このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
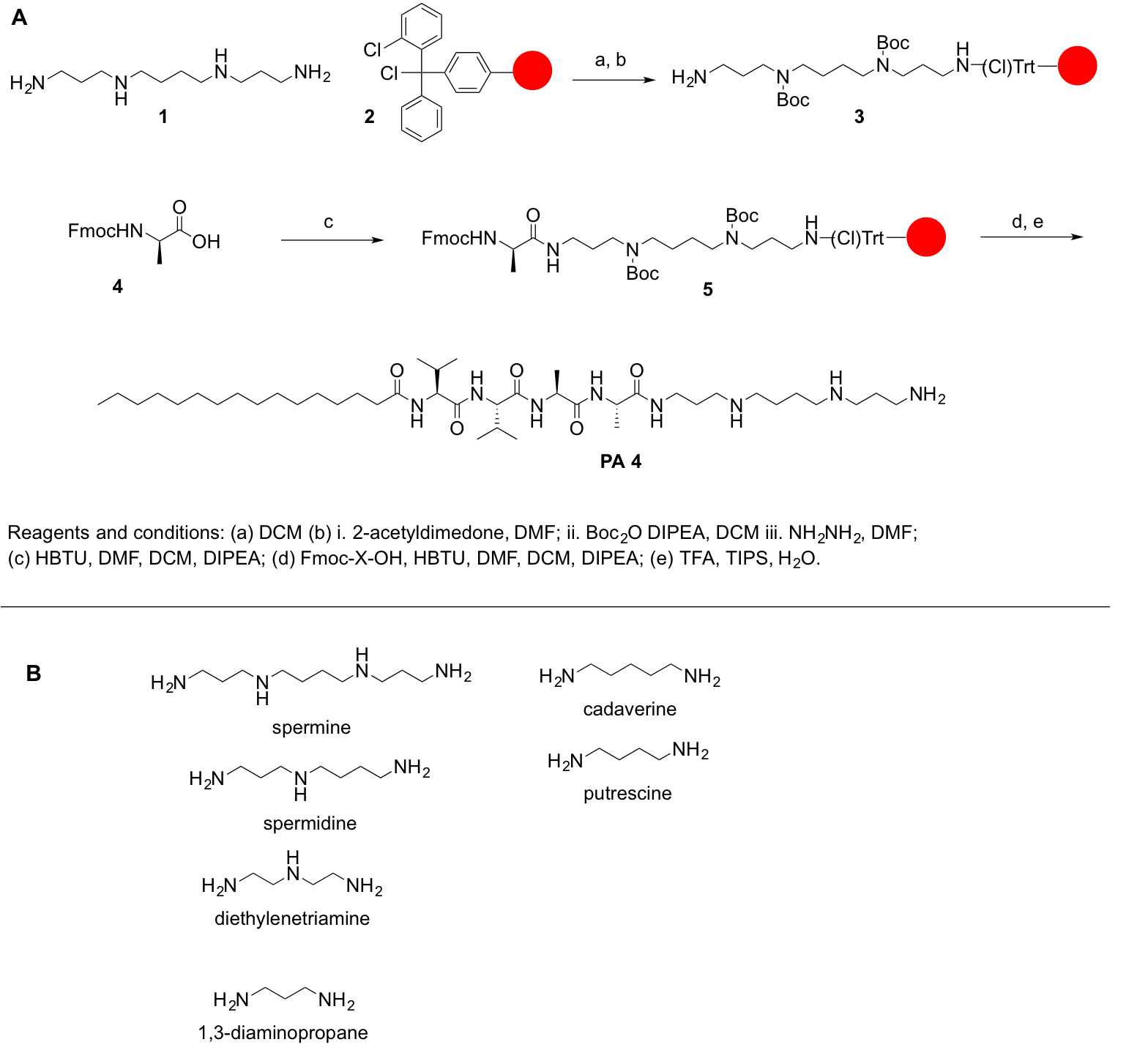
自己組織化ペプチドのポリアミンによる両親媒性物質 (Ppa の) と関連生体材料の合成のための安易なプロトコル
要約
両親媒性ペプチドのポリアミン ベース (Ppa の) の合成は、これらの事後対応型の機能をマスクするグループを保護するための賢明な使用を必要とする複数のアミン窒素の存在のために重要な課題です。本稿では自己組織化分子のこれらの新しいクラスの準備のための安易な方法をについて説明します。
要約
ポリアミンによるペプチド両親媒性 (Ppa の) は、自己組織化両親媒性ペプチド両親媒性物質 (PAs) に生体材料関連の新しいクラスです。伝統的な PAs では、荷電アミノ酸 (リジン、アルギニン)、脂質セグメントに直接接続されている、または中性のアミノ酸のリンカー領域を含めることができますグループを可として所有しています。PAs のペプチッド シーケンスのチューニングは多様な形態をもたらすことができます。同様に、Ppa は疎水性セグメントと中性アミノ酸を有するがまた水 (親水性) グループを可としてポリアミンの分子が含まれています。PAs の場合と同様に、Ppa を小さな棒、ツイスト ナノリボンと融合ナノ-シート、水に溶けるなどを含む、多様な形態に自己組み立てるもできます。ただし、単一のポリアミンの分子にプライマリとセカンダリの両方のアミンの存在は、Ppa を合成する場合、重要な課題を生じます。本稿では Ppa の固相ペプチド合成 (SPP) を使用しての簡易合成を達成するために、文献の判例に基づく単純なプロトコルを紹介します。このプロトコルは、PAs と他の類似システムの合成を拡張できます。また樹脂や同定、精製から胸の谷間に必要な手順を示します。
概要
自己組織化ペプチド両親媒性物質 (PAs) は、通常次のセグメントから成る生体材料のクラス: (a) 親水性頭部、(b) リンカーの地域、および (c) 疎水性尾。文献で説明されているほとんどの PAs では、あるいは極性アミノ酸残基1,2,3,4から成る親水性頭部を所有しています。PAs は、ドラッグデリバリー、病気診断、再生医療など5など生物医学アプリケーションの広い範囲を発見しました。そのアミノ酸配列に基づく、PAs は、ナノ球状ミセル ナノ フィラメントなどのさまざまなを形成できます。我々 は最近のハイブリッド ポリアミンによるペプチド媒、Ppa の6と呼ばれるクラスを報告しています。その混ぜ合わせたヘッド グループに関連する形態、自己組織化の動力学、これら生体の代謝の分解が見つかりました。また、PPA ナノ構造はテストされた濃度で哺乳類細胞 (MiaPaCa2、HeLa 細胞線) に向けて毒性を示さなかった。PPA ベース ナノキャリアは、魅力的なドラッグデリバリー車: (1) ポリアミン吸収及び代謝は癌細胞で増加する示されている、(2) カチオン性ナノ構造体はエンドソーム脱出7、8を達成することが高い循環と、セルと PA; と比較して異なる代謝プロファイルが必要 (3) 内にあるレジデンスにつながるたとえば、彼らは人間の体は、プロテアーゼの方安定する (ただし彼らのアミン酸化酵素など、他の酵素に多分敏感)9,10。また、Ppa は、多様な形態と物理化学的特性、ナノ粒子剛性、および長さと個々 の PPA 分子6の料金に応じてアセンブリ動態に発見されています。ここで、合成、識別、そして PAs 又は同様のハイブリッド ペプチド分子の調製にも適用できる Ppa の浄化のための詳しいプロトコルについて述べる。
ポリアミンが市販の保護されたフォームで一般的でないため、記載されているアミノ酸と他の分子、それらの活用の大切はポリアミンの第一次および二次アミンを保護するため、合成ステップの保護を達成するために。このプロトコルの全体的な目標は、アミノ酸にポリアミンの活用の簡単な方法を提供します。ポリアミンがないカルボン酸官能基;したがって、スケート リンク アミドまたは王樹脂に結合されることはできません。代わりに、2 chlorotrityl 塩化物などの樹脂は合成プロトコルに適しています。PPA 合成の主な課題は、第一次および二次アミン官能基の存在です。我々 の目的はカップリング反応を許可する無料のポリアミンをプライマリのアミノ グループを維持しながら、ポリアミンの第二級アミンを保護されています。反応は、各結合と脱保護手順の後仕事を容易にするために固相ペプチド合成 (SPP) の原則に従ってしっかりサポートで行われました。次のプロトコルは、Ppa の手動および自動合成 (ただし、いくつかの手順の検証は自動化されたシステムに挑戦すること) です。自動化されたシンセサイザーまたはマイクロ ウェーブ合成装置の助けを借りて、これらの分子の合成することができますアウト実施も (自動または半自動)。反応機構は、図 1に要約されています。

図 1:(A) A の一般的な反応スキーム Ppa の合成。ために使用することができます (B) の代表的なポリアミン合成 Ppa のここで説明します。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}
プロトコル
1 Ppa の合成一般的なプロトコル
- 合成 (通常モル) のスケールを計算します。このスケールは、ターゲットの PPA 量の質量に基づいています。SPP の反応効率はアミノ酸の増加とともに徐々 に減少するに留意してください。したがって、正確な反応効率は計算することは困難です。
- 樹脂の荷重によると使用される樹脂の重量を計算します。ロードはコンテナーまたは樹脂の分析のプロトコルとモル/g で表されます。樹脂の重量を計算する次の数式を使用できます。
- 2 chlorotrityl 塩化樹脂を慎重に比較検討 (我々 の場合、読み込みは ~0.85 モル)
- 以下の仕様のガラスフリット合成容器に樹脂を配置: 容量: 50 mL、フリット ディスク-25 mm、気孔率-媒体。
- 樹脂にジクロロ メタン (DCM) の 15 ミリリットルを追加します。
- 可変的な速度の機械的シェーカー (フラスコ シェーカー/攪拌機) に合成容器を貼るし、撹拌を最大化する (または水平方向に) 45 ° の角度に船を回します。
- うねりを樹脂ビーズを許可する 15 分の腫れを高める反応収量分子拡散とアクティブなサイトへのアクセスを容易にするため結合効率の向上します。
- 樹脂 (スペルミン、スペルミジン、Diethyelenetriamine、1, 3-ジアミノ プロパン、等) の目的のポリアミンの 8 同等 (0.25 モル スケールの 2 モル) を追加し、5 h の反応することができます。
注: 低い反応期間は、収量を減らすでしょう。 - カイザー テスト (材料表参照) 樹脂にポリアミンの成功した結合を確認します。第一級アミンの成功した結合は、遊離アミンに対応するバイオレット/ブルー色を生成します。
- 反応混合物一晩11を揺することによって無水メタノール (15 mL) に溶解 1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl チェンジ (Dde) の 4 の同等を使用して第一次アミン グループを保護します。
注: 短い反応時間収量が減少します。 - カイザーのテストを実行します。樹脂ビーズから青い色の不在はの巧妙な保護を確認します。第一次アミンの失敗した保護の場合に、前の手順を繰り返します。
- ドレイン、DCM、DCM と DMF (2:1、15 mL) の混合物で二回樹脂を洗ってください。
- ディ tert ディ-炭酸ブチル (Boc) DCM (20 mL) での 20 の同等 (5 モル) を溶解し、3 時間続行する反応させます。
- クロラニルを行う二次アミンの完全な保護を確認する (材料の表を参照) をテストします。肯定的なテスト (保護) は、無色または薄い黄色の色を生成します。無料第二級アミンは第一級アミンを与える赤い色、緑/青の暗い色を与えます。
- 溶剤の混合物を排出し、DCM と DMF (2:1、15 mL) の混合物で二回洗います。
- ヒドラジンの 2% 溶液を加えて DMF (10 mL)、1 h に振る。
- カイザーは第一次アミンの成功した脱保護を確認するテストします。
-
目的のアミノ酸をあなたが書き込み/描画それらの逆の順序で追加します。ペプチド C テルミニに N テルミニ駅から描かれていますが、逆に、N と C の合成たとえば、次のペプチドを必要とする PPA のコア - GLFD-、続くフェニルアラニン (F)、ロイシン (L)、および最終的にグリシン (G) アスパラギン酸 (D) を追加。
注: ほとんどのアミノ酸次のカップリング カクテルは読み込まれるポリアミン樹脂への愛着のために適切です。- Fmoc 保護保護アミノ酸の 4 相当 (1 モル)、2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) の 3.95 同等物、diisopropylethyl アミン (DIPEA) の 15 の同等物をミックスします。
- DCM と DMF (1:1、15 mL) の混合物でそれらを溶解し、(完全な解散) まで 2 分のカクテルを超音波します。
- アミノ酸のカルボン酸の活性化を確保するための追加の 3-5 分を待ちます。
- 容器に反応混合物を追加します。室温で 2-4 h の反応を行います。
注: 我々 は発見した HBTU 効率的な代替として次 (通常アミノ酸とカップリング剤の量を減らすが必要): DIC/Oxima, HATU PyBOP ゅ。 - カイザーは成功した結合を確認するテストします。
- DMF の 4-メチル ピペリジン (10 mL) の 20% 溶液を追加することによってアミノ酸から fmoc 保護グループを保護解除。それを行う 15 分間反応混合物を振るたびに、二度。
注: fmoc 保護の除去のための効率的な代替は、ピペラジン/DBU12です。- 追加の間に DCM (15 mL) 洗浄を実行します。
- カイザーはアミノ酸の解消の巧妙な保護を確認するテストします。
- 4-メチルのすべてのトレースを削除する徹底的に樹脂を洗浄します。DMF (10 mL)、各洗浄 5 分の持続で 2 回樹脂を洗って、最後に dcm (10 mL) 10 分。
- カップリングを連続して繰り返すことによって残りのアミノ酸はすべてを追加し、解き放つ保護手順を実行します。
-
最後に、すべての必須アミノ酸を結合後ポリアミン ペプチド構造の最後のアミノ酸疎水性尾を共役します。
- DCM と DMF の混合物 (1:1、20 mL) に溶解し HBTU の 9.5 同等物、DIPEA 12 相当必要なカルボン酸の機能を 10 の同等物を追加します。
注: いくつかの尾は、n-メチルピロリドン (NMP) など他の溶剤や添加溶媒 (通常 DCM の大きい %) の別の割合を必要がありますよりも留意してください。 - (我々 は尾を完全に溶解に時間がかかることを見た) 解散まで 5-10 分のカクテルを超音波照射容器に追加。
注: 複数の反応部位を含むの尾、必要がありますそれらを結合する前に保護されます。 - 最高の収穫のための一晩を実行することをお勧めしても少なくとも 5 時間 (疎水性尾結合) この反応を実施します。
- DCM と DMF の混合物 (1:1、20 mL) に溶解し HBTU の 9.5 同等物、DIPEA 12 相当必要なカルボン酸の機能を 10 の同等物を追加します。
2. PPA 胸の谷間しっかりサポートから
この手順の目的は、樹脂から、アミノ酸およびポリアミン残基から Boc 保護グループを削除する PPA の胸の谷間。
- DMF (2 分 8 mL) と 2 回 DCM (8 mL、各時間 5 分) の樹脂を洗浄します。各加算の前に船から溶剤を排出します。最後の洗浄を実行すると、15 分間の真空下で樹脂を乾燥します。
- 次の混合比を使用して胸の谷間ソリューションを準備: 28:1:1 トリフルオロ酢酸 (TFA): H2O: トリイソプロピル シラン (ヒント)。15 mL カクテル胸の谷間の H2O の 0.5 mL とヒントの 0.5 mL に TFA、14 mL を追加します。
- 樹脂にこの胸の谷間のソリューションを追加し、室温で 2-4 h に振る。
- 25 ソリューションを収集または 50 mL の丸底フラスコ。
- TFA真空中で40 ° C での混合物を加熱しながら減圧下でロータリーエバポレーターを使用して 1-2 mL に集中 (PPA の分解を避けるために 55 ° C を上回るない)。
- 蒸発後に、無水コールド エーテル (15 mL) を含む丸底フラスコに (滴下) 得られた TFA 溶液を追加します。これはすぐに、PPA を沈殿させます。
- また、TFA は集中して元のフラスコに無水コールド エーテル (5 mL) を追加します。
- 追加の固体を回復し、2.6 のステップからエーテル溶液を組み合わせてこのフラスコを超音波照射します。
注: 転送のピペットは、DCM の体積が小さく、洗浄できます。ゲル状の物質を形成する傾向があるために、プロセス中に DMF を行わずに済みます。 - 冷蔵庫内 (カバー) フラスコを置き、それは沈殿物を最大化するために一晩立ってみましょう。
- 漏斗フィルター焼結ディスクを使用して真空ろ過で沈殿させた材料を収集します。理想的な細孔径は、罰金または媒体です。
- 沈殿物の任意の残留有機物を削除する冷たいエーテル (5-10 mL) で二回洗います。
3. MALDI 乾燥アンド ドロップ メソッドを使用して粗生成物の同定
-
肯定的なモードでの解析のマトリックスの準備:
- マトリックス支援レーザー脱離イオン化 (MALDI) 質量分析法による分子量分析を開始するには、微量遠心チューブに 10 ~ 20 mg α-シアノ-4-ヒドロキシケイ皮酸を追加します。
- 0.1% トリフルオロ酢酸 (TFA) を水/アセトニ トリル (1:1、1.0 mL) のソリューションを追加します。ボルテックスによって徹底的にミックスします。
注: この行列は正荷電ペプチド使用する必要があります。MALDI 負モードのような溶媒添加剤として 0.1% アンモニアと 9 aminoacridine を含むマトリックスが必要です。 - MALDI のステンレス ターゲット板に 1-2 μ L のサンプルを追加し、空気のサンプルを乾燥します。行列の 1-2 μ L を追加し、それを再び乾燥します。
注: プレートは、特定の場所を探すために役立つターゲット プレートに沿ってグリッド円形のマーキングを持っています。
- 自分のアイデンティティを確認する MALDI 楽器のサンプルを分析します。エレクトロ スプレー イオン化 (ESI) は、Ppa の本人確認のための有効な代替手段です。
4. 精製分取用逆相高速液体クロマトグラフィー (HPLC) を用いた Ppa の浄化
-
アセトニ トリルと水の最小限の PPA 沈殿を溶解します。
- 親指のルールとしてアセトニ トリルと水 5 mL 未満で乾燥粗 PPA の 100 mg を溶解します。疎水性 Ppa 溶解するアセトニ トリルの大きい割合を必要があります、場合も溶剤量が多い一般に、Ppa (表 1) の純充満によって。
- 完全な分解が行われない場合は追加 1 %tfa (ACN または H2O)。また、他の互換性のある溶媒をジメチルスルホキシド、メタノール、イソプロパノールなどの微量を追加することが可能だ (5% に自分のコンテンツを制限する)。
| 溶剤 | Ppa の正荷電 | Ppa の荷電 | ||
| 0.1 水に % TFA | 0.1 水に % NH3 | |||
| 0.1 %acn で TFA | 0.1 ACN で % NH3 | |||
表 1: 溶媒システム。Ppa を積極的に負に帯電の溶媒系を提案する.
- 48% の Amp1 10 s パルスで 20 分や超音波風呂で 2-3 h ホーン超音波発生装置を使用して PPA を超音波照射します。
-
その後、0.20 μ m のポリテトラフルオロ エチレン (PTFE) シリンジ フィルターを用いたろ過続いて 0.45 μ m シリンジ フィルターを使用してフィルターします。PPA ソリューションは、はっきりとすべての粒子状材料の無料にする必要があります。
- ろ過が困難である場合は、長期間の超音波します。長期の保管はして集計したり、ゲル化に再超音波処理と、おそらく、再濾過が必要とするを引き起こす可能性があります、PPA ソリューションは精製されたすぐに注射器ろ過後を強くお勧めします。
- 中、浄化後、HPLC グレード溶剤または 0.25 μ m シリンジ フィルター/膜濾過するもののいずれかを使用します。
注: Ppa を浄化するために最も一般的な溶媒条件グラデーションで H2O と ACN で構成されます。 - このプロトコルの標準的な逆相高速液体クロマトグラフィー装置を使用します。それは、溶出グラデーション プログラマ、バイナリ溶剤配信システム、220、254 nm、およびプログラム可能な分数コレクターで検出可能な UV 検出器を含める必要があります。最大流量は 50 mL/分にする必要があります。
- 使用、C-18 逆相カラム分離のために。精製されている固体の質量に応じて次のディメンションの列を使用できます、ネット担当 PPA (表 2)。
| PPA の料金 | 粒子サイズ | 列のサイズ | 原油の PPA の質量 |
| + ve の荷電 | 5 μ m | 150 × 30 mm | 170 mg |
| -ve 荷電 | 5 μ m | 150 × 30 mm | 170 mg |
| + ve の荷電 | 5 μ m | 150 x 21.2 mm | 90 mg |
| -ve 荷電 | 5 μ m | 150 x 21.2 mm | 90 mg |
表 2: 列を提案した:列のサイズ、粒子サイズと C18 の注入量最大負荷容量の逆相 HPLC カラム
- PPA を注入すると、ボリュームの HPLC の注入ループのボリュームと列の両方の容量によって決まります。表 3 (40 分の溶出時間) で見られる設定によると高速液体クロマトグラフィーの勾配法を実行します。
| 時間 | 溶剤 (アセトニ トリル) | 溶剤 B (水) | 流量 (mL/分) |
| 0 | 5% | 95% | 流量は、充填カラムとそのサイズによって異なります。 |
| 2 | 5% | 95% | |
| 35 | 95% | 5% | |
| 38 | 100% | 0% | |
| 40 | 5% | 95% |
テーブル 3: 提案されたグラデーション:期間にわたって水 vs アセトニ トリルの相対組成を示す逆位相勾配を提案しました。流量は、列の仕様に依存します。
-
MALDI を使用して様々 なテスト チューブに収集した分数を分析し、どの管 (または管) PPA が含まれますを決定します。これは、各チューブは、分子量を研究することによって評価されます。
- 分析 HPLC に分数の純度を確認してください。220 で UV 検出器を搭載した高速液体クロマトグラフィー グラデーション システムを使用して nm。
- 不純物が存在する場合は、高速液体クロマトグラフィーによる浄化のサイクルを行います。列コンバーターのオンライン ソフトウェアを使用して分析の高速液体クロマトグラフィーで使用する分取 HPLC 用上記のグラデーションをスケール ダウンできます。各分画に純粋な物理特性や生物学的評価のための ≥95% であります。
- 大丸底フラスコまたは蒸発のフラスコ画を収集し、すべてのアセトニ トリルと水のほとんどを削除します。PPA の最終的な解決策は、10 mL 以上にする必要があります。
- この濃縮液を 50 mL の遠心管に転送します。Ppa が洗剤のような物質であることをお勧めします。したがって、溶媒の蒸発の間に泡が生成されます。エチルアルコール添加が気泡を減少することがわかりました。
- 掃除に集中します、集中。真空蒸着中に大量の PPA は、コンテナーの壁に従っているかもしれません。繰り返し HPLC 水フラスコの壁を洗浄によってこれを回復します。PPA の集中と、やすの合計量は、30 mL 以上にする必要があります。
- 水溶液 50 mL のチューブの内部に配置します。
-
フラッシュは、この PPA ソリューションを液体窒素で凍結します。
注: より速く、エアカーテンで崇高な小さな氷の結晶の凍結の結果をフラッシュします。さらに、遅い凍結には、自己組織化超分子構造の形成が可能です。別の方法として (しかし望ましい以下) が徐々 に-80 ° C のフリーザーで凍結するかを使用してドライアイス + アセトン混合物を凍結するには- エアカーテンで遠心管を配置すること、前に穿孔またはサンプルを脱出し、冷凍ユニットで収集取得する水分をできるようにチューブのキャップを緩めます。設定範囲-54 ° C 0.016 mbar し凍結乾燥ユニットを設定します。
注: 別のオプション キャップをはずし、開口部の上に (ゴムバンド) とワイプを配置することです。また、我々 は、角度でサンプルを凍結または氷を小さな部分に分割では、表面積の増加のためのより速くより凍結乾燥を発見しました。 - 固体は、溶融を開始、有機溶媒 (通常アセトニ トリル) の痕跡が存在可能性があります。凍結のステップを繰り返し、凍結乾燥の状態を評価します。
- 融解が解決しない場合は、2 つの潜在的なソリューションがあります。
- 分割 (管に配置する) を 2 つの部分でサンプルで水で希釈し、再び凍結します。大量の有機溶剤の機器を損傷する可能性がありますのでこのソリューションを多くの場合使用しないでください。
- または、フラスコにサンプルを配置し、さらに真空下で溶媒を蒸発させます。通常の液体の形態にあるサンプルを lyophilizing バンプや PPA 化合物の損失につながります。
- エアカーテンで遠心管を配置すること、前に穿孔またはサンプルを脱出し、冷凍ユニットで収集取得する水分をできるようにチューブのキャップを緩めます。設定範囲-54 ° C 0.016 mbar し凍結乾燥ユニットを設定します。
5. Ppa のストレージ
- キャップ締め、適切なラベルが付いたチューブをパラフィルムでシールが-20 ° C に、Ppa を格納します。
- 使用する前に、Ppa の水蒸気の結露を避けるために真空チャンバー内温度に PPA を戻します。
結果
合成・精製と物理化学的または生物学的評価の前は、Ppa の大衆は純度確認分析 HPLC を用いて、再チェックをお勧めします。材料解析または生物学的評価、Ppa の必要がありますの純度 > 95%。図 2は、HPLC トレース (上) と MALDI スペクトル (下) 製品の存在を確認することを示します。高速液体クロマトグラフィー分析システム (AUC) 曲線の下の領域を...
ディスカッション
ここで説明されているプロトコルは、Ppa の PAs として井戸と関連ペプチドを用いた分子 (ハイブリッド PA ペプチド) などを合成する使用ことができます。によるペプチドの合成では、簡単な手順ですが、生体ホーミングの分子を含むペプチドの合成が特に困難になることができます。スペルミン、スペルミジン、diethyelenetriamine等のようなポリアミンは、対象化、がん細胞1...
開示事項
著者は宣言する利害の衝突があります。
謝辞
このプロジェクト資金が供給されたネブラスカ大学医療センター (スタートアップ資金、MC S);5P20GM103480、NIH COBRE (T. Bronich) とアメリカの化学社会、PRF # 57434-DNI7(MC-S)。
資料
| Name | Company | Catalog Number | Comments |
| 2-Chlorotrityl chloride resin | AappTec | RTZ001 | |
| SynthwareTM synthesis vessel | Aldrich | SYNP120050M | |
| Dichloromethane | Acros | AC406920250 | Fisher Sci. Catalogue # |
| Wrist Shaker | Boekel Scientific | 401000-2 | |
| Kaiser test kit | Sigma-Aldrich | 60017 | |
| 2-[(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl-amino]-ethanol | Sigma-Aldrich | CDS004772 | |
| Anhydrous Methanol | Acros | AC610981000 | Fisher Sci. Catalogue # |
| Chloranil test kit | TCI | TCC1771-KIT | VWR Catalogue # |
| Di-tert butyl di-carbonate | Acros | AC194670250 | Fisher Sci. Catalogue # |
| Dimethylformamide | Fisher Scientific | BP1160-4 | |
| Hydrazine | Acros | AC296815000 | FIsher Sci. Catalogue # |
| (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate) | p3biosystems | 31001 | |
| 4-methyl piperidine | Acros | AC127515000 | FIsher Sci. Catalogue # |
| Trifluoroacetic Acid | AappTec | CXZ035 | |
| Triisopropyl Silane | Sigma-Aldrich | 233781 | |
| Ether | Fisher Scientific | E138-1 | |
| α-Cyano-4-hydroxycinnamic acid | Sigma-Aldrich | C8982 | |
| 9-Aminoacridine | Sigma-Aldrich | 92817 | |
| Fisherbrand Syringe Filters: PTFE Membrane | Fisher Scientific | 09-730-21 |
参考文献
- Cui, H., Pashuck, E. T., Velichko, Y. S., Weigand, S. J., Cheetham, A. G., Newcomb, C. J., Stupp, S. I. Spontaneous and x-ray-triggered crystallization at long range in self-assembling filament networks. Science. 327, 555-559 (2010).
- Pashuck, E. T., Cui, H., Stupp, S. I. Tuning supramolecular rigidity of peptide fibers through molecular structure. Journal of the American Chemical Society. 132, 6041-6046 (2010).
- Stupp, S. I., Zha, R. H., Palmer, L. C., Cui, H., Bitton, R. Self-assembly of biomolecular soft matter. Faraday Discussions. 166, 9-30 (2013).
- Conda-Sheridan, M., Lee, S. S., Preslar, A. T., Stupp, S. I. Esterase-activated release of naproxen from supramolecular nanofibres. Chemical Communications. 50, 13757-13760 (2014).
- Mata, A., Palmer, L., Tejeda-Montes, E., Stupp, S. I. Design of biomolecules for nanoengineered biomaterials for regenerative medicine. Nanotechnology in Regenerative Medicine. , 39-49 (2012).
- Samad, M. B., Chhonker, Y. S., Contreras, J. I., McCarthy, A., McClanahan, M. M., Murry, D. J., Conda-Sheridan, M. Developing Polyamine-Based Peptide Amphiphiles with Tunable Morphology and Physicochemical Properties. Macromolecular bioscience. 17, (2017).
- Nel, A. E., Mädler, L., Velegol, D., Xia, T., Hoek, E. M., Somasundaran, P., Klaessig, F., Castranova, V., Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nature Materials. 8, 543 (2009).
- Gujrati, M., Malamas, A., Shin, T., Jin, E., Sun, Y., Lu, Z. -. R. Multifunctional cationic lipid-based nanoparticles facilitate endosomal escape and reduction-triggered cytosolic siRNA release. Molecular Pharmaceutics. 11, 2734-2744 (2014).
- Zhu, Y., Li, J., Kanvinde, S., Lin, Z., Hazeldine, S., Singh, R. K., Oupický, D. Self-immolative polycations as gene delivery vectors and prodrugs targeting polyamine metabolism in cancer. Molecular Pharmaceutics. 12, 332-341 (2014).
- Planas-Portell, J., Gallart, M., Tiburcio, A. F., Altabella, T. Copper-containing amine oxidases contribute to terminal polyamine oxidation in peroxisomes and apoplast of Arabidopsis thaliana. BMC Plant Biology. 13, 109 (2013).
- Nash, I. A., Bycroft, B. W., Chan, W. C. Dde - A selective primary amine protecting group: A facile solid phase synthetic approach to polyamine conjugates. Tetrahedron Letters. 37, 2625-2628 (1996).
- Ralhan, K., KrishnaKumar, V. G., Gupta, S. Piperazine and DBU: a safer alternative for rapid and efficient Fmoc deprotection in solid phase peptide synthesis. RSC Advances. 5, 104417-104425 (2015).
- Casero, R. A., Marton, L. J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nature Reviews Drug Discovery. 6, 373 (2007).
- Wuts, P. G. M., Greene, T. W. . Protection for the Amino Group. In Greene's Protective Groups in Organic Synthesis. , 696-926 (2006).
- Palasek, S. A., Cox, Z. J., Collins, J. M. Limiting racemization and aspartimide formation in microwave-enhanced Fmoc solid phase peptide synthesis. Journal of Peptide Science. 13, 143-148 (2007).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved