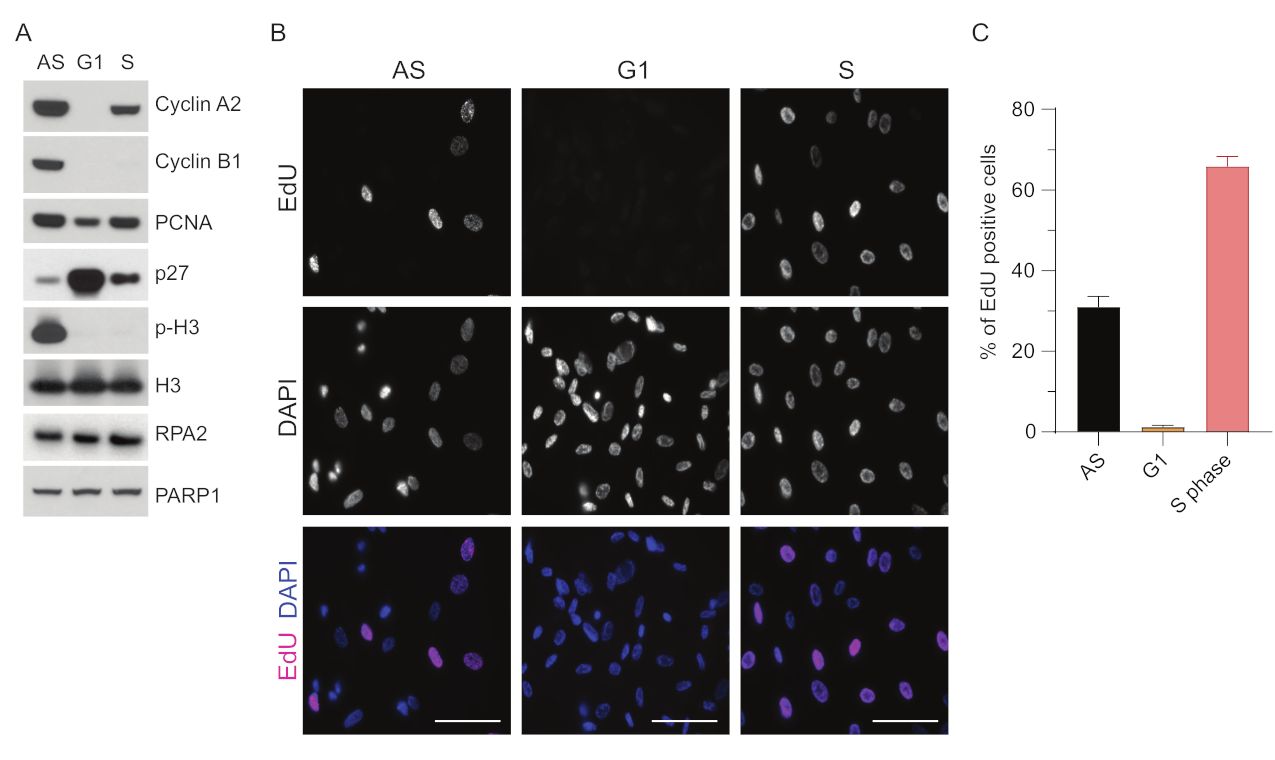
Per superare i limiti del rilevamento di ssDNA in G1, abbiamo utilizzato RPA2, che migliora sia la specificità che l'intensità del rilevamento dei focolai di ssDNA35. Per ottenere una sincronizzazione precisa delle cellule, abbiamo utilizzato cellule RPE1 che possono essere affamate di siero in modo efficiente e sincronizzate nella fase G0. Possono quindi essere indotti a rientrare nel ciclo cellulare mediante l'aggiunta di siero dopo la privazione del siero. Per confermare l'efficienza di sincronizzazione, abbiamo etichettato le cellule con EdU e il loro contenuto di DNA con ioduro di propidio. Abbiamo inoltre raccolto risultati qualitativi e quantitativi tramite citometria a flusso (Figura supplementare S1A). I dot-plot mostrano che dopo 72 ore di fame sierica, ~98% delle cellule sono in fase G0. Dopo l'aggiunta di terreni contenenti siero per 6 ore, le cellule rientrano nel ciclo cellulare (come si vede dall'aumento dei livelli di p27 nella Figura 1A), avendo ~ 97% di cellule in G1, mentre hanno solo il <1% di cellule in fase S e <2% di cellule in fase G2 (Figura supplementare S1A). Dopo 20-28 ore di aggiunta di siero alle cellule, queste passano gradualmente attraverso la fase S, come mostrato dai grafici della citometria a flusso (Figura supplementare S1A). Questo protocollo di sincronizzazione cellulare fornisce una popolazione G1 pura ~97% (6 ore dopo l'aggiunta di siero dopo 72 ore di fame sierica). Per convalidare ulteriormente l'efficienza della sincronizzazione, abbiamo confrontato l'espressione dei marcatori del ciclo cellulare dopo il rilascio di siero utilizzando il western blotting (Figura 1A e Figura supplementare S1B) e, in parallelo, abbiamo eseguito un test di incorporazione EdU per visualizzare la replicazione del DNA. La colorazione EdU evidenzia anche l'efficienza di sincronizzazione e la mancanza di replicazione del DNA in fase G1 (Figura 1B,C).
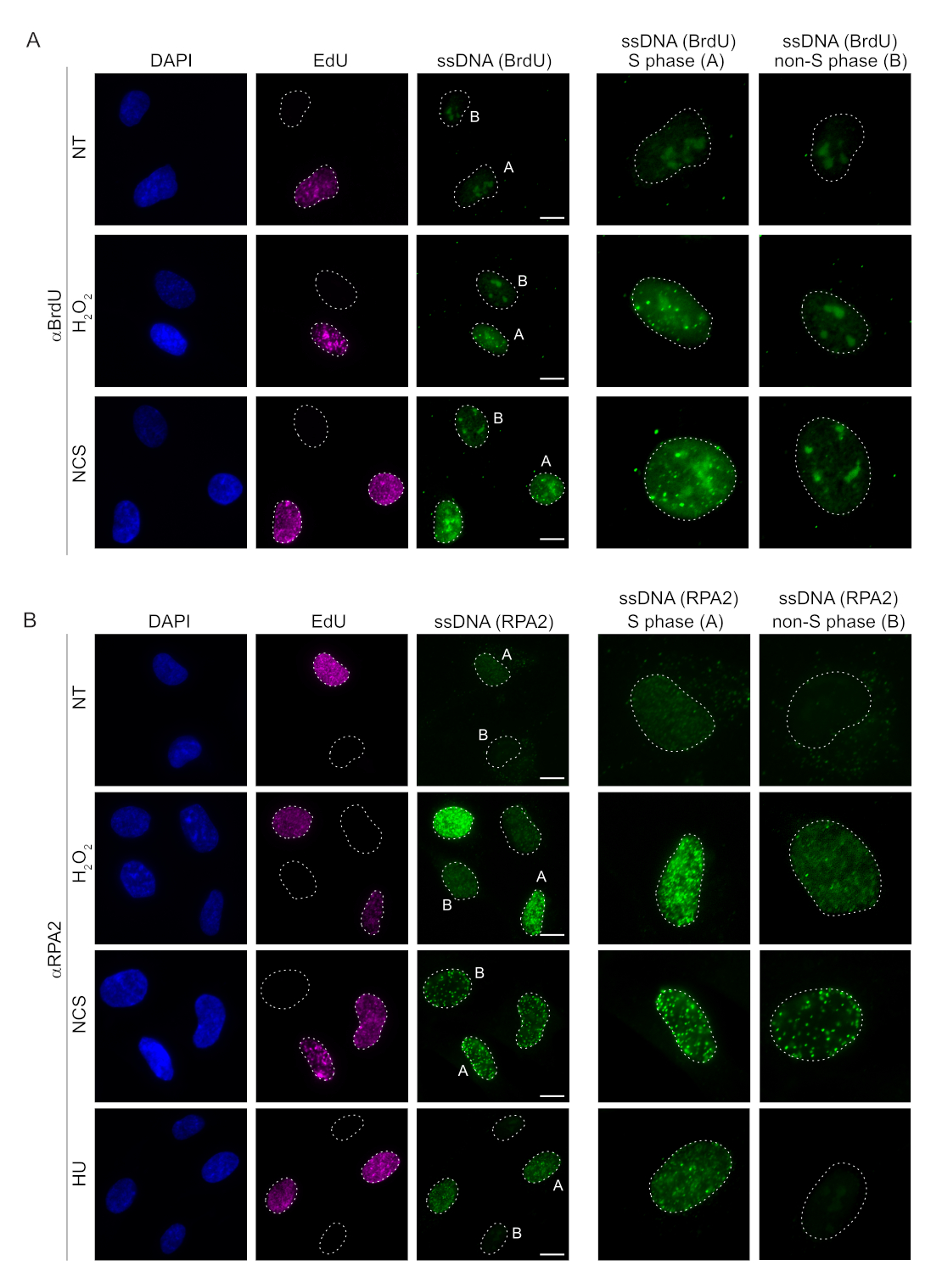
I metodi convenzionali per rilevare l'ssDNA nelle cellule di mammifero si basano sul rilevamento di BrdU nell'ssDNA. La Figura 2A mostra che dopo il trattamento con H2O2 e neocarzinostatina (NCS), i focolai di BrdU erano rilevabili solo nelle cellule in fase S, mentre nessun focolaio di ssDNA era rilevabile nelle cellule in fase non S. La colorazione degli anticorpi BrdU ha anche mostrato una notevole colorazione di fondo nucleolare che poteva essere rilevata in tutti i nuclei, indipendentemente dalla fase del ciclo cellulare o dai trattamenti applicati. Utilizzando il protocollo click EdU qui descritto, non siamo stati in grado di rilevare focolai co-localizzanti di EdU e BrdU, che è evidente nei campioni non trattati della Figura 2A. Per escludere completamente qualsiasi segnale BrdU derivante dalla cross-reattività, abbiamo evitato la marcatura EdU e abbiamo piuttosto utilizzato la ciclina A2 come marcatore S-G2. Tuttavia, la colorazione della ciclina A2 non ha permesso la pre-estrazione di CSK e, in questa condizione, non abbiamo visto alcun focolaio di BrdU, anche dopo stress genotossico (Figura supplementare S2A). Ciò evidenzia il fatto che la pre-estrazione di CSK è necessaria per la colorazione dell'ssDNA a base di anti-BrdU. Come controllo, abbiamo testato la colorazione degli anticorpi BrdU in condizioni di denaturazione. Questo apre il DNA per esporre il BrdU incorporato, che rivela che BrdU è stato incorporato uniformemente (Figura supplementare S2B).
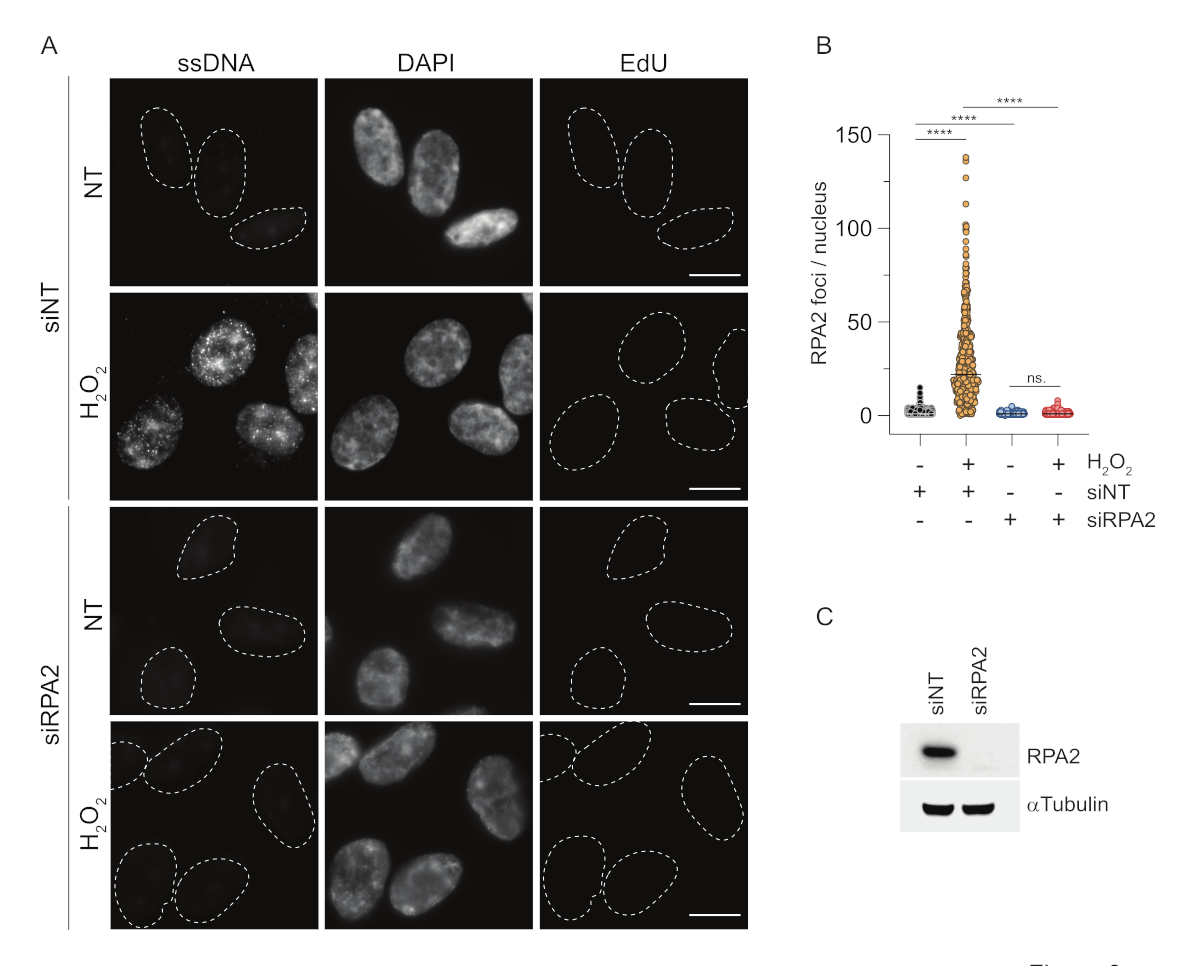
Al contrario, la colorazione di RPA2 mostra la formazione di focolai dipendenti da NCS e H2O2 non solo nella fase S ma anche in altre fasi del ciclo cellulare (Figura 2B). Come controllo, abbiamo anche trattato le cellule con HU, che provoca solo l'accumulo di ssDNA nelle cellule in fase di replicazione. Come previsto, abbiamo rilevato un aumento del segnale solo dopo il trattamento con HU con l'anticorpo RPA2 nelle cellule EdU-positive, evidenziando la specificità di questo approccio. L'anticorpo RPA2 è anche in grado di rilevare la formazione naturale di ssDNA durante la replicazione in assenza di stress genotossico esogeno (Figura 2B). La natura altamente sensibile dell'anticorpo RPA2 ci ha spinto a provare a utilizzarlo nella fase G1 in cui la colorazione convenzionale di BrdU non è riuscita a rilevare alcun segnale in seguito a stress genotossico (Figura supplementare S2C). La Figura 3A mostra che la formazione di focolai di ssDNA dopo il trattamento con H2O2 era rilevabile quando si utilizzava un anticorpo anti-RPA2, anche in G1. C'è stato un aumento significativo del numero di focolai di RPA2 in questi nuclei dopo il trattamento con H2O2 (Figura 3B). Questi focolai erano specifici per RPA2 poiché il silenziamento di RPA2 aboliva il segnale IF (Figura 3A,B). La Figura 3C e la Figura supplementare S1C mostrano l'efficienza del silenziamento RPA2 in queste celle. Rispetto ai metodi convenzionali, la rilevazione dell'ssDNA basata su RPA2 è altamente sensibile e la sua applicazione può quindi essere estesa alle cellule in fase G1.

Figura 1: Efficienza di sincronizzazione delle cellule RPE1 dopo la fame sierica. (A) Gli immunoblot mostrano i livelli proteici indicati nelle cellule RPE1 sincronizzate in fase Asincrona, G1 e S. (B) Le immagini rappresentative mostrano cellule RPE1 asincrone, sincronizzate in fase G1 e S che sono state esposte a 10 μM EdU per 30 minuti prima della fissazione e visualizzate mediante reazione Click-IT. Il DAPI è stato utilizzato per contrastare il DNA nucleare. Barre della scala = 50 μm. (C) Il grafico mostra la percentuale di cellule EdU-positive sulla popolazione cellulare totale valutata da DAPI. La barra di errore rappresenta l'errore standard della media, e i numeri di nuclei analizzati sono stati i seguenti: AS n = 219, G1 n = 630, S n = 437. Abbreviazioni: RPE1 = cellule epiteliali pigmentate retiniche immortalizzate hTERT; AS = asincrono; EdU = 5-etinil-2'-deossiuridina; DAPI = 4',6-diamidino-2-fenilindolo. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Rilevamento di ssDNA con anticorpo BrdU o anticorpo RPA2 in caso di danno al DNA. (A) Le immagini rappresentative illustrano i focolai di ssDNA utilizzando αBrdU (verde), le cellule della fase S sono evidenziate da EdU (viola) e DAPI è stato utilizzato per contrastare il DNA nucleare (blu). Le cellule RPE1 sono state mantenute in 10 μM BrdU per 48 ore prima di qualsiasi trattamento aggiuntivo. Dopo 48 ore, le cellule sono state pulsate con 10 μM EdU per 30 minuti, seguite da un trattamento di H2O2 (250 μM) per 1 ora o Neocarzinostatina (0,5 μg/mL) per 4 ore. Le cellule sono state fissate dopo la pre-estrazione di CSK. Una linea tratteggiata bianca indica il bordo di ciascun nucleo. Barra della scala = 5 μm. Nei pannelli a destra sono rappresentate immagini ingrandite dei nuclei della fase S o non S indicati. (B) Immagini rappresentative illustrano i focolai di ssDNA utilizzando l'anticorpo αRPA2 (verde). Le cellule della fase S sono evidenziate da EdU (viola) e DAPI è stato utilizzato per contrastare la colorazione del DNA nucleare (blu). Le cellule RPE1 sono state pulsate con 10 μM EdU per 30 minuti seguite da 1 ora H2O2 (250 μM), 4 ore di idrossiurea (2 mM) o 4 ore di NCS (0,5 μg/mL). Le cellule sono state fissate dopo la pre-estrazione di CSK. Una linea tratteggiata bianca indica il bordo di ciascun nucleo. Barra della scala = 10 μm. Nei pannelli a destra sono rappresentate immagini ingrandite dei nuclei della fase S o non S indicati. Abbreviazioni: ssDNA = DNA a singolo filamento; BrdU = 5-bromo-2'-desossiuridina; DAPI = 4',6-diamidino-2-fenilindolo; RPE1 = cellule epiteliali pigmentate retiniche immortalizzate hTERT; EdU = 5-etinil-2'-deossiuridina; NCS = Neocarzinostatina; HU = idrossiurea. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Rilevamento di focolai di ssDNA in fase G1 utilizzando l'anticorpo RPA2. (A) Le cellule RPE1 sono state trasfettate con siRNA mirati a RPA2 o a un controllo siRNA non mirato, e successivamente sincronizzate in G1 e marcate a impulsi con 10 μM EdU per 30 minuti prima di trattarle con H2O2 (250 μM) per 1 ora dove indicato. Il DAPI è stato utilizzato per contrastare il DNA nucleare. Le cellule sono state fissate dopo la pre-estrazione di CSK. Una linea tratteggiata bianca indica il bordo di ciascun nucleo. Barra di scala = 5 μm. (B) Le misurazioni per il numero di fuochi/nuclei RPA2 sono state effettuate da due esperimenti indipendenti. Durante l'analisi sono state prese in considerazione solo le cellule EdU-negative. Le linee rappresentano il valore medio sui grafici. Per l'analisi statistica è stato eseguito un test ANOVA non parametrico (Kruskal-Wallis). Le stelle indicano P < 0,0001. Il numero di nuclei analizzati è stato il seguente: siNT no H2O2 n = 513, siNT H2O2 n = 603, siRPA2 no H2O2 n = 266, siRPA2 H2O2 n = 536. (C) L'efficienza del knockdown del siRNA è dimostrata nell'immunoblotting. Abbreviazioni: siNT = controllo siRNA non mirato; BrdU = 5-bromo-2'-desossiuridina; DAPI = 4',6-diamidino-2-fenilindolo; RPE1 = cellule epiteliali pigmentate retiniche immortalizzate hTERT; EdU = 5-etinil-2'-deossiuridina. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Quantificazione dei focolai di ssDNA utilizzando le Figi. Passaggi dettagliati nelle Figi che mostrano come valutare i numeri dei focolai RPA2 nel nucleo. (A-E) La creazione di una maschera nucleare utilizzando il canale DAPI. (F-H) Soglia per identificare i singoli focolai di ssDNA nucleare dal segnale di fondo. Abbreviazioni: ssDNA = DNA a singolo filamento; DAPI = 4',6-diamidino-2-fenilindolo. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Tampone citoscheletrico (CSK) | |
| TUBI pH 7.0 | 10 milioni di metri |
| NaCl | 100 mM |
| EDTA pH 8 | 1 millimetro |
| MgCl2 | 3 milioni di minuti |
| D-saccarosio | 300 mM |
| Tritone X-100 | 0.20% |
| Cocktail di inibitori della fosfatasi | 1 compressa per 10 mL |
| Cocktail di inibitori della proteasi | 1 compressa per 10 mL |
| diluito in ddH2O | n/a |
| Tampone di lavaggio | |
| Tritone X-100 | 0.05% |
| diluito in PBS | n/a |
| Tampone di permeabilizzazione | |
| Tritone X-100 | 0.50% |
| diluito in PBS | n/a |
| Soluzione di fissaggio | |
| Paraformaldeide | 3.60% |
| Tritone X-100 | 0.05% |
| diluito in PBS | n/a |
| Buffer di blocco | |
| Albumina sierica bovina (BSA) | 5% |
| Tritone X-100 | 0.10% |
| diluito in PBS | n/a |
| Cocktail di reazione Click-iT Plus | |
| 1x tampone di reazione Click-iT | 435 ml |
| Soluzione Alexa Fluor PCA | 5 mL |
| Premiscelato protettivo CuSO 4-rame | 10 ml |
| 1x additivo tampone Click-iT | 50 ml |
| Volume totale | 500 ml |
Tabella 1: Composizione dei tamponi utilizzati in questo protocollo.
Figura supplementare S1. (A) Le cellule RPE1 sono state sincronizzate con la fase G0 utilizzando la fame di siero per 72 ore e successivamente rilasciate in diverse fasi del ciclo cellulare reintroducendo il siero. I dot plot mostrano le cellule nelle fasi G0/G1, S o G2/M, dove le ore indicano il tempo dopo la re-aggiunta del siero dopo la fame di siero. Il grafico a destra mostra la percentuale di celle G0/G1, S e G2/M in ciascuna condizione. L'analisi FACS è stata effettuata utilizzando un kit di proliferazione cellulare disponibile in commercio utilizzando EdU e ioduro di propidio secondo le raccomandazioni del produttore. (B) Scansioni western blot non ritagliate per la Figura 1. I numeri mostrano i marcatori di peso molecolare in kDa. PARP1 è stato utilizzato come controllo di carico e caricato sul gel che è stato sviluppato anche contro CCNA2, p27 (ulteriormente spogliato per PCNA) e pH3 (S10) (ulteriormente spogliato per H3) tagliando la membrana. CCNB1 e RPA2 sono stati caricati su un gel separato, utilizzando la stessa quantità di lisato proteico per garantire la comparabilità. (C) Scansioni western blot non ritagliate per la Figura 3. I numeri mostrano i marcatori di peso molecolare in kDa. Abbreviazione: EdU = 5-etinil-2'-deossiuridina. Fare clic qui per scaricare il file.
Figura supplementare S2: (A) Immagini rappresentative illustrano i focolai di ssDNA utilizzando l'anticorpo BrdU (verde); Le celle della fase S sono evidenziate dalla ciclina A2 (rosso); e DAPI è stato usato per contrastare il DNA nucleare (blu). Le cellule RPE1 sono state mantenute in 10 μM BrdU per 48 ore prima di un ulteriore trattamento. Dopo 48 ore, le cellule sono state trattate con H2O2 (250 μM) per 1 ora o Neocarzinostatina (0,5 μg/mL) per 4 ore prima della fissazione. Una linea tratteggiata bianca indica il bordo di ciascun nucleo. Barra di scala = 5 μm. (B) Colorazione BrdU delle cellule RPE1 con e senza condizione di denaturazione. Le cellule asincrone RPE1 sono state pretrattate con 10 μM di BrdU per 48 ore. Barra della scala = 10 μm. (C) Le misurazioni per il numero di fuci/nuclei BrdU sono state effettuate da due esperimenti indipendenti in cellule RPE1 sincronizzate G1. Durante l'analisi sono state prese in considerazione solo le cellule EdU-negative. Le linee rappresentano il valore medio sui grafici. Per l'analisi statistica è stato eseguito un test ANOVA non parametrico (Kruskal-Wallis). La 'ns' indica una differenza non significativa. Il numero di nuclei analizzati è stato il seguente: NT n = 52, NCS n = 105, H2O2 n = 82. Abbreviazioni: siNT = controllo siRNA non mirato; BrdU = 5-bromo-2'-desossiuridina; DAPI = 4',6-diamidino-2-fenilindolo; RPE1 = cellule epiteliali pigmentate retiniche immortalizzate hTERT; NCS = Neocarzinostatina. Fare clic qui per scaricare il file.
Video supplementare S1: Registrazione dello schermo dell'analisi dei focolai RPA2 basata sulle Figi. Fare clic qui per scaricare il file.
Discussione
Il mantenimento di una coltura cellulare sana e priva di micoplasmi è fondamentale per tutti gli esperimenti sopra descritti. Le cellule RPE1 hanno un forte attaccamento alla plastica trattata con colture tissutali in normali terreni di coltura; tuttavia, le loro caratteristiche di legame diminuiscono significativamente se mantenute in condizioni prive di siero. Inoltre, per catturare immagini ad alta risoluzione dei focolai di ssDNA al microscopio, le cellule devono essere placcate su un vetro di copertura di 0,17 mm di spessore, che non è abbastanza idrofilo per supportare il corretto attacco delle cellule RPE1. Senza cellule adeguatamente appiattite e distribuite uniformemente, è molto difficile visualizzare i singoli focolai di ssDNA. Pertanto, è fondamentale scegliere il materiale di rivestimento appropriato (ad esempio, vitronectina) e lasciare un tempo adeguato (6-12 ore) affinché le cellule si diffondano e si attacchino dopo averle rilasciate in fase G1.
Una parte impegnativa del protocollo consiste nell'ottenere cellule RPE1 sincronizzate G1 omogenee. Ciò richiede due passaggi critici. Innanzitutto, per un'efficiente carenza di siero, le cellule devono essere tripsinizzate, lavate accuratamente con PBS e seminate direttamente su nuove piastre di coltura tissutale utilizzando terreni privi di siero. Il lavaggio delle cellule direttamente nelle piastre di coltura tissutale per rimuovere il siero non produrrà un'efficiente sincronizzazione G0. In secondo luogo, quando si rilasciano le cellule in fase G1, le cellule devono essere nuovamente tripsinizzate e seminate su piastre di coltura tissutale fresche. Allo stesso modo, la semplice modifica del terreno e l'aggiunta di terreno di coltura contenente siero alle cellule non comporterà un ingresso sincrono di G1. Inoltre, per un corretto ingresso di G1, la densità di semina delle cellule sui vetri di copertura rivestiti deve essere a determinati livelli di confluenza. Mentre la perfetta sincronizzazione cellulare è generalmente irraggiungibile, questo protocollo di sincronizzazione qui descritto fornisce una popolazione G1 pura ~97%. La densità di semina consigliata per RPE1 su un vetrino coprioggetto di 12 mm di diametro è ~4 × 104 per acquisire un campo visivo omogeneo per l'imaging, con una confluenza di circa il 70%. Una maggiore densità di semina provoca il distacco e il "peel-off" delle cellule dopo l'estrazione di CSK e si tradurrà in un segnale di fondo più elevato durante l'acquisizione dell'immagine.
Per ridurre qualsiasi segnale di fondo e ottenere un rapporto segnale/rumore favorevole, è essenziale un lavaggio accurato dopo l'incubazione degli anticorpi primari e secondari. Poiché devono essere applicate numerose fasi di lavaggio, è anche essenziale evitare che il pozzetto si secchi durante ogni fase di lavaggio. Riduciamo al minimo questo artefatto applicando un minimo dello 0,05% di Triton X-100 in tutte le fasi di lavaggio e incubazione. Una volta che i pozzi si sono asciugati, le celle hanno mostrato un rapporto segnale/rumore alterato; Questo porta a un modello simile a un mosaico al microscopio e potrebbe interferire con la valutazione. L'acquisizione di immagini Z-stack combinata con la deconvoluzione può aiutare a catturare i fuochi in diversi piani focali per migliorare l'analisi.
I metodi convenzionali si basano sul rilevamento di BrdU incorporato in condizioni non denaturanti. Questi metodi, tuttavia, dipendono dal pretrattamento delle cellule con alti dosaggi di BrdU per almeno 1-2 giorni (o tempo equivalente a un ciclo cellulare completo nella linea cellulare utilizzata) per garantire un'incorporazione genomica uniforme. Indesiderabilmente, un'ampia incorporazione di BrdU può causare interferenze nel ciclo cellulare36. Per affrontare queste limitazioni, questo metodo utilizza RPA2 endogeno per rilevare i focolai di ssDNA. Questo approccio non richiede l'incorporazione di BrdU guidata dalla replicazione, ma può essere utilizzato anche nelle cellule post-mitotiche. Poiché non è necessaria un'ampia incorporazione di BrdU, ciò consente di risparmiare tempo e riduce la complessità sperimentale. Utilizzando la colorazione RPA2 per visualizzare l'ssDNA, possiamo utilizzare la 2′-deossi-5-etiniluridina (EdU) e la click-chemistry per marcare la replicazione del DNA evitando la possibile crossreattività degli anticorpi BrdU contro EdU 27,37,38. Particolare attenzione deve essere prestata per mascherare adeguatamente l'EdU incorporata durante la reazione di clic in modo che gli anticorpi BrdU non reagiscano in modo incrociato con l'EdU27,39.
Infine, un importante vantaggio dell'utilizzo di RPA2 al posto di BrdU è semplicemente quello di avere un rapporto segnale/rumore superiore rispetto alla colorazione di BrdU al di fuori della fase S. Abbiamo scoperto che la colorazione BrdU non denaturante e la sua capacità di visualizzare l'ssDNA sono limitate alla fase S anche nelle cellule in replicazione (Figura 2). L'anticorpo BrdU si lega solo al BrdU sufficientemente esposto negli allungamenti di ssDNA. Il legame delle proteine di riparazione, tra cui RPA2, ai tratti di ssDNA può sopprimere o ostacolare l'esposizione sufficiente di BrdU nell'ssDNA. Abbiamo anche scoperto che la pre-estrazione di CSK è necessaria per la visualizzazione dell'ssDNA utilizzando l'anticorpo BrdU. Ciò è possibile perché le tracce di ssDNA non sono accessibili per l'anticorpo senza rimuovere da esse i componenti proteici leggermente legati.
Tuttavia, ci sono alcune limitazioni associate a questo protocollo. Un limite dell'utilizzo di RPA2 per il rilevamento di ssDNA è la necessità di ottimizzare la fase di pre-estrazione CSK. L'RPA2 in eccesso non legato deve essere lavato via dal DNA prima di fissare le cellule. Da un lato, la sottoestrazione porta a un elevato background a causa della frazione proteica RPA2 che non è legata all'ssDNA. D'altra parte, l'estrazione eccessiva porterà alla perdita del segnale. Per il rilevamento di BrdU, questa non è una variabile poiché BrdU è stabilmente incorporato nel DNA e non può essere lavato via dalla pre-estrazione. Pertanto, è necessario considerare attentamente il tempo di pre-estrazione CSK, la quantità di Triton X-100 nel tampone, il volume e la temperatura alla quale viene eseguita la pre-estrazione. La preestrazione CSK limita anche l'uso delle dimensioni del nucleo per discriminare le cellule G0/G1 dalle cellule S/G2.
Inoltre, non possiamo escludere la possibilità che parte del segnale che proviene da RPA2 provenga dal suo legame con altri interattori proteici leganti la cromatina. Bisogna anche considerare la specificità di specie dell'anticorpo RPA2. L'anticorpo utilizzato in questo protocollo è in grado di riconoscere l'RPA2 umano, il topo, il ratto, il criceto e la scimmia. Un'altra limitazione di questo approccio è che non tutte le linee cellulari possono essere affamate di siero per la sincronizzazione G0. La maggior parte delle linee cellulari tumorali può bypassare i checkpoint del ciclo cellulare e proliferare anche in terreni privi di siero. Sebbene la fame di siero sia benefica, poiché non provoca danni al DNA, è necessario monitorare attentamente la loro efficienza di sincronizzazione cellulare per assicurarsi che venga raggiunto un corretto arricchimento della fase del ciclo cellulare. Per le cellule che non rispondono alla deprivazione sierica, devono essere presi in considerazione altri metodi di sincronizzazione cellulare (ad esempio, shake off mitotico, inibizione di CDK1 per l'arresto di G2 o tecniche non invasive come l'elutriazione centrifuga). Un altro metodo possibile è l'utilizzo dell'imaging ad alto contenuto per misurare il contenuto di EdU e DNA nucleare per la profilazione del ciclo cellulare delle cellule asincrone31. È necessario considerare le implicazioni dell'utilizzo di metodi di sincronizzazione alternativi per evitare interferenze con l'analisi a valle. Ad esempio, l'uso del doppio blocco di timidina o dell'afidicolina, spesso utilizzata in letteratura, provocherà stress da replicazione e danno al DNA40.
Lo studio dei meccanismi di riparazione del DNA continua ad essere un punto focale di discussione nel campo del cancro e della biologia cellulare. Il protocollo qui presentato offre un approccio prezioso per la preparazione delle cellule, consentendo la visualizzazione e l'analisi quantitativa dell'ssDNA in seguito all'esposizione ad agenti dannosi per il DNA. In particolare, questo protocollo evidenzia l'utilizzo della proteina legante l'ssDNA, RPA2, dimostrando la sua elevata specificità per visualizzare piccole quantità di focolai di ssDNA evitando reattività crociate indesiderate in tutte le fasi del ciclo cellulare. L'utilizzo di RPA2 conferisce numerosi vantaggi, in particolare per i ricercatori che mirano ad analizzare le cellule nella fase G1 del ciclo cellulare. Questo protocollo prende in considerazione diverse limitazioni e affronta le preoccupazioni relative all'interferenza del segnale, al rumore di fondo indesiderato e alla reattività incrociata quando si utilizza la colorazione RPA2 o BrdU per rilevare l'ssDNA.
Divulgazioni
Gli autori non hanno interessi contrastanti da dichiarare.
Riconoscimenti
Gli autori ringraziano Michele Pagano per il suo supporto e le sue utili intuizioni, Ashley Chui e Sharon Kaisari per la lettura critica del manoscritto, e Jeffrey Estrada e Vilma Diaz per il loro continuo supporto. Questo lavoro è stato sostenuto da un supplemento sulla diversità al National Institutes of Health Grant GM136250.
Materiali
| Name | Company | Catalog Number | Comments |
| Alpha-tubulin antibody | Sigma-Aldrich | T6074 | primary antibody (1:5,000) |
| Axio Observer Inverted Microscope | Zeiss | na | microscope |
| Bis-Tris Plus Mini Protein Gels, 4-12% | Invitrogen | NW04127BOX | Western Blot |
| Bovine Serum Albumin | Jackson ImmunoResearch | 001-000-162 | blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Sigma-Aldrich | B5002-100MG | nucleotide analogue |
| BrdU antibody BU1/75 | Abcam | ab6326 | primary antibody (1:500) |
| CellAdhere Dilution Buffer | Stemcell Technologies | 07183 | coating reagent |
| Click-iT Plus EdU Flow Cytometry Assay Kits | Invitrogen | C10632 | flow cytomery |
| Click-iT Plus EdU Cell Proliferation Kit for Imaging, Alexa Fluor 647 dye | Thermo Fisher Scientific | C10640 | click-reaction kit |
| cOmplete ULTRA Protease inhibitor tablets | Sigma-Aldrich | 5892791001 | reagent |
| Countess 3 Automated cell counter | Thermo Scientific | AMQAX2000 | cell counter |
| Coverslip | neuVitro | GG12PRE | tissue culture |
| Cyclin A2 antibody | Santa Cruz Biotechnology | sc-271682 | primary antibody (1:1,000) for IF and WB |
| Cyclin B1 antibody | Santa Cruz Biotechnology | sc-245 | primary antibody (1:5,000) |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D2650-100ML | vehicle control |
| DMEM, high glucose, with HEPES | Gibco | 12430051 | cell culture medium for RPE cells |
| DPBS, no calcium, no magnesium | Gibco | 14190144 | the PBS used throughout the protocol |
| D-Sucrose | Thermo Fisher Scientific | bp220-1 | reagent |
| Eclipse Ti2 Series Epifluorescent Microscope | Nikon | na | microscope |
| EdU (5-Ethynyl-2'-deoxyuridine) | Thermo Fisher Scientific | C10637 | nucleotide analog |
| Falcon 24-well plate | Corning | 351147 | tissue culture |
| Falcon Cell Culture Dishes 100 mm | Corning | 353003 | tissue culture |
| Fetal Bovine Serum, heat inactivated | Gibco | 16140071 | media supplement |
| Fiji (ImageJ) | NIH | version 1.54f | software and algorithms |
| FxCycle PI/RNase Staining Solution | Invitrogen | F10797 | PI staining |
| Goat anti-mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 555 | Thermo Fisher Scientific | A21422 | secondary antibody (1:1,000) |
| Goat anti-rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A48262 | secondary antibody (1:1,000) |
| Histone H3 antibody | Abcam | ab1791 | primary antibody (1:10,000) |
| hTERT RPE1 | ATCC | CRL-3216 | cell line |
| Hydrochloric acid | Sigma-Aldrich | H1758-100ML | reagent |
| Hydrogen peroxide 30% soultion | Sigma-Aldrich | H1009-100ML | reagent |
| Hydroxyurea,98% powder | Sigma-Aldrich | H8627-5G | reagent |
| Invitrogen Ultra Pure 0.5 M EDTA pH 8.0 | Thermo Fisher Scientific | 15-575-020 | reagent |
| Lipfectamine RNAiMAX Transfection Reagent | Invitrogen | 13778150 | transfection reagent |
| Magnesium chloride solution 1 M | Sigma-Aldrich | M1028-100ML | reagent |
| MycoFluor | Thermo Fisher | M7006 | Mycoplasma Detection Kit |
| Neocarzinostatin from Streptomyces carzinostaticus | Sigma-Aldrich | N9162-100UG | reagent |
| NuPage MES SDS Running Buffer (20x) | Invitrogen | NP0002 | Western Blot |
| onTARGETplus Human RPA2 siRNA | Dharmacon | L-017058-01-0005 | siRNA |
| p27 antibody | BD Biosciences | 610241 | primary antibody (1:1,000) |
| Paraformaldehyde aqueous solution (32%) | Electron Microscopy Sciences | 50-980-494 | fixative |
| PARP1 antibody | Cell Signaling Technology | 9542S | primary antibody (1:1,000) |
| PCNA antibody | Cell Signaling Technology | 13110S | primary antibody (1:2,000) |
| Penicillin-Streptomycin | Gibco | 15140163 | media supplement |
| pH3 antibody | Cell Signaling Technology | 3377S | primary antibody (1:2,000) |
| PhosSTOP phosphatase inhibitor tablets | Sigma-Aldrich | 4906837001 | reagent |
| PIPES Buffer 0.5 M solution, pH 7.0 | Bioworld | 41620034-1 | reagent |
| Precision Plus Protein Kaleidoscope Prestained Protein Standards | Bio-Rad | 1610395 | Western Blot |
| Prism | GraphPad | version 10 | statistical analysis and graph |
| ProLong Diamond Antifade Mountant | Thermo Scientific | P36961 | mounting media |
| Reduced serum media (Opti-MEM) | Gibco | 31985070 | used for transfection |
| Rpa32/rpa2 antibody (mouse) | EMD Millipore | NA19L | primary antibody (1:1,000) for WB |
| Rpa32/rpa2 antibody (rat) | Cell Signaling Technology | 2208S | primary antibody (1:1,000) for IF |
| Sodium Chloride solution (5 M) | Sigma-Aldrich | S5150 | reagent |
| Sodium Pyruvate (100 mM) | Gibco | 11360070 | media supplement |
| Sodium tetraborate decahydrate | Sigma-Aldrich | B3535-500G | reagent |
| Thermo Scientific Pierce DAPI Nuclear Counterstain | Thermo Scientific | 62248 | nucleic acid stain |
| Thymidine,powder | Sigma-Aldrich | T1985-1G | reagent |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | detergent |
| Trypsin-EDTA (0.5%), no phenol red | Gibco | 1540054 | cell dissociation agent |
| Vitronectin XF | Stemcell Technologies | 07180 | coating reagent |
| ZE5 Cell Analyzer | Bio-Rad | na | flow cytomery |
Riferimenti
- Hakem, R. DNA-damage repair; the good, the bad, and ugly. EMBO J. 27 (4), 589-605 (2008).
- Gutierrez, R., O'Connor, T. R. DNA direct reversal repair and alkylating agent drug resistance. Cancer Drug Resist. 4 (2), 414-423 (2021).
- Krokan, H. E., Bjoras, M. Base excision repair. Cold Spring Harb Perspect Biol. 5 (4), a012583 (2013).
- Marteijn, J. A., Lans, H., Vermeulen, W., Hoeijmakers, J. H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 15 (7), 465-481 (2014).
- Li, G. M. Mechanisms and functions of DNA mismatch repair. Cell Res. 18 (1), 85-98 (2008).
- Hustedt, N., Durocher, D. The control of DNA repair by the cell cycle. Nat Cell Biol. 19 (1), 1-9 (2016).
- Yang, W., Gao, Y. Translesion and repair DNA polymerases: diverse structure and mechanism. Annu Rev Biochem. 87, 239-261 (2018).
- Bhat, D. S., et al. Therapeutic disruption of RAD52-ssDNA complexation via novel drug-like inhibitors. NAR Cancer. 5 (2), zcad018 (2023).
- Gupta, P., Saha, B., Chattopadhyay, S., Patro, B. S. Pharmacological targeting of differential DNA repair, radio-sensitizes WRN-deficient cancer cells in vitro and in vivo. Biochem Pharmacol. 186, 114450 (2021).
- Pena-Diaz, J., et al. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol Cell. 47 (5), 669-680 (2012).
- Schroering, A. G., Edelbrock, M. A., Richards, T. J., Williams, K. J. The cell cycle and DNA mismatch repair. Exp Cell Res. 313 (2), 292-304 (2007).
- Scully, R., Panday, A., Elango, R., Willis, N. A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 20 (11), 698-714 (2019).
- Escribano-Diaz, C., et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 49 (5), 872-883 (2013).
- Genschel, J., Modrich, P. Mechanism of 5'-directed excision in human mismatch repair. Mol Cell. 12 (5), 1077-1086 (2003).
- Hu, J., et al. Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J Biol Chem. 288 (29), 20918-20926 (2013).
- Huertas, P., Jackson, S. P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 284 (14), 9558-9565 (2009).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in DNA replication with putative roles in cancer. Int J Mol Sci. 20 (1), 74 (2018).
- Symington, L. S. End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb Perspect Biol. 6 (8), a016436 (2014).
- Liu, Y., et al. DNA polymerase beta and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J Biol Chem. 280 (5), 3665-3674 (2005).
- Wold, M. S., Kelly, T. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc Natl Acad Sci U S A. 85 (8), 2523-2527 (1988).
- Wienholz, F., Vermeulen, W., Marteijn, J. A. Amplification of unscheduled DNA synthesis signal enables fluorescence-based single cell quantification of transcription-coupled nucleotide excision repair. Nucleic Acids Res. 45 (9), e68 (2017).
- Wold, M. S. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 66, 61-92 (1997).
- Chen, R., Wold, M. S. Replication protein A: single-stranded DNA's first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays. 36 (12), 1156-1161 (2014).
- Kang, Y., et al. Alteration of replication protein A binding mode on single-stranded DNA by NSMF potentiates RPA phosphorylation by ATR kinase. Nucleic Acids Res. 51 (15), 7936-7950 (2023).
- Kilgas, S., Kiltie, A. E., Ramadan, K. Immunofluorescence microscopy-based detection of ssDNA foci by BrdU in mammalian cells. STAR Protoc. 2 (4), 100978 (2021).
- Madabhushi, R., Pan, L., Tsai, L. H. DNA damage and its links to neurodegeneration. Neuron. 83 (2), 266-282 (2014).
- Liboska, R., Ligasova, A., Strunin, D., Rosenberg, I., Koberna, K. Most anti-BrdU antibodies react with 2'-deoxy-5-ethynyluridine -- the method for the effective suppression of this cross-reactivity. PLoS One. 7 (12), e51679 (2012).
- Biehs, R., et al. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol Cell. 65 (4), 671-684.e5 (2017).
- Cruz-Garcia, A., Lopez-Saavedra, A., Huertas, P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 9 (2), 451-459 (2014).
- Ercilla, A., et al. Physiological tolerance to ssDNA enables strand uncoupling during DNA replication. Cell Rep. 30 (7), 2416-2429.e7 (2020).
- Lezaja, A., et al. RPA shields inherited DNA lesions for post-mitotic DNA synthesis. Nat Commun. 12 (1), 3827 (2021).
- Mukherjee, B., Tomimatsu, N., Burma, S. Immunofluorescence-based methods to monitor DNA end resection. Methods Mol Biol. 1292, 67-75 (2015).
- Ochs, F., et al. 53BP1 fosters fidelity of homology-directed DNA repair. Nat Struct Mol Biol. 23 (8), 714-721 (2016).
- Raderschall, E., Golub, E. I., Haaf, T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc Natl Acad Sci U S A. 96 (5), 1921-1926 (1999).
- Forment, J. V., Walker, R. V., Jackson, S. P. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytometry A. 81 (10), 922-928 (2012).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Sci Rep. 6, 19567 (2016).
- Aten, J. A., Bakker, P. J., Stap, J., Boschman, G. A., Veenhof, C. H. DNA double labelling with IdUrd and CldUrd for spatial and temporal analysis of cell proliferation and DNA replication. Histochem J. 24 (5), 251-259 (1992).
- Podgorny, O., Peunova, N., Park, J. H., Enikolopov, G. Triple S-phase labeling of dividing stem cells. Stem Cell Reports. 10 (2), 615-626 (2018).
- Cappella, P., Gasparri, F., Pulici, M., Moll, J. Cell proliferation method: click chemistry based on BrdU coupling for multiplex antibody staining. Curr Protoc Cytom. Chapter 7, (2008).
- Ligasova, A., Koberna, K. Strengths and weaknesses of cell synchronization protocols based on inhibition of DNA synthesis. Int J Mol Sci. 22 (19), 10759 (2021).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati