
Method Article
Aprimorada Redução Representação Bisulfite Sequencing para a Avaliação de metilação de DNA na resolução de pares de bases
* Estes autores contribuíram igualmente
Neste Artigo
Resumo
Enhanced Reduced Representation Bisulfite Sequencing is a method for the preparation of sequencing libraries for DNA methylation analysis based on restriction enzyme digestion combined with cytosine bisulfite conversion. This protocol requires 50 ng of starting material and yields base pair resolution data at GC-rich genomic regions.
Resumo
Padrão de metilação do ADN de mapeamento é fortemente estudada em tecidos normais e doentes. Uma variedade de métodos tem sido estabelecido para interrogar os padrões de metilação da citosina em células. Representação reduzida do genoma bissulfito sequenciamento completo foi desenvolvido para detectar padrões de pares de bases resolução metilação da citosina quantitativos em loci genômico rico em GC. Isto é conseguido através da combinação da utilização de uma enzima de restrição seguida por conversão de bissulfito. Reforçada reduzida Representação Bissulfito Sequencing (ERRBS) aumenta os loci genómico biologicamente relevante coberto e foi usado ao perfil metilação da citosina no ADN de humano, rato e outros organismos. ERRBS inicia com enzimas de restrição para gerar fragmentos de ADN de baixo peso molecular para utilização na preparação da biblioteca. Estes fragmentos são submetidos a construção da biblioteca de padrão para a próxima geração de sequenciação. Conversão Bisulfite de citosinas não metiladas anteriores da amplificati definitivana etapa permite a resolução de base quantitativa dos níveis de metilação de citosina em loci genômica coberto. O protocolo pode ser concluído no prazo de quatro dias. Apesar de baixa complexidade nas três primeiras bases sequenciadas, bibliotecas ERRBS produzir dados de alta qualidade ao usar uma pista controle seqüencial designado. Mapeamento e análise bioinformática é então realizada e produz dados que podem ser facilmente integrado com uma variedade de plataformas de todo o genoma. ERRBS podem utilizar pequenas quantidades de material de entrada tornando viável para processar amostras clínicas humanas e aplicável em uma variedade de aplicações de pesquisa. O vídeo produzido demonstra etapas críticas do protocolo ERRBS.
Introdução
A metilação do DNA em citosina (5-metilcitosina) é uma marca epigenética crítico em células de mamífero para uma variedade de processos biológicos, incluindo, mas não se limitando a impressão, inactivação do cromossoma X, desenvolvimento, e da regulação da expressão do gene 1-8. O estudo dos padrões de metilação do DNA em distúrbios malignos e outros determinou padrões específicos de doenças e contribuiu para o entendimento da patogênese da doença e potenciais descobertas biomarcadores 17/09. Há muitos protocolos que interrogam o epigenoma para estado de metilação do DNA. Estes podem ser divididos em baseada em afinidade, à base de enzima de restrição e ensaios baseados em bissulfito de conversão que utilizam plataformas de microarray de sequenciação ou a jusante. Além disso, existem alguns protocolos que colmatar estas categorias gerais, incluindo, mas não limitado a, combinado análise de restrição Bisulfite 18 e Redução Representação Bisulfite Sequencing (RRBS 19).
RRBS foi originalmente descrito por Meissner et al. 19,20. O protocolo introduzido um passo para enriquecer regiões genómicas rico em GC, seguida de sequenciação bissulfito, o que resultou em dados quantitativos de resolução de base de par que é de custo eficaz 21,22. As regiões ricas em GC são alvo da enzima de restrição MspI (C ^ CGG), e metilação da citosina é resolvido por conversão bissulfito de citosinas (desaminação de citosinas não modificados para uracilo), seguido de reacção em cadeia da polimerase (PCR). RRBS coberto a maioria dos promotores de genes e ilhas de CpG em uma fracção da sequência necessária para um genoma inteiro; no entanto RRBS tinha cobertura limitada de margens CPG e outras regiões intergênicas de relevância biológica. Vários grupos publicaram atualizado protocolos RRBS desde o relatório original que melhorar a metodologia e cobertura resultante dessas regiões genômicas 23-25. Reforçada Representação Redução Bisulfite sequmentavam (ERRBS) inclui modificações de preparação da biblioteca e um alinhamento de dados alternativo de aproximação 26 quando comparado com RRBS. ERRBS resultou em um maior número de CpGs representadas nos dados gerados e aumento da cobertura de todas as regiões genômicas interrogados 26. Este método tem sido utilizado para resolver padrões de metilação de ADN em paciente humanos e outros espécimes animais 26-30.
O protocolo descrito ERRBS ofertas detalhes sobre todos os passos necessários para a realização e os dados foram gerados utilizando ADN humano representativo (amostras foram obtidas a partir previamente relatados, identificada de 31 amostras de pacientes, e uma amostra de medula óssea CD34 + de um dador humano normal). O protocolo inclui um processo automatizado de selecção do tamanho, o que reduz o tempo de processamento por amostra e permite uma maior precisão na selecção do tamanho da biblioteca. O protocolo combina uma série de técnicas de biologia molecular estabelecidas. ADN de elevado peso molecular é digerido wom uma enzima de restrição metilação-insensitive (MspI), seguido pelo final do reparo, A-tailing, e ligadura de adaptadores metilados. Selecção do tamanho dos fragmentos rico em GC é seguido por conversão bissulfito e a amplificação por PCR antes da sequenciação. Conversão Bisulfite tem sido descrito anteriormente 32 e revisão detalhada de análise e aplicações de dados está além do escopo deste artigo, no entanto recomendações e referências são incluídos para uso dos leitores. O protocolo pode ser realizada ao longo de quatro dias, e é passível de pequena entrada (50 ng ou menos) quantidades de material. O protocolo como descrito produz dados com elevada cobertura por sítio CpG suficiente não só para a região do sítio de metilação e determinações diferenciais, mas também para a detecção de polimorfismo epigenética como descrito por Landan, et ai. 33.
Protocolo
Todos os procedimentos executados são aprovados pela Escola de Medicina da Institutional Animal Care e do Comitê Use Universidade de Indiana e siga Instituto Nacional de diretrizes de saúde.
1. Técnica Cirúrgica
- Manter a técnica asséptica durante esse procedimento usando luvas estéreis, instrumentos e um campo cirúrgico estéril acordo com as diretrizes do NIH 25. Esterilizar ferramentas antes de iniciar a cirurgia por autoclavagem-los (ver Tabela de Specific Reagentes / Equipamentos para a lista completa). Use um esterilizador de contas de vidro para esterilizar instrumentos durante a operação.
2. Anestesia e Preparação
- Anestesiar o rato em uma caixa de anestesia com uma mistura de 0,9 L / min de oxigénio e 2,5% de isoflurano utilizando um sistema de isoflurano vaporizador veterinária. Certifique-se de que o mouse não responder às mudanças na posição do corpo antes de removê-lo da caixa.
- Aplicar pomada oftálmica para o mouOs olhos de si para protegê-los de secar.
- Desligue o fluxo de gás a partir da caixa para o cone do nariz. Posicione o mouse diretamente em seu lado esquerdo em uma almofada aquecida coberta com um bloco cirúrgico e papel absorvente banco com o seu nariz e boca dentro do cone. Monitorar continuamente o ritmo de respiração do mouse e taxa e ajustar os níveis de isoflurano, conforme necessário (entre 2,5-3% de isoflurano) para manter um nível adequado de anestesia, e usar o aperto reflexo toe para confirmar sedação total.
3. Abordagem Cirúrgica
- Harmonizar e concentrar o estereoscópio com o campo cirúrgico. Ajustar o cone do nariz e fita-lo para baixo de modo que ele está posicionado ao longo da borda do campo visual.
- Com o rato encontra-se no seu lado esquerdo, fixe a ponta da orelha direita para o cone de nariz, expondo a área atrás da orelha, onde a incisão será feita. Certifique-se de que a veia auricular posterior viaja horizontalmente através da orelha. Note-se que o posicionamento correto de tele animal ea gravação do ouvido são cruciais, a fim de encontrar rapidamente o nervo facial.
- Molhar a pele em e atrás da orelha com 70% de etanol e raspar o local da cirurgia usando uma lâmina de barbear ou bisturi. Pré-molhar a pele torna a depilação mais fácil nesta localização anatômica.
- Limpar a pele com uma solução de iodo, tais como Betadine limpeza cirúrgica (7,5% de povidona-iodo), seguido de etanol a 70%. Repetir esta lavagem mais duas vezes para desinfectar completamente a área.
- Para determinar onde fazer a incisão, rastrear a veia auricular posterior da orelha caudalmente para a área posterior à protuberância orelha. Com uma tesoura primavera, fazer uma incisão 4 milímetros 2 - 3 mm posterior à protuberância.
- Dissecar através da gordura subcutânea e fáscia usando dissecção romba. Evitar corte directo com as tesouras pois os vasos ou o tecido muscular pode ser facilmente danificado.
- Se ocorrer sangramento, faça pressão no local da cirurgia com um cotonete estérildurante pelo menos 30 seg. Se ocorrer perda de líquido significativo, injetar o mouse por via intraperitoneal com até 0,5 ml de solução salina estéril 0,9% usando uma agulha G 25 ou 27.
- Use vários marcos importantes, o nervo acessório espinhal, canal auditivo, e anterior do músculo digástrico (descrito abaixo), para localizar o nervo facial. Dissecar em torno destes pontos de referência até os ramos do nervo facial são visualizados. O nervo aparece como uma estrutura sólido branco significativo quando se revela e uma camada de fáscia adere a ele as estruturas subjacentes.
- Encontre o nervo acessório espinhal, que viaja a partir da porção caudal do crânio na inervação do músculo trapézio, uma vez que a gordura subcutânea e fáscia foram dissecados. O nervo facial é profundo do nervo acessório espinhal.
- Encontre o canal auditivo cartilaginoso que parece branco perolado e pode ser visto rostral ao nervo facial.
- Encontre o ventre muscular do músculo digástrico anterior, que se situa no topo de e caudal ao nervo facial.
- Quando os principais ramos do nervo facial são visualizados, rastreá-las dorsal para encontrar sua origem a partir do forame estilomastóideo. Usando uma pinça de ponta fina Dumont # 5/45 para manter o local da cirurgia aberta, avançar as dicas de tesoura primavera seguinte caminho do nervo, em seguida, passar a pinça dorsal para manter a área recém-avançado aberto.
- Visualize o tronco do nervo facial com o zigomático, bucal e ramos mandibulares marginais neste momento.
NOTA: O ramo temporal será encontrado mais perto do forame. Os ramos do nervo mandibular marginais nas suas partes superior e inferior mais perto da maxila, assim os ramos do nervo não será visível a este nível.- Se a execução de uma secção do nervo, estabilizar o nervo suavemente com a pinça de ponta fina e cortar o nervo com a tesoura primavera. Evite aplicar muita tração para o nervo com a pinça para evitar avulsing o nervo do tronco cerebral. Empurreos tocos de distância um do outro, ou cortar e remover uma porção do nervo distal para assegurar que não pode ocorrer a reconexão.
- Se a execução de uma lesão por esmagamento, utilize Dumont # 5/45 fórceps para comprimir o nervo por 30 segundos usando constante pressão para cortar todos os axônios, em seguida, repita esta paixão com um segundo ângulo perpendicular ao primeiro local do esmagamento. Evite aplicar quantidades variáveis de pressão durante a 30 seg esmagamento, caso contrário, o prejuízo será inconsistente entre animais.
4. Encerramento e Recuperação
- Reposicionar a gordura e músculos sobre as estruturas subjacentes.
- Harmoniza as bordas da incisão e fechar a ferida utilizando um grampo de feridas 7,5 milímetros. Suturas ou cola também são aceitáveis para o fechamento da ferida. Analgésicos pós-cirúrgicas podem ser fornecidos no momento.
- Remova a fita da orelha do mouse. Desligue o fluxo de isoflurano e permitir que o mouse para respirar oxigênio puro por 30 segundos a 1 minuto. Place o rato em uma gaiola vazia, sem roupa de cama para se recuperar da anestesia.
- Quando o mouse é recuperado, analisar seu comportamento em busca de sinais de confirmação de paralisia facial. Os bigodes será paralisado e em ângulo de volta para o rosto, o nariz será desviado, eo olho não piscará em resposta a um sopro de ar.
- Animais Casa conjuntamente após a cirurgia se eles são do sexo feminino. Evite alojar ratos machos, em conjunto, porque eles são mais agressivos e tendem a remover forçosamente clips ferida do seu cagemate, o que leva à infecção. Fornecer analgésicos pós-cirúrgicas, neste momento, se necessário.
- Monitorar os ratos uma vez por dia durante vários dias após a operação para garantir que nenhuma infecção ou outra complicação ocorre no pós-operatório. Remover agrafos 7-10 dias após a cirurgia, caso não tenham caído por conta própria.
- Aplicar lubrificante pomada no olho afectado diariamente para evitar complicações na córnea, ou até que o reflexo de piscar de olhos é rerecobertos ou até que a eutanásia.
Resultados
A Figura 1 apresenta uma visão geral do ERRBS, destacando etapas fundamentais, que são explicados ao longo do protocolo descrito. ERRBS bibliotecas foram preparadas utilizando 50 ng de ADN de entrada.
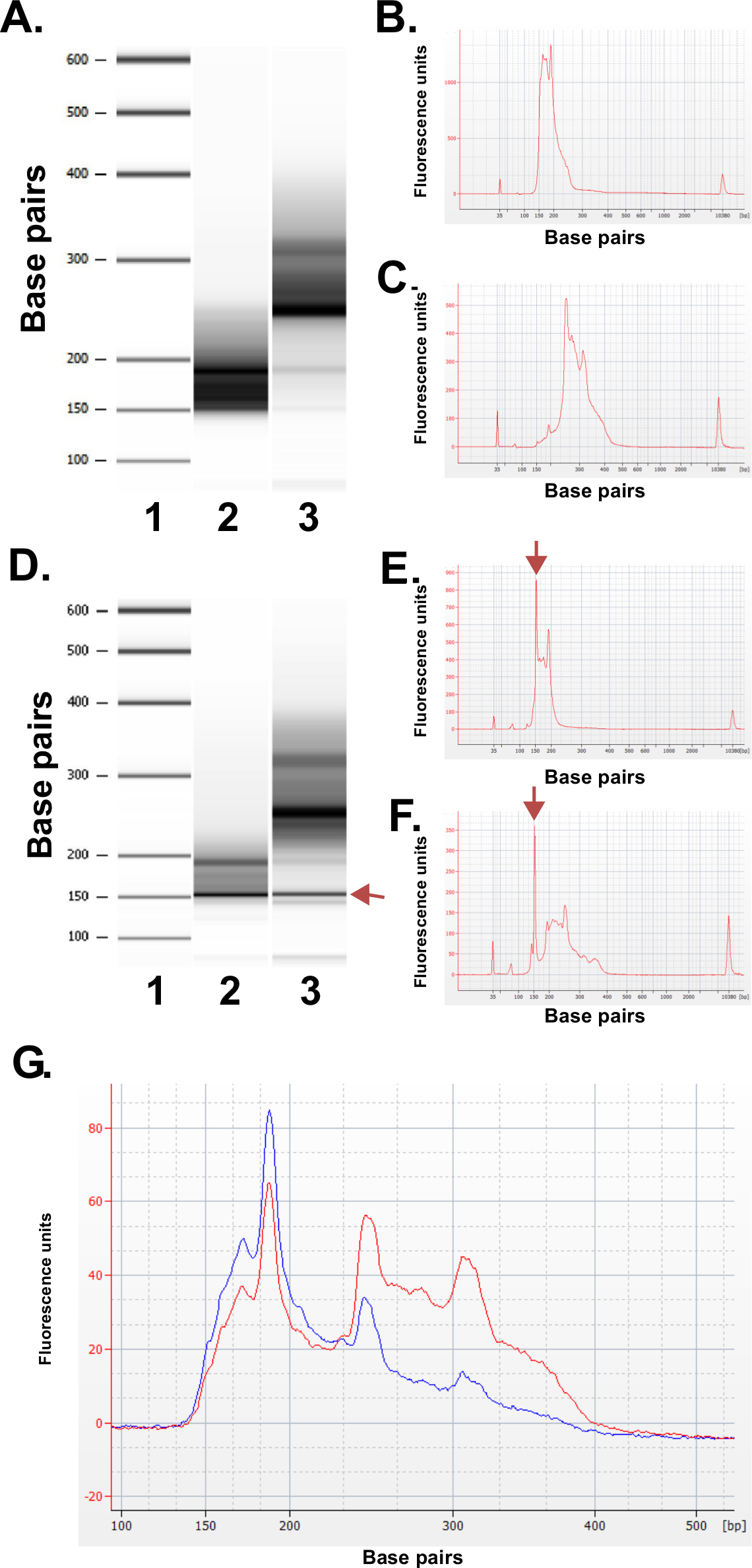
Avaliar a qualidade das bibliotecas preparadas. Biblioteca produção rotineiramente produz fracções de tamanhos 150-250 pb e 250-400 pb (Figura 3A-C). As pequenas diferenças entre as distribuições de tamanho de biblioteca amostras são esperados. Note-se que em ambas as fracções da biblioteca mais baixas e mais altas não são muito intensas tamanhos de ADN, indicativo de enriquecimento de uma sequência particular. MspI resultados de digestão no enriquecimento de uma família de sequências repetitivas de ADN presente no genoma humano de 190 pb, 250 pb e 310 pb nas bibliotecas ERRBS. Estes três repetições representar uma assinatura característica de uma biblioteca ERRBS 20 (ver Figuras 3A-C e 3G). Bibliotecas representativos foram sequenciados em uma seqüência de próxima geraçãor utilizando-end único lê. Ao carregar na concentração recomendada biblioteca em um Illumina HiSeq 2500 sequenciador, são esperados densidades de cluster de 500,000-700,000 por mm 2. Neste densidade de agrupamento, 81,6% ± 3,14% (n = 81) dos aglomerados de passar filtro (Figura 4A). Devido ao baixo final complexidade das inserções de biblioteca (MspI local de reconhecimento: C ^ CGG), os valores de intensidade e índices de qualidade gravados durante o seqüenciamento são altamente variáveis nos primeiros três bases (Figura 4B-C), no entanto, se uma pista de controlo independente está incluído (ver discussão), 85% das bases terá pontuação de 30 ou maior qualidade (valores Q30; Figura 4D).
Alinhamento de dados e determinação metilação da citosina como descrito nos dados de resolução rendimentos protocolo de pares de bases (Tabela 7). Para o genoma humano, uma seqüência de execução single-51 leia-ciclo de uma biblioteca ERRBS em uma pista de um HiSeq 2500 em modo de alto rendimento normally gera 153.194.882 ± 12918302 total de lê-se que após a filtragem de qualidade e adaptador de aparar rendimentos 152.231.183 ± 13189678 lê para entrada no pipeline de análise. Eficiência média de mapeamento para uma biblioteca ERRBS é tipicamente 62,95% ± 5,92%, com representação de 3.183.594 ± 713.547 CpGs com uma cobertura mínima per CpG de 10x e um rendimento médio per CpG de 84,94 ± 16,29 (n = 100).
O protocolo ERRBS é passível de multiplexação (veja o arquivo Suplementar 1: adaptação protocolo para seqüenciamento multiplexado). Os dados do sequenciamento representante é executado é resumidos na Figura 5 Dados de corridas de sequenciação multiplexados (51 de ciclo único de leitura run sequenciamento;. N = 128 para duas bibliotecas por pista; n = 11 para três bibliotecas por pista; n = 11 para quatro bibliotecas por pista) foram comparados a um sequenciamento pista cheia de uma biblioteca ERRBS (51 de ciclo único de leitura é executado sequenciamento; n = 100), bem como redução da resolução, uma única pista para SIMULAÇÃOe 50%, 33% e 25% de leituras por pista (2, 3, 4 e multiplexagem amostra por pista, respectivamente; n = 3). À medida que o número de leituras por amostra diminui com o factor de multiplexagem, o número de CpGs cobertos com uma cobertura mínima de 10x e a cobertura por CpG também diminui (Figura 5 e Tabela 8). Taxas de conversão média de locais não-CpG esperados são 99,85% ± 0,04% (n = 400). Baixas taxas de conversão de 99% pode indicar menos de conversão bissulfito ideal que pode resultar em altos índices de níveis de metilação falsos.
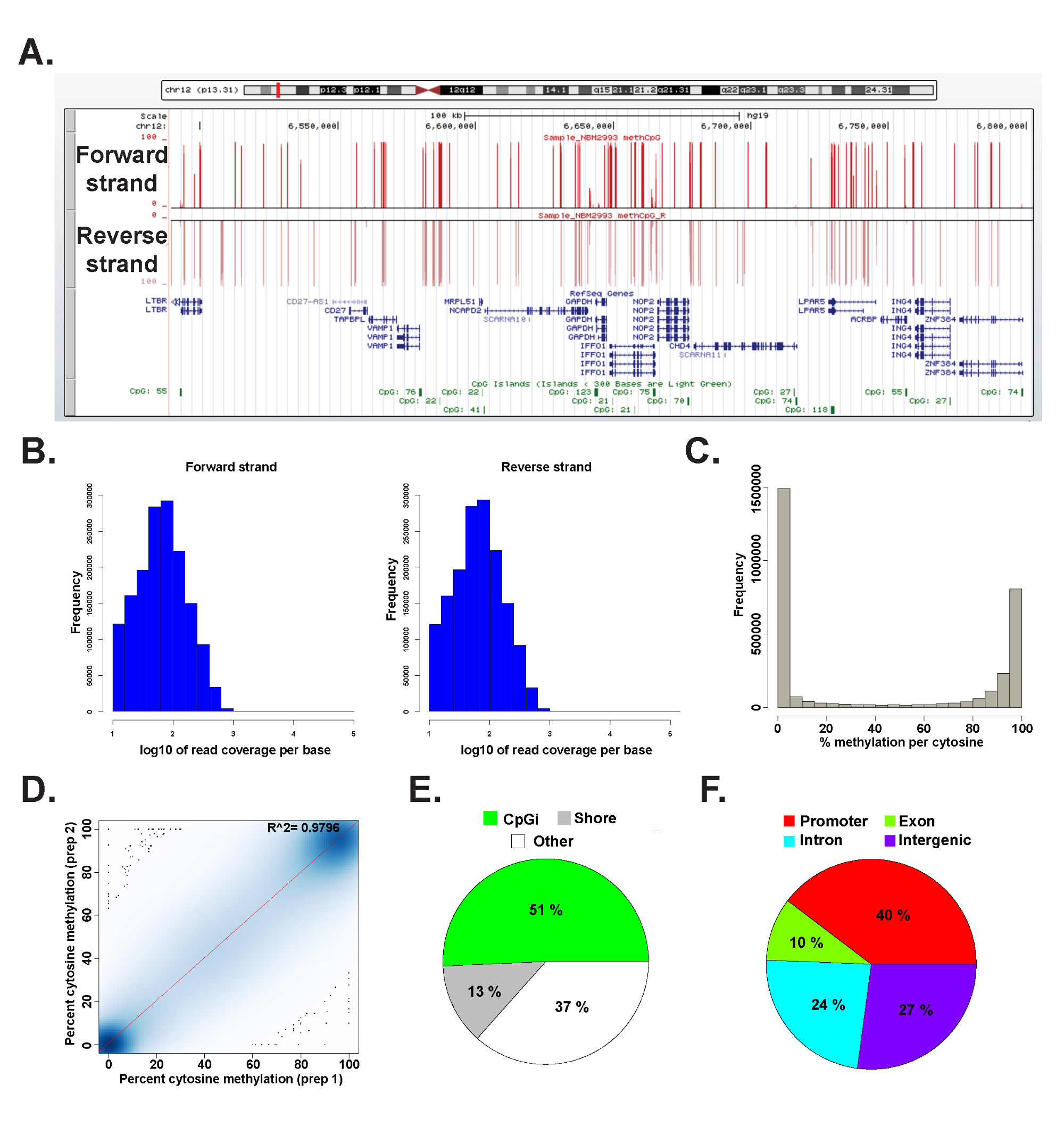
Os dados de uma biblioteca ERRBS preparado a partir de um DNA genómico humano representante foi analisada em R 2.15.2 45 usando o pacote methylKit 26 (ver arquivo de código Suplementar 1 para detalhes de comando). Os dados podem ser visualizados em navegadores genoma vulgarmente utilizadas (Figura 6A). A metilação da citosina dados é igualmente derivada de ambas as cadeias (Figura 6B) e varia a todaespectro de potenciais níveis de metilação da citosina (Figura 6C). Análise de repetições técnicas de um representante rendimentos amostra de ADN humano alta concordância entre os resultados de dados (Figura 6D) e abrange CpGs em um amplo espectro de loci genômica (Figura 6E e F e, como descrito anteriormente 26). Enquanto réplicas técnicos irão produzir grandes valores de R 2 (superior a 97%), réplicas biológicas produzirá valores de R 2 que variam 0,92-0,96 26, e comparando diferentes tipos de células humanas produzirá valores de R 2 mais baixa do que 0,86 (dados não mostrados).

Figura 1: Gráfico das etapas do protocolo ERRBS fluxo. Gráfico representa etapas, que podem ser concluídos em um dia de trabalho tradicional. * Indica um potencial ponto de pausa (siga imediatamente ing ligadura limpar e antes de seleção de tamanho, protocolo passo 5) em que as amostras podem ser congeladas a -20 ° C antes de prosseguir com a duração do protocolo.

Figura 2: protocolo de seleção Size. (A) Captura de tela de configurações usadas no protocolo ERRBS Pippin Prep (ver seção de protocolo 5.1.2 - 5.1.6): (1) Selecione o tipo de cassete. (2) Selecione o padrão a ser usado. (3) Selecione o modo de coleta para cada pista. (4) Entre as faixas coleção pb. (5) Salve o protocolo (B) Estágios de extração manual do gel usado na seção de protocolo 5.2:. (1) escadas gel visualizadas. (2) Marcado Tamanhos para seleção de tamanho usando uma lâmina de barbear. (3) Imagem de amostras excisadas (fração menor: 150-250 bp e maior fração: 250-400 pb)."> Por favor, clique aqui para ver uma versão maior desta figura.

Figura 3: resultados do controlo de qualidade para bibliotecas representativos ERRBS preparados a partir de amostras de DNA humano, utilizando uma máquina Bioanalyzer. (A) Gel-like imagem que mostra uma escada padrão (1), menor fração de biblioteca (135-240 fração bp de Pippin Prep); 2) e a fracção da biblioteca mais elevada (fracção 240-410 pb do Pepino Prep); . 3) (B) Bioanalyzer electroferograma da fracção biblioteca inferior esperado (C) Bioanalyzer electroferograma da fracção biblioteca superior esperado. D -. F) Os dados representativos a partir de uma biblioteca de qualidade pobre prep. Imagem do tipo gel (D) da escada padrão (1), fracção biblioteca inferior (2) e a fracção biblioteca superior (3). A banda de 150 pb marcada com um arrow indica quantidades excessivas de adaptador. Electroferograma da parte inferior (E) e as fracções da biblioteca superiores (F) com os picos de adaptador em excesso de 150 pb (marcados com setas). (G) Bioanalyzer electroferograma de uma biblioteca ERRBS reunidas para sequenciação. Traço vermelho representa uma biblioteca agrupada de alta qualidade com igualdade de representação de frações superiores e inferiores. Traço azul representa uma biblioteca de pool não adequada para a sequenciação devido a uma falta de igualdade de representação das frações superiores e inferiores. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Seqüenciamento paradas por um representante ERRBS 51 de sequenciação de ciclo único de leitura de execução em um seqüenciador HiSeq 2500 em alto rendimentomodo. (A) as densidades de cluster (K / mm 2 = 1.000 aglomerados por milímetro quadrado; azul). E densidades de cluster passando filtro (verde) em duas pistas com bibliotecas ERRBS (B) intensidades típicas observados nos primeiros 30 ciclos em uma pista com um biblioteca ERRBS. Note-se a assinatura CGG da digestão MspI nas intensidades dos três primeiros ciclos. (C) Percentagem de bases com um índice de qualidade de 30 ou mais (%> Q30) para cada ciclo em uma pista ERRBS. (D) a distribuição do escore de qualidade para todos os ciclos em uma pista ERRBS. Azul = menos de Q30, Verde = maior ou igual a Q30. Nesta faixa, 84,7% das bases apresentaram escores de 30 ou superior qualidade.

Figura 5: Sequenciação resultados de saída. Os diagramas de caixa de dados experimentais de amostra multiplexados e única por faixa sequenciamento ru ns (apresentado como caixas verdes) e de dados provenientes de uma redução da resolução simulado de corridas de sequenciação de três ERRBS bibliotecas (apresentado como caixas azuis; amostrados cinco vezes para cada corrida seqüenciamento) a partir de 51 de sequenciação de ciclo único de leitura corre O fator de multiplexação corresponde. o número de bibliotecas ERRBS sequenciado por pista. 1 = inteiro pista ou 100% de leituras e representa os dados a partir de uma única biblioteca ERRBS por pista; 2 = 50% de pista e representa os dados de duas bibliotecas ERRBS por pista; 3 = 33% de uma pista e representa os dados a partir de três bibliotecas ERRBS por pista; e, 4 = 25% de uma pista e representa dados de quatro ERRBS bibliotecas por faixa. (A) A contagem de ler, ou o número de sequências analisadas, por fator de multiplexação. (B) O número de CpG do coberto pelos dados de sequenciamento por multiplexação fator. (C) A cobertura média per CpG por fator de multiplexação._blank "> Clique aqui para ver uma versão maior desta figura.

Figura 6: Dados representativos de uma biblioteca ERRBS preparado a partir de DNA genómico humano (A) da Universidade da Califórnia, Santa Cruz (UCSC) navegador genoma 43 imagem de dados representativos de uma pista ERRBS seqüenciamento.. A barra de escala do eixo y representa 0-100% de metilação em cada citosina coberto com um mínimo de 10x. A pista costume superior representa a vertente para a frente e menor faixa personalizada representa a cadeia reversa. É mostrado chr12:.. 6,489,523-6,802,422 (hg19) inclusive de genes RefSeq e ilhas CpG dentro desta região genômica histogramas (B) distribuição de cobertura CpG juntamente frente e costas reverter em um CD34 humano + medula óssea amostra representativa (C) Distribuição histogramade níveis de metilação CpG ao longo dos dois fios de uma CD34 humano + medula óssea amostra representativa. (D) curva de correlação de níveis de metilação CpG de uma réplica técnico representante de uma amostra de ADN humano. chart (E) Pie ilustrando as proporções de CpGs cobertas de ERRBS que anotação de ilhas CpG (verde claro), o CPG margens (cinza) e outras regiões (branco) em uma amostra representativa a partir de ADN genómico humano. chart (F) Pie ilustrando as proporções de CpGs cobertas de ERRBS que anotados para promotores de genes (vermelho ), exons (verde), íntrons (azul) e regiões intergênicas (roxo). Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
| Reagente | Volume | Comentário |
| T4 ADN ligase 10x Tampão de Reacção | 10 ul | |
| Trifosfato desoxinucleotídeo (dNTP) Solution Mix | 4 ul | Mistura de 10 mM de cada nucleotídeo |
| T4 ADN polimerase | 5 ul | 3000 unidades / ml |
| ADN Polimerase I Grande (Klenow) Fragmento | 1 ul | 5000 unidades / ml |
| Quinase de polinucleótido T4 | 5 ul | 10000 unidades / ml |
| Água livre de DNase | 45 ul |
Tabela 1: reagentes de reacção. End reparação nomes e quantidades de reagentes usados na reacção de reparação final (protocolo passo 2.1).
| Reagente | Volume | Comentário |
| Tampão de reacção 10x | 5 ul | por exemplo, 2 NEBuffer |
| 1 mM de 2'-desoxiadenosina 5'-trifosfato (dATP) | 10 ul | |
| Fragmento Klenow (3 '→ 5' exo) | 3 ul | 5000 unidades / ml |
Tabela 2: A-tailing. Reagentes de reacção nomes de reagentes e quantidades usadas na reacção-tailing A (protocolo passo 3.1).
| Reagente | Volume | Comentário |
| 15 mM adaptadores recozidos em água livre de DNase | 3 ul | Adaptador de 1.0 e PE PE adaptador de 2,0; ver Tabela 4 para sequências e referência |
| T4 DNA ligase 10x Tampão de Reacção | 581; l | |
| T4 DNA ligase | 1 ul | 2.000.000 unidades / ml |
| Água livre de DNase | 31 ul |
Tabela 3: Adaptador reagentes reacção de ligação nomes de reagentes e quantidades utilizadas na reação adaptador de ligação (protocolo passo 4.2)..

Tabela 4:. Os oligos utilizados no protocolo ERRBS Lista de oligos utilizados durante todo o protocolo ERRBS na reacção de ligação (protocolo passo 4) e as etapas de amplificação por PCR (protocolo de passo 7).
| Reagente | Volume | Comentário |
| 10x FastStart High Fidelity Reaction Buffer with cloreto de magnésio 18 mM | 20 ul | |
| DNTP 10 mM Solução Mistura | 5 ul | |
| 25 uM de iniciadores de PCR PE 1.0 | 4 ul | Ver Tabela 4 |
| 25 uM de iniciadores de PCR PE 2.0 | 4 ul | Ver Tabela 4 |
| FastStart High Fidelity Enzyme | 2 ul | 5 unidades / mL FastStart Taq DNA polimerase |
| Água livre de DNase | 125 ul |
Tabela 5: reagentes de reacção de PCR nomes de reagentes e as quantidades utilizadas na reacção de amplificação por PCR (protocolo passo 7.1)..
| Protocolo passo | Reagente / detail protocolo | Quantidade de DNA de entrada | ||
| 5-10 ng | 25 ng | 50 ng | ||
| 1 | Enzima MspI | 1 ul | 2 ul | 2 ul |
| Volume de reação MspI digest | 50 | 100 | 100 | |
| 4 | Adaptadores em reacção de ligação | 1 ul | 2 ul | 3 ul |
| Volume de reacção de ligação | 20 ul | 25 ul | 50 ul | |
| 5 | Protocolo de seleção Tamanho | Só gel manual | Pippin Prep ou gel Manual | Pippin Prep ou gel Manual |
| 7 | PCR concentração do primário | 25 uM | 25 uM | 10 uM durante 14 ciclos; 25 uM para 18 ciclos |
| Número de ciclos de PCR | 18 | 18 | 14-18 | |
Tabela 6: Protocolo. Modificações passo para a entrada de quantidades de materiais que vão 5-50 ng Várias etapas durante todo o protocolo exigir a modificação das quantidades de reagentes usados para gerar bibliotecas de qualidade de diversas quantidades de matérias-primas. Mudanças a quantidades importantes de reagentes estão incluídos aqui. Ajustar o volume de água e tampão nas reacções em conformidade.
| Chr | Base | Costa | Cobertura | freqC | freqT |
| Chr1 | 10564 | R | 366 | 85,52 | 14.48 |
| Chr1 | 10571 | F | 423 | 91.25 | 8,75 |
| Chr1 | 10542 | F | 432 | 91,2 | 8,8 |
| Chr1 | 10563 | F | 429 | 94.64 | 5,36 |
| Chr1 | 10572 | R | 366 | 96.99 | 3.01 |
| Chr1 | 10590 | R | 370 | 88.11 | 11.89 |
| Chr1 | 10526 | R | 350 | 92 | 8 |
| Chr1 | 10543 | R | 368 | 92,93 | 7,07 |
| Chr1 | 10525 | F | 433 | 91,92 | 8,08 |
| Chr1 | 10497 | F | 435 | 88,74 | 11.26 |
Tabela 7: dados ERRBS representativos. Após o alinhamento de dados e citosina determinação metilação, dados par de bases é obtida Para cada CpG coberto, o protocolo de alinhamento como descrito irá determinar a genômica coordenada (colunas: chr = cromossomo, Base e Strand)., A taxa de cobertura do locus específico (Coverage ), e a taxa de citosina detecção com timidina como percentagem (freqC e freqT respectivamente).
| Número de bibliotecas ERRBS por pista | Número médio de alinhados exclusivamente lê | O número médio de CpGs coberta | Cobertura por CpG média |
| 1 | 152231184 ± 13189678 | 3.183.594 ± 713.547 | 85 ± 16 |
| 2 | 77680837 ± 7657058 | 2674823± 153494 | 49 ± 9 |
| 3 | 49938156 ± 2436865 | 2.552.186 ± - 76624 | 39 ± 2 |
| 4 | 34457208 ± 4441686 | 1.814.461 ± 144.339 | 28 ± 4 |
Tabela 8:. Parâmetros representativos de seqüenciamento bibliotecas individuais e multiplexados ERRBS É mostrado dados por faixa de 51 de ciclo único de leitura corridas de sequenciamento: média e desvios-padrão dos alinhado exclusivamente lê, número de CpGs cobertas e cobertura por site CpG obtido a partir de sequenciamento único bibliotecas ERRBS por pista (n = 100), dois ERRBS bibliotecas por pista (n = 128), três ERRBS bibliotecas por pista (n = 11), e quatro ERRBS bibliotecas por pista (n = 11).
Discussão
Os dados de resolução rendimentos de pares de bases de protocolo apresentada de metilação da citosina em regiões genômicas de interesse biológico. O protocolo como escrito é optimizada para 50 ng de material de partida, no entanto, ele pode ser adaptado para tratar uma variedade de material de entrada (5 ng ou mais) 26. Isto irá necessitar de ajustes de algumas das etapas do protocolo como pode ser visto na Tabela 6. As bibliotecas são passíveis de ERRBS emparelhado sequenciação extremidade e uma cobertura adicional genómico também pode ser conseguida por sequenciação lê mais do que 51 ciclos. Seqüenciamento multiplexado vai oferecer um protocolo de menor custo por amostra, no entanto, isso vai resultar em uma cobertura reduzida por site CpG representado nos dados (Figura 5 e Tabela 8), e não vai render profundidade suficiente de cobertura para realizar análises que requerem alta cobertura por sítio CpG (por exemplo, como descrito por Landan et ai. 33). Finalmente, este protocolo (ou qualquer Protoco à base de bissulfitol) não pode distinguir entre metil-citosina e de citosina hidroximetil-46,47. No entanto, os dados gerados podem ser integrados com outro protocolo resulta 48,49 para delinear as diferentes modificações, e outras modificações citosina recentemente relatado 50, eles devem ser de interesse.
Bibliotecas de alta qualidade irá aparecer como mostrado na Figura 3A-C, e uma vez reunidas para sequenciação origina um traço tal como mostrado na Figura 3G (traço vermelho) representando contribuições molares iguais de ambas as fracções da biblioteca. Falha preparação da biblioteca pode resultar de qualquer passo durante o processo. Se DNA degradado é processada que irá resultar em bibliotecas que não são enriquecidas em fragmentos MspI e, portanto, em baixa cobertura CpG utilizando os parâmetros de sequenciamento descritos neste protocolo. Se uma enzima é não-funcional ou excluídas inadvertidamente uma das reacções, o protocolo não irá produzir a biblioteca esperado. Se a rea ligaduracção é ineficiente, adaptadores estão em uma concentração mais elevada do que o esperado, e / ou a concentração de iniciadores utilizado é um reagente limitante para as etapas finais de amplificação, pode ocorrer falha na biblioteca. Adaptadores excesso (visto como um picos em ~ 150 pb em resultados Bioanalyzer; Figura 3D-F) na biblioteca também irá interferir com a sequenciação devido à aglomeração indiscriminada tanto da biblioteca e adaptadores em excesso. Embora uma tal biblioteca pode sequenciar aparentemente normalmente, uma porção significativa da lê será meramente sequências adaptadoras. Se os adaptadores em excesso são observados em uma biblioteca, o melhor é repetir a preparação biblioteca se o material está disponível usando material de entrada ideal para o adaptador relações de quantidades. Finalmente, para assegurar a eficiência de amplificação por PCR das bibliotecas, as fracções da biblioteca mais baixas e mais altas são mantidas como amostras separadas em toda a conversão de bissulfito e passos de enriquecimento de PCR. Não fazer isso gera eficiência diferencial de amplificação durante a PCR reacção das fracções superiores e inferiores (como visto na Figura 3G azul traço) e o potencial para a representação desigual do respectivo locus genómico coberto em cada fracção da biblioteca durante a sequenciação. O utilizador pode optar por incluir um passo de PCR quantitativo imediatamente após a conversão para além de bissulfito de titulação de ciclos de PCR ideais necessárias para amplificar as bibliotecas sendo gerado.
ERRBS protocolo de preparação biblioteca tem vários passos-chave em que são recomendados reagentes específicos. No passo final de reparação, o uso de uma mistura de dNTP quatro nucleótidos permite a fim de reparação de quaisquer produtos que não contenham a saliência CG, tais como os resultantes de MspI actividade enzimática estrela e fragmentos de ADN cortados presentes na amostra original do ADN. Isto resulta numa melhoria representação de CpG nos resultados. No passo de ligação é crítica a utilização de uma ligase de alta concentração (2.000.000 unidades / ml) e adaptadores metilados para assegurar que a ligadura Reactiem é eficiente e que a conversão de bissulfito não influencia as sequências essenciais para o alinhamento preciso do adaptador de dados. No passo de PCR, usando uma polimerase capazes de amplificar fragmentos de DNA rico em GC tratados com bissulfito é necessária para uma elevada especificidade. Finalmente, para garantir a eliminação dos adaptadores em excesso e os iniciadores, SPRI purificação talão (por exemplo: Agencourt AMPure XP) é recomendado em vez de ensaios baseados em colunas de ligação e os isolamentos de produtos de PCR.
A fim de gerar dados de alta qualidade, é importante para garantir a conversão eficiente bissulfito. O controle apresentou oferece ao usuário a capacidade de determinar a eficiência da conversão antes da sequenciação. Como uma alternativa, um ADN não-humana, tais como DNA de lambda pode ser usado como um controlo interno (espeta-in). Devido às diferenças de espécies, este tipo de controlo pode ser directamente incluído na sequenciação a jusante (por exemplo, tal como utilizado por Yu, et ai. 34). No entanto, se o pico de i-ins utilizados, que não pode ser usado para determinar a eficiência de conversão antes da sequenciação, a menos biblioteca amplificada exclusivamente e sequenciado antes da sequenciação biblioteca de forma independente. As taxas de conversão são determinadas com base no estado de metilação em locais que não de CpG. Isto pode não ser adequado para utilização no contexto de alta metilação da citosina no contexto não CpG (por exemplo, células estaminais embrionárias) e amostras paralelas ou qualquer outro meio de avaliar a eficiência de conversão pode ser utilizado para este fim.
Há algumas ressalvas para o endereço que são exclusivos para o seqüenciamento de bibliotecas ERRBS. As primeiras três bases das fracções da biblioteca sequenciados são quase uniformemente não aleatória devido ao local de reconhecimento do MspI corte (C ^ CGG; ver Figura 4B, C). Isso resulta na possibilidade de perda de dados significativa devido à baixa qualidade lê resultantes de uma má localização do cluster apesar de aparente alta densidade de agrupamento durante o seqüenciamento. Para superar essa barreira,incluir uma biblioteca de alta complexidade em uma pista independente (controle phix ou outro tipo de biblioteca) como uma pista de controle dedicado. Bibliotecas alta complexidade têm extremidades contendo uma representação equilibrada de A, C, T e G nas quatro primeiras bases sequenciadas. Faixas de controlo adequados incluem bibliotecas como RNA-seq, ChIP-seq, seqüenciamento do genoma inteiro, ou um controle oferecido pelo fabricante de máquinas de seqüenciamento (eg v3 Controle phix). Quando designado como uma pista de controlo para o respectivo prazo de sequenciação, que pode servir como base para a geração de matriz que é utilizado durante as primeiras quatro bases da sequenciação para detectar posições de fragmentação. A maior qualidade lê capturado elevará a cobertura média por local CpG em 5,2 (n = 4). Alternativamente, esta dificuldade técnica, também podem ser ultrapassadas pela utilização de uma abordagem de sequenciação escuro como previamente descrito 23. Outros critérios de sequenciamento seguir os procedimentos operacionais padrão por protocolos do fabricante. Finalmente, a cobertura por CpG chosen para análise de dados será guiado pelo usuário e, em parte, pelas questões biológicas de interesse. Limite de cobertura de 10x oferece uma abordagem de análise de alta cobertura, no entanto, este limite pode ser reduzido que deveria ser de interesse.
Uma discussão completa de análise de dados ERRBS está além do escopo deste artigo, no entanto, diferencialmente cytosines e regiões metilados pode ser determinada utilizando ferramentas de código aberto 31,51-53. Análise considerações adicionais e abordagens têm sido bem descrita 54,55, eo leitor é encorajado a procurar a literatura para instrumentos mais adequados para a análise planejada.
Comparado a outros métodos publicados, ERRBS oferece um protocolo de quatro dias, que quando realizada como rendimentos descritas altas taxas de reprodutibilidade. Ele foi validado em comparação com o padrão ouro MassARRAY EpiTYPER 26, é para dados de alta cobertura de baixo custo, e é adaptável para diversos materiais de entradaquantidades (favoráveis para processamento de amostras clínicas e outros tipos de células de baixa frequência) e as abordagens de sequenciamento. Ele oferece resolução de pares de bases em loci biologicamente relevante e pode ser usado em integrativa analisa com outras técnicas de perfil fator de todo o genoma de transcrição, a remodelação da cromatina, marcas epigenéticas e outras modificações citosina de interesse. Uso de dados ERRBS em tais estudos podem contribuir para uma abordagem molecular abrangente e permitir alta analisa dimensional no estudo de modelos biológicos e doenças humanas.
Divulgações
Os autores não têm conflitos de interesse a divulgar.
Agradecimentos
We thank all the authors of the original ERRBS report. We thank Mame Fall for technical assistance. We acknowledge the Weill Cornell Medical College Epigenomics Core for technical services and assistance. The work was supported by a Sass Foundation Judah Folkman Fellowship, an NCI K08CA169055 and ASH-AMFDP12005 to FGB, NIH R01HG006798 and R01NS076465, funding from the Irma T. Hirschl and Monique Weill-Caulier Charitable Trusts and STARR Consortium (I7-A765) to CEM, and an LLS SCORE grant (7006-13) to AMM.
Materiais
| Name | Company | Catalog Number | Comments |
| MspI | New England Biolabs | R0106M | 100,000 units/ml |
| NEBuffer 2 | New England Biolabs | B7002S | Reaction buffer for MspI enzyme; protocol step 1.2 |
| Phenol solution | Sigma-Aldrich | P4557 | Equilibrated with 10 mM Tris HCl, pH 8.0; see safety and handling instructions at http://www.sigmaaldrich.com/catalog/product/sigma/p4557 |
| Chloroform | Sigma-Aldrich | C2432 | See safety and handling instructions at http://www.sigmaaldrich.com/catalog/product/sial/c2432 |
| Glycogen | Sigma-Aldrich | G1767 | 19-22 mg/ml |
| NaOAc | Sigma-Aldrich | S7899 | 3 M, pH 5.2 |
| Ethanol | Sigma-Aldrich | E7023 | 200 proof, for molecular biology |
| Buffer EB | Qiagen | 19086 | 10 mM Tris-Cl, pH 8.5 |
| tris(hydroxymethyl)aminomethane (Tris) | Sigma-Aldrich | T1503 | prepare a 1 M, pH 8.5 solution |
| Tris- Ethylenediaminetetraacetic acid (TE) | Sigma-Aldrich | T9285 | Dilute to 1x buffer solution per manufacturer's recommendations |
| T4 DNA Ligase Reaction Buffer | New England Biolabs | B0202S | 10x concentration |
| Deoxynucleotide triphosphate (dNTP) Solution Mix | New England Biolabs | N0447L | 10 mM each nucleotide |
| T4 DNA Polymerase | New England Biolabs | M0203L | 3,000 units/ml |
| DNA Polymerase I, Large (Klenow) Fragment | New England Biolabs | M0210L | 5,000 units/ml |
| T4 Polynucleotide Kinase | New England Biolabs | M0201L | 10,000 units/ml |
| QIAquick PCR Purification Kit | Qiagen | 28104 | Used for DNA product purification in protocol step 2.3 |
| 2'-deoxyadenosine 5'-triphosphate (dATP) | Promega | U1201 | 100 mM |
| Klenow Fragment (3'→5' exo-) | New England Biolabs | M0212L | 5,000 units/ml |
| MinElute PCR Purification Kit | Qiagen | 28004 | Used for DNA product purification in protocol step 3.3 |
| T4 DNA Ligase | New England Biolabs | M0202M | 2,000,000 units/ml |
| Methylation Adapter Oligo Kit | Illumina | ME-100-0010 | |
| Agencourt AMPure XP | Beckman Coulter | A63881 | Used in protocol sections that implement magnetic bead purification steps (steps 4.3 and 8.2). Equilibrate to room temperature before use. |
| Pippin Prep Gel Cassettes, 2% Agarose, dye-free | Sage Science | CDF2010 | with internal standards |
| Certified Low Range Ultra Agarose | Bio-Rad | 161-3106 | |
| Tris-Borate-EDTA (TBE) buffer | Sigma-Aldrich | T4415 | |
| Ethidium bromide solution | Sigma-Aldrich | E1510 | 10 mg/ml |
| 50 bp DNA Ladder | NEB | N3236S | |
| 100 bp DNA Ladder | NEB | N3231S | |
| Gel Loading Dye, Orange (6x) | NEB | B7022S | |
| Scalpel Blade No. 11 | Fisher Scientific | 3120030 | |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | |
| EZ DNA Methylation Kit | Zymo Research | D5001 | Used in protocol step 6.2 |
| EZ DNA Methylation-Lightning Kit | Zymo Research | D5030 | Alternative for step 6.2 |
| Universal Methylated Human DNA Standard | Zymo Research | D5011 | Used as bisulfite conversion control |
| FastStart High Fidelity PCR System | Roche | 03553426001 | |
| Qubit dsDNA High Sensitivity Assay Kit | Life Technologies | Q32854 | A fluorescence-based DNA quantitation assay; used in protocol steps 1.1, 9.1 and 10.1 |
| DynaMag-2 Magnet | Life Technologies | 12321D | |
| High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 | |
| 2100 Bioanalyzer | Agilent Technologies | ||
| PhiX Control v3 | Illumina | FC-110-3001 | |
| HiSeq 2500 | Illumina | ||
| Pippin Prep | Sage Science | ||
| Qubit 2.0 Fluorometer | Life Technologies | Q32872 | |
| TruSeq SR Cluster Kit v3-cBot-HS | Illumina | GD-401-3001 | |
| TruSeq SBS Kit v3-HS | Illumina | FC-401-3002 | |
| TruSeq RNA Sample prep | Illumina | RS-122-2001 | Barcoded adapters used for multiplexing libraries; See Supplemental file for multiplexing protocol. |
| Microcentrifuge | |||
| Vortex Mixer | |||
| Dry Block Heater | |||
| Thermal Cycler | |||
| Water Bath | |||
| Gel electrophoresis system | |||
| Electrophoresis power supply | |||
| Gel doc | |||
| UV or blue light transilluminator |
Referências
- Jones, P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 13 (7), 484-492 (2012).
- Barlow, D. P. Genomic imprinting: a mammalian epigenetic discovery model. Annual Review Of Genetics. 45, 379-403 (2011).
- Thiagarajan, R. D., Morey, R., Laurent, L. C. The epigenome in pluripotency and differentiation. Epigenomics. 6 (1), 121-137 (2014).
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 447 (7143), 425-432 (2007).
- Hartnett, L., Egan, L. J. Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis. 33 (4), 723-731 (2012).
- Smith, Z. D., Meissner, A. DNA methylation: roles in mammalian development. Nat Rev Genet. 14 (3), 204-220 (2013).
- Li, E., Bestor, T. H., Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 69 (6), 915-926 (1992).
- Okano, M., Bell, D. W., Haber, D. A., Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 99 (3), 247-257 (1999).
- Feinberg, A. P. Phenotypic plasticity and the epigenetics of human disease. Nature. 447 (7143), 433-440 (2007).
- Bock, C. Epigenetic biomarker development. Epigenomics. 1 (1), 99-110 (2009).
- Laird, P. W. The power and the promise of DNA methylation markers. Nat Rev Cancer. 3 (4), 253-266 (2003).
- How Kit, A., Nielsen, H. M., Tost, J. DNA methylation based biomarkers: practical considerations and applications. Biochimie. 94 (11), 2314-2337 (2012).
- Mikeska, T., Bock, C., Do, H., Dobrovic, A. DNA methylation biomarkers in cancer: progress towards clinical implementation. Expert Review Of Molecular Diagnostics. 12 (5), 473-487 (2012).
- Gyparaki, M. T., Basdra, E. K., Papavassiliou, A. G. DNA methylation biomarkers as diagnostic and prognostic tools in colorectal cancer. Journal of Molecular Medicine. 91 (11), 1249-1256 (2013).
- Figueroa, M. E., et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 17 (1), 13-27 (2010).
- Heyn, H., Mendez-Gonzalez, J., Esteller, M. Epigenetic profiling joins personalized cancer medicine. Expert review of Molecular Diagnostics. 13 (5), 473-479 (2013).
- Kulis, M., Esteller, M. DNA methylation and cancer. Advances in Genetics. 70, 27-56 (2010).
- Xiong, Z., Laird, P. W. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 25 (12), 2532-2534 (1997).
- Meissner, A., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33 (18), 5868-5877 (2005).
- Gu, H., et al. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 6 (4), 468-481 (2011).
- Bock, C., et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 28 (10), 1106-1114 (2010).
- Harris, R. A., et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 28 (10), 1097-1105 (2010).
- Boyle, P., et al. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol. 13 (10), R92(2012).
- Chatterjee, A., Rodger, E. J., Stockwell, P. A., Weeks, R. J., Morison, I. M. Technical considerations for reduced representation bisulfite sequencing with multiplexed libraries. Journal of Biomedicine & Biotechnology. 2012, 741542(2012).
- Lee, Y. K., et al. Improved reduced representation bisulfite sequencing for epigenomic profiling of clinical samples. Biological Procedures Online. 16 (1), 1(2014).
- Akalin, A., et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 8 (6), e1002781(2012).
- Hatzi, K., et al. A Hybrid Mechanism of Action for BCL6 in B Cells Defined by Formation of Functionally Distinct Complexes at Enhancers and Promoters. Cell Reports. 4 (3), 578-588 (2013).
- Will, B., et al. Satb1 regulates the self-renewal of hematopoietic stem cells by promoting quiescence and repressing differentiation commitment. Nature Immunology. 14 (5), 437-445 (2013).
- Lu, C., et al. Induction of sarcomas by mutant IDH2. Genes Dev. 27 (18), 1986-1998 (2013).
- Kumar, R., et al. AID stabilizes stem-cell phenotype by removing epigenetic memory of pluripotency genes. Nature. 500 (7460), 89-92 (2013).
- Li, S., et al. An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinformatics. 14, Suppl 5. S10(2013).
- Patterson, K., Molloy, L., Qu, W., Clark, S. DNA methylation: bisulphite modification and analysis. Journal of Visualized Experiments. (56), 3170(2011).
- Landan, G., et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat Genet. 44 (11), 1207-1214 (2012).
- Yu, M., et al. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat Protoc. 7 (12), 2159-2170 (2012).
- Goecks, J., Nekrutenko, A., Taylor, J., Galaxy, T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11 (8), R86(2010).
- Dorff, K. C., et al. GobyWeb: simplified management and analysis of gene expression and DNA methylation sequencing data. PLoS One. 8 (7), e69666(2013).
- Roehr, J. T., Dodt, M., Ahmed, R., Dieterich, C. Flexbar − flexible barcode and adapter processing for next-generation sequencing platforms. MDPI Biology. 1 (3), 895-905 (2012).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal, North America. 17 (1), 10-12 (2011).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Needleman, S. B., Wunsch, C. D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 48 (3), 443-453 (1970).
- Krueger, F., Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27 (11), 1571-1572 (2011).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Res. 12 (6), 996-1006 (2002).
- Thorvaldsdottir, H., Robinson, J. T., Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics. 14 (2), 178-192 (2013).
- Team, R. C. R. A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. ISBN 3-900051-07-0, http://www.R-project.org (2012).
- Nestor, C., Ruzov, A., Meehan, R., Dunican, D. Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. BioTechniques. 48 (4), 317-319 (2010).
- Huang, Y., et al. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 5 (1), e8888(2010).
- Yu, M., et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 149 (6), 1368-1380 (2012).
- Song, C. X., et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 153 (3), 678-691 (2013).
- Ito, S., et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 333 (6047), 1300-1303 (2011).
- Akalin, A., et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13 (10), R87-1186 (2012).
- Stockwell, P. A., Chatterjee, A., Rodger, E. J., Morison, I. M. DMAP: Differential Methylation Analysis Package for RRBS and WGBS data. Bioinformatics. 30 (13), 1814-1822 (2014).
- Sun, D., et al. MOABS: model based analysis of bisulfite sequencing data. Genome Biol. 15 (2), R38(2014).
- Bock, C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 13 (10), 705-719 (2012).
- Rivera, C. M., Ren, B. Mapping human epigenomes. Cell. 155 (1), 39-55 (2013).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados