
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Fluorescência Baseado Técnica Extensão Primer para determinar Transcriptional pontos de partida e de clivagem Sites de RNases
Neste Artigo
Resumo
We here describe a fluorescence based primer extension method to determine transcriptional starting points from bacterial transcripts and RNA processing in vivo using an automated gel sequencer.
Resumo
A fluorescência baseada extensão do iniciador (FPE) é um método molecular para determinar pontos de partida da transcrição ou locais de processamento de moléculas de RNA. Isto é conseguido através de transcrição reversa do ARN de interesse, utilizando iniciadores marcados com fluorescência específicos e análise subsequente dos fragmentos de ADNc resultantes por electroforese em gel desnaturante de poliacrilamida. Simultaneamente, uma reacção de sequenciação de Sanger tradicional é executado sobre o gel para mapear as extremidades dos fragmentos de ADNc correspondentes às suas bases exactas. Em contraste com 5'-RACE (Amplificação Rápida de Extremidades de ADNc), no qual o produto deve ser clonados e sequenciados vários candidatos, a maior parte dos fragmentos de ADNc gerados por extensão do iniciador pode ser detectado em simultâneo uma corrida de gel. Além disso, todo o procedimento (a partir de transcrição reversa para análise final dos resultados) pode ser completada em um dia de trabalho. Ao utilizar os iniciadores marcados com fluorescência, a utilização de reagentes perigosos isótopo radioactivo marcadopode ser evitada e os tempos de processamento são reduzidos como produtos podem ser detectadas durante o processo de electroforese.
No protocolo seguinte, nós descrevemos um método de extensão do iniciador fluorescente in vivo para detectar de forma fiável e rápida extremidades 5 'dos RNAs para deduzir pontos de partida da transcrição e locais de processamento de RNA (por exemplo, por componentes do sistema de toxina-antitoxina) em S. aureus, E. coli e outras bactérias.
Introdução
A extensão do iniciador 1 é um método molecular para determinar as extremidades 5 'de moléculas de RNA específicas até uma resolução de uma base. A vantagem de outros métodos, tais como 5'-RACE (amplificação rápida de extremidades de ADNc) é o tempo de resposta rápido e a capacidade de analisar facilmente uma mistura de diferentes comprimentos de moléculas de RNA.
Este método funciona por sujeição de moléculas de ARN para reacções de transcrição reversa, utilizando iniciadores fluorescentes específicos, gerando fragmentos de cDNA de determinados comprimentos. Estas moléculas de cDNA são executados juntamente com as reacções de sequenciação de Sanger tradicional 2 em géis desnaturantes de poliacrilamida e podem ser detectadas pela sua fluorescência devida à utilização de iniciadores marcados com fluorescência. Os comprimentos dos fragmentos de cDNA são então avaliadas por comparação com a escada de sequenciação, permitindo que o mapeamento dos terminais 5 'de RNA.
Tradicionalmente, as reacções de extensão de iniciadores são utilizados em conjuntocom isótopos radioactivos para detectar moléculas de cDNA em películas de raios-X. Devido aos riscos de saúde, problemas de eliminação de resíduos e facilidade de manuseamento, os protocolos mais recentes utilizam fluorescência para a detecção da extensão do iniciador com sequenciadores automatizados, embora a sua sensibilidade é ligeiramente inferior. Usando primers marcados com fluorescência, o procedimento da marcação recorrentes podem ser omitidos, como iniciadores fluorescentes são estáveis por um longo tempo (mais de um ano em nossas mãos).
O método aqui descrito utiliza um sequenciador automatizado de gel, mas com ligeiras modificações, sequenciadores capilares podem também ser utilizados para a separação e detecção de cDNA 3. A natureza paralela de análise de gel faz com que seja possível detectar mesmo uma pequena quantidade de clivagem ou processamento de RNA. Outra vantagem é a elevada resolução deste método, como clivagem ou mesmo um tratamento de base terminal pode ser detectado.
No que diz respeito à detecção de clivagem de RNA ou de transformação, typically dois diferentes tipos de extensões de primer são distinguidos. Em um caso, o tratamento enzimático é realizado in vitro utilizando ARN purificado e enzima purificada, enquanto que no outro caso, o processamento é realizado in vivo, e o ARN resultante é purificado. Em ambos os casos, o ARN é sujeito a uma extensão de primer efectuada in vitro, no entanto, dependendo da fonte de RNA, o método é chamado tanto uma extensão do iniciador in vivo, in vitro ou in. No protocolo apresentamos aqui, nos concentrarmos apenas na extensão do iniciador in vivo, por causa da facilidade de utilização (não há proteínas purificadas necessário) e a possibilidade de determinar pontos de partida da transcrição e processamento ao mesmo tempo. No entanto, in vitro extensões de primer são, em princípio, criado da mesma maneira e este protocolo pode servir como um ponto de partida.
O método ilustrado aqui pode ser aplicado a muitas espécies de bactérias, desde que eles são passíveis de altapureza e elevado rendimento de preparação de ácidos nucleicos.
A pesquisa em nosso laboratório centra-se no âmbito regulatório da toxina-antitoxina (TA) Sistemas de 4,5, um campo em que o método de extensão do primer é usado extensivamente. TA-sistemas são pequenos elementos genéticos presentes nos genomas procarióticos que consistem de uma proteína tóxica e estável endogenamente activa e uma proteína ou RNA antitoxina na maior parte instável, que neutraliza a toxicidade 6,7. A actividade da toxina é por vezes exercida pela inibição da replicação, síntese da parede celular ou outros mecanismos, mas na maioria das vezes pela actividade de ARNase 8,9. Tipicamente, a RNase especificidade é determinada através da realização de testes diferentes, um dos quais é o método de extensão do primer. Reacções de extensão de iniciadores são adequados para esta aplicação, como uma mistura de fragmentos de comprimento completo e clivados podem ser simultaneamente analisadas para determinar suas extremidades 5 '. Usando uma mistura de in vitro e in vivo em extensões de iniciadores, otoxina de clivagem específico de RNase, por exemplo, a especificidade de sequência pode ser determinada 10-13.

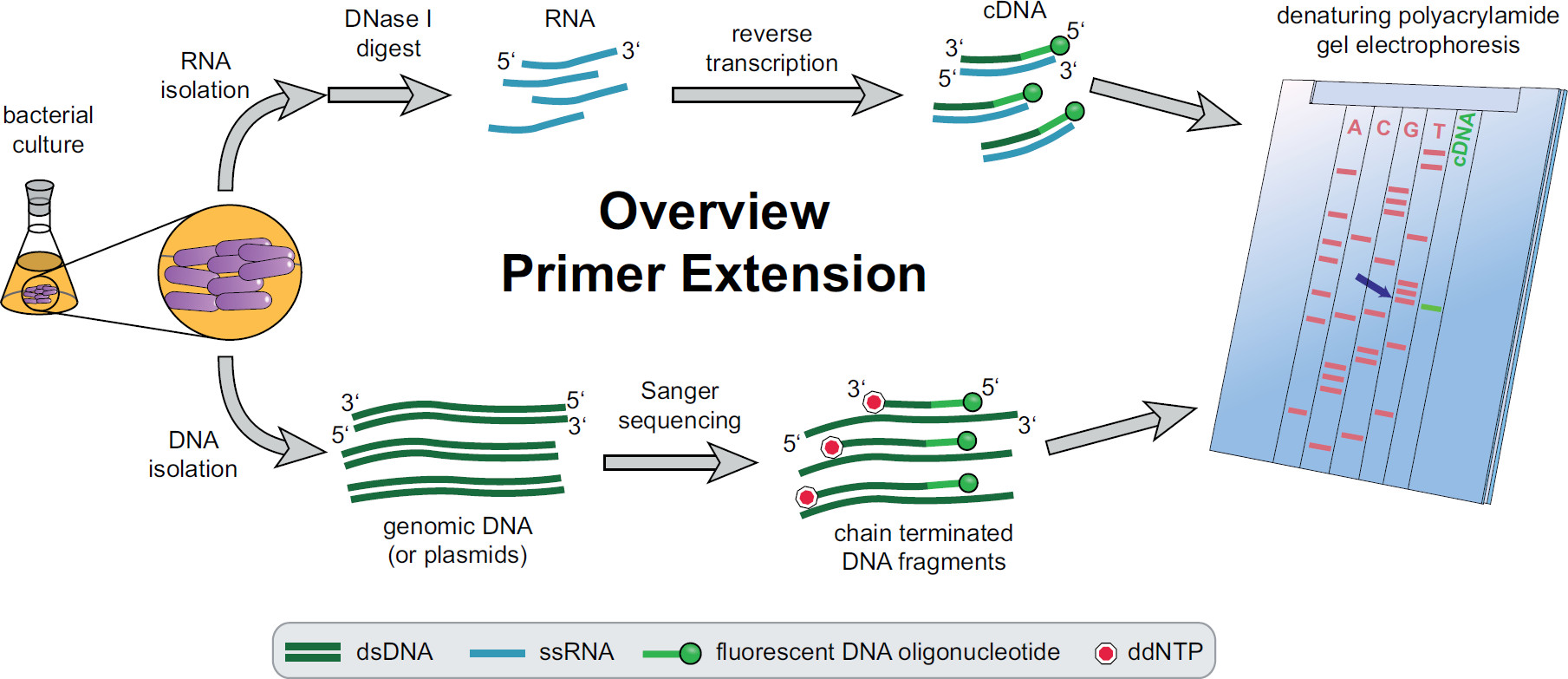
Figura 1. Visão geral do procedimento de extensão do primer. As culturas bacterianas são incubados e tratados de acordo com as necessidades experimentais. O RNA total é extraído das células, tratadas com ADNase I para remover os vestígios de ADN e submetidos a uma reacção de transcrição inversa utilizando iniciadores fluorescentes específicos de ADN alvo produzindo cDNA. O ADN genómico ou plasmídeos são extraídos e subsequentemente utilizada para a fluorescentes reacções de sequenciação de Sanger para comparação do tamanho com fragmentos de cDNA. Produtos de extensão dos iniciadores são executados juntamente com os produtos de sequenciação de Sanger sobre um gel de poliacrilamida desnaturante de ureia e analisadas com um microscópio a laser e automatizado. A base de seqüenciamento que se alinha com a banda de cDNA é a last base da extremidade 5 'de cDNA (seta azul). Mais informações em Fekete, et al. 3 Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
Uma visão geral de todo o processo de extensão do primer pode ser encontrada na Figura 1. Resumidamente, as células bacterianas são cultivadas, colhidas, o sedimento de células lisadas e o RNA extraído. RNA purificado é então tratado com ADNase I para remover vestígios de moléculas de ADN, que possam actuar como modelos para a transcriptase reversa. Iniciadores fluorescentes específicos são adicionados ao ARN, hibridados com a região de interesse e subsequentemente objecto de transcrição reversa, resultando numa única cadeia de ADN complementar (cDNA). Uma escada de sequenciação é criado por sequenciação de Sanger tradicional utilizando iniciadores fluorescentes e separados num gel de poliacrilamida desnaturante a par dos fragmentos de cDNA de extensão de iniciadores. A resultantegel é analisado por comparação das bandas fluorescentes, permitindo a identificação de terminais 5 'de interesse. Pontos de partida de transcrição e locais de processamento são, então, avaliados individualmente pela comparação de seqüências.
Protocolo
1. High Yield RNA Preparação
- Isolamento de ARN
NOTA: As elevadas concentrações de ARN total são necessários para a reacção de extensão do iniciador. Kits de coluna spin normalmente não produzem a quantidade de ARN necessária (~ 5-16 ug em 5 ul de volume). Portanto, recomenda purificação utilizando o método de extração de tiocianato-fenol-clorofórmio guanidina ácido, descrito abaixo.
NOTA: O fenol é cancerígeno, tóxico e corrosivo. Por favor, leia as folhas de dados de segurança e usar sob um exaustor com proteção adequada!- Crescer ou tratar as células bacterianas (S. aureus ou E. coli neste exemplo) como desejado e colheita por 10 minutos de centrifugação a 4.600 x g e 4 ° C. Nota: Tipicamente nós colher um total de OD 600 de 20 - 70. Os sedimentos celulares podem ser armazenadas durante várias semanas a -20 ° C.
- Ressuspender o sedimento de células em 1 ml de solução de tiocianato-fenol-clorofórmio guanidínio ácido e transferir para um2 ml parafuso copo contendo 0,5 ml de 0,1 milímetros pérolas de vidro de zircônio / sílica.
- Lyse as células três vezes em uma batedeira de preparação rápida / talão de 6,5 m / s por 30 s por três rodadas, resfriamento as amostras em gelo durante 5 minutos após cada ensaio. Nota: amostra homogeneizada pode ser armazenado a -80 ° C durante várias semanas.
- Incubar lisado durante 5 min à temperatura ambiente e, em seguida, adicionar 200 ul de clorofórmio.
- Sacudir vigorosamente ou vortex a amostra por 30 segundos para extrair RNA.
- Incubar à temperatura ambiente durante 3 min, em seguida, centrifuga-se durante 15 min a 13.000 - 15.000 x g e 4 ° C. Nota: Os solventes são separados em uma fase orgânica inferior (rosa, contém proteínas), uma interfase (branco, contém ADN) e uma fase aquosa superior (claro, contém RNA).
NOTA: A partir deste passo sobre o uso de apenas RNase reagentes livres e utensílios de plástico! - Prepare fresco livre de RNase 1,5 ml tubos de reação, etiqueta de forma adequada e adicionar cerca de 500 mL de 100% RNase livre isopropanol cada (utilizar aproximadamente o mesmo volume como a fase aquosa no tubo anterior).
- Segurar o tubo em ângulo e transferir a fase aquosa (cerca de 500 ul) para os tubos preparados utilizando RNase pontas livres. Não perturbe a interfase.
- Precipitar o ARN invertendo diversas vezes e incubando durante 10 minutos à temperatura ambiente.
- Centrifugar as amostras durante 15 minutos a 13.000 - 15.000 × g e 4 ° C e remover o sobrenadante por pipetagem ou aspiração (bomba de jato de água e mamadeira). Não perturbar o pellet de RNA branco transparente na parte inferior.
- Adicionar 1 ml de etanol 70-80% RNase livre (não vortex) para lavar. Nota: o ARN em etanol podem ser armazenadas durante várias semanas a -20 ° C.
- Centrifugar durante 5 minutos a 7.500 x g e 4 ° C e desprezar o sobrenadante por pipetagem ou de preferência aspiração.
- Air pellet RNA seco para 15-30 min sob a coifa. Não overdry, caso contrário, pelotas pode ser difícil de dissolver.
- Ressuspender o sedimento em 50 mL de RNase livre DDH 2 O ou tampão de armazenamento RNA.
- Medir a concentração de RNA com um espectrofotômetro microvolume UV-Vis ou uma tina de quartzo (e fotômetro convencional) e proceder à DNase I digestão.
- ADNase I digestão de ARN para remover vestígios de ADN
Observação: Uma vez que o ADN pode actuar como um modelo espúrio na reacção de transcrição reversa (extensão do iniciador), ele deve ser removido a partir da amostra. Vários métodos para a remoção de DNA a partir de soluções de ARN estão disponíveis que geralmente dependem de digestão de DNase. Um método simples, mas eficaz e de custo eficiente para a remoção de DNA está indicado a seguir.- Pré-aqueça o banho de água a 37 ° C.
- Misturar os compostos listados na Tabela 1 em um tubo de 1,5 ml de reacção.
- Incubar a mistura durante 1 hora a 37 ° C num banho de água, e, em seguida, seguir diretamente para a extracção com fenol / clorofórmio. Nota: inativação Calor da DNase I não é recomendado, pois isso pode degradar o RNA.
- Extracção com fenol / clorofórmio, o ARN com DNase I, após digestão
NOTA: O ARN devem ser purificados para remover os nucleótidos livres, fragmentos de ADN e os componentes do tampão a partir da digestão com DNase I. Extracção com fenol / clorofórmio permite uma elevada recuperação e concentração da amostra de RNA e, por conseguinte, está indicado a seguir. Outros métodos de purificação de RNA também pode ser usado, desde que satisfaçam esses requisitos.- Dividir a DNase 500 ul I digestão misturar em duas amostras de 250 ul em 2 ml tubos de reacção.
- Adicionar 1 volume (250 uL) de / solução ácida P C / I (em água saturada de fenol, clorofórmio e isopentanol, proporção de 25: 24: 1, pH 4,5 - 5).
NOTA: / C / I solução P é cancerígeno, tóxico e corrosivo. Por favor, leia as folhas de dados de segurança e usar sob um exaustor com proteção adequada! - Vigorosamente vórtice ou coloque em uma plataforma vórtex para 1-3 min.
- Centrifugar durante 30 minutos a 13.000 - 15.000 x g e 4 ° C.
- Recolher a fase superior (aquosa) e transferir para um tubo fresco (250 ul).
- Adicione 1/9 do volume (28 ul) de 3 M de acetato de sódio pH 5,2.
- Adicionar 2,5-3 volumes de etanol puro (700 ul).
- Misturar em vortex e logo lugar a -80 ° C durante 30 min ou a -20 ° C durante 2-3 horas. Se necessário, armazenar o ARN O / N à temperatura de -20 ° C.
- Centrifugar durante 30 - 60 min a 13.000 - 15.000 x g e 4 ° C.
- Remover o sobrenadante por pipetagem ou aspiração.
- Lavar pelete por adição de 1 ml de etanol a 70% sobre o sedimento. Não amostra vórtice.
- Centrífuga amostra durante 5 minutos a 13.000 - 15.000 x g e 4 ° C.
- Remover o sobrenadante por pipetagem ou aspiração.
- Air pellet seco sob a coifa. Armazenar o sedimento a -20 ° CO / N se necessário.
- Dissolve-se o pellet em 30 ul tratada com DEPC H2O em vortex durante 2 min e usar esta solução para dissolver o pellet do segundo tubo correspondente por amostra (30 solução ul por um par de extração).
- Medir a concentração de RNA, e garantir que ele seja superior a 1 mg / mL para uso em extensão do primer de mRNAs expressos em média.
- Se necessário, o ARN armazenar a -20 ° C durante várias semanas até meses.
2. Primer Extension Reaction
- Projeto Primer
NOTA: Ao projetar primers para uma cartilha de extensão experiência, obedecer as diretrizes gerais de desenho de primers PCR (consulte o manual que acompanha o sequenciador gel automatizado para mais seção de informações e discussão neste artigo).- Especificamente, assegurar que os iniciadores (i) não contêm pistas de bases, (ii) possuem um G ou C na extremidade 3 ', (iii) têm um GC equilibrada: AT proporção, (iv) tem uma temperatura de recozimento de cerca de 55-60 ° C e (v) se ligam, pelo menos, 50 pb, 100 pb melhor a jusante da região de interesse para receber imagens claras.
- Reacção de extensão do iniciador
NOTA: A reacção de extensão do iniciador (síntese de ADNc) requer grandes quantidades de ARN de modelo. Se as quantidades de RNA utilizadas são escolhidas para baixo, o sinal pode ser muito baixo para detectar! Portanto, recomendamos a purificação do ARN, tal como descrito acima.
NOTA: CUIDADO: Use RNase reagentes livres e plásticas !!!- Pré-aqueça o termo-reciclador a uma temperatura de 95 ° C e levar a cabo todos os passos de incubação em mais um termo-reciclador para facilidade de uso e reprodutibilidade.
- Misturar os compostos da Tabela 2, num tubo de PCR para cada amostra de RNA.
- Desnaturar as amostras durante 1 min a 95 ° C.
- Colocar os tubos em gelo e frio durante 5 min para hibridar RNAs e iniciadores.
- Defina a máquina de PCR a 47 ° C.
- Enquanto isso preparar a mistura principal de transcrição reversa, tal como descrito na Tabela 3.
- Adicionar 4 ul de mistura principal de transcrição reversa para cada RNA hibridadaamostra.
- Incubar os tubos durante 1 hora a 47 ° C. Nota: A temperatura óptima para AMV RT é de 42 ° C, as temperaturas mais elevadas no entanto ajudar a superar estruturas secundárias das moléculas de RNA.
- Parar a reacção por aquecimento das amostras a 95 ° C durante 2 min.
NOTA: Formamide é corrosivo, tóxico e pode ser prejudicial para o feto. Por favor, leia as folhas de dados de segurança material, manusear com cuidado e usar proteção adequada! - Adicionar 6 ul de corante de carga de formamida (95% (v / v) de formamida desionizada, 10 mM de EDTA, 0,05% (w / v) de azul de bromofenol) e armazenamento de O / N a duas semanas à temperatura de -20 ° C no escuro.
3. Preparação da Escada Sequencing
NOTA: A reação escada sequenciamento requer ou quantidades moderadas de plasmídeos ou grandes quantidades de DNA genômico. Sempre que possível, o uso de plasmídeos na sequência de reacção é recomendado devido à facilidade de isolamento e de alta sigintensidade nal. Em outros casos, que rotineiramente usar um método de Marmur 5,14 adoptada para preparar DNA genómico a partir de E. coli e S. aureus células sem a necessidade de utilizar fenol. Em princípio, qualquer método que produz elevadas quantidades e pureza de ADN genómico pode ser utilizado.
- Isolamento de DNA genómico
- Cresça 10 ml de E. coli ou S. células aureus O / N em LB, BM 5 ou meio TSB.
- Células colheita por centrifugação durante 10 min a 4600 × g num tubo Falcon de 15 ml.
- Pelota foi ressuspensa em 2 ml de tampão P1 como encontrado em alguns kits de preparação de mini (50 mM Tris-HCl pH 8,0, 10 mM de EDTA, 100 ug / ml de RNase A).
- Lisar células para 45 - 60 min com 20 - 40 ul lisostafina (0,5 mg / ml, de armazenamento a -20 ° C). Nota: Para E. células de E. coli o pré-tratamento enzimático pode ser omitidas ou lisozima utilizados.
- Adicionar 100 ul de solução saturada de SDS-(em 45% de etanol) à suspensão e incubate durante 5 min a 37 ° C.
- Adicionar 650 mL 5 M NaClO 4 e as células brevemente vórtice.
NOTA: O clorofórmio é um potencial cancerígeno. Por favor, leia as folhas de dados de segurança e usar sob um exaustor com proteção adequada !!! - Adicionar 3 ml de clorofórmio / isopentanol (24: 1 ratio) à mistura e agitar durante pelo menos 60 seg. Nota: O líquido deve se transformar em uma emulsão branca homogénea.
- Centrífuga amostra durante 10 min a 4.600 x g e RT para separar as fases.
- Transferir cuidadosamente a fase límpida superior (aquosa) para um tubo fresco. Se a solução estiver turva, repetir a extracção de clorofórmio / isopentanol. Medir o volume da solução de DNA e preparar um tubo fresco com 2 volumes de etanol (100%).
- Lentamente decantar ou pipetar a solução de DNA em etanol contendo o tubo. Nota: DNA deve precipitar, bobinas densas como transparentes no fundo ou quando completamente desidratado como um aglomerado branco flutuante.
- Recuperar o DNA usando ganchos feitos a partir de pipetas de Pasteur de vidro (Figura 2) e lava-se cada uma das amostras duas vezes por imersão em um tubo individual de 1 ml de etanol de 70%.
- Coloque os ganchos na posição vertical em um rack e secar o sedimento por 60 min. Se necessário, o ADN armazenar seco durante vários dias à TA.
- Dissolve-se o ADN por romper os ganchos de vidro coberto de ADN e colocando em um tubo de 2,0 ml de reacção contendo 100 - 500 ul ddH 2 O. Ajustar o volume para uma concentração final do DNA 1000 - 1500 ng / mL. Para uma reacção de sequenciação, utilizam 10-18 ug de ADN genómico.

Figura 2. Instruções sobre a forma de criar uma cana de pesca de ADN. Segurar a ponta de uma pipeta de Pasteur de vidro para a chama de um bico de Bunsen. Isto faz com que o vidro para começar a derreter após vários segundos, criando um pequeno gancho em tele termina. Remover rapidamente do fogo e deixe esfriar por 1 min.
- Plasmídeo Isolamento
- Preparar os plasmídeos utilizando estojos de mini-preparação padrão e dissolvem-se em tampão de eluição (10 mM Tris-Cl, pH 8,5). Dependendo do tamanho do plasmídeo, usam 100 - 500 ng de plasmídeo de uma escada de sequenciação.
- Reação Sanger Sequencing
NOTA: O abaixo um protocolo simples, que utiliza um kit de sequenciação com iniciador marcado por fluorescência 7-desaza-dGTP que funciona bem para a finalidade de extensões de iniciador. Consulte o manual do kit de seqüenciamento para obter informações detalhadas. Por favor note que a reacção de sequenciação deve usar o mesmo iniciador como a reacção de extensão do iniciador para criar produtos com o mesmo comprimento.- Misturar 12 ul de ADN genómico (~ 10 - 15 ug) com 1 ul de DMSO e 1 ul iniciador marcado de modo fluorescente (2 pmol / ul).
- Para cada um dos quatro ul de misturas de reacção de sequenciação (A, C, G ou T), adicionar 3 ul de DNA / DMSO / mistura de iniciadores.
- Coloque as amostras em uma máquina PCR, e executar o seguinte programa de PCR: 95 ° C durante 2 min; 35 ciclos de 95 ° C durante 20 seg, 54 ° C durante 20 seg, 70 ° C durante 30 seg; manter a 4 ° C para sempre.
- Após a corrida, remover as amostras a partir da máquina, adicionar 6 ul de corante de carga e armazenar em gelo (a curto prazo) ou a -20 ° C durante vários dias a semanas.
4. Setup Gel e Aparelho Run
NOTA: informação pormenorizada sobre a forma como o aparelho é montado gel de sequenciação, o gel é preparado e como o gel é executado pode ser encontrada no protocolo do fabricante.
- Preparativos
- Prepare 10x TBE como indicado na Tabela 4.
- No dia da execução do gel preparar 1 L de tampão 1 x TBE com ultrapura ddH 2 O.
- Prepare a 10% (w / v) de APS. Nota: Pode ser armazenado em alíquotas de 200 uL a -20 ° C durante vários meses, mas a actividade pode diminuir ao longo do tempo <./ Li>
- Assembléia de gel câmara de moldagem
- Evite o pó ea sujidade entre as placas de vidro. Superfícies de trabalho, portanto, completamente limpas usando lenços umedecidos.
- Limpar um par de chapas de 25 centímetros de vidro usando toalhas de papel descartáveis e água destilada em ambos os lados e, em seguida, isopropanol para o lado interior das placas de vidro.
- Coloque 0,25 milímetros espaçadores na placa de vidro traseira e baixar a placa de vidro dentada na parte superior (Figura 3).
- Conecte os trilhos de gel para ambos os lados das placas de vidro com a extremidade entalhada e os pilotos entrada trilho viradas para cima e apertar botões levemente.

Figura 3. Vista explodida das placas de vidro de electroforese de gel. Placas de vidro deve ser usado direcionalmente. Cuidar para enfrentar o lado interior das placas de vidro para o interior e o exterioro outro para o exterior.

Figura 4. Vista de um aparelho de gel montado. Depois de injectar a solução de gel, do bolso do espaçador é colocado na solução entre as placas de vidro. A placa de fundição é, então, deslizou entre a placa de vidro da frente e os trilhos de gel e protegidas pelo apertar os botões ferroviários.
- Lançando o gel
NOTA: a acrilamida não polimerizada é neurotóxico! Por favor, leia as folhas de dados de segurança e usar com a proteção adequada !!!- Adicionar os compostos listados na Tabela 5 para uma proveta e misturar utilizando uma barra de agitação e um agitador magnético.
- Imediatamente após a adição de APS e TEMED, levar até a solução de gel em uma seringa de 50 ml e coloque um filtro de 0,45 nm na ponta.
- Ou segurar a borda superior da placa de vidro com uma mão ou colocar o sandwich em um casting gel se para criar uma inclinação angular 10-20 °.
- Dispensar lentamente a solução do gel de entre as placas de vidro enquanto se move continuamente a ponta da seringa a partir de um lado para o outro e parar uma vez que a solução de gel se encontra com o fim de fundo.
- Mover-se para o lado ou remover completamente as bolhas formadas usando um gancho de bolha.
- Deslize o espaçador bolso gel (0,25 mm) entre as placas de vidro na extremidade entalhada, mergulhe na solução de gel e corrigir, anexando a placa de casting.
- Parafusos do trilho superior Aperte levemente (Veja a Figura 4 para aparelho totalmente montado).
- Vamos conjunto gel para 1-2 horas.
- Remova a placa de elenco e bolso espaçador e limpar o bolso de sal e resíduos de gel.
- Enxágüe com DDH 2 O e limpe o excesso de solução com papéis de seda.
- Executando e visualizando o gel
NOTA: Os geles de sequenciação são directamente sujeitos a electroforese em gel de o gerador de imagens, enquantoa fluorescência é detectada simultaneamente através de um microscópio de laser. Em contraste com a electroforese em gel convencional, onde o gel é executado em primeiro lugar e, em seguida, coradas e visualizados, a unidade de detecção é fixo e examina as bandas em tempo real, à medida que passam a laser. Abaixo um procedimento para o software de coleta de dados ImagIR no OS / 2 está delineado, que pode ser adotado para as versões mais recentes. Para mais informações consulte o manual do usuário.- Deslize o suporte do tanque de reserva para os trilhos de gel sobre as placas de vidro da frente e apertar os botões.
- Coloque gel no tanque de gel inferior do gerador de imagens de gel automatizado contra a placa de aquecimento e fixar fazendo deslizar o piloto entrada trilho nos suportes de aparelhos.
- Preencha tampão TBE 1x para as câmaras de amortecimento em gel inferior e superior, feche a câmara tampão inferior e ligar a câmara tampão superior à energia usando o cabo de alimentação.
- Se estiver presente, limpar o bolso gel de sal-resíduos por pipetagem repetidamente tampão no bolso.
- Feche a câmara de depósito de inércia superior usando a tampa do tampão de topo.
- Feche a porta da máquina e ligar o gerador de imagens e computador e iniciar o software de coleta de Base de Dados ImagIR.
- Crie um novo arquivo de projeto (File-> New ...), digite um nome de arquivo do projeto, selecione as faixas de laser apropriados (700 ou 800 nm) e confirme com OK.
- Selecione Opções> ganho Auto ... no menu de imagem no topo, clique em Auto para iniciar a medição de ganho automático e aceitar as configurações clicando em OK.
- Concentre-se o laser, selecionando Opções> Foco ... no menu de controle do scanner, clicando no botão Auto e aceitar as configurações clicando em OK.
- Repita o procedimento de ganho automático para ajustar-se à região recentemente focado.
- Configuração de controle do scanner de acordo com essas configurações: 2.000 V, 35 mA, 45 W, 45 ° C, filtro de digitalização: 3, Velocidade de digitalização: 3.
- Prerun o gel vazia por 20 min (selecione ON tensão e pressione ).
- No entanto, aquecer a escada de sequenciação e os produtos de extensão de iniciadores numa máquina de PCR durante 2 min a 90 ° C, em seguida, arrefecer em gelo.
- Pare a eletroforese, abra o seqüenciador automático gel e retire a tampa do tanque de reserva superior.
- Inserir o dente de tubarão-pente, entre as placas de vidro e ligeiramente perfurar o gel com a dentes de tubarão (ver Figura 5).

Figura 5. Close-up vista de gel com pente de dentes de tubarão. Amostra (roxo) é aplicada entre os dentes de tubarão.
- Pipeta ou 1 - 2 ul de produtos de extensão do primer ou reacções de escada de sequenciação em cada bolsa de gel (formado pelos dentes de tubarão).
- Se são necessários nem todos os bolsos, encher os bolsos vazios com corante para evitar acomportamento corrida consistente.
- Feche a porta do tanque de armazenamento e do sequenciador gel.
- Comece eletroforese e ligue a laser (Select Tensão ON e Laser ON e pressione ).
- Pare de eletroforese uma vez a região de interesse passou a laser.
Resultados
Como representado na Figura 6, uma reacção de extensão do iniciador pode ser utilizado para determinar os pontos de partida da transcrição de transcritos de interesse e pode ajudar a deduzir regiões promotoras (tipicamente identificados por elementos de -10 e -35). O nível superior (mais longa) fragmento de cDNA representa o "fim do ARNm 5 e, portanto, pode ser facilmente mapeada quando comparada com a escada de sequenciação.
Discussão
Extensão do iniciador fluorescente é um método simples e rápido para a determinação extremidades 5 'dos RNAs, quer para identificação TSP- ou processamento de RNA secundário. Devido ao uso de iniciadores fluorescentes, as reacções podem ser configurar e executar, sem precauções de segurança adicionais (ao contrário do caso de iniciadores marcados radioactivamente). Como as amostras são detectadas por fluorescência, que pode ser convertido em imagem, enquanto a electroforese está em progresso ...
Divulgações
The authors have nothing to disclose that would present a conflict of interest.
Agradecimentos
We thank Anne Wochele for her assistance in the laboratory and Vera Augsburger for help with the automated gel sequencer. We thank the Deutsche Forschungsgemeinschaft for funding by grants BE4038/2 and BE4038/5 within the “priority programmes” SPP1316 and SPP1617.
Materiais
| Name | Company | Catalog Number | Comments |
| AMV Reverse Transcriptase (20-25 U/µl) | NEB / Roche | NEB: M0277-T / Roche: 10109118001 | |
| DNase I (RNase free) | Ambion (life technologies) | AM2222 | |
| FastPrep-24 Instrument | MPBio | 116004500 | |
| Fluorescently labeled primers | Biomers | n/a | 5’ DY-681 modification of “ordinary” DNA oligonucleotides. Compatible dyes such as the LICOR IRDye 700/800 are also available from other suppliers such as IDTdna. |
| Li-Cor 4200 Sequencer incl. ImagIR Data collection software | Li-Cor | Product discontinued | |
| NanoDrop 2000 | Thermo Scientific | ||
| Nuclease free water | Ambion (life technologies) | AM9915G | |
| Plasmid mini preparation kit | QIAGEN | 12125 | |
| RapidGel-XL-40% Concentrate | USB | US75863 | |
| RNA STORAGE BUFFER | Ambion (life technologies) | AM7000 | |
| Roti-Aqua-P/C/I | Carl Roth | X985.3 | Alternative: “Acid-Phenol:Chloroform, pH 4.5 (with IAA, 125:24:1)” from Ambion (AM9720) |
| SUPERase•In RNase Inhibitor | Ambion (life technologies) | AM2696 | |
| Thermo Sequenase fluorescently labelled primer cycle sequencing kit with 7-deaza-dGTP | GE Healthcare | RPN2538 | |
| TRIzol reagent | life technologies | 15596-026 | |
| Zirconia/Silica Beads 0.1 mm | BioSpec | 11079101z |
Referências
- Simpson, C. G., Brown, J. W. Primer extension assay. Methods Mol. Biol. 49, 249-256 (1995).
- Sanger, F., Nicklen, S., Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74, 5463-5467 (1977).
- Fekete, R. A., Miller, M. J., Chattoraj, D. K. Fluorescently labeled oligonucleotide extension: a rapid and quantitative protocol for primer extension. Biotechniques. 35, 90-94 (2003).
- Schuster, C. F., et al. Characterization of a mazEF Toxin-Antitoxin Homologue from Staphylococcus equorum. J. Bacteriol. 195, 115-125 (2013).
- Nolle, N., Schuster, C. F., Bertram, R. Two paralogous yefM-yoeB loci from Staphylococcus equorum encode functional toxin-antitoxin systems. Microbiology. 159, 1575-1585 (2013).
- Yamaguchi, Y., Park, J. H., Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 45, 61-79 (2011).
- Schuster, C. F., Bertram, R. Toxin-antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 340, 73-85 (2013).
- Goeders, N., Van Melderen, L. Toxin-antitoxin systems as multilevel interaction systems. Toxins. 6, 304-324 (2014).
- Yamaguchi, Y., Inouye, M. mRNA interferases, sequence-specific endoribonucleases from the toxin-antitoxin systems. Progress in molecular biology and translational science. 85, 467-500 (2009).
- Park, J. H., Yamaguchi, Y., Inouye, M. Bacillus subtilis MazF-bs (EndoA) is a UACAU-specific mRNA interferase. FEBS Lett. 585, 2526-2532 (2011).
- Zhu, L., et al. et al.Staphylococcus aureus MazF specifically cleaves a pentad sequence, UACAU, which is unusually abundant in the mRNA for pathogenic adhesive factor SraP. J. Bacteriol. 191, 3248-3255 (2009).
- Zhu, L., et al. The mRNA interferases, MazF-mt3 and MazF-mt7 from Mycobacterium tuberculosis target unique pentad sequences in single-stranded RNA. Mol. Microbiol. 69, 559-569 (2008).
- Fu, Z., Donegan, N. P., Memmi, G., Cheung, A. L. Characterization of MazFSa, an endoribonuclease from Staphylococcus aureus. J. Bacteriol. 189, 8871-8879 (2007).
- Marmur, J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3, 208-218 (1961).
- Emory, S. A., Belasco, J. G. The ompA 5' untranslated RNA segment functions in Escherichia coli as a growth-rate-regulated mRNA stabilizer whose activity is unrelated to translational efficiency. J. Bacteriol. 172, 4472-4481 (1990).
- Cole, S. T., Bremer, E., Hindennach, I., Henning, U. Characterisation of the promoters for the ompA gene which encodes a major outer membrane protein of Escherichia coli. Mol. Gen. Genet. 188, 472-479 (1982).
- Schleifer, K. H., Kilpper-Bälz, R., Devriese, L. Staphylococcus arlettae sp. nov., S. equorum sp. nov. and S. kloosii sp. nov.: Three New Coagulase-Negative, Novobiocin-Resistant Species from Animals. Syst. Appl. Microbiol. 5, 501-509 .
- Yu, H., Goodman, M. F. Comparison of HIV-1 and avian myeloblastosis virus reverse transcriptase fidelity on RNA and DNA templates. J. Biol. Chem. 267, 10888-10896 (1992).
- Ying, B. W., Fourmy, D., Yoshizawa, S. Substitution of the use of radioactivity by fluorescence for biochemical studies of RNA. RNA. 13, 2042-2050 (2007).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados