
Method Article
蛍光活性化細胞選別により確立された精製 GABAergic または Glutamatergic ニューロンの初代細胞培養
要約
このプロトコルは、生後マウスまたはラットの新皮質および海馬からの蛍光 GABAergic または glutamatergic ニューロンの精製および培養のための細胞選別ベースの方法を記載している。
要約
このプロトコルの全体的な目標は、GABAergic または glutamatergic ニューロンのいずれかから派生した精製されたニューロン培養を生成することである。精製されたニューロンは、インビトロで16日間にわたって定義された培地で培養することができ、電気生理学的、形態学的および生存分析を含む解離した培養物に典型的に実施されるあらゆる分析に従順である。これらの培養物の主な利点は、グリア細胞または他のニューロンタイプから生じるような複雑な外的影響がない場合に特定の細胞型を選択的に研究することができることである。しかし、精製された細胞を用いて実験を計画する際には、ニューロンが成長と生存のためにグリアの調整を受けた培地に強く依存することに注意することが重要です。さらに、glutamatergic ニューロンは、シナプス伝達の確立のためのグリア分泌因子にさらに依存する。そこで我々は、非接触配列におけるニューロンとグリア細胞の共培養の方法についても述べる。これらの方法を用いて、GABAergic と glutamatergic ニューロンネットワークの開発の間の主な違いを特定しました。したがって、精製されたニューロンの培養を研究することは、神経系がどのように発達し機能するかについての理解を深める大きな可能性を秘めている。さらに、精製された培養物は、薬理学的薬剤の直接作用、増殖因子、または特定の細胞型に対する遺伝的操作の結果を調べるために有用であり得る。より多くのトランスジェニック動物が利用可能になるにつれて、関心のある追加の特定の細胞タイプにラベルを付け、ここに記載されたプロトコルがそれらの適用可能性および電位において成長することを期待する。
概要
細胞選別は、細胞の不均一な混合物から関心のある生細胞を単離するための強力なツールです。細胞は、サイズと形状の基準だけでなく、蛍光マーカー1,2の発現に基づいて並べ替えることができます。多くの場合、蛍光色素共役抗体は、細胞特異的な表面抗原3,4を標的として、異なる細胞タイプにラベルを付けるために使用されます。あるいは、トランスジェニック動物またはウイルス送達系は、細胞特異的プロモーター5、6の下で蛍光色素を発現するように設計されている。歴史的に、トランスジェニックツールおよび動物の開発は、費用と時間を要した。最近では、コストの削減と技術的な問題の減少によって、使用できるレポーターラインの数が劇的に増加しました。トランスジェニックツールおよびレポーター動物の利用可能性が成長し続けるにつれて、蛍光ベースの細胞選別法の有用性および適用可能性も同様である。
我々は、最近、細胞培養7に備えて一次ニューロンを精製するためにトランスジェニック動物からの細胞選別を日常的に適用することができることを実証した。ラットまたはマウスのいずれかから細胞を選別することにより、蛍光ニューロンを分離および培養することができ、GABAergic または glutamatergic ニューロンのいずれかで特異的に蛍光レポータータンパク質を発現することが可能である6、8、9。これらの精製されたニューロン培養を研究することにより、GABAergic および glutamatergic ニューロンがシナプス伝達7の確立のためのグリア分泌因子に依存する方法における重要な相違を同定することができた。さらに、精製されたニューロンとのグリア細胞の共培養によって、グリア細胞がニューロン10、11の増殖および生存において果たす重要な役割を示す以前の観察にも拡張することができた。このように、細胞選別と細胞培養を組み合わせることで、特定のニューロンタイプの開発だけでなく、その機能に対するグリア細胞の影響を調べることができました。
ここでは、トランスジェニックマウスまたはラットの皮質および海馬からの GABAergic および glutamatergic ニューロンの精製および培養のためのプロトコルをここに提示する。また、Geissler et al.12から適応した、精製ニューロンおよびグリア細胞の非接触共培養のための方法も提示する。精製された GABAergic 培養物を生成するために、我々は、VGAT-ヴィーナス-ウィスターラット8または VGAT-ヴィーナスマウス13から蛍光ニューロンを選別し、強化された黄色蛍光タンパク質変異体 (Venus) を選択的に発現する > 95% の皮質 GABAergic ニューロン.精製された glutamatergic 培養物を生成するために、NexCre から蛍光ニューロンを選別しました。Ai9 マウス6,9は、皮質 glutamatergic ニューロンにおいて tdTomato を発現する。全体の選別および培養手順は 3-4 h 内で行うことができ、電気生理学的、形態学的および細胞生存分析に適した数百の複製培養物を生成するために使用することができる。この方法は、単純で再現性があり、対象の細胞タイプに対して純度 97% を超える精製ニューロン培養を生成することができます。
プロトコル
すべての手順および動物の維持は、地方からの許可の存在下で、動物の保護に関する制度的ガイドライン、ドイツ動物福祉法および欧州評議会指令 86/609/EEC に従って行われました。権威 (LaGeSo ベルリン、T-0215/11)。
注:このプロトコルは、単一のトランスジェニックマウスまたはラット pup からの精製されたニューロンの培養物 (生後1日0〜 2) を記載する。すべての技術は、無菌条件下で実行されるべきです。すべての解決は0.2 μ m フィルターを使用して殺菌されるべきである (材料のテーブルを見なさい)。ガラスカバースリップおよび解剖用具は185° c で 3 h のために熱滅菌されるべきである。
1. ポリ L リジンをカバースリップコーティングガラス
- 5 mL アリコートの200μ L/mL カバースリップ (PLL) 溶液を除霜し、ガラスを調製します。この原液を20μ g/mL に希釈するには、45 mL の純射出グレードの水を加えます。フィルター-新しい 50 mL の円錐形の管に解決を殺菌しなさい。このチューブを「PLL 滅菌」としてラベル付けします。
注:室温で保存された水を使用して、除霜プロセスをスピードアップします。 - 滅菌 PLL 溶液に100滅菌、丸形、12mm ガラスカバースリップを追加します。攪拌分、2 ~ 3 分のチューブを使用して、コーティングも確実にします。この方法でカバースリップを1時間塗ります。
注:彼らは信頼性の高い培養面を提供し、24ウェル細胞培養プレートのウェルに容易に適合するように、ドイツ製の丸いガラスカバースリップ (直径 = 12 ミリメートル) が好ましいです。 - 40分の PLL コーティングの後、2枚のティッシュペーパーを取り、それらをフローキャビネットに平らに置きます。70% のエタノールを使用して紙を滅菌し、その後平らにしてしわを取り除き、乾燥させます。
- カバースリップガラスを1時間 PLL でコーティングした後、過剰な PLL を除去し、滅菌インジェクショングレードの水を加えます。余分な PLL を除去できるように、カバースリップを 2 ~ 3 秒間静かに攪拌します。このすすぎ手順を2回繰り返します。
- 余分な水を除去し、その後、滅菌ティッシュペーパーにカバースリップを移します。慎重に湾曲した鉗子を使用して、各カバースリップを分離し、乾燥するために残します。
注:容易に分離できないカバースリップは破棄することができる。また、分離を助けるために少量の水を添加することもできます。 - 乾燥したら、カバースリップを24ウェル培養プレートに移します。
注:カバースリップは、解剖プロセスを開始する前に約30分 ~ 1 時間の準備をする必要があります。プレートは、必要になるまで37° c および 5% の co2 でインキュベーター内に保存することができます。
2. 海馬と皮質組織の解離
- 細胞培養液の調製
- 神経組織の解離のために、最初に解凍 40 mL アリコートの細胞培養バッファー (組成 [mM]: 116 NaCl、5.4 KCl、26 NaHCO3、1.3 いいえ2PO4、1 MgSO4· 7h2o、1 CaCl2· 2h2o、0.5 EDTA ·2Na ·2O および 25 D-グルコース、pH = 7.4)。12 mL の細胞培養緩衝液を 15 mL の円錐管に測定します。「BSA」としてラベルを付けます。異なる 15 mL チューブに 5 mL の細胞培養バッファーを測定します。「パパイン」としてラベルを付けます。37° c で15分間、両方のチューブを水浴中でインキュベートします。
- フィルター-残りの細胞培養バッファーを滅菌し、「バッファー-無菌」としてラベル付けし、必要になるまで4° c で保管する。
- ウシ血清アルブミン (BSA) の 120 mg を秤量し、「BSA」という標識のチューブに添加する。パパインの 7 mg を計量し、「パパイン」というラベルのチューブに追加します。両方のチューブを水浴に15分間戻す。
注:パパインパウダーの転送を容易にするために計量船に少量のバッファーを追加すると便利です。 - すべての粉体が溶解したら、各溶液を新鮮な 15 mL の円錐管にフィルター消毒します。チューブのパパインのために、フィルタリングされた溶液を「パパイン滅菌」としてラベル付けします。チューブ BSA の場合、滅菌 BSA 溶液を3つのチューブに分割し、それぞれに 4 mL の BSA 溶液を入れます。各チューブに「BSA-滅菌」とラベルを付け、「1」、「2」、または「3」のいずれかにします。すべてのチューブを水浴に戻し、必要になるまで37° c でインキュベートし続けます。
- 組織解剖のための準備
- 35 mm の濾紙 (材料のテーブルを参照) の1つの部分を取り、70% エタノールで殺菌しなさい;100 mm ペトリ皿の蓋をして、フローキャビネットに乾燥させておきます。
- 、海馬および皮質の解剖のために必要なはさみ、鉗子、メスおよびスパチュラ (材料のテーブルを参照) をレイアウトします。
- 2 35 mm ペトリ皿を置き、フローキャビネットの中央のアクセス可能な場所に滅菌フィルターペーパーを含む 100 mm ペトリ皿を配置する。
- トランスジェニック NexCre を収集する。解剖される Ai9 および VGAT ヴィーナスマウス仔。野生型同腹仔からの蛍光仔を識別するために、適切な励起および発光フィルタ (材料の表を参照) を備えた蛍光灯を使用してください。
- 動物を解剖する直前に、35 mm のペトリ皿を冷やされた、無菌の細胞培養緩衝液で満たしてください。
注:1つのペトリ皿は、解剖間の工具を洗浄するためのものであり、他は海馬および皮質を収集するためのものである。
- 組織解離
- 細胞培養バッファーが注がれるとすぐに、斬るは、大きく鋭いはさみを使用して生後1日 0-2 トランスジェニックマウスまたはラット pup を使用し、頭部をフローキャビネットに移すために無菌の 100 mm ペトリ皿を用いる。鉗子、はさみ、スパチュラを使用して、トランスジェニック・ pup の脳を慎重に滅菌フィルター紙に移します。
- タイプ22のメスを使用して小脳を分析し、2つの半球を分離する。横方向に半球をロールバックするために、小さなスパチュラを使用してください。各半球で、皮質がフィルターペーパーと接触するように、穏やかに押し下げます。静かに海馬と皮質を明らかにするために中脳の領域を移動します。
注:半球またはセルを押し下げて押しつぶされるようにするには、あまり多くの圧力をかけないように注意してください。 - 各半球から海馬と皮質を分離するために、メスまたはスパチュラを使用してください。この組織を、冷却細胞培養緩衝液を含む 35 mm ペトリ皿に移動させる。
注:複数の動物を解剖する場合は、滅菌細胞培養バッファーを氷上の 50 mL 円錐管に保存する。それぞれの解剖の後、組織を冷却された細胞培養バッファーに切開した。 - 皮質-海馬組織の解剖を成功させた後、滅菌パパイン溶液を水浴からフローキャビネットに移す。3 mL パスツールのピペットを使用して、摘出した海馬および皮質を滅菌 35 mm ペトリ皿の蓋に移します。タイプ22メスの平らな部分を使用して、組織が小片になるまで、十字運動でチョップ。
- 少量のパパインソリューションを使用して、ペトリ皿の蓋から切り刻まれたティッシュを滅菌パパインチューブに移す。パパインチューブを水浴に戻し、37° c で25分間組織をインキュベートします。
- パパインインキュベーションの間、完全な蛍光培地溶液を構成する。1 mL アリコートの B27、0.5 mL アリコートの Glutamax と 0.5 mL アリコートのペン喉頭炎を解凍します。アリコート 48.5 mL の休止状態は、50 mL の円錐形のチューブに低蛍光媒体を備えています。B27、Glutamax、ペン喉頭炎をソリューションに追加します。ソリューションをよく振ってから、フィルター消毒を行い、「休止状態の完全なメディア」 (日付とイニシャルを含む) としてラベルを付けます。37° c で水浴中でインキュベートします。
- パパインインキュベーション後、パパインチューブと滅菌 BSA チューブをフローキャビネットに移します。1 mL パスツールピペットを使用して、パパインチューブから BSA チューブ1に皮質-海馬組織のみを除去します。
注:過剰なパパインソリューションの転送を最小限に抑えるように注意してください。
- 細胞の解離
- 組織の大きな塊を破壊するために、1 mL のパスツールピペットを使用して組織を数回 triturate します。これに続いて、ファインチップパスツールピペットを用いて組織を7回 triturate。組織を30秒間放置しておくと、組織の大きな部分が堆積物になります。
- 30秒後、下 1 mL の溶液および組織を bsa-チューブ1から BSA チューブ2に移す。Triturate は、ファインチップパスツールピペットを使用して、BSA チューブ2を数回にわたって組織を再利用します。
- からむ後、BSA-チューブ1から組織および溶液の下部1ml を再び移したが、これは BSA-チューブ3になる。Triturate は、ファインチップパスツールピペットを使用して、BSA チューブ3の組織を数回繰り返します。
- からむ後、チューブ2および3からすべての溶液および組織を BSA チューブ1に移す。Triturate はさらに2-3 回、3分間 3000 x gで遠心分離します。
- 遠心分離中に、完全な神経基礎メディア (NBA メディア) を構成します。アリコート48.5 の NBA メディアを 50 mL の円錐形のチューブに、水浴で37° c でインキュベートします。このチューブに「NBA のみ」とラベルを付けます。さらに、B27 の1ml のアリコートを、室温で Glutamax の 0.5 mL アリコートおよび 0.5 mL アリコートのペン喉頭炎を解凍します。
- 遠心分離後、ペレット化した組織から上清を慎重に除去し、細胞を 2 mL の完全な培地で再懸濁させる。P1000 ピペットを使用して、組織を20回 triturate します。
注:あまりにも活発に細胞をピペットにしないように注意してください。あまりにも多くの圧力が適用されると、細胞が損傷することがあります。 - 1 mL パスツールピペットを使用して、30μ m の細胞篩を通した細胞懸濁液をポリスチレンサンプルチューブに濾過します。
注:細胞懸濁液が細胞篩を直ちに通過しない場合、流れを開始するために無菌の手袋を用いて篩の上を押す必要があるかもしれない。この重要なステップは、セルソーターの目詰まりを防止します。 - 選別した後にニューロンを収集するには、必要な数のポリプロピレンチューブに、ピペットで300μ l のフルメディアを使用して収集チューブを準備します。
- セル懸濁液を選別するためにセルソーターに移動する準備が整いました。サンプルの希釈が必要な場合には、セル懸濁液、余分な回収チューブを取り、完全にメディアをスペアにしてください。
3. 精製 GABAergic または Glutamatergic ニューロンの細胞選別
注:選別中の細菌汚染の可能性を最小限にするために、ソーティングのサンプルチューブを 70% エタノールでソートする前に少なくとも5分間洗浄します。詳細な選別パラメータは、機器によって異なり、基本的考察は以下の通りである。
- 最初のセルソート中に、野生型動物からの細胞を選別して比較することによって、未標識細胞からのバックグラウンド蛍光のレベルを確立する。
- ソートする蛍光セルタイプごとに、適切な励起および発光フィルタを選択します。488 nm または 531 nm の励起波長をそれぞれ使用して、金星と TdTomato タンパク質を励起します。530/40 と575/30 の発光フィルタセットを介して放出された光を検出します。
- 細胞のコレクションのための細胞の選別機に完全な媒体の300μ l を含んでいるポリプロピレンのコレクションの管を、加えなさい。
- 高純度の場合、明るい標識の蛍光セルをソートします (例えば、ソートされたセルのドット・プロットの場合は図 1bを参照してください)。
- 並べ替えの後、最適な結果を得られるように、選別されたセルの純度検定を実行して純度レベルを推定します。並べ替え後のセルの通常の回復率は 70 ~ 80% の間であることに注意してください。
- 各コレクションチューブにソートされたセルの数を書き留めます。この値は、セルソーティング装置によって与えられます。
注:セルソーティングは、70μ m ノズル (70 psi シース液圧) または100μ m ノズル (20 psi シース液圧) を使用して行うことができます。
4. ソートニューロンの培養
- 細胞選別の後、採取した細胞を、1ml のパスツールピペットを用いて2ml の丸い底面遠心管に移します。細胞ペレットを形成するために、3000 x gで3分間遠心します。
注:細胞ペレットの位置を特定するのを助けるために、配向を外側に向いている蓋のヒンジで丸い底部遠心管を形成し、これは、細胞ペレットが遠心管の背面 (すなわち、ヒンジとしての管の同じ側) 上に構成することを可能にする。 - 遠心分離の間に、B27、0.5 mL の Glutamax および 0.5 mL の喉頭炎を予温められた NBA メディアの 48.5 mL に追加します。ソリューションをよく振ってから、滅菌とラベルを「NBA コンプリート・メディア」 (日付とイニシャルを含む) としてフィルタリングします。必要になるまで水浴に戻ります。
- 遠心分離後、1 mL パスツールピペットを使用して上清を別の遠心管に転写する (細胞ペレットが乱された場合には上清を保持する)。1000細胞/μ l の細胞密度を達成するために予め温められた完全な NBA メディアの必要量で細胞ペレットを再懸濁させる。 P200 または P1000 ピペットを使用して、セルを上下にピペットで再懸濁します。
- 解離した細胞の存在を確認するために、4倍または10倍対物レンズを使用して、顕微鏡下で細胞溶液を確認してください。あるいは、少量の細胞溶液をカバースリップ上にピペットで載せ、顕微鏡下で確認する。多数の細胞が存在する場合、ステップ4.3 から上清を廃棄することができる。
- 細胞をめっきする前に、中速で2〜3秒間渦をしても細胞懸濁液を確保する。ボルテックスに続いて、10μ l の細胞懸濁液を各カバースリップの中心に迅速にピペットで当てる。細胞をピペットで戻した後、各プレートをインキュベーター (5% CO2/37 ° c) に迅速に戻り、1時間インキュベートします。
注:複数の24ウェルプレートにセルを追加する場合は、プレート間のセルのボルテックスによってもセル密度を確認します。 - 1時間後、予温めた完全な NBA のメディアの500μ l で細胞をフィードし、インキュベーターに戻ります。
注:細胞を供給する際には、細胞の離脱を避けるために、ウェルの側に優しく培地をピペットで送ることが重要です。短い実験 (< 16 日) のために、上記の密度での給餌は必要ありません。より大きい密度の細胞をめっきする場合、より頻繁な供給間隔が必要になることがある (考察を参照)。
5. Astrocyte に濃縮したグリアのサポート文化
注:グリア培養14の産生および細胞培養挿入物の使用は、以前に12に記載されている。簡単に説明すると、astrocyte 豊かなグリア培養物は、皮質-海馬組織 (髄膜を取り除いたもの) から得られる。P2 – P5) は、20μ g/mL の PLL 被覆6ウェルプレートで1週間培養されたもので、血清添加 DMEM 培地である。
- 精製されたニューロンおよびグリア細胞を共培養するためには、必要な数の細胞培養挿入物 (0.4 μ m の細孔の大きさ—材料の表を参照) に合流してグリア細胞を継代する。細胞を通過させるために、培養器からコンフルエントなグリア細胞を含む6ウェルプレートを、2ウェルを洗浄し、2回、2 mL のプレ加温 (37 ° c) リン酸緩衝生理食塩水 (PBS) を用いた。
- PBS 溶液を吸引し、P1000 ピペットを使用して、予加温された (37 ° c) トリプシンを 1 mL 追加します (1: 250)/EDTA (0.25%/0.02%)それぞれの井戸へのソリューション。
- 6ウェルプレートをインキュベーターに戻し、細胞剥離のために 2 ~ 3 分ごとに確認します。
- 有意な細胞剥離が発生したら、細胞および細胞溶液を微細チップパスツールピペットを使用して穏やかに上下にピペットで、個々の細胞のさらなる剥離および分離を助ける。
- セル溶液を 15 mL の円錐管に移し、3000 x gで3分間遠心分離します。
- P1000 ピペットを 5 mL の予温めた (37 ° c) NBA の完全なメディアで再懸濁し、細胞ペレットが溶液になるまで静かにピペットで動かします。
- Haemocytometer または自動セルカウンタを使用してセル数を実行します。
注:培養7日後、約 750000-1000000 の細胞は、6ウェルプレートの各ウェルから収穫することができます。 - プレート4万は、グリア細胞を各細胞培養液に500μ l 液滴 (80 細胞/μ l) で挿入する。
- 細胞を1時間接着させた後、滅菌鉗子を使用して、グリアで覆われた細胞培養液を、精製したニューロンを含む24ウェルプレートに移します。余分なメディアを細胞培養液の表面から取り除きます (約300μ l)。
注:気泡が細胞培養インサートの下に閉じ込められることがよくあるので、これらの気泡を取り除くためにインサートを静かに傾ける必要があるかもしれません。より多くのグリア細胞が細胞培養挿入物に添加される場合、さらなる洗浄および遠心分離ステップは、過剰なトリプシンを除去することを必要とし得るが、これは細胞生存に有害であり得る。
結果
トランスジェニックマウスまたはラット皮質からの蛍光ニューロンの解離および細胞選別を、約3〜4時間で行うことができる。結果は、非常に純粋な蛍光ニューロンの集団であり、16日間培養で増殖させることができる。
精製培養物を生成するために、トランスジェニック動物を、適切なフィルターセットを有する蛍光灯 (蛍光性新生児 VGAT ヴィーナスおよび NexCre の例) を使用して最初に同定した。Ai9 マウスの仔を図 1aに示す)。トランスジェニック動物の同定に続いて、解剖した組織を解離し、そして最も強い蛍光ニューロンを選別して、精製された細胞集団を作製した。NexCre の蛍光強度ドットプロットの例;FACS 中に得られた Ai9 および VGAT ヴィーナスマウスニューロンを、図 1bに示す。最適化されると、個々の P0-2 NexCre の皮質および海馬から50万および80万細胞の間で収穫することができる。Ai9 または VGAT ヴィーナスマウス。細胞は、600のイベント/秒以上の速度で並べ替えることができる。細胞純度の推定値は、DAPI を用いて核マーカーとして行った (図 1c)。陽性細胞を強く選別することにより、97% 以上の純度を日常的に7個達成することができる。
成功した選別の後、めっきされたニューロンは、形状が丸く現れ、滑らかな膜を有し、インビトロで約1時間後に神経突起芽を見るべきである (図 2a、B)。インビトロで7日間によって、いくつかの細胞死が明らかであるかもしれないが、生存細胞はすべての培養条件下に存在すべきである (図 2c、D)。インビトロでの 12 ~ 16 日間において、細胞全体のパッチクランプ記録およびビオシチン15を用いて、精製されたニューロンの形態学的および電気生理学的発達を調査することができる (図 3)。精製された培養物の分析は、glutamatergic と GABAergic ニューロンの両方が、細胞体から軸索およびデンドライトを拡張することができたことを明らかにしている (図 3a, B) と応答してアクション電位を生成する能力を保持するsuprathreshold 脱分極性現在の注射 (図 3c, D)。しかし特に、精製後、GABAergic ニューロンのみが大量の自発的シナプス伝達を受け、glutamatergic ニューロンはグリア細胞が存在しない場合にシナプス伝達を非常にわずかに受ける (図 3e、F)7.
以前に報告したように、精製培養の細胞は、並べ替えられていない培養物よりも不十分であり、非常に小さなデンドライトと軸索があります。これらの欠損を克服するために、グリア細胞12,14を用いて精製された培養物の開発を支援するための適応および応用方法を行った。我々のグリア支持培養物 (DIV7) の細胞構成を図 4aに示す。これらの培養物は、主としてグリア線維性酸性タンパク質 (GFAP) 陽性アストロサイトを含んでいたが、分化分子のいくつかのクラスター (CD11b) 陽性ミクログリアおよびミエリン塩基性タンパク質 (MBP) 陽性のオリゴデンドロサイトを含んでいた。DIV 7 の後、これらの細胞を細胞培養インサートに継代して、精製ニューロンの非接触サポートを提供することができます (図 4b、C)。グリア-ニューロンの共培養の分析により、約4万のグリア細胞は、精製 glutamatergic と GABAergic ニューロンの両方の長期生存と成長を改善するのに十分であったことが明らかになった (図 4d、E)7。さらに、電気生理学的解析は、glutamatergic ニューロンが、グリア細胞と共培養され、非常に活発であり、かつネットワーク活性が強く増加していたことを明らかにした (glutamatergic ニューロンを支持したグリアの割合は、作用の破裂を発火するポテンシャル:62%;n = 28;図 4f、G)。したがって、グリアの支持は、ニューロンの成長と生存を促進するだけでなく、シナプス伝達を促進し、glutamatergic 培養におけるネットワーク活動を確立するためにも重要である。

図 1: glutamatergic と GABAergic ニューロンの精製(A) NexCre の TdTomato の導入遺伝子発現からの蛍光シグナルを示す画像;VGAT ヴィーナスマウス (下段) に Ai9 マウス (上段) 及び金星。スケールバー = 5 mm。 (B) 皮質の TdTomato と金星の蛍光を NexCre から分離した細胞の強度散布図。Ai9 マウス (上段) および VGAT ヴィーナスマウス (下段)。強力な蛍光 TdTomato またはヴィーナスニューロンが選別のために選択されました (ゲーティングボックスで示される)。(C、左)ソートされた TdTomato (上) および金星 (下) 陽性ニューロンの共焦点画像。(C、右)DAPI で共染色した細胞を示す結合画像 (青色より疑似カラー)。TdTomato 蛍光は内因性であり、固定にもかかわらず強いままである (マゼンタ疑似カラー)。Venus 発現は、GFP およびアレクサ Fluor-488-共役二次抗体 (緑色の疑似カラー) に対する一次抗体の組み合わせを用いて増強される。この図の大規模なバージョンを表示するには、ここをクリックしてください。
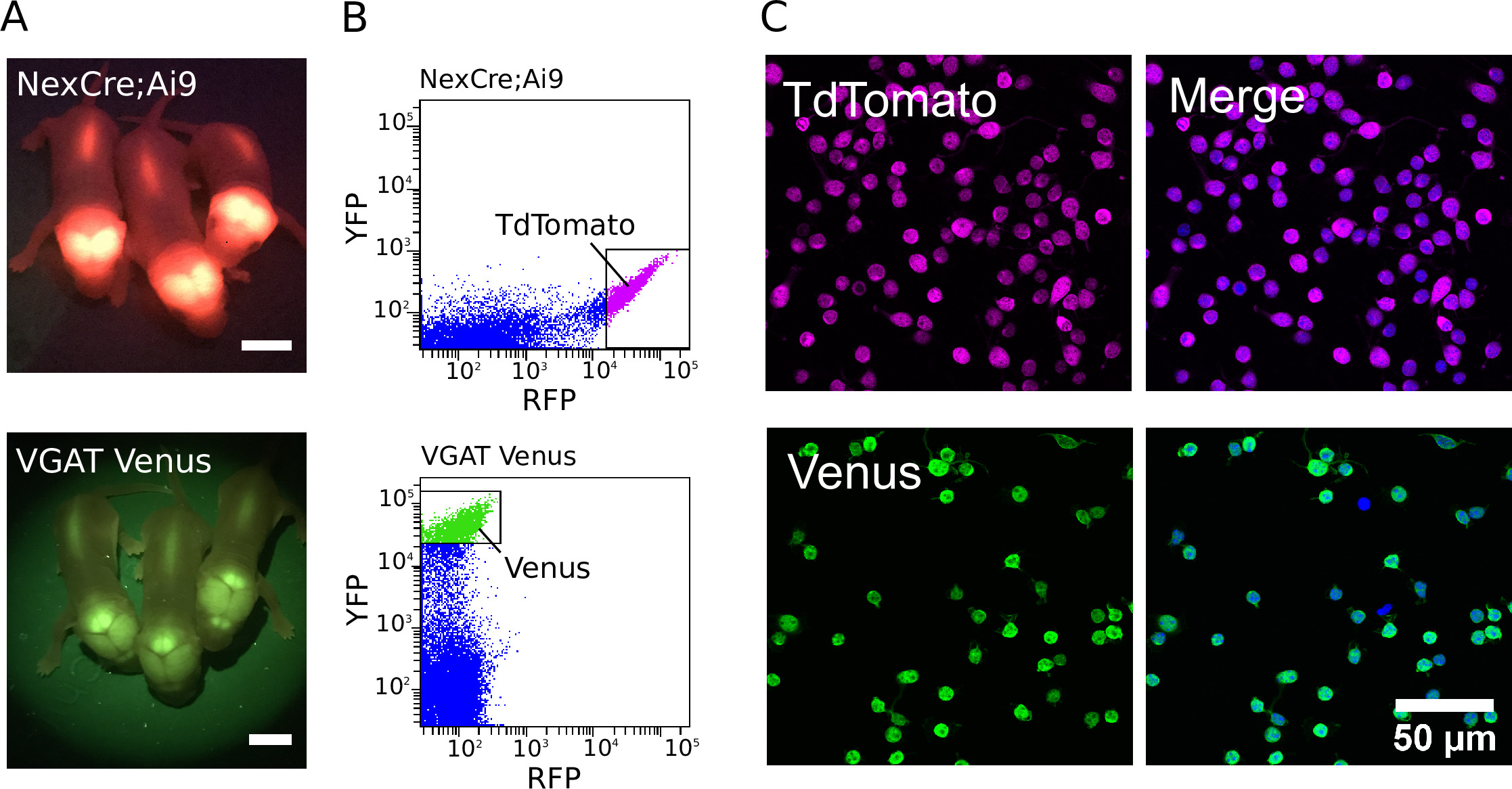{kind=link}

図 2: 精製 glutamatergic または GABAergic ニューロンの細胞培養(A) 赤外線 (明視野) 画像を結合し、TdTomato (左) と金星 (右) 陽性ニューロンから蛍光シグナルを重畳して1時間インビトロで培養した。白い矢印は成長している神経突起の位置を示す。(B) TdTomato (左) と金星 (右) 陽性ニューロンの共焦点画像をインビトロで1時間培養した。(C, D)TdTomato (C) および金星陽性ニューロン (D) の明視野 (上部) および結合明視野および蛍光画像 (下段) は、インビトロで7日間培養した (DIV)。白い矢は死んだ細胞からの凝縮した核の位置を示す。色付き矢印は蛍光標識ニューロンを示します。この図の大規模なバージョンを表示するには、ここをクリックしてください。
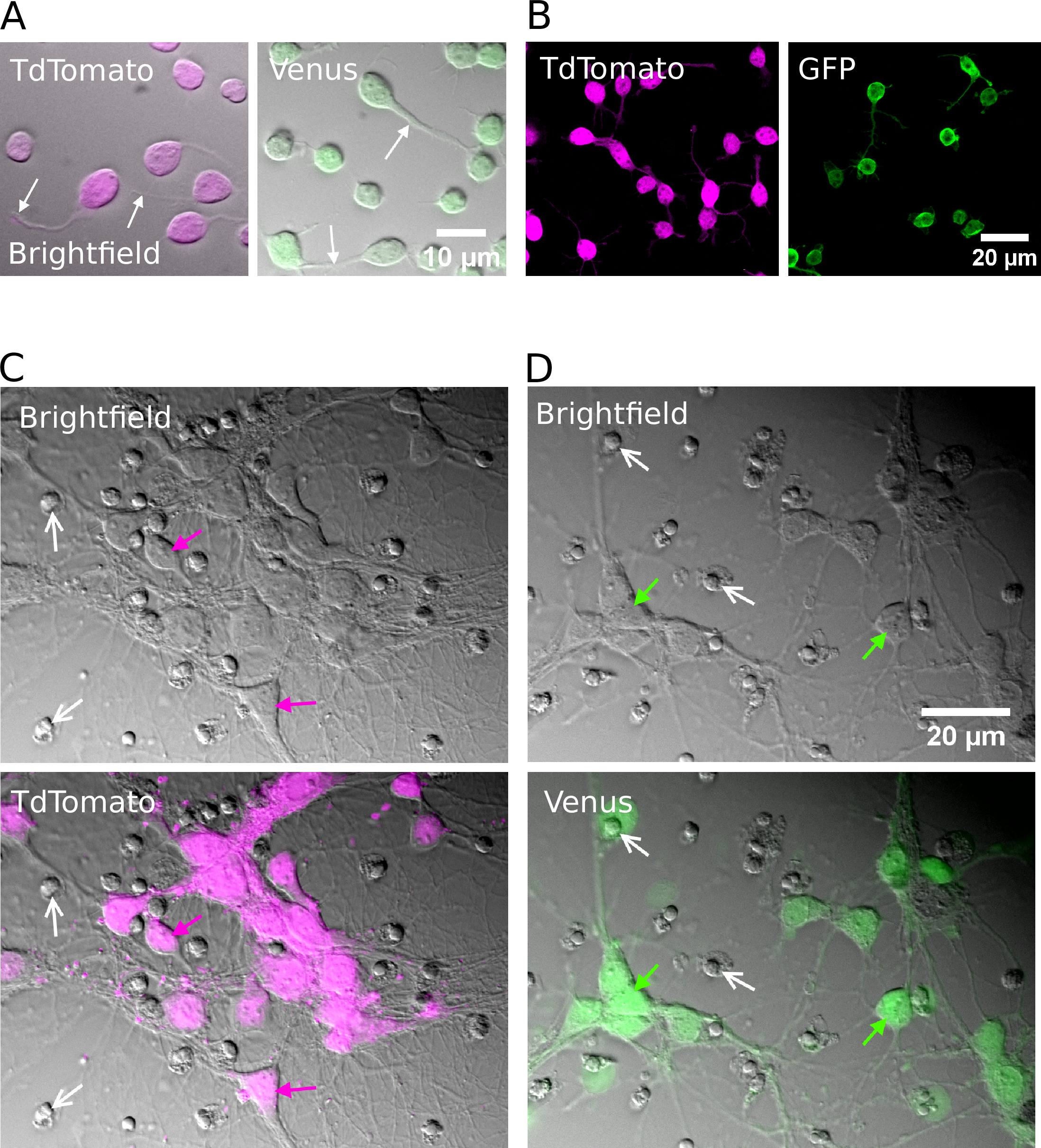{kind=link}

図 3: 精製されたニューロンの形態学的、電気生理学的およびシナプス特性。(A、B)TdTomato (A) と金星陽性ニューロン (B) の共焦点像は、それぞれ DIV 13 および DIV 14 にある。細胞は、神経突起の可視化を支援するために反転ルックアップテーブルを使用して黒色で提示されます。インセットは、同定されたニューロンによって発現される蛍光シグナルを示す。(C, D)TdTomato (C) および Venus 陽性ニューロン (D) から hyperpolarizing (200 pA、20 pA ステップ) および脱分極性 (200 pA) の電流パルス、全細胞パッチクランプ記録による電圧応答。インセットは、対応する記録されたニューロンを示します。各セルの静止膜電位は、記録トレースの左側に表示されます。(E、F)TdTomato (E) および Venus 陽性ニューロン (F) から得られた代表的な電圧-クランプ録音 (10 s)。自発的興奮性シナプス後事象 (EPSCs) は、TdTomato 陽性ニューロンから記録し、50 mV の保持電位で維持した。自発的に発生する阻害シナプス後事象 (IPSCs) を、0 mV の保持電位に維持した Venus 陽性ニューロンから記録した。この図の大規模なバージョンを表示するには、ここをクリックしてください。
{kind=link}

図 4: グリア共培養によるニューロン発達の支援(A) グリア線維性酸性タンパク質 (GFAP、左)、分化分子 (CD11b、中) およびミエリン塩基性タンパク質 (MBP、右) に immunolabeled グリア培養の共焦点画像。グリア細胞は、DIV 7 について培養した。(B) 細胞培養物の画像は、非接触配置で精製されたニューロンとのグリア細胞を共培養するために用いられる。(C) ニューロンとグリアを共培養するために用いられる空間的配置の概略図である。(D, E)TdTomato (D) と金星陽性ニューロン (E) の共焦点画像を、グリア支持の欠如 (左) または存在 (右) で14日間にわたって増殖させた。(F, G)TdTomato (F) および Venus 陽性ニューロン (G) からの電流クランプ記録 (60 s) を、グリア支持の不在 (上) または存在 (底) で14日間培養した。ニューロンは、それらの安静時膜電位で記録された (各記録トレースの左側に提示される)。この図の大規模なバージョンを表示するには、ここをクリックしてください。
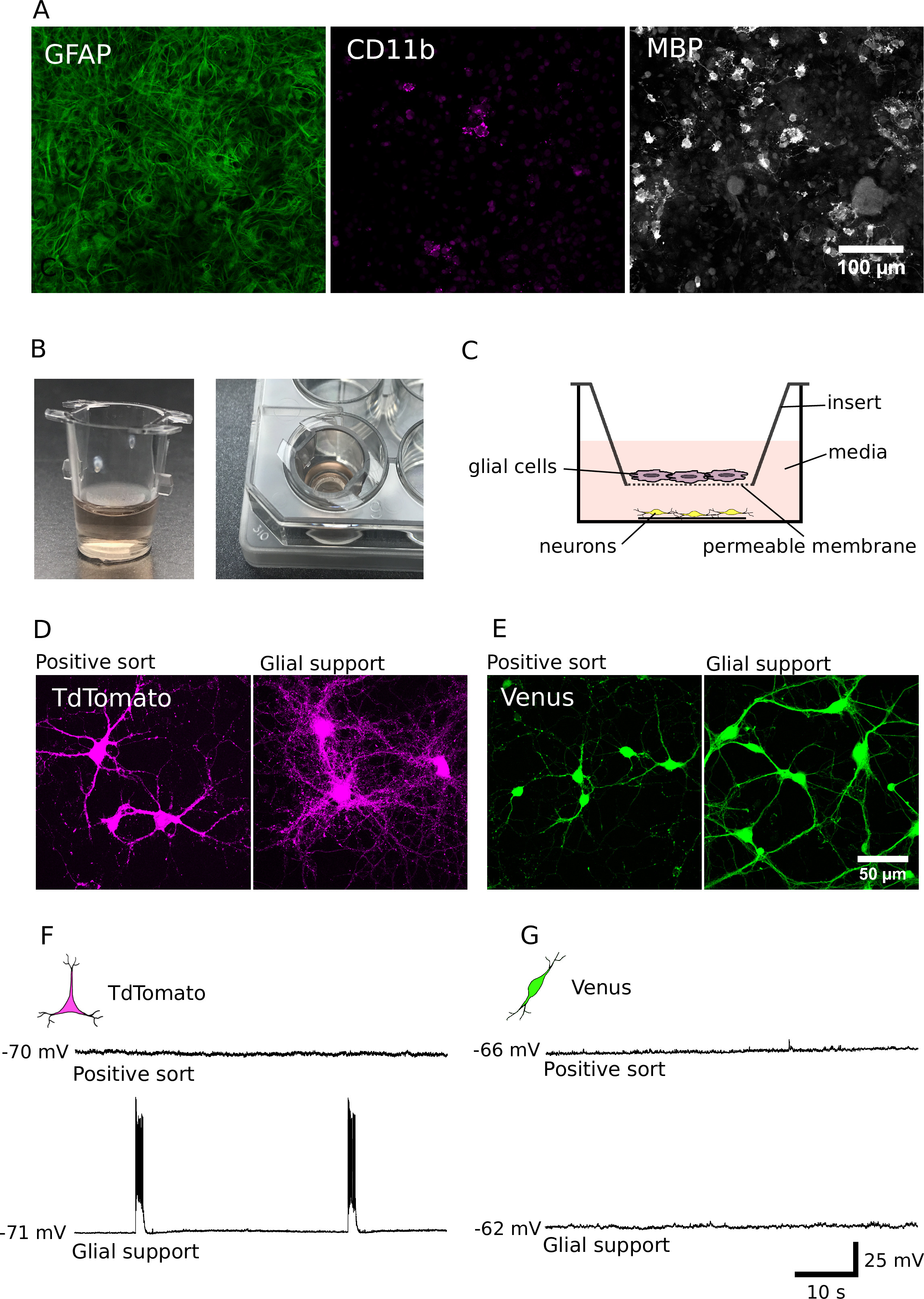{kind=link}
ディスカッション
ここでは、一次ニューロンの選別と培養を組み合わせて、精製された神経細胞培養を生成する方法について述べる。この方法は、典型的な主に解離したニューロン培養プロトコルよりも約1時間長くかかりますが、特定のニューロンタイプを含む何百もの反復培養を生成することができます。精製されたニューロンは、少なくとも16日間分離して成長することができ、軸索およびデンドライトを延長し、反復する運動電位の列車を発射することができる (図 2および図 3)。重要なことに、これらの細胞は、電気生理学的、形態学的および生存の分析を含む、通常の主要なニューロンの解離と同じ実験分析に従うことができる。これらの精製された培養物を用いた作業の主な利点は、特定の細胞型の開発を単独で研究することができることである。また、精製培養の開発を支援するために、グリア細胞と精製したニューロンを共培養するためのプロトコールを提示している。前に示したように、グリア細胞との精製されたニューロンの共培養は、それらの生存を改善し、増殖およびネットワーク形成7を促進することができる (図 4)。そこで、我々は、グリアニューロン相互作用の研究をさらに進めるべき方法の組み合わせをここに提示し、関心のある他の細胞タイプの間の発達および相互作用を研究するのに有用であることを証明することができる。
精製 glutamatergic ニューロンの培養に関する研究は、グリア細胞が神経ネットワークおよびシナプス形成の発達を促進する因子を分泌する方法に関する基礎的な洞察を明らかにした16,17,18.一般に、特定のニューロン型を精製するための方法は、GABAergic ニューロンではなく glutamatergic ニューロンの開発を研究するためにより首尾よく適用されている。このことが、この2つの細胞の種類の理解に格差をもたらしています。GABAergic と glutamatergic ニューロンは、解剖学、生理学、発達の起源に関して大きく異なるため、GABAergic ニューロンを自らの権利で研究し、その機能と生理学をよりよく理解することが重要です。ここで提示されたプロトコルを用いて、GABAergic および glutamatergic ニューロンがシナプス伝達7の確立のためのグリア分泌因子に依存する方法における重要な相違を以前に同定した。このプロトコルを公開することにより、他の人がニューロンとグリア細胞間の重要な相互作用についてさらに洞察を得ることを願っています。
このプロトコルでは、異なるトランスジェニックげっ歯類の GABAergic または glutamatergic ニューロンを精製するために使用したフローサイトメトリーベースの細胞分類法について述べる。Venus 陽性 GABAergic ニューロンは、VGAT ヴィーナスマウス13またはラット8および TdTomato 陽性 glutamatergic ニューロンから選別され、NexCre から選別された。Ai9 マウス (NexCre9および Ai9 レポーターライン6から元々飼育される、Turko et al., 20187) を参照のこと。近年、技術の進歩により、トランスジェニック動物の生成が著しく容易になっている。このように、蛍光分子を発現する動物の利用可能性は、多くの異なる細胞タイプにおいて、急速に成長してきた。これは、順番に、蛍光活性化細胞選別の使用および適用性を増加させた。対象となる細胞を単離するための代替方法は現在16、19、20存在するが、それらは自然発生表面に対する適切な抗体の利用可能性に依存することによって幾分妨げられている抗原。これは、既に利用可能である多くの細胞特異的トランスジェニックレポーター動物から細胞を選別するために既に使用することができる蛍光ベースの細胞選別方法と比較すると、その汎用性を制限する。それにもかかわらず、最適化された場合、抗体ベースの選別法は迅速かつ高収量であり、軸索およびデンドライトがそのままで細胞を精製できるため、細胞解剖学をより良好に保存することができる。したがって、抗体選別法は、ソート戦略を決定する際に考慮されるべきである。最終的に、関心のある細胞タイプ、細胞が選別される年齢、トランスジェニック動物または表面抗原の利用可能性および細胞数が最も適切な選別戦略を選択する際の決定要因となるであろう。
蛍光ベースの細胞選別は、細胞を精製するための簡単で再現性のある方法ですが、細胞の品質を維持するためには、プロトコルの特定の段階で注意する必要があります。例えば、各遠心分離ステップに続いて、細胞ペレットが可能な限り迅速に再懸濁され、細胞が正常に回収されたことを確認することが重要である。時折、細胞ペレットは、上清を除去する際に妨げられ得る。したがって、遠心分離ステップの間に、広範囲の細胞損失を除外するために顕微鏡下の細胞の存在をチェックすることが推奨される。細胞めっきに続いて、細胞は、摂食前に少なくとも1時間接着することを許されるべきである。細胞があまりにも早く供給されると、いくつかの細胞がカバースリップから外れてしまい、それによって培養密度が低下することがある。培養中の1時間後、細胞の健康を評価することが賢明である。細胞が正常に見えない (健康な細胞の例が図 2aに示されている) 場合、または有意な細胞死がある場合、これは、解離または選別手順の間の問題を示す可能性がある。さらに重要な考慮事項は、任意の細胞型を培養する際に、細胞培養培地を管理する際に注意を要することである。NBA メディアは、pH インジケータ22として機能するフェノールレッドが含まれています。メディアの色が黄色くなりすぎると、pH が酸性になりすぎることを示します。メディアがピンク色になると、溶液のアルカリ性が強すぎることを示します。長期間開いているストックソリューションは、特に円錐管に等分メディアは、時間の経過とともによりアルカリ性になる傾向がある。したがって、新鮮な完全 NBA メディアを毎週作成し、2週間ごとにメディアを完成させることをお勧めします (中程度のバッファーは大気条件下で、より安定している必要があります)。これらの点を考慮して、細胞選別および細胞培養装置へのアクセスを備えた任意の実験室において、精製細胞培養を確立することが可能でなければならない。
私たちの実験の大部分では、単一の動物から精製された培養物を製造しました。しかし、生化学的分析のために細胞を精製する場合、ソートのために複数の動物を一緒にプールする必要があるかもしれません。私たちは、上記のプロトコル (データは示されていません) を使用して8つの胚マウス (胚日13匹) まで正常に選別しました。しかし、より多くの動物を選別する場合、組織量の増加に対応するために、パパインと BSA ソリューションの両方の量を増やす必要があるかもしれません (手順 2.1.1-2.1.4 で説明)。さらに、より多くの細胞が培養物にめっきされる場合、より頻繁な供給スケジュールが必要とされ得る。出発点として、細胞は調節された媒体の100μ l を除去し、新鮮で完全な NBA の媒体の200μ l を加えることによって7日ごとに毎日供給することができる。ニューロンの生存性のために, より多くの動物を並べ替える場合, できるだけ多くのソート時間を最小限に抑えるために考えられるべきである.これは、多くの場合、精製細胞を効率的に使用するために下流分析を慎重に最適化する必要があります。私たちは日常的にマウスニューロンを600イベント/秒、最大500000–800000細胞 (生後 0 ~ 2) の割合でソートしています。しかし、これはソート速度と条件を徹底的に最適化することができませんでした。したがって、ソート速度および歩留まりのさらなる改善が可能であるべきである。
精製されたニューロンは、生存のためにグリアに調節される培地を必要とする。これは、ニューロンおよびグリア細胞を別々に培養することによって以前に実証されており、グリアを調節した培地10でニューロンを処置する前に。実験では、細胞培養プレート内に配置された半透過性の細胞培養インサートでグリア細胞を培養することにより、精製ニューロンを支持することを選択しました。この方法は、グリア由来の細胞外マトリックスタンパク質およびニューロン12との相互作用を研究するために首尾よく適用されている。非接触であるが、この方法によって提供される継続的な支持体は、細胞の別個の培養物と比較した場合の多くの利点を有する。最も顕著なのは、ニューロンを有するグリア細胞の共培養は、細胞培養培地の連続的な調節およびコンディショニングを可能にする、インビボの状況により密接に類似している。加えて、細胞の連続的な共培養は、ニューロンとグリアとの間の潜在的なフィードバックシグナル伝達を可能にするが、これは分離された培養物では不可能である。私たちのプロトコルでは、必要に応じて、連続的な共培養法を容易に省略することができ、グリアに調整された媒体を有するニューロンの古典的な治療を行うことができる。
要約すると、ここで提示されるプロトコルは、彼らが独自の精製された細胞培養実験を確立することができる強固な基盤を読者に提供することを目的とする。我々は、トランスジェニック動物及びウイルス構築物の利用可能性が当面の間増加し続けることを予測する。したがって、蛍光に基づく細胞選別技術は、さらに広く用いられ、かつ有用となる可能性が高い。
開示事項
著者らは、競合する金銭的利益はないと宣言している。
謝辞
筆者は、ジェニー・キルシュとアナ Teichmüller が、フローサイトメトリー・コア施設、ドイツ Rheuma ・ Forschungszentrum、ベルリンで提供されている優れた技術サポートに感謝したい。私たちは、生存分析の彼の助けのためのジエの歌に感謝したいと思います。また、プロトコルの批判的な読書のための蛍光マウスとキリスト教のエブネルの画像をキャプチャする彼女の助けのためにリタ Loureiro に感謝したいと思います。VGAT トランスジェニックラットは、宮脇博士によって提供された pCS2 ・ヴィーナスを用いて、日本の岡崎自然科学研究所において、柳川氏、平林、川口氏によって作製した。この作品は、ドイツの研究評議会によってサポートされました (Forschungsgemeinschaft, DFG EXC 257 IV).
資料
| Name | Company | Catalog Number | Comments |
| Neural Basal A media (NBA) | ThermoFisher Scientific | 10888022 | Cell Culture Buffer |
| B27 | ThermoFisher Scientific | 17504001 | Culture supplement |
| Glutamax | ThermoFisher Scientific | 35050-038 | Culture supplement |
| Penicillin-Streptomycin (10,000 U/mL) | ThermoFisher Scientific | 15140-122 | Antibiotic |
| Poly-L-Lysine | SIGMA | P1399 | Coverslip coating |
| Papain | SIGMA | P4762-1G | Enzyme |
| Bovine Serum Albumin | SIGMA | A3294-100G | Serum |
| Hibernate A low fluorescence media | Brain Bits Ltd | HALF | Cell Transport media |
| Dulbeccos Modified Eagles Medium (DMEM) | Biochrom | F0435 | Glial Culture Buffer |
| Fetal Calf Serum (FCS) | Biochrom | S0115 | Serum |
| Trypsin/EDTA | Biochrom | L2163 | Enzyme |
| Fine Tip Pasteur Pipette | Neo Labs | - | Used for trituration of cells |
| 24-well plates | BD | 353047 | Culture plate |
| 50 mL Falcon tubes | BD | 352070 | - |
| 15 mL Falcon tubes | BD | 352096 | - |
| Glass coverslips: 12 mm round | Roth | P231.1 | - |
| 35 mm Petri dish | Corning | 353001 | - |
| 100 mm Petri dish | Corning | 353003 | - |
| 30 µm CellTrics Cell Sieve | sysmex | 04-004-2326 | To remove cell clumps before cell sorting |
| Round bottom polystyrene tubes | BD | 352054 | Transport tube for sorted cells |
| Round bottom polypropylyne tubes | BD | 352063 | Collection tube for sorted cells |
| Cell culture inserts – 0.4 µm transparent PET | Falcon | 353 095 | For the co-culture of neurons and glia |
| Extra fine Bonn Scissors | Fine Scientific Tools | 14084-08 | To remove overlying skin and bone of mice |
| Extra narrow Scissors | Fine Scientific Tools | 14088-10 | To remove overlying skin and bone of rats |
| Forceps | Fine Scientific Tools | 11242-40 | To hold the head in place |
| Spatula (130 mm long/5 mm tip width) | Fine Scientific Tools | 3006.1 | To remove the brain to filter paper |
| Scalpel Blades | Swan-Morton | #0308 | To mechanically dissociate neural tissue |
| Haemocytometer (Neubauer Imroved) | Optik Labor | - | To cell count dissociated cells |
| Fluorescent Miners Goggles (excitation light) | Biological Laboratory Equipment Maintenance and Servce Ltd (BLS) | FHS/LS-1G | To excite TdTomato |
| Fluorescent Miners Goggles (emission filter) | Biological Laboratory Equipment Maintenance and Servce Ltd (BLS) | FHS/EF-4R2 | Td Tomato compatible emission filter |
| Fluorescent Miners Goggles (excitation light) | Biological Laboratory Equipment Maintenance and Servce Ltd (BLS) | FS/ULS-02B2 | To excite Venus |
| Fluorescent Miners Goggles (emission filter) | Biological Laboratory Equipment Maintenance and Servce Ltd (BLS) | FS/TEF-3GY1 | Venus compatible emission filter |
| Mouse-Anti-GFP primary antibodies | UC Davis | 75-132 | To enhance Venus signal following fixation |
| Mouse-Anti-GFAP primary antibodies | SIGMA | G-3893 | The identification of reactive astrocytes |
| Rabbit-Anti-CD11b primary antibodies | Southern Biotech | 1561-15 | The identification of microglial cells |
| Rabbit-Anti-MBP primary antibodies | Millipore | AB980 | The identification of oligodendrocyes |
参考文献
- Radcliff, G., Jaroszeski, M. J. Basics of flow cytometry. Methods in Molecular Biology. 91, 1-24 (1998).
- Feher, K., Kirsch, J., Radbruch, A., Chang, H. D., Kaiser, T. Cell population identification using fluorescence-minus-one controls with a one-class classifying algorithm. Bioinformatics. 30 (23), 3372-3378 (2014).
- Sergent-Tanguy, S., Chagneau, C., Neveu, I., Naveilhan, P. Fluorescent activated cell sorting (FACS): a rapid and reliable method to estimate the number of neurons in a mixed population. Journal of Neuroscience Methods. 129 (1), 73-79 (2003).
- Adan, A., Alizada, G., Kiraz, Y., Baran, Y., Nalbant, A. Flow cytometry: basic principles and applications. Critical Reviews in Biotechnology. 37 (2), 163-176 (2017).
- Guez-Barber, D., et al. FACS purification of immunolabeled cell types from adult rat brain. Journal of Neuroscience Methods. 203 (1), 10-18 (2012).
- Madisen, L., et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature Neuroscience. 13 (1), 133-140 (2010).
- Turko, P., Groberman, K., Browa, F., Cobb, S., Vida, I. Differential Dependence of GABAergic and Glutamatergic Neurons on Glia for the Establishment of Synaptic Transmission. Cerebral Cortex. , (2018).
- Uematsu, M., et al. Quantitative chemical composition of cortical gabaergic neurons revealed in transgenic venus-expressing rats. Cerebral Cortex. 18, 315-330 (2008).
- Goebbels, S., Bormuth, I., Bode, U., Hermanson, O., Schwab, M. H., Nave, K. -. A. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 44, 611-621 (2006).
- Banker, G. A. Trophic interactions between astroglial cells and hippocampal neurons in culture. Science. 209, 809-810 (1980).
- Lindsay, R. M. Adult rat brain astrocytes support survival of both NGF-dependent and NGF-insensitive neurones. Nature. 282 (5734), 80-82 (1979).
- Geissler, M., et al. Primary hippocampal neurons, which lack four crucial extracellular matrix molecules, display abnormalities of synaptic structure and function and severe deficits in perineuronal net formation. Journal of Neuroscience. 33 (18), 7742-7755 (2013).
- Wang, Y., et al. Fluorescent labeling of both gabaergicand glycinergic neurons in vesicular GABA transporter (VGAT)-venus transgenic mouse. Neuroscience. 164 (3), 1031-1043 (2009).
- Höltje, M., et al. Role of Rho gtpasein astrocyte morphology and migratory response during in vitro wound healing. Journal of Neurochemistry. 95 (5), 1237-1248 (2005).
- Booker, S. A., Song, J., Vida, I. Whole-cell Patch-clamp Recordings from Morphologically- and Neurochemically-identified Hippocampal Interneurons. Journal of Visualized Experiments. , e51706 (2014).
- Pfrieger, F. W., Barres, B. A. Synaptic efficacy enhanced by glial cells in vitro. Science. 277 (5332), 1684-1687 (1997).
- Ullian, E. M., Sapperstein, S. K., Christopherson, K. S., Barres, B. A. Control of synapse number by glia. Science. 291, 657-661 (2001).
- Christopherson, K. S., et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 120, 421-433 (2005).
- Berghuis, P., et al. Brain-derived neurotrophic factor controls functional differentiation and microcircuit formation of selectively isolated fast-spiking gabaergic interneurons. European Journal of Neuroscience. 20 (5), 1290-1306 (2004).
- Liddelow, S. A., et al. Activated microglia induce neurotoxic reactive astrocytes via Il-1α, tnfα, and c1q. Nature. 541, 481-487 (2017).
- Tomlinson, M. J., Tomlinson, S., Yang, X. B., Kirkham, J. Cell separation: Terminology and practical considerations. Journal of Tissue Engineering. 4, (2013).
- Brewer, G. J., Torricelli, J. R., Evege, E. K., Price, P. J. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. Journal of Neuroscience Research. 35 (5), 567-576 (1993).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved