
Method Article
Quantitative Mass Spectrometric Profiling de proteomas de células do cancro da derivadas do líquido e tumores sólidos
Neste Artigo
Resumo
In-depth analyses of cancer cell proteomes facilitate identification of novel drug targets and diagnostic biomarkers. We describe an experimental workflow for quantitative analysis of (phospho-)proteomes in cancer cell subpopulations derived from liquid and solid tumors. This is achieved by combining cellular enrichment strategies with quantitative Super-SILAC-based mass spectrometry.
Resumo
Análises aprofundadas de proteomas de células de câncer são necessários para elucidar pathomechanisms oncogênicos, assim como para identificar alvos potenciais drogas e biomarcadores de diagnóstico. No entanto, os métodos para caracterização proteômica quantitativa de tumores derivados do paciente e em particular as suas subpopulações celulares são muito insuficientes. Aqui nós descrevemos um experimental set-up que permite a análise quantitativa de proteomas de subpopulações de células de câncer derivada de tumores líquidos ou sólidos. Isto é conseguido através da combinação de estratégias de enriquecimento celulares com espectrometria de massa baseia-Super-SILAC seguido por análise quantitativa dos dados de bioinformática. Para enriquecer subconjuntos celulares específicos, tumores líquidos são primeiro immunophenotyped por citometria de fluxo seguido de FACS-ordenação; para os tumores sólidos, por captura de laser de microdissecação é usado para purificar as subpopulações celulares específicos. Num segundo passo, as proteínas são extraídas das células purificadas e posteriormente combinados com um específico para o tumor,SILAC marcado padrão espiga em que permite a quantificação da proteína. A mistura de proteína resultante é submetido a electroforese em gel ou filtro ou Aided Preparação da Amostra (FASP) seguido por digestão tríptica. Finalmente, os péptidos trípticos são analisados utilizando um espectrómetro de massa de quadrupolo-Orbitrap híbrido, e os dados obtidos são processados com pacotes de software de bioinformática, incluindo MaxQuant. Por meio do fluxo de trabalho aqui apresentado, até 8000 proteínas podem ser identificados e quantificados em amostras derivadas de pacientes, e os perfis de expressão de proteínas resultantes podem ser comparados entre pacientes para identificar as assinaturas de proteómica ou de diagnóstico potenciais alvos de drogas.
Introdução
Proteômica baseada em espectrometria de massa surgiu e agora é uma disciplina amplamente utilizado em biologia celular e investigação biomédica translacional. Os avanços técnicos no campo, foi possível estudar os processos celulares complexos em linhas de células e modelos animais, como milhares de proteínas podem ser identificadas em um único experimento de espectrometria de massa 1,2. Progresso semelhante foi feita por análise de várias modificações pós-traducionais tais como a fosforilação ou ubiquitinação, embora isto geralmente requer fluxos de trabalho à medida para o enriquecimento e a análise dos dados para cada tipo de modificação 2,3. Além disso, o envolvimento das estratégias de rotulagem químicas e metabólicas, incluindo SILAC, permite a quantificação precisa relativa de proteínas e PTMs, tornando este método particularmente atraentes para a descoberta de processos celulares básicos, diagnóstico biomarcadores e alvos potenciais drogas em cancro humano 4.
No entanto,vários desafios precisam ser superados em relação à análise proteômica dos cânceres humanos primários 5. Em primeiro lugar, as amostras de cancro humano apresentam frequentemente um elevado grau de heterogeneidade celular que é devido à presença de vários tipos de células que pertencem ao microambiente tumoral, incluindo células do sistema imunológico e fibroblastos. Em segundo lugar, a evolução clonal leva a diversidade genética dentro de si os tumores, resultando na existência de várias subpopulações celulares com propriedades funcionais distintas. De acordo com o conceito atual de algumas células-tronco do câncer, sendo as forças motrizes do desenvolvimento e progressão do câncer, análise proteômica dessas subpopulações celulares (funcionalmente altamente relevantes) é esperado para ser de grande importância para uma melhor compreensão dos mecanismos oncogênicos relevante para a aplicação clínica 6. Em terceiro lugar, os perfis de expressão de proteínas quantitativos de grandes conjuntos de amostras são muitas vezes necessários para a identificação de Biomar clínico fiávelkers; esta não pode ser conseguida por marcação química, tais como iTRAQ 7, enquanto que as estratégias de marcação metabólica - contando, como o fazem, com a proliferação celular 4 - não têm aplicação. Em quarto lugar, a maioria das amostras de tumores sólidos que estão disponíveis são fixados em formalina, o que complica a análise de espectrometria de massa proteoma devido à formação de ligações cruzadas de proteína 8. Por fim, a maioria dos fluxos de trabalho existentes proteômica requerem quantidades significativas de processamento de amostras e aquisição de dados, tornando a análise dos números de amostras relevantes para a investigação clínica difícil, e chamando para novo fluxo de trabalho paradigmas 9,10.
Para enfrentar esses obstáculos, desenvolvemos uma montagem experimental que combina ou célula de citometria de fluxo de classificação 11 ou captura a laser microdissection 12,13 para o enriquecimento celular com uma estratégia de Super-SILAC 14 a introduzir uma norma interna global para análise por espectrometria de massa global. Ao utilizar o método aqui descrito, é possível quantificar até 8.000 proteínas em amostras de tumores humanos individuais derivadas de cancros, quer líquido ou sólido.
Protocolo
Todos os experimentos com tecidos ou sangue amostras humanas devem ser aprovados por um comitê de ética e conduzida de acordo com todas as orientações dadas na votação ética.
1. Cellular Enriquecimento
- Tumores líquidos / separação de células activadas por fluorescência.
- Isolar células mononucleares de medula óssea ou amostra de sangue através da realização de lise de eritrócitos com cloreto de amónio ou por gradiente de densidade de Ficoll centrifugação.
- Para a lise de eritrócitos, combinar um ml de amostra com 4 ml de solução de cloreto de amónio. Incubar a solução em gelo durante 5 - 10 min. Adicionar 40 ml de salino tamponado com fosfato (PBS) + 2% de soro fetal de vitelo (FCS) e centrifugar durante 5 minutos a 400 xg e 4 ° C.
- Para Ficoll centrifugação em gradiente, camada de 10 ml de amostra em 15 ml de FicollHypaque para formar duas camadas distintas (cuidado para não perturbar as camadas). Centrifuga-se a 400 xg durante 30 min à temperatura ambiente (15 - 25 ° C) cuidadosamente e colher os mononuclearescélulas recolhidas na interfase.
- Girar as células e levá-los até em PBS + 2% FCS. Centrifuga-se a 400 xg durante 5 min e tomar-se em 1 ml de PBS + 2% FCS. Tomar uma alíquota como controle negativo e uma alíquota para cada corante fluorescente usado na mancha final.
- Incubar com anticorpos marcados por fluorescência úteis para isolar a população de células leucémicas em diluição, de acordo com as instruções do fabricante em gelo durante 30 min. Também manchar as alíquotas utilizados para a compensação com cada um dos anticorpos individualmente. Lavar duas vezes com PBS + 2% de FCS.
NOTA: Os anticorpos utilizados foram de ratinho anti-humano contra CD117-PE (clone 95C3), CD34-FITC (clone 8G12) e CD33-PE (clone P67.6). - Células filtrar através de 35 um filtro de células, o ideal é usar tubos com filtro de células integradas no cap.
- Adicionar mancha de viabilidade celular (por exemplo, 7-AAD). Classificar células usando um separador de células adequado 11 (Figura 2). Recolher as células em Isc Modificado por Dulbecco de ove Médio (IMDM) contendo 10% de FCS.
- Isolar células mononucleares de medula óssea ou amostra de sangue através da realização de lise de eritrócitos com cloreto de amónio ou por gradiente de densidade de Ficoll centrifugação.
- Os tumores sólidos / captura de Laser microdissection.
- Para as experiências descritas, foram utilizadas amostras de FFPE de espécime de câncer de pulmão. Para esta finalidade tecido de cancro de pulmão foi imediatamente fixado em formalina a 4% tamponado após ressecção cirúrgica e pedaços de cerca de 3 x 2 x 1 cm em caso embebidos em parafina para análise posterior.
- Os cortes de 5-10 mm de espessura do embebido em parafina (FFPE) amostra fixado em formol com um micrótomo. Montagem de secções em lâminas de membrana coberta por película e seco a 37 ° C durante 1 h. Desparafinar e hidratar as secções montadas por incubação sucessiva em xileno, etanol absoluto, 70% e água, durante 1 min cada.
- Corar as secções com hematoxilina durante 20 segundos e, em seguida, enxaguar com água da torneira. Recolha a população de células de interesse através de um sistema de captura de microdissecção a laser (ver também 12).
- Tumores líquidos.
- Centrifuga-se a suspensão de células a partir do passo 1.1.5 a 400 xg, 4 ° C durante 4 minutos e desprezar o sobrenadante. Lavar duas vezes com 500 ul de PBS frio e centrifugação durante 5 min a 400 xg e 4 ° C
- Adicionar 40 ul de tampão de lise por 10 6 células e incubar durante 15 min em gelo (10 5 células (aprox. 10 ug de proteína total) deve ser o número mínimo de células). Centrifuga-se o lisado a 14.000 xg, a 4 ° C durante 10 minutos e transferir o sobrenadante (lisado celular limpo) para um novo tubo de reacção. Desprezar o pelete.
- Tumores sólidos.
- Adicionar 60 ul de tampão de lise do tecido com o tecido microdissecados e incubar durante 15 min em gelo, recolher o fluido por centrifugação curta e transferir a suspensão para um novo tubo de reacção. Sonicar o lisado em gelo durante 3 min.
- Adicionar 15 ul de 20% de dodecilsulfato de sódio (SDS) para uma alcançar um co final de SDSncentration de 4%. Incubar o tecido microdissecados a 99 ° C num bloco de aquecimento durante 1 hora e agitada a 600 rpm. Centrifuga-se o lisado a 16.000 x g, 18 ° C durante 10 minutos e transferir o sobrenadante para um novo tubo.
3. Estabelecimento de um SILAC de Spike-in Quantificação Padrão (Super-SILAC padrão)
NOTA: O padrão de quantificação consiste em uma mistura de proteínas marcadas com SILAC derivado 4-6 linhas de células que correspondem ao tipo de tumor de interesse. Para atingir a máxima sobreposição entre o proteoma de referência marcado com SILAC e do proteoma derivado de tumor, uma análise do componente principal deve ser realizada antes que as linhas de células são seleccionadas para o padrão de quantificação 14.
- Análise de componentes principais (PCA).
- Para determinar os padrões de linhas de células de expressão de proteínas, cultivar cerca de dez linhas de células diferentes em meios de cultura de células apropriado. Lisam as células como descritono passo 2.1. Prepare os lisados celulares para espectrometria de massa como descrito na etapa 6.1.
- Analisar o padrão de cada linha celular expressão da proteína em um de alta resolução, o espectrômetro acoplado à cromatografia líquida de massa (nanoLC-MS / MS).
- Analisar os dados brutos resultantes usando uma MaxQuant mercadorias livres e realizar uma análise principal-componente utilizando o software associado, como Perseus 16,17.
- Selecione 4-6 linhas celulares que mostram a maior diversidade entre os seus perfis de expressão de proteínas e usá-los para gerar o padrão spike-in Super-SILAC.
- SILAC-rotulagem e verificação de etiquetas.
- Cultivar as linhas celulares seleccionadas para, pelo menos, cinco ciclos de células em meio de cultura de células apropriado SILAC, em que a arginina e a lisina são marcados com isótopos estáveis de carbono e de azoto (meio SILAC) 4.
- Lise das células, tal como descrito no passo 3.1 e preparar lisados celulares por espectrometria de massa como descrito na etapa 6.1.Medir a eficácia de incorporação de rotulagem SILAC por nanoLC-MS / MS. Analisar os dados em bruto resultantes com MS MaxQuant e determinar a eficácia do processo de rotulagem SILAC. Isto é conseguido através da contagem do número de péptidos identificados na sua marcada (pesado) e as suas formas endógenas (luz), e o cálculo da razão (pesado) / (luz pesada +). Eficiência de marcação deve ser superior a 98%.
- Combinação de proteomas SILAC e validação.
- Cultive e expandir linhas de células seleccionadas de acordo com instruções do fabricante em meio SILAC apropriado. Lise das células, tal como descrito no passo 2.1 para tumores líquidos ou como descrito no passo 3.2 para tumores sólidos e determinar a concentração de proteína para cada lisado celular.
- Misture quantidades equimolares de proteína de cada linha celular e dividir a mistura em aliquota. Snap-congelar as alíquotas e armazenar a -80 ° C até que a medição. Para uma experiência que você precisa 20 - 50 mg de padrão Super-SILAC. Tome cuidado para preparar o standard em excesso como alterar o padrão dentro de uma série de experiências devem ser evitados.
4. Medição da concentração de proteína e Spike-in
- Tumores líquidos.
- Usando o ensaio de quantificação de proteína determinar as concentrações de proteína dos respectivos lisados celulares e o padrão Super-SILAC.
- Misture quantidades iguais de lisado celular foi afastada e o padrão Super-SILAC e, subsequentemente, adicionar o dodecil sulfato de lítio (LDS) tampão (25% do volume da amostra) e agente redutor (10% do volume da amostra).
- Aquece-se a solução resultante em um bloco de aquecimento a 72 ° C durante 10 min. Opcionalmente, armazenar as proteínas desnaturadas resultantes à temperatura de -80 ° C.
- Tumores sólidos.
- Para a medição da concentração de proteína num leitor de placas, misturar um padrão de albumina de soro bovino (BSA) a diluição série solução, o lisado e o padrão Super-SILAC apropriado com uma proteína comercialmente disponívelssay em uma placa de 96 poços, e agita-se durante 1 min, incubar durante tempo indicado e medir a absorvância, tal como indicado pelo fabricante.
NOTA: A alta concentração de SDS e ditiotreitol (DTT) no lisado é um problema para a maioria dos ensaios disponíveis para a determinação da concentração de proteína. - Misture quantidades iguais de lisado clarificado e o padrão Super-SILAC com 200 ul de ureia na unidade de filtro e centrifugar a 14000 xg durante 30 min a 20 ° C. Não use mais de 50 ul de ligado clarificado e o padrão Super-SILAC. Evitar a temperaturas abaixo de 15 ° C, de modo que a ureia não se cristalizar.
- Para a medição da concentração de proteína num leitor de placas, misturar um padrão de albumina de soro bovino (BSA) a diluição série solução, o lisado e o padrão Super-SILAC apropriado com uma proteína comercialmente disponívelssay em uma placa de 96 poços, e agita-se durante 1 min, incubar durante tempo indicado e medir a absorvância, tal como indicado pelo fabricante.
5. A separação da amostra e Proteína Digest
- Tumores líquidos.
- Separados 30 - 100 ug de proteína total por pista num 4 - gradiente de gel de SDS-PAGE a 12%. Proteínas mancha com Coomassie Blue O / N. Retire o excesso de corante Coomassie de duas lavagens subsequentes com água.
- Corte cada pista a partir do gel e dividi-loem 23 fatias de tamanho igual, independentemente do padrão de coloração gel. Processe as fatias de gel em separado, cada uma em 0,6 ml em frasco de polipropileno.
- Lavam-se as fatias de gel com água e metanol / água (50:50, v / v), reduzir com DTT 10 mM por incubação durante 30 min a 56 ° C. Alquilar as fatias de gel com 55 mM de iodoacetamida (IAA), por incubação de 60 min à temperatura ambiente no escuro.
- Entre os passos de manipulação de amostra, lavar com fatias de acetonitrilo durante 15 minutos e seco num SpeedVac para remover o excesso de solvente e melhorar a absorção da solução de reagente.
- Realizar de clivagem de protease através da hidratação fatias de gel foram secas com a quantidade mínima de solução de tripsina porcina (12,5 ng / mL em 0,025 M de bicarbonato de amónio aquoso) durante 16 horas a 37 ° C.
- Adicionar 10 ul de água a fatia de gel e incubar durante 15 min a 37 ° C. Adicionar 80 ul de acetonitrilo e incubar durante 15 min a 37 ° C. Centrifugar a 15.800 xg, por 1 min. Recolher o sobrenadante e armazenar em um separadas 0,6 mltubo.
- Adicionar 65 ul de 5% de ácido fórmico a solução, vortex e incubar durante 15 min a 37 ° C. Adicionar 65 ul de acetonitrilo e incubar durante 15 min a 37 ° C. Centrifugar a 15.800 xg por 1 min. Recolhe-se o sobrenadante e adicionar-se ao sobrenadante da etapa anterior. Evapora-se o sobrenadante combinado até à secura num concentrador de vácuo.
- Tumores sólidos.
- Para remover SDS de lisado tecido usar o seguinte protocolo FASP, também descrito como na referência 15.
- Depois da primeira centrifugação descrito no passo 5.2.3 adicionam-se mais 200 ul de ureia 8 M sobre o filtro, e ainda de centrifugação a 14000 xg durante 20 min a 20 ° C. Descartar o filtrado por escoamento.
- Adicionar 100 ul de IAA e misturar num termomisturador a 600 rpm durante 1 min. Incubar o filtro durante 20 min a 20 ° C no escuro. Centrifuga-se o filtro a 14000 xg durante 10 min a 20 ° C.
- Adicione 100 ml de ureia para o filtro e centrifuge a 14.000 xg, durante 15 min a 20 ° C. Repita este passo mais uma vez.
- Adicionar 100 ul de NH 4 HCO 3 ao filtro e centrifugar a 14000 xg durante 10 min a 20 ° C. Repita esta etapa duas vezes.
- Adicionar 40 ul de NH 4 HCO 3 + 1 ml (= 0,4 ug) de tripsina e misturar em termomisturador a 20 ° C durante 600 rpm, 1 min. Incubar o O filtro / N numa câmara húmida a 37 ° C. Transferir o filtro para novos tubos de recolha.
- Centrifuga-se o filtro a 14000 xg durante 10 min a 20 ° C. Adicionar 50 ul de NH 4 HCO 3 e centrifugar o filtro a 14000 g durante 10 min a 20 ° C. Armazenar os péptidos resultantes à temperatura de -20 ° C até à medição espectrométrica de massa.
- Para a medição da concentração de péptido, 50 uL de distribuir o fluxo de passagem e que resulta de uma série de diluições apropriadas de triptofano puro em uma placa de 96 poços. Medir a fluorescência do triptofano. Converter a concentração resultante detriptofano a concentração de péptido, como 0,1 ug de triptofano corresponde a 9 ug de proteína.
6. Análise de Cromatograf ia Líquida e Espectrometria de Massa
- Redissolver peptídeos em tampão de carga de 30 ul durante 5 minutos num banho de ultra-sons. Girar para baixo numa centrífuga a 15.800 xg durante 1 min e pipetar a solução límpida em um frasco de auto-amostrador MS.
- Injectar 5 ul de amostra por análise usando o amostrador automático do sistema nanoLC-MS / MS. Concentre-se e peptídeos dessalinizar on-line em uma fase reversa C18 pré-coluna (0,15 mm de diâmetro x 20 mm com 5 mm de tamanho de poros do material C18) montado em qualquer uma configuração de coluna ventilada ou uma configuração pré-coluna de comutação.
- Péptidos separados numa fase inversa C18 Microcoluna (0,075 mm Dl x 200 mm de auto-embalada com 3 mm ou menor tamanho de poros do material numa coluna C18 de nanospray auto-pack tais como PicoFrit, utilizando um gradiente de 90 min de 5> acetonitrilo a 35% vs 0,1% de ácido fórmico. aquosa a 300 nl /min. Transferência eluente em um quadrupolo híbrido / espectrômetro de massa Orbitrap através de uma fonte de íons nanospray.
- Analisar peptídeos usando um método de dados Top15 dependente aquisição (MS m / z gama 350 -. 1600, alvo resolução 70.000 FWHM, alvo AGC 1 x 10 6, tempo de enchimento máximo 60 ms MS / MS começar a massa 100, alvo resolução 17.500 FWHM, AGC Meta 2 x 10 5, tempo de preenchimento máximo 60 ms MS / MS limite de 3 x 10 4, incluem estados cobram 2 -. 5, Normalizado Collision Energia NCE 25%, dinâmica de exclusão de 15 segundos).
Análise 7. Dados
- Para análise dos dados usar o software MaxQuant 16 disponíveis gratuitamente. Um protocolo detalhado para a análise dos dados é descrito em bioinformática 16,17. Para a análise, combinar arquivos de dados brutos de todas as fatias de uma pista SDS-PAGE em um único experimento 17.
Resultados
Perfilamento Proteomic de tumores líquidos e sólidos a partir de pacientes é uma abordagem promissora para a descoberta de novos biomarcadores preditivos e de diagnóstico. No entanto, o procedimento de preparação da amostra e análise de espectrometria de massa são um desafio, devido à complexidade das amostras e a necessidade de quantificar a proteína preciso em grandes conjuntos de amostras. O procedimento experimental descrito acima começa com o isolamento de células de interesse, quer por escolha de células activado por fluorescência ou por captura a laser microdissecação.
Para esta finalidade, as células de aspirados de medula óssea derivadas de doentes ou de amostras de sangue foram coradas com anticorpos marcados com fluorescência contra marcadores de superfície definidos de interesse antes de triagem baseada em FACS e lise celular.
Para enriquecer tipos celulares a partir de cortes de tecidos, estes têm que ser primeiramente montadas em lâminas adequadas para permitir a captura a laser microdissection. Para esta finalidade t montarele secções de tecido em lâminas de membrana apropriados e utilizar uma captura por laser microdissector de acordo com as instruções do fabricante. A seleção das áreas e células de interesse requer um conhecimento adequado sobre o histomorfologia de tecido normal e neoplásico. As células extraídas são então transferidos para um tubo de recolha apropriado. As experiências iniciais deve ser realizada a fim de definir a quantidade de tecido necessário para a extracção de uma quantidade suficiente de proteína (10 pg de proteína mínima total) para cada novo tecido de interesse. Cada amostra microdissecados é tratada com 60 ul de tampão de lise do tecido. Para a lise eficiente, células de amostras de tecido têm de ser ultra-sons durante 3 minutos e após a adição de 15 ul de SDS amostras são incubadas num termomisturador a 99 ° C durante 1 h para remover as reticulações da proteína induzida por formalina. Centrifugação vai finalmente facilitar a recolha do ligado celular desmarcada.
Para cada doença de interest (líquido e tumores sólidos) a Super-SILAC padrão quantificação adequada tem de ser estabelecido 14. Um padrão adequado devem representar mais de 90% das proteínas expressas nas amostras de interesse. Para a preparação de um padrão de quantificação, as linhas celulares relacionadas ao câncer de interesse deve primeiro ser analisados por espectrometria de massa para determinar os seus perfis de expressão de proteínas e análise de componentes principais, posteriormente, deve ser realizada. 4 - 6 linhas celulares diferentes, com padrões de expressão diferencial da proteína deve ser escolhido e SILAC-arginina marcada com "pesadas" e lisina. Eficiência de marcação deve ser verificada por nanoLC-MS / MS e deve ser maior do que 98%. Em seguida, as linhas celulares deverão ser lisadas na mesma maneira como a respectiva amostra derivada de paciente e, subsequentemente, quantidades equimolares de proteína de cada amostra e o padrão espiga em SILAC deve ser misturado. Um passo crítico para a quantificação de proteína precisa de amostras clínicas of interesse é a mistura precisa de quantidades de proteína equimolares das amostras derivadas de pacientes e o SILAC spike-in padrão de quantificação. Para determinar a concentração de proteína de lisados derivados de células classificadas, vários ensaios de quantificação de proteína pode ser utilizado. Para os lisados celulares a partir de células derivadas de tecido é necessário um ensaio que pode lidar com altas concentrações de SDS e DTT no lisado. As concentrações de proteína das amostras derivadas de pacientes e o padrão espiga em SILAC deve ser medido antes de cada vez combinação com as respectivas amostras clínicas, a fim de assegurar a mistura correcta, que é fundamental para fazer mesmo centenas de amostras comparáveis.
Depois de a mistura de quantidades iguais de digestão espiga em amostras padrão e de lisado, obtidos a partir de tumores líquidos são misturados com LDS, aquecidas num termomisturador a 72 ° C durante 10 min e, subsequentemente, submetidas a 1D-PAGE e em gel do separadas proteínas com tripsina. As amostras obtidas a partir detecidos são liberados a partir de SDS e digeridas com tripsina usando a abordagem FASP, conforme estabelecido pela Wisniewski et al. 15
Os péptidos trípticos obtidos podem ser quantificados por medição da fluorescência de triptofano, que é de interesse especial para amostras de tecidos como a quantidade de péptidos libertados do filtro difere entre 15% e 75% em comparação com as quantidades de proteína inicialmente carregados no filtro. Para este efeito, podemos medir a fluorescência do triptofano. A comparação com uma série de diluições de triptofano apropriado permite a quantificação dos peptídeos, como 1,1 ug de triptofano corresponde a cerca de 100 ug de péptido. Se necessário, as amostras derivadas de processamento especialmente FASP pode ser pré-purificado em qualquer estágio dicas comerciais ou caseiros embalados com (RP-C18) de material de fase inversa C18 antes de o colocar no sistema nanoLC / MS / MS 18.
A análise por espectrometria de massa é realizada num elevado-Resolução, sistema de espectrometria de massa de alta sensibilidade. Em resumo, as amostras de péptidos são dessalinizados e preconcentrados sobre uma pré-coluna RP-C18, e separado numa coluna analítica RP-C18 acoplado directamente ao espectrómetro de massa. Para atingir profundidade suficiente de análise, que empregam ou uma combinação de SDS-PAGE prefraccionamento proteína com 40 min gradientes RP-C18 (para tumores líquidos) ou injeções individuais com longas 2-3 hr gradientes RP-C18 (para tumores sólidos tratados pela FASP ). MS espectros foram adquiridos com resolução de 70.000 FWHM ou melhor, para permitir a quantificação precisa de pares SILAC por integração dos picos de perfis cromatográficos. Para a identificação de proteínas, um método de aquisição dependente dos dados Top15 é usado para gerar um grande número de péptido espectros MS / MS para a identificação de proteínas e de péptidos.
A partir dos dados brutos resultantes, identificação e quantificação de proteínas são alcançados pelo sistema de dados com o software de procura MaxQuant contra um UniProt Knowledgebase banco de dados Human Proteome seqüência completa 17 anos. Em MaxQuant software (versão actual 1.5.0.25) são péptidos identificados a partir da adquirida espectro MS / MS pelo seu fragmento de péptido correspondente contra espectros in silico derivado da base de dados de sequência de proteína. Ao mesmo tempo, um precursor de ião perfis isotópicos são extraídos em torno dos seus tempos de retenção cromatográficos, e as suas áreas de pico integradas são utilizados para a quantificação relativa da luz: pares de péptidos pesados gerados por meio de rotulagem SILAC. Identidades de péptidos e as intensidades relativas são então atribuídos às propriedades correspondentes de proteína. Software Perseus (atual versão 1.5.0.15) é então usado para realizar uma avaliação mais aprofundada estatística jusante dos resultados do processamento MaxQuant, incluindo amostra-a-amostra comparações, PCA e de agrupamento hierárquico.
Utilizando a montagem experimental descrita nós identificaram e quantificaram-se a 8.000 proteínas de tão pouco como 30 ug de proteína totalderivadas de tumores líquidos.
Até 2500 proteínas a partir de amostras de tumores sólidos podem ser identificados e quantificados numa abordagem proteómica espingarda com um gradiente de LC de apenas 2 horas, permitindo a análise de centenas de amostras clínicas de um tempo relativamente curto.

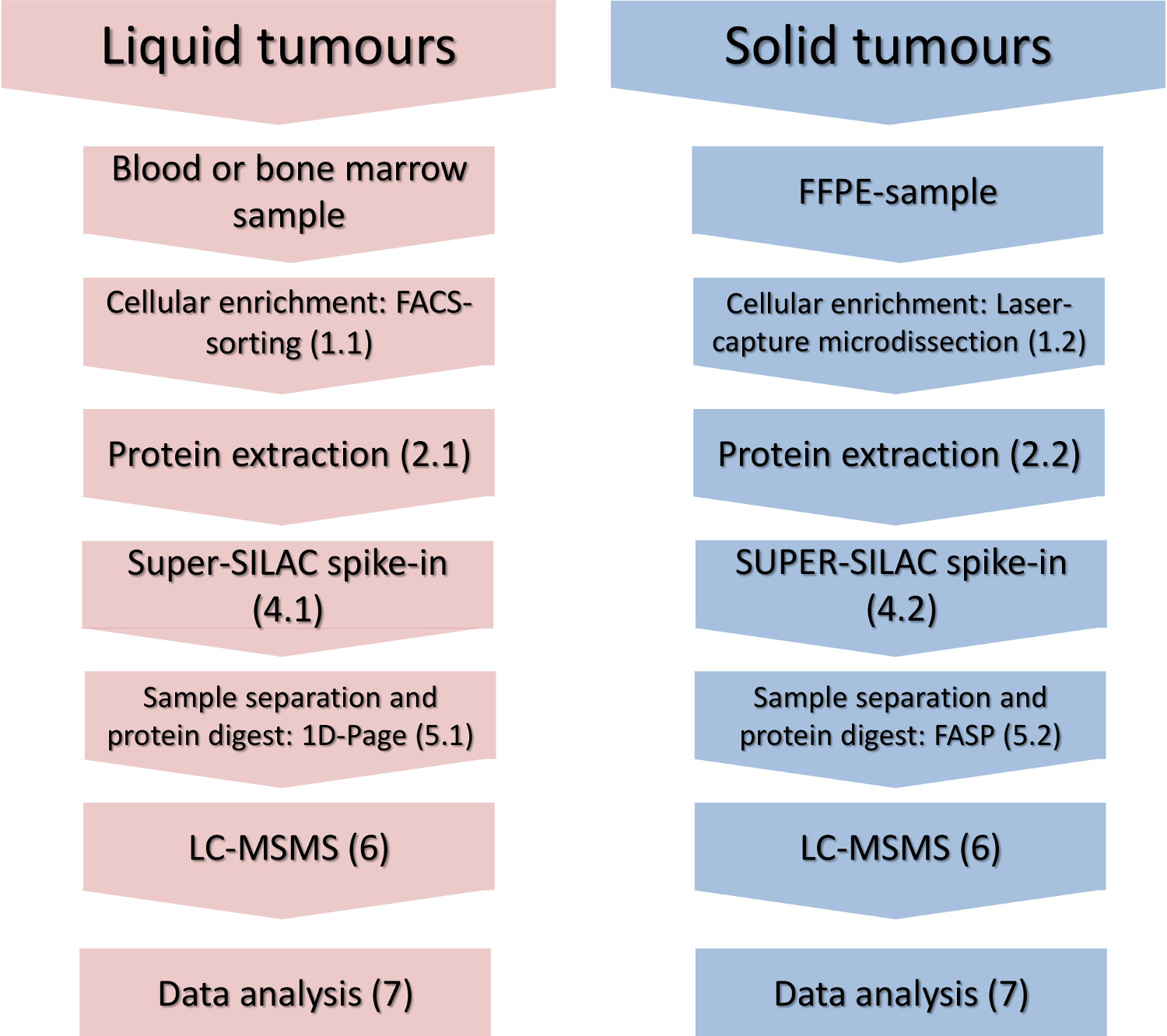
Figura 1. fluxo de trabalho Experimental. As principais etapas de enriquecimento celular-subconjunto, isolamento de proteínas, spike-in do padrão de quantificação e análise por espectrometria de massa são mostrados para os tumores sólidos e líquidos. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}

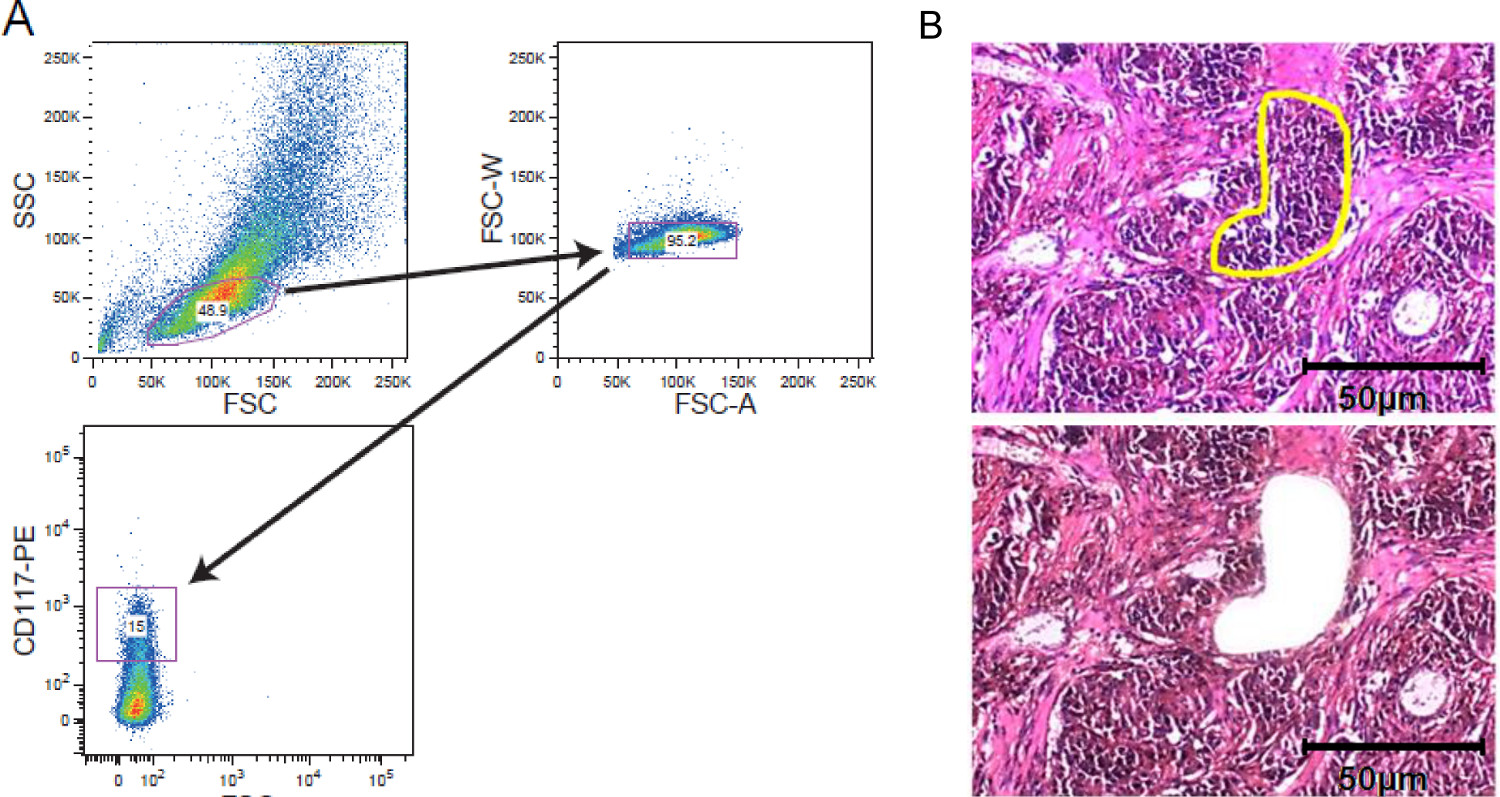
Figura 2. Cellular strategi enriquecimentoes. FACS triagem e LCM. (A) a estratégia gating Exemplar retratando gating em uma população de células leucêmicas manchada de CD117 adquirida por um separador de células. (B) microdissection captura Laser de tecido do tumor sólido. As secções de tecido de 5 - 10 mm de espessura foram montadas em lâminas de membrana coberta por película antes da coloração. Região de interesses foi selecionado manualmente para captura a laser microdissection. São mostrados seções antes microdissection com a região de interesse marcado amarelo e depois microdissection. A barra de escala de 50 mm está incluído na figura. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3. A análise de agrupamento de proteomas de células humanas de câncer. Análise de agrupamento não supervisionado de prperfis de expressão otein de tumores sólidos e líquidos foi realizada utilizando a plataforma computacional Perseus. AC - Adenocarcinoma, SCC - carcinoma de células escamosas, SCC-Met - metástases carcinoma de células escamosas de cabeça e pescoço, R - replicar técnico.
Discussão
Perfis de espectrometria de massa de proteomas-celulares de cancro derivadas de pacientes é necessária para a descoberta de novos biomarcadores preditivos de diagnóstico / bem como para se obter uma melhor compreensão da biologia das células do cancro, que pode por sua vez conduzir à identificação de novos alvos potenciais drogas. No entanto, tais análises de espectrometria de massa são altamente desafiador, em particular porque várias questões pré-analíticos têm de ser resolvidos, se se quiser obter resultados robustos e biologicamente relevantes.
O fluxo de trabalho experimental descrito aqui permite a caracterização proteômica quantitativa de proteomas derivados de subpopulações celulares de ambos os tumores sólidos e líquidos. É necessário efectuar o enriquecimento inicial de células tumorais, quer por FACS com base nas células-sortingor microdissecação para evitar a contaminação por células do microambiente do tumor. Além disso, estas técnicas permitem isolar subpopulações celulares de interesse. Estudos com células-biológico recentes têm demôniostrated que determinadas subpopulações celulares têm propriedades de iniciação do tumor e são, portanto, altamente relevante para a patogênese do câncer 19,20. Como espectrometria de massa tornou-se mais sensível nos últimos anos, as análises proteômicas quantitativos são viáveis para as pequenas quantidades de proteína que podem ser derivadas de alguns milhares de células, tornando possível para se concentrar em populações de células funcionalmente relevantes.
A configuração aqui apresentado pode ser utilizado para identificar e validar novos biomarcadores de diagnóstico em amostras de FFPE. Por isso, promete bea ferramenta útil para a melhoria do diagnóstico clínico, a data ainda há uma falta de biomarcadores moleculares em número e em qualidade adequada para vários tipos de câncer. Exemplos importantes de diagnósticos diferenciais difíceis, para os quais biomarcadores estão faltando, são a discriminação entre câncer de pulmão primário de metástases no pulmão, carcinoma colangiocelular intrapancreatic e adenocarcinoma do pâncreas, assim como diferemciação de neurofibroma entre tumores benignos e de bainha de nervo periférico altamente malignas. Além disso, nós e outros demonstraram que a elucidação quantitativa de assinaturas proteomic pode ser útil no estudo da biologia celular de cancro em geral, e para revelar biomarcadores preditivos da resposta terapêutica em doentes com cancro 21.
Duas desvantagens atuais do método aqui apresentado são a exigência de ampla amostra de transformação manual e a demanda de nanoLC-MS / MS tempo de aquisição. Enquanto o primeiro pode ser abordada, movendo a preparação da amostra para por exemplo, formatos de 96 poços e usando processamento robótico, este último vai exigir uma mudança na estratégia de aquisição de espectrometria de massa. Uma vez que os subconjuntos de proteínas-alvo foram identificadas, que pode estar associada com, por exemplo, classificação de tumores, prevemos a concepção de métodos de espectrometria de massa-alvo que fornecem leituras quantitativas para estes subconjuntos com um esforço bastante reduzido de separação, eportanto, com a correspondente redução do tempo de aquisição. Se o tempo de aquisição necessária requerida pode ser reduzida 24-36 hr (tumores líquidos) ou 3 h (tumores sólidos) para, por exemplo, 1 hora usando espectrometria de massa e a separação de péptidos unidimensional simples, então o ganho em rendimento resultante alvo Pode ser utilizado para aumentar significativamente o número de réplicas biológicas e técnicas examinados, com a melhoria correspondente na significância dos resultados de quantificação. Abordagens de espectrometria de massa visados já foi demonstrado ser uma ferramenta adequada para a verificação de associados a cancro da proteína biomarcadores candidatos-22, e têm sido desenvolvidos para um ponto onde eles são promissores para a validação ou mesmo como uma potencial ferramenta para o uso clínico de rotina 23 , 24.
Divulgações
Os autores não têm qualquer conflito de interesses ou outros assuntos para divulgar.
Agradecimentos
The authors thank Uwe Plessmann, Monika Raabe und Silvia Münch for technical support.
Materiais
| Name | Company | Catalog Number | Comments |
| 660 nm Kit | Thermo scientific | 22662 | |
| Cell culture medium depleted of arginine and lysine | Thermo Scientific | 88421 | |
| Coomassie Brilliant Blue R-250 staining solution | Bio Rad | 161-0436 | |
| Dialyzed fetal calf serum (FCS) | PAA | A15-107 | |
| Diffuser caps for microdissection | MMI | 50202 | |
| FACS-sorter | BD | FACSAria III | |
| Ionic Detergent Compatibility Reagent | Thermo scientific | 22663 | |
| Laser-capture microdissector | MMI | cell cut plus | |
| LDS buffer | Life Technologies | NP0009 | |
| Membrane slides for microdissection | MMI | 50103 | |
| Microcon YM-30 | Millipore | MRCF0R030 | |
| NuPAGE 4-12% Bis-Tris Mini Gels | Life Technologies | NP0335PK2 | |
| Picofrit Self-Pack Columns | New Objective | PF360-75-15-N-5 | Mass Spectrometry Column/Emitter |
| Reducing agent | Life Technologies | NP0007 | |
| Reprosil-Pur LC/MS/MS Column stationary phase | Dr. Maisch | 120 C18-AQ, 3 µm | |
| Reprosil-Pur LC/MS/MS Precolumn stationary phase | Dr. Maisch | 120 C18-AQ, 5 µm | |
| SILAC-labeled arginine | Eurisotop | CLM-2265-H-0.1 | |
| SILAC-labeled lysine | Eurisotop | DLM-2640-0.25 | |
| Trypsin, NB Sequencing Grade | Serva | 3728301 | for in-gel digests |
| Trypsin, Sequencing Grade | Promega | V5111 | for in-solution digests |
| Buffer and solutions | |||
| Cell lysis buffer: 150 mM NaCl, 50 mM Tris/HCl pH 7.8, 5 mM NaF, 0.5% NP40, 0.1% laurylmaltoside, Roche complete protease inhibitor, 1 mM Na3VO4 | |||
| Tissue lysis buffer: 100 mM Tris/HCl pH 7.8, 0.1 M DTT | |||
| Urea: 8 M urea in 0.1 M Tris-HCl, pH 8.5 | for FASP-protocoll | ||
| IAA: 0.05 M iodoacetamide, 8 M urea, 0.1 M Tris-HCl, pH 8.5 | for FASP-protocoll | ||
| 0.05 M NH4HCO3 | |||
| 10 mM dithiothreitol (DTT) in 0.1 M ammonium bicarbonate | for in-gel digest | ||
| 55 mM iodoacetamide (IAA) in 0.1 mM ammonium bicarbonate | for in-gel digest | ||
| 5% aqueous formic acid. |
Referências
- Walther, T. C., Mann, M. Mass spectrometry-based proteomics in cell biology. J Cell Biol. 190 (4), 491-500 (2010).
- Lenz, C., Urlaub, H. Separation methodology to improve proteome coverage depth. Expert Rev Proteomics. 11 (4), 409-414 (2014).
- Olsen, J. V., Mann, M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol Cell Proteomics. 12 (12), 3444-3452 (2013).
- Ong, S. E., et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 1 (5), 376-386 (2002).
- Jimenez, C. R., Verheul, H. M. Mass spectrometry-based proteomics: from cancer biology to protein biomarkers, drug targets, and clinical applications. Am Soc Clin Oncol Educ Book. , e504-e510 (2014).
- Tang, D. G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 22 (3), 457-472 (2012).
- Evans, C., et al. An insight into iTRAQ: where do we stand now. Anal Bioanal Chem. 404 (4), 1011-1027 (2012).
- Ostasiewicz, P., Zielinska, D. F., Mann, M., Wisniewski, J. R. Proteome, phosphoproteome, and N-glycoproteome are quantitatively preserved in formalin-fixed paraffin-embedded tissue and analyzable by high-resolution mass spectrometry. J Proteome Res. 9 (7), 3688-3700 (2010).
- Malmström, J., Picotti, P., Aebersold, R. Perspectives of targeted mass spectrometry for protein biomarker verification. Curr Opin Chem Biol. 13 (5-6), 518-525 (2009).
- Gillet, L. C., et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 11 (6), (2012).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. 41, (2010).
- Edwards, R. A. Laser capture microdissection of mammalian tissue. J Vis Exp. 8, 309(2007).
- Liu, N. Q., et al. Proteomics pipeline for biomarker discovery of laser capture microdissected breast cancer tissue. J Mammary Gland Biol Neoplasia. 17 (2), 155-164 (2012).
- Geiger, T., et al. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat Protoc. 6 (2), 147-157 (2011).
- Wisniewski, J. R. Proteomic sample preparation from formalin fixed and paraffin embedded tissue. J Vis Exp. (79), (2013).
- Cox, J., Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 26 (12), 1367-1372 (2008).
- Cox, J., et al. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc. 4 (5), 698-705 (2009).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2 (8), 1896-1906 (2007).
- Sarvi, S., et al. CD133+ cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res. 74 (5), 1554-1565 (2014).
- Shlush, L. I., et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 506 (7488), 328-333 (2014).
- Schaab, C., et al. Global phosphoproteome analysis of human bone marrow reveals predictive phosphorylation markers for the treatment of acute myeloid leukemia with quizartinib. Leukemia. 28 (3), 716-719 (2014).
- Hüttenhain, R., et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 4 (142), 142ra94(2012).
- Burgess, M. W., et al. Simplified and efficient quantification of low-abundance proteins at very high multiplex via targeted mass spectrometry. Mol Cell Proteomics. 13 (4), 1137-1149 (2014).
- Boja, E. S., et al. Analytical Validation Considerations of Multiplex Mass Spectrometry-based Proteomic Platforms for Measuring Protein Biomarkers. J Proteome Res. , (2014).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados