
Method Article
Strand DNA Deslocamento Portas para redes de reação de Execução químicos derivados do plasmídeo
Neste Artigo
Resumo
This protocol describes a method for deriving DNA strand displacement gates from plasmids and testing them using fluorescence kinetics measurements. Gates can be modularly composed into multi-component systems to approximate the behavior of formal chemical reaction networks (CRN), demonstrating a new use for CRNs as a molecular programming language.
Resumo
DNA nanotechnology requires large amounts of highly pure DNA as an engineering material. Plasmid DNA could meet this need since it is replicated with high fidelity, is readily amplified through bacterial culture and can be stored indefinitely in the form of bacterial glycerol stocks. However, the double-stranded nature of plasmid DNA has so far hindered its efficient use for construction of DNA nanostructures or devices that typically contain single-stranded or branched domains. In recent work, it was found that nicked double stranded DNA (ndsDNA) strand displacement gates could be sourced from plasmid DNA. The following is a protocol that details how these ndsDNA gates can be efficiently encoded in plasmids and can be derived from the plasmids through a small number of enzymatic processing steps. Also given is a protocol for testing ndsDNA gates using fluorescence kinetics measurements. NdsDNA gates can be used to implement arbitrary chemical reaction networks (CRNs) and thus provide a pathway towards the use of the CRN formalism as a prescriptive molecular programming language. To demonstrate this technology, a multi-step reaction cascade with catalytic kinetics is constructed. Further it is shown that plasmid-derived components perform better than identical components assembled from synthetic DNA.
Introdução
A previsibilidade de bases de Watson-Crick tem permitido nanotecnologia de DNA dinâmico a emergir como uma forma programável para projetar dispositivos moleculares com propriedades dinâmicas 1,2. Em particular, o deslocamento cadeia de DNA -, uma reação de hibridização competitiva programável - provou ser um poderoso mecanismo de engenharia de sistemas dinâmicos de DNA. Em uma reação de deslocamento de cadeia de DNA, um oligonucle�ido de entrada desloca um "output" vertente anteriormente vinculado a partir de um parceiro de ligação complementar. Várias tais reações podem ser encadeados em cascatas de reação de várias etapas com um alto grau de controle sobre a ordem eo tempo de reação individual etapas 3. DNA cascatas de deslocamento de cadeia foram usadas para criar circuitos digitais e analógicos moleculares 4-7, nanoestruturas comutáveis 8-10, motores moleculares autónomas 11-15, e amplificadores catalíticos não covalentes 13,16-21. Além disso, DNA dispositivos que utilizam reações de deslocamento de cadeia pode ser simulado e projetado para diversas aplicações utilizando software 22-24 desenho assistido por computador.
Atualmente, o DNA sintetizado quimicamente serve como o principal material para a nanotecnologia de DNA. No entanto, os erros no processo de síntese de DNA, e os oligonucleótidos imperfeitos resultantes, são acreditados para limitar o desempenho de dispositivos de ADN dinâmicas, fazendo com que as reacções laterais erradas. Por exemplo, as reacções de "vazamento" pode resultar na libertação de um oligonucleótido de saída, mesmo na ausência de uma reacção de disparo. Tais efeitos são mais óbvias nas cascatas de reacção autocatalítica onde mesmo uma quantidade mínima de vazamento inicial irá eventualmente resultar na activação completa da cascata 19,20. Por outro lado, as reacções muitas vezes não conseguem alcançar o nível esperado de activação porque alguns componentes não provocam, mesmo na presença de 7,25 a entrada pretendida. Para fazer com que o desempenho de DNA com base emnanodispositivos comparáveis com os seus homólogos à base de proteínas biológicas, tais modos de erro precisa ser reduzido drasticamente.
Plasmídeos bacterianos ou outro ADN biológica pode servir como uma fonte de ADN altamente puro relativamente barato para aplicações de nanotecnologia. Grandes quantidades de ADN pode ser gerado por replicação em bactérias e as capacidades inerentes de revisão dos sistemas vivos garantir a pureza do ADN resultante. De fato, vários trabalhos recentes têm reconhecido a utilidade potencial de DNA biológico para aplicações de nanotecnologia 21,26-28. No entanto, a natureza totalmente de cadeia dupla de DNA de plasmídeo foi medida proibida a sua utilização como um material para a fabricação de dispositivos de ADN dinâmicas, que consistem tipicamente de vários oligonucleótidos e contêm ambos os domínios de cadeia dupla e de cadeia simples. Em um recente artigo 29 esta questão foi abordada e uma nova arquitetura portão DNA que consiste principalmente de DNA de cadeia dupla entalhado (ndsDNA) foi introduzird.
É importante ressaltar que os sistemas de portões ndsDNA pode ser concebido que perceber a dinâmica especificados por qualquer rede de reação química formais (CRN) 29. portões ndsDNA poderia, assim, ser utilizados, em princípio, para criar sistemas dinâmicos que apresentam oscilações e caos, biestabilidade e memória, lógica booleana ou comportamentos algorítmicos 30-38. Por exemplo, Ref. 29 demonstraram uma CRN três reacção que fornecida uma implementação molecular de um protocolo de "consenso", um tipo de algoritmo de computação distribuída 29,39,40. Este trabalho demonstrou pela primeira vez uma nova utilização para o formalismo CRN como uma "linguagem de programação" para sintetizar rapidamente sistemas moleculares funcionais (Figura 1A).
Aqui, um protocolo detalhado para derivar portões ndsDNA de ADN de plasmídeo é fornecida. Primeiro é uma revisão do processo de concepção sequência. Depois segue-se uma explicação de como oligonucleótidos sintéticos que contêmas sequências de porta são clonados em plasmídeos e sequência verificada e amplificado através de cultura bacteriana. Em seguida, é mostrado como ndsDNA portas podem ser derivados a partir dos plasmídeos por tratamento enzimático (ver Figura 2). Por fim, um método para testar o comportamento portão usando a cinética de fluorescência os ensaios é descrita.
Mecanismo de reacção
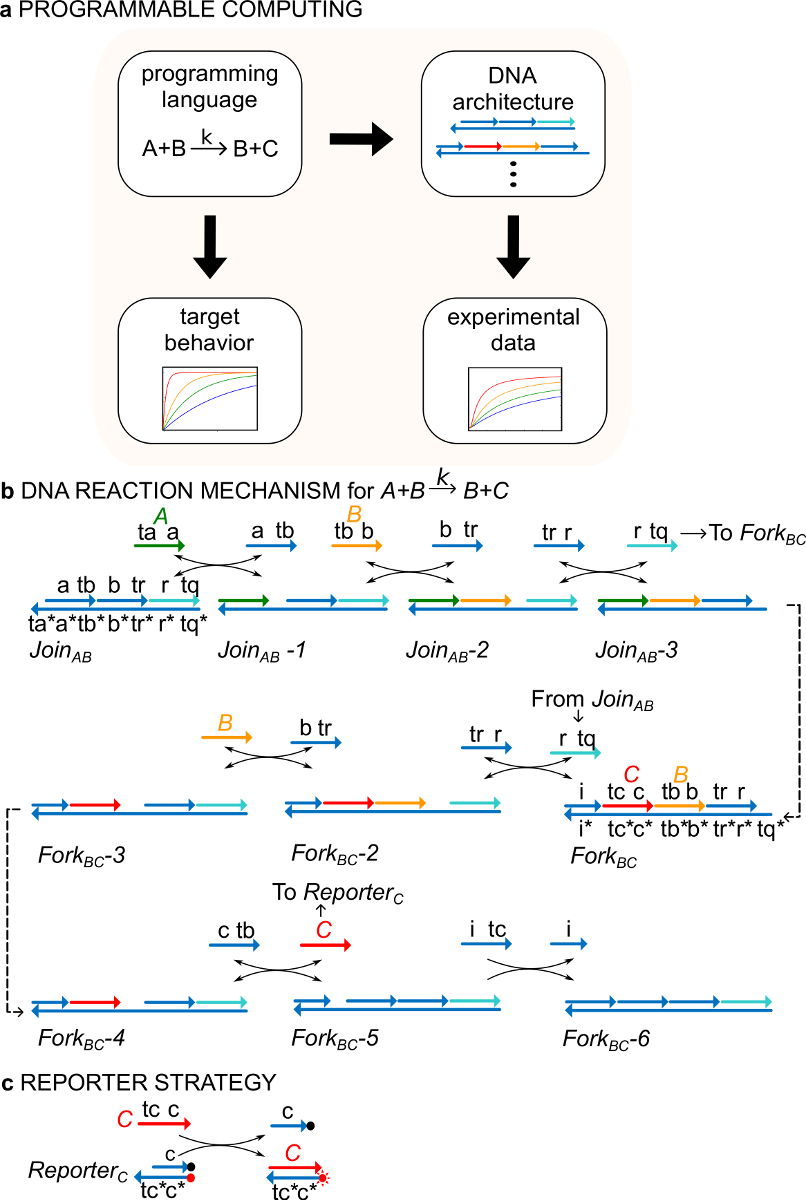
Como um exemplo, o protocolo centra-se na reacção química catalítica de A + B> B + C. As espécies de A, B, e C ("sinais", Figura 1B) correspondem todas a uma molécula diferente de cadeia simples de ADN. As sequências destas moléculas são completamente independentes e os fios não reagem um com o outro directamente. As sequências de todos os sinais têm dois domínios funcionais diferentes, ou seja, subsequências que agem em conjunto em reacções de deslocamento de cadeia: 1) um ponto de apoio de domínio curto (etiquetas TA, TB, TC) que é usado para a iniciação da cadeia deslocamento reaction, e 2) um domínio longo (etiquetas a, b, c) que determina a identidade do sinal.
Interações entre fios de sinal são mediados por ADN (ndsDNA) Complexos de porta de cadeia dupla com entalhe (chamados Adira AB e Fork BC) e as espécies de cadeia simples auxiliares (, , e ). A reacção de formal de A + B> B + C é executada através de uma série de passos de reacção de deslocamento de cadeia, em que cada passo reaccional expõe um ponto de apoio para uma reacção subsequente (Figura 1B). Neste exemplo, os sinais A e B são inicialmente livre em solução enquanto o sinal de C está ligado ao portão forquilha. No final da reacção, B e C estão em solução. De modo mais geral, os sinais que estão vinculados a um portão estão inativos, enquanto os sinais que são livres em solução são ativos, isto é, eles podem participar de uma reação de deslocamento de cadeia comouma entrada. O curso do tempo da reacção é seguido usando uma estratégia repórter fluorescente (Figura 1C). Em trabalho anterior 29, demonstrou-se que este mecanismo de reacção, não só realiza a estequiometria correcta, mas também a cinética da reacção alvo.
Protocolo
1. Sequência de design
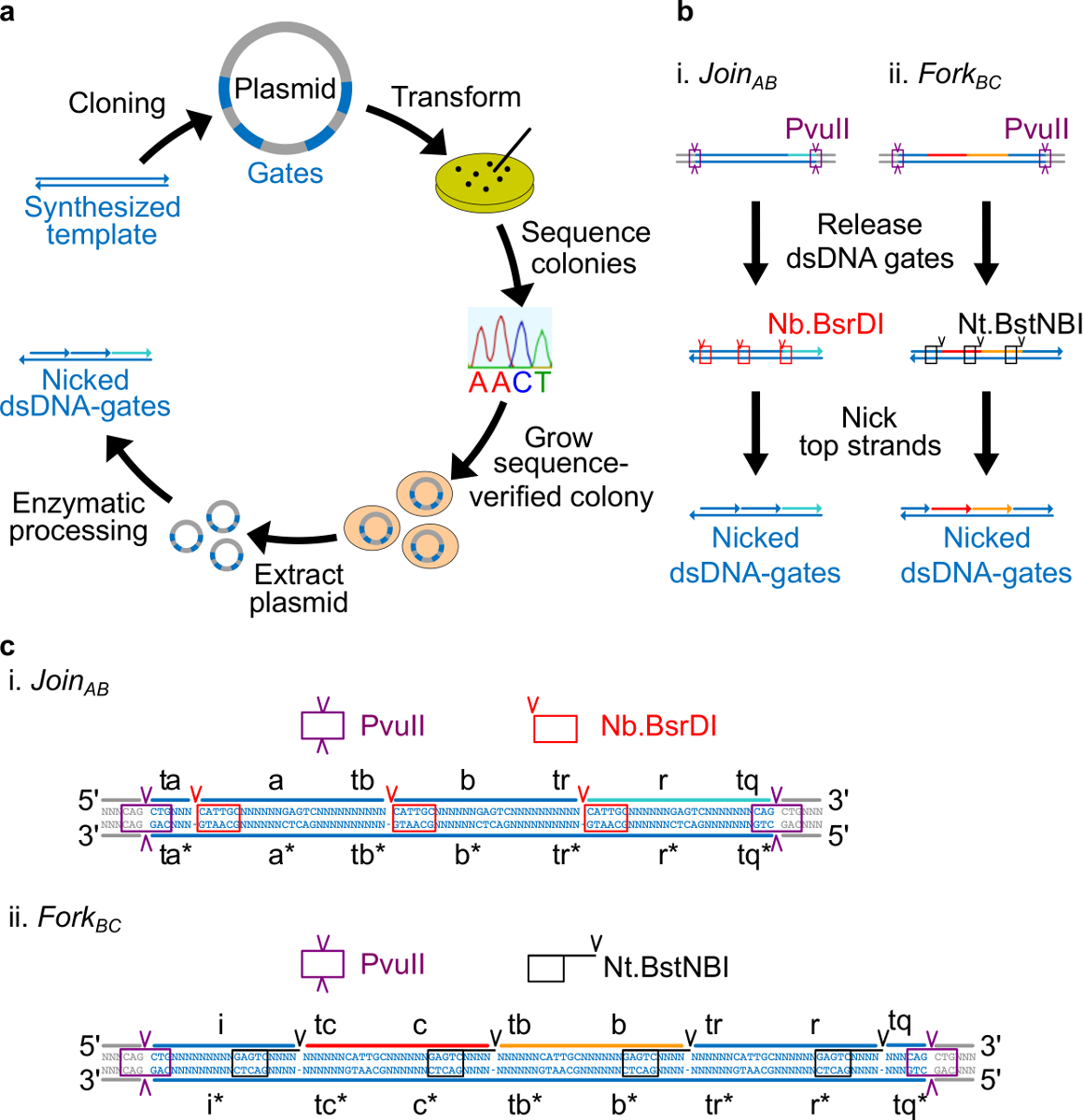
Nota: visão geral do design Sequência: Nesta seção, a estratégia para a criação de portas de DNA derivados de plasmídeo é descrito. Locais de enzimas colocados em cada extremidade das portas, para permitir a libertação de portas de cadeia totalmente dupla após a digestão. Nicking locais são então colocados de tal forma que as enzimas criar nicks na cadeia de cima para criar os portões finais ndsDNA. Finalmente, as restantes sequências são escolhidas de tal modo que os domínios independentes são ortogonais uma à outra e não exibem a estrutura secundária.
- Coloque o local nicking Nt.BstNBI quatro nucleótidos de distância a partir da extremidade 3 'de cada domínio de comprimento (a, b, c, r, e i). Coloque o local nicking Nb.BsrDI na extremidade 5 'de cada domínio longo (a, b, c e r. Note-se que o domínio I não tem qualquer local nicking Nb.BsrDI). A Figura 2C mostra a vista detalhada da sequência Junte-se AB e BC Fork portões.
- Coloque local de restrição PvuII em ambas as extremidades de portões ndsDNA de modo que a digestão com PvuII pode liberar as portas de plasmídeos (ver figura 2C).
- Conceber outras sequências não restringidas por dois princípios seguintes: (a) fios não deve apresentar estruturas secundárias (estruturas de ADN pode ser prevista utilizando NuPack 41), e (b) todos os domínios devem ser ortogonais para minimizar a diafonia.
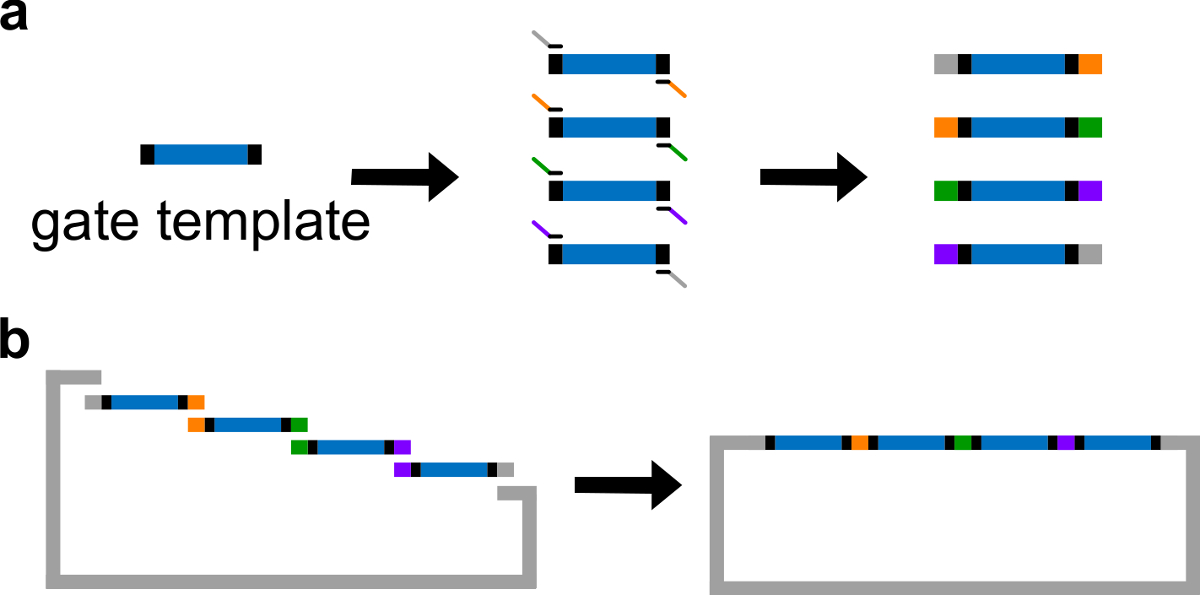
- Coloque sequências ndsDNA no centro de um molde de portão. Coloque sequências espaçadoras 30-40 pb aleatórios em ambas as extremidades do molde portão, cada espaçador serve como um local de ligação único para a seguinte reacção em cadeia da polimerase (PCR).
2. Clonagem de NdsDNA portas em plasmídeos
Nota: Esta seção descreve o método de clonagem Gibson para inserção de 4 cópias do portão em um backbone plasmídeo.
- Modelos de porta Pedido ndsDNA como blocos genómico de cadeia dupla a partir de um fabricante de ADN (sequências molde de porta são mostradosna Tabela 1; fios ocorrer em portões ndsDNA são mostrados na Tabela 2; sequências nível do domínio são mostrados na Tabela 3).
- Depois de receber o ADN ordenada, girar os tubos contendo blocos genómicos em 10,000-14,000 xg durante 1 min para garantir que todo o ADN é seco no fundo do tubo.
- Ressuspender as genómicas blocos secos em água isenta de ADNase para atingir uma concentração final de 10 ng / ul.
Observação: Como alternativa, o DNA pode ser ressuspenso usando 1x Tris ácido etilenodiaminotetracético (EDTA) tampão (tampão TE: Tris 10 mM e EDTA 1 mM, pH 8,0). No entanto, o EDTA é um agente quelante para catiões bivalentes e pode inibir a PCR. - Gerar 4 fragmentos de porta com diferentes regiões de sobreposição através de um PCR com um padrão de alta-fidelidade DNA polimerase (ver Figura 3A). As sequências dos iniciadores estão detalhados na Tabela 4 (temperatura de fusão destes iniciadores é de 62 ° C).
- Executar um gel de agarose a 2% a 140 V durante 30 min à temperatura ambiente (para um protocolo de electroforese em gel de agarose detalhada ver 42) e cortar as bandas correspondentes a cada fragmento de PCR amplificado para fora do gel. Em seguida, purificar as fatias de gel utilizando um kit de extracção de gel (por favor, referir-se aos Materiais) seguindo as instruções do fabricante.
- Digerir um número elevado de cópias de plasmídeo espinha dorsal (ver materiais) com PvuII e PstI-HF-HF a 37 ° C durante 1 h (ver Tabela 5) de acordo com o protocolo do fabricante. PvuII-HF e PstI-HF são enzimas de restrição de alta fidelidade, o que reduz drasticamente cortes inespecíficos.
- Executar um gel de agarose a 1,5% e cortar a espinha dorsal linearizado (tipicamente executar o gel a 140 V durante 30-40 min à temperatura ambiente). Em seguida, extrair o ADN a partir da fatia de gel utilizando o kit de extracção de gel de acordo com as instruções do fabricante.
- Realizar Gibson montagem 43 com o vector linearizado e fragmentos de PCR purificados (ver Tabela 6 e Figura 3B ) a 50 ° C durante 1 h.
- Transformar o produto do passo de montagem Gibson 2,8 em Escherichia coli (E. coli) e em uma placa Lisogenia Broth (LB) contendo antibióticos placa de agar de ampicilina (a uma concentração de 100 ug / mL). Execute transformação através da eletroporação ou um método do choque térmico 44,45, e usar o apropriado E. estirpe de E. coli. Por exemplo, usar E. coli estirpe JM109 para a transformação de choque térmico, e usar DH5ct electrocompetentes E. células de E. coli para electroporação.
Nota: A espinha dorsal do plasmídeo utilizado contém uma cassete de resistência à ampicilina. Se estiver usando um marcador de seleção diferente, usar os antibióticos apropriados em vez de ampicilina.
3. Cultura bacteriana Amplification e Controle de Qualidade
Nota: Esta seção descreve a produção em massa e isolamento de plasmídeos contendo os portões de DNA após controle de qualidade.
- Escolha uma única colôniaa partir da placa de Ampicilina selectiva do passo 2.9 e incubar uma cultura de 3 ml de meio enriquecido contendo antibióticos ampicilina (a uma concentração de 100 ug / mL). Marque a colónia de tal modo que pode ser novamente utilizada nos passos subsequentes experimentais. Incubar a cultura a 37 ° CO / N com agitação vigorosa (200-300 rpm). Tipicamente, incubar durante 16-24 horas.
- Extrai-se a ADN de plasmídeo a partir da cultura bacteriana usando um kit Mini-prep seguindo as instruções do fabricante.
- Medir o DNA de plasmídeo purificado utilizando um espectrofotómetro de acordo com as instruções do fabricante. Gamas de rendimento típico de 50-1.000 ng / mL.
- Obter o ADN plasmídico extraído sequenciados através do envio de uma amostra a empresa sequenciação de ADN. Iniciadores de sequenciação devem ser localizado a cerca de 100 nucleótidos a montante e a jusante da região a ser sequenciada; o iniciador de sequenciação para o plasmídeo (ver materiais de plasmídeo) tem a seguinte sequência: ATTACCGCCTTTGAGTGAGC.
Nãote: Se houver erro de seqüência ou de recombinação no inserida ndsDNA portões, selecione uma colônia diferente da placa do passo 2.9. Siga os passos de 3,1-3,4 para verificar se as sequências dos portões inseridos estão corretos. - Depois de verificar que as sequências estão correctas, escolher a colónia correspondente da placa selectiva de ampicilina (do passo 2.9), e incubar uma cultura de 800 ml de Terrific Broth (TB) contendo antibióticos ampicilina (a uma concentração de 100 ug / mL). Crescer a cultura a 37 ° C durante 16-24 horas com agitação vigorosa (200-300 rpm). TB é particularmente bem adequado para a produção do plasmídeo de elevado rendimento.
Nota: Em alternativa, LB também poderia ser utilizado para crescer as bactérias, embora o rendimento de plasmídeo pode ser um problema. - Purifica-se o ADN utilizando um kit Maxi-prep seguindo as instruções do fabricante.
- Siga o passo 3,3-3,4 para verificar se as sequências estão corretas. Se qualquer recombinação ocorreu, consulte a seguinte nota. Caso contrário, passe para o passo 4.
Nota: Um possível problema aqui é que várias cópias de portões inseridos no plasmídeo pode recombinar devido ao reparo do DNA. Para resolver este problema, use um E. estirpe de E. coli sem a proteína recA (uma proteína relacionada com a reparação do ADN), tal como JM109 ou DH5a para transformar um plasmídeo previamente verificada-sequência (ou seja, sem quaisquer erros de sequência e de recombinação). Em seguida, escolher uma colónia desta placa e verificar a sequência do plasmídeo através do envio de uma amostra a empresa sequenciação de ADN.
4. enzimática Processamento
Nota: Esta secção descreve o processo para a digestão de plasmídeos de tal modo que eles são cortados e com entalhe nas localizações correctas e pronto para ser utilizado para experiências de cinética.
- Digerir o ADN do plasmídeo purificado a partir do passo 3.7 com a enzima de restrição PvuII-IC durante 1 h a 37 ° C (ver Tabela 7). Tipicamente digerir o plasmídeo com 4 unidades de PvuII-HF por 1 mg de plasmídeo. Alta fidenzimas de restrição Elity são recomendados para uso, pois reduzem drasticamente cortes inespecíficos.
- Realizar precipitação com etanol na amostra.
- Adicionar 2 volumes equivalentes de etanol absoluto arrefecido em gelo para a amostra.
- Incubar a mistura a -80 ° C durante pelo menos 1 h (esta mistura também pode sentar-se, a -80 ° C durante S / N).
- Centrifugar a 10,000-14,000 xg à temperatura de 0 ° C durante 30 min.
- Remover o sobrenadante.
- Adicionar 1000 mL de etanol a 95% temperatura ambiente com a amostra, e inverter 10-15 vezes.
- Centrifugar a 10,000-14,000 xg, a 4 ° C durante 10 min.
- Remover o sobrenadante e o ar seco no banco durante 10-20 min.
- Ressuspender os sedimentos de DNA num volume apropriado de nuclease livre H2O (tipicamente 100-200 uL). A adição de mais de 200 uL irá geralmente fazer a amostra demasiado diluída para utilização em experiências de cinética.
- Medir o DNA ressuspenso usando um espectrofotómetro seguindo o minstruções de ABRICANTE.
- Digest juntar portões com nicking Nb.BsrDI enzima a 65 ° C durante 1 hora utilizando 4 unidades de enzima por 1 ug de plasmídeo (ver Tabela 8); digerir forquilha portões com nicking Nt.BstNBI enzima a 55 ° C durante 1 hora utilizando 8 unidades de enzima por 1 ug de plasmídeo (ver Tabela 9).
Nota: Passo 4.2 remove um tampão de digestão de enzimas e ajuda a concentrar-se as portas para a cinética experiências. Passo 4.2 pode ser ignorada para se juntar portões porque tanto a enzima de restrição PvuII-HF e enzima nicking Nb.BsrDI compartilham o mesmo tampão de digestão. No passo 4.2.8, nuclease livre H2O é utilizado em vez de TE porque o EDTA é um agente quelante para catiões divalentes e podem inibir as enzimas de restrição que necessitam destes iões para funcionar.
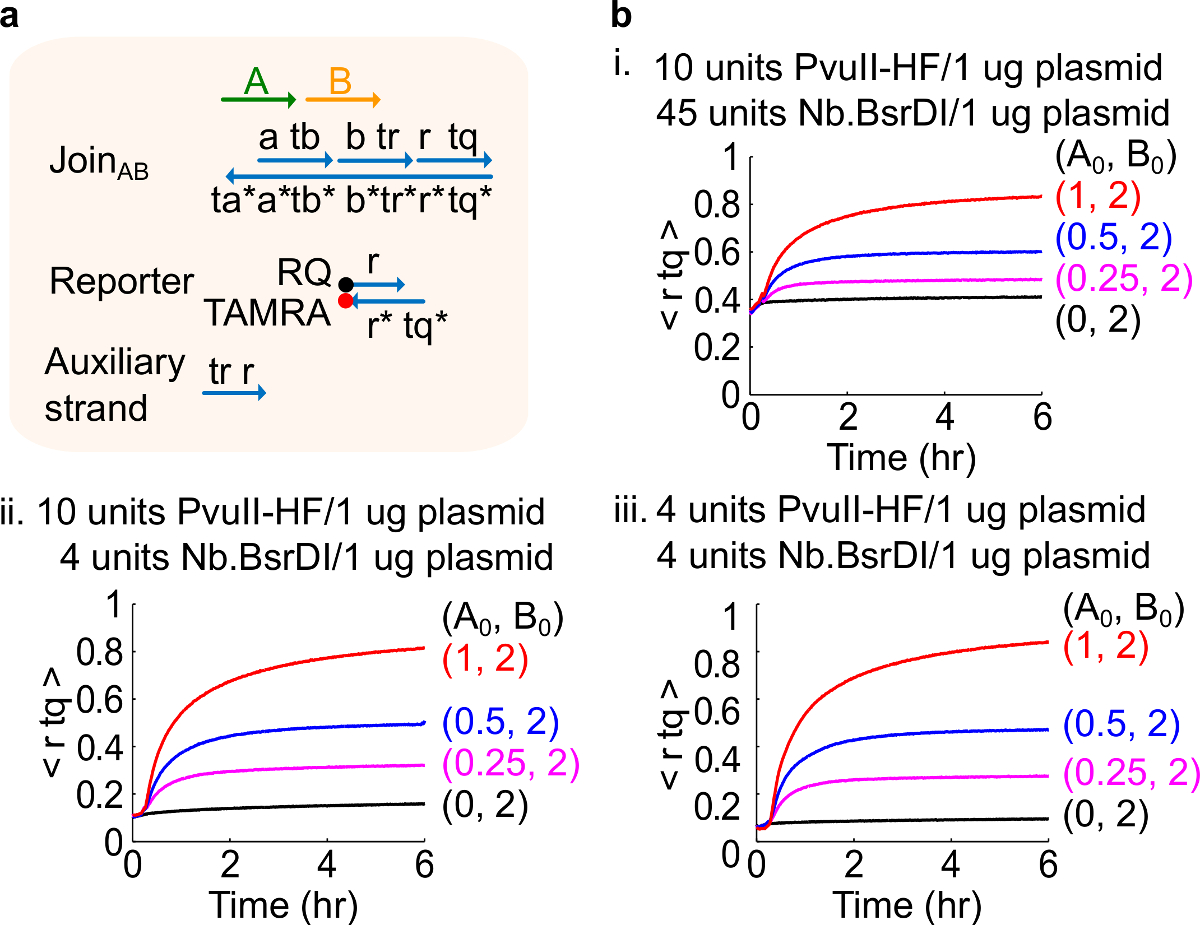
Nota: A adição de quantidades excessivas de enzimas pode conduzir a quantidades elevadas de vazamento circuito inicial (Figura 4), o que é provavelmente causado por excesso de digestão 46. Esse problema can ser endereçada através da optimização das quantidades de enzimas (ver Figura 4). Gama típica de enzimas é de 1-10 unidades / 1 ug de plasmídeo.
5. Preparação de oligonucleótidos de cadeia simples
Nota: Esta secção descreve o protocolo para a ressuspensão e quantificar o ADN de cadeia simples sintetizado quimicamente (ssDNA) que vai ser utilizado para os cordões e fios de sinal auxiliares. Para as sequências de cadeia ver Tabela 10. Note-se que o protocolo seguinte é um exemplo da preparação de 10 uM de ADNcs. Outras concentrações de ADNcs podem ser preparados de modo semelhante.
- Depois de receber os oligos de ADN fabricante, girar os tubos contendo ADN em 10,000-14,000 xg durante 1 min para garantir que todo o ADN é seco no fundo do tubo.
- Ressuspender o ADN utilizando 1x Tris ácido etilenodiaminotetracético (EDTA) tampão (tampão TE: Tris 10 mM e EDTA 1 mM, pH 8,0) para alcançar uma concentração final de 100 uM. Paraexemplo, ressuspender 8 nmol de ADN em 80 ul de tampão TE.
- Misturar 10 ul de DNA a 100 uM com 90 ul de água molecular num tubo de microcentrífuga, que deve alcançar uma concentração final de 10 uM.
- Medir a concentração exacta da amostra de ADN, utilizando um espectrofotómetro de acordo com as instruções do fabricante. O protocolo a seguir dá um exemplo de como a concentração de ADN pode ser medido.
- Anule o espectrofotômetro com 2 l de água molecular.
- Medir a absorvância a 260 nm (A 260) da amostra de ADN. Use a seguinte equação para calcular a concentração de ações.
Nota: A concentração da amostra é M = coeficiente 260 / extinção. O coeficiente de extinção pode ser encontrada na especificação folha de dados do fabricante ADN.
6. Preparação da Repórteres fluorescentes
Nota: Esta seção descreve oProtocolo para a preparação Repórter C, Outros repórteres fluorescentes podem ser montados de forma similar.
- Ordem de cromatografia líquida de alta resolução (HPLC) os oligonucleótidos purificados ROX- (a cadeia de cima do Repórter C) e -rq (a cadeia de baixo do Repórter C) a partir do fabricante de ADN (ver Tabela 10 para sequências ).
- Depois de receber o oligonucleotídeos sintetizados, ressuspender e quantificar amostras conforme explicado na Etapa 5.
- Misturar o topo repórter e fios de fundo (ou seja, ROX- e -rq) em 1x tampão Tris-Acetato-EDTA (TAE) com Mg 2+ 12,5 mM (ver a Tabela 11 para a receita detalhada ). Note-se que aqui a 30% o excesso de fio quencher rotulado -rq é adicionado para montar o repórter, o que garante que todas as vertentes marcado com fluoróforo apagaram-se mesmo com estequiometria imperfeito.
- Recozer o complexo de repórter C utilizando um termociclador, arrefecimento de 95 ° C a 20 ° C a uma velocidade de 1 ° C / min. As amostras podem ser armazenadas a 4 ° C.
7. As medições de fluorescência
Nota: A secção descreve um protocolo geral para as medições de cinética de fluorescência (ver Figura 5 para o procedimento experimental), e este protocolo irá ser utilizado nos passos 8, 9, e 10. De referir ainda que este protocolo é para a utilização de um espectros luorimetro. Alternativamente, estas experiências também poderia ser realizado num leitor de placas embora as variações de sensibilidade, bem-a-poço e falta de controlo da temperatura em experiências a longo prazo pode ser um problema.
- Definir o controlador de temperatura a 25 ° C, e esperar que a temperatura estabilize. Utilizando um controlador de temperatura pode reduzir a variabilidade no sinal que podem resultar da variação da temperatura.
- Definir parâmetros adequados foas medições de cinética r no software de aquisição de dados do espectros. Configurações exemplo detalhado são como se segue:
- Defina a largura da fenda de 2,73 nm para ambas excitação e emissão monocromadores.
- Defina o tempo de integração para 10 segundos para cada ponto de tempo de 60 segundos. Defina o tempo total de medição de 24 horas.
- Definir os comprimentos de onda de excitação / emissão para coincidir com os fluoróforos utilizadas na experiência. Comprimentos de onda Exemplo são como se segue: ROX (588 nm / 608 nm), e TAMRA (559 nm / 583 nm).
- Adicionar Nuclease livre H2O e 10x tampão de Tris-acetato-EDTA contendo 125 mM de Mg 2+ (10x TAE / Mg2 +) para uma célula de quartzo sintético. Veja as Tabelas 12, 13 e 14, por exemplo, volumes de usar.
- Adicionar cadeias poli-T para alcançar uma concentração final de 1 uM ~ (ver Quadro 12, 13, e 14 para volumes), e em seguida, vortex o sintéticocélulas de quartzo para 10-15 seg. Geralmente, as pontas da pipeta irá não se ligam especificamente a DNA. A adição de elevadas concentrações de fios de poli-T pode reduzir este erro de ligação não específica.
- Adicionar repórteres e fios auxiliares. Ver Tabela 12, 13, e 14, por exemplo, os volumes para usar. Note-se que para a calibração repórter, não há necessidade de fios auxiliares.
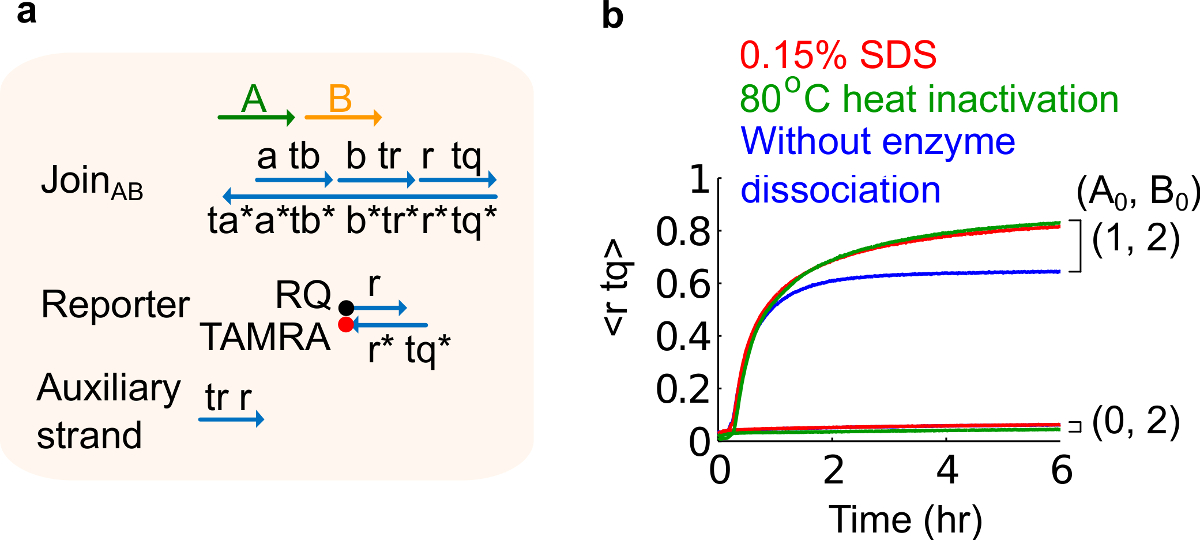
- Adiciona-se 10% de sulfato de dodecilo de sódio (SDS) para atingir uma concentração final de 0,15% de SDS. Nota: SDS é utilizado para dissociar os portões de enzimas derivado do plasmídeo por enzimas podem interferir com a reacção de deslocamento de cadeia (ver Figura 6). SDS é recomendado aqui, em vez de desnaturação de enzimas para evitar a dissociação e recombinação incorreta de fios de portão, que podem afetar adversamente a função do circuito.
- [Saltar este passo para a calibração repórter.]
- Adicionar juntar e portões garfo (ver Tabela 13, e 14 para volumes)para a célula de quartzo sintético e misturar a solução pipetando-o para cima e para baixo, pelo menos, 20 vezes (não vortex a cuvete porque as soluções vórtex com SDS pode resultar em bolhas que irão afectar as medições de cinética de fluorescência).
- Além disso, mover-se para os seguintes passos da medição tão rapidamente quanto possível, porque a reacção de vazamento inicia imediatamente após a adição de junção e da forquilha portas para a célula de quartzo sintético.
- Coloque as células de quartzo sintéticos para a câmara de um espectrofluorímetro.
- Inicie a medição de cinética.
- Depois de 5 minutos de medição, adicionar fios de entrada (ver Tabela 12, 13, e 14 para volumes) para a célula de quartzo sintético e misturar a mistura reaccional pipetando-o para cima e para baixo, pelo menos, 20 vezes. Note-se que a amostra deve ser misturada com cuidado para evitar bolhas. Execute esta etapa, enquanto o programa de aquisição de dados está em pausa para evitar medir sinais desencadeada pela externoclaro.
- Grave a cinética da reacção até atingir o estado estacionário. As cinéticas de reacção são apresentadas no computador.
8. Calibre fluorescentes Repórteres
Nota: Esta seção descreve o protocolo para fazer curvas de calibração de repórteres fluorescentes. As curvas de calibração irá ser utilizada para converter unidades de fluorescência arbitrárias ao sinal de concentração molar.
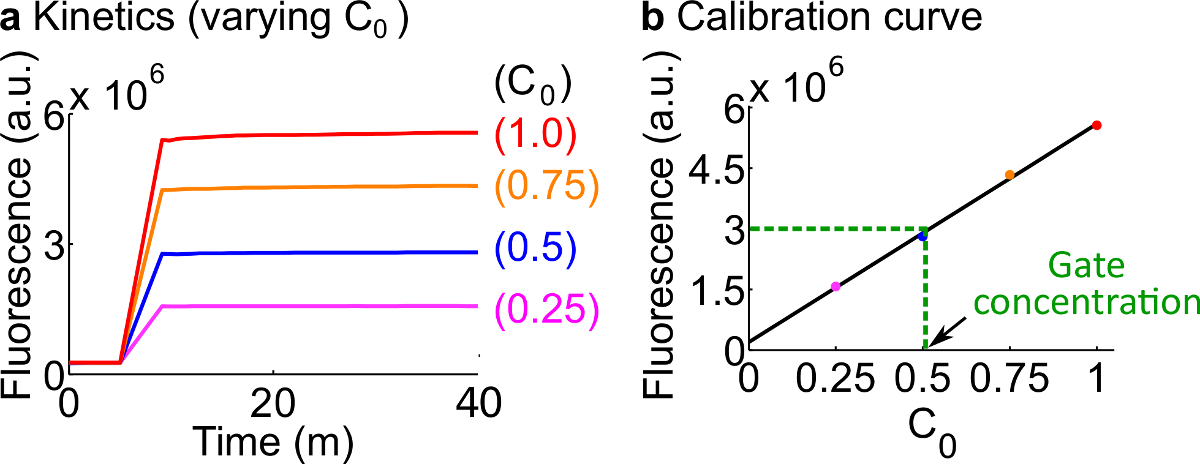
- Calibrar repórteres fluorescentes seguindo o protocolo descrito no Passo 7. A utilização dos volumes de reagentes e tampões, como resumido na Tabela 12 A concentração padrão para este exemplo é de 50 nM (1x).; repórteres estão em 3x; entrada é 1x. Para os casos em que a entrada são a 0,25x, 0,5x, 0,75x, ajustar o volume livre de nuclease H2O correspondentemente para manter o volume final de cada reacção de 600 ul para ser. Um exemplo de dados são mostrados na Figura 7A.
- Fazer uma curva de calibração doRepórter C por um ajustamento linear dos valores de fluorescência finais contra a concentração inicial do sinal de C (Uma curva de calibração de exemplo é mostrado na Figura 7B). Esta curva de calibração pode ser usado para converter unidades de fluorescência arbitrárias à sua concentração de sinal correspondente.
9. Quantificar a concentração de plasmídeo derivado de ndsDNA portas
Nota: Todos os lotes processados independentemente dos portões ndsDNA plasmídeo derivado de resultados com um rendimento diferente de portas funcionais, e esta secção descreve um protocolo para a quantificação da concentração de portões ndsDNA derivado de plasmídeo.
- Quantificar a concentração de portões ndsDNA derivado de plasmídeo seguindo o protocolo descrito no Passo 7. A utilização dos volumes de reagentes tal como resumido na Tabela 13 Nota:. A Tabela 13 descreve um exemplo de receita forquilha BC quantificação Junte-se. AB e outras portas pode ser realizada de forma semelhante, mas usando diferentes vertentes de entrada, fios auxiliares e jornalistas.
- Converter o valor de fluorescência final medida neste ensaio a uma concentração de sinal C, utilizando a curva de calibração a partir do passo 8.2. Em seguida, volta a calcular a concentração portão ndsDNA. Por exemplo, um valor de fluorescência final para o experimento de quantificação porta corresponde a 25 nM sinal C (0,5x) com base na curva de calibração na figura 7B. Uma vez que o estoque de forquilha aC é diluído 40 vezes nesta reacção, a concentração de estoque da porta garfos BC é 1 uM.
10. Medições Cinética para a reacção A + B-> B + C
Nota: Esta secção descreve um protocolo para testar a realização de ADN de uma reacção química usando formal de medições de cinética de fluorescência.
- Porcinética do formulário de medição seguindo o protocolo descrito na Etapa 7. Utilização volumes de reagentes e tampões, como resumido na Tabela 14.
Resultados
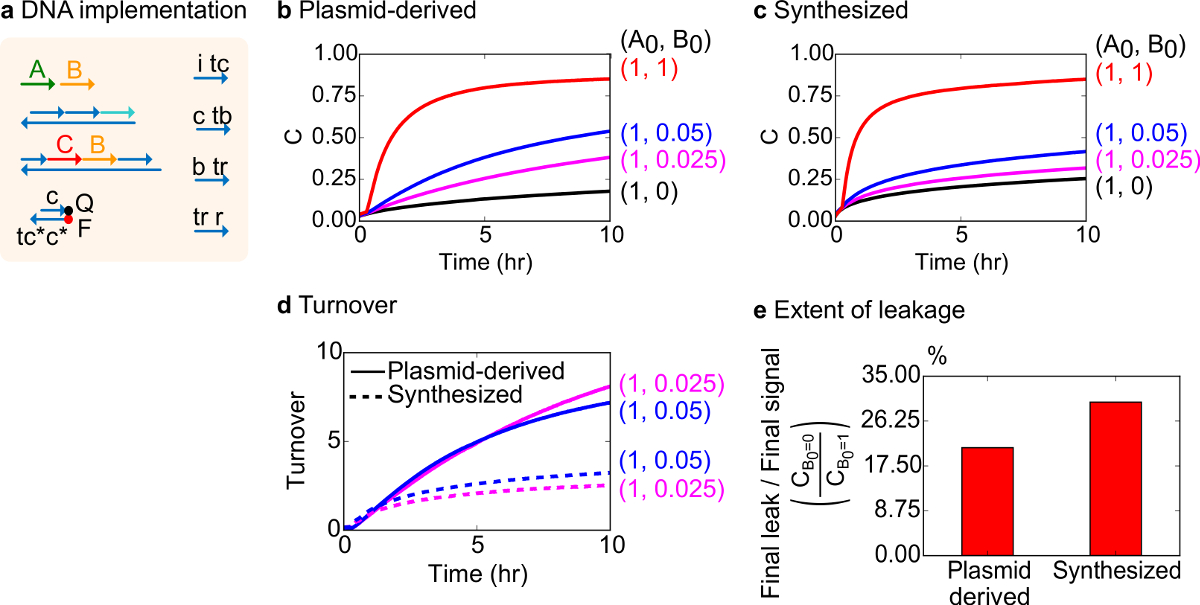
Para um teste funcional, uma implementação de ADN da reacção catalítica biomolecular (isto é, A + B> B + C) foi criada. O desempenho de portas derivado do plasmídeo foi comparado com portas montadas a partir de ADN sintético. Reações catalíticas são um bom teste para portão pureza porque um portão defeituoso pode irreversivelmente armadilha um catalisador, provocando um efeito desproporcional sobre a quantidade de produto produzido 18,19. Ao mesmo tempo, uma pequena fuga de reacção resultando na libertação sem trigger do sinal catalítica irá ser amplificado linearmente, que conduz a um sinal de erro desproporcionada. Os dados experimentais para portas e sintetizados derivados do plasmídeo são mostrados na Figura 8B e 8C, respectivamente. Nas experiências, a concentração de cadeia sinal A é fixo, enquanto a quantidade do sinal catalítica B é variada. O sinal C é usado para ler o progresso da reacção, sem interromper o catalíticaciclo. Catálise pode ser observado nos dados uma vez que as reacções se aproximar conclusão, mesmo com quantidades de catalisador B muito mais pequenas do que a quantidade de A. Uma vez que não foi adicionado SDS para experiências feitas com o sistema de sintetizados, a velocidade da reacção (que pode ser afectada pela adição de SDS) não é comparada e é o foco analítico em vez de ciclos catalíticos (como detalhado abaixo).
Uma análise mais aprofundada do volume catalítico desta reacção foi conduzida. O volume de negócios é definida como a quantidade de sinal C produzido para cada catalisador B em um determinado momento. Especificamente, o volume foi calculado a partir dos nossos dados experimentais dividindo o vazamento subtraído do sinal C pela quantidade inicial de catalisador B adicionado. Para um sistema catalítico ideal, este número de turnover deve aumentar linearmente com o tempo e seja independente da quantidade de catalisador, desde que o substrato não é limitativo. Em um sistema real, portões defeituosos podem desativar gato alysts, e o volume irá atingir um valor máximo, se não mesmo todos substrato disponível é convertido no produto. O valor máximo volume de negócios indica quantos tipos de pisos (sinal a) Um catalisador (sinal B) pode converter antes de se tornar inativado. Aqui, observa-se que o sistema sintetizado desvia do aumento linear ideal de rotação muito mais cedo do que o sistema de plasmídeo derivado de acontecer, indicando apreensão do catalisador através de uma reacção secundária indesejável (Figura 8D). A comparação volume de negócios só é mostrado para baixas concentrações, porque em altas concentrações de catalisadores, todas as portas serão acionados e solte sinal C. O circuito de vazamento também é comparado, e observa-se que a proporção de sinal de vazamento usando portas derivado do plasmídeo é de cerca de 8% a menos de que o uso de portões sintetizados após 10 h de reacção (Figura 8E).
iles / ftp_upload / 53087 / 53087fig1.jpg "/>
Figura 1. (A) CRNs servir como uma linguagem de programação prescritiva. Redes de reacção de ADN podem ser manipulados para aproximar a dinâmica de um CRN formal de implementação de DNA (B) de uma instrução química exemplo:. A + B-> B + C. Cadeias de DNA são desenhados como linhas com setas na extremidade 3 'e * indica complementaridade. Todos os sinais vertentes A (, verde), B (, laranja), e C ( c>, vermelho) são consistiu de um domínio toehold (rotulado como ta, tb, e tc) e um domínio de identidade (rotulado como a, b, e c). A reação bimolecular A + B-> B + C requer dois complexos multi-stranded Junte-se AB e BC Fork, e quatro vertentes auxiliares , , e . A reacção prossegue através de sete passos de deslocamento da cadeia, onde cada passo inícios com toehold ligação (C) Estratégia Reporter.. A reacção é seguida através de um repórter em que a cadeia inferior é marcado com um fluoróforo (ponto vermelho) ea cadeia de cima está ligada a um inibidor (ponto negro). Devido à co-localização de o fluoróforo e o extintor, fluorescência repórter é extinta no repórter intacta. O sinal C pode substituir a cadeia de cima do repórter, o que leva a um aumento de fluorescência. (Este valor foi modificado a partir Ref 29). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2. portas (A) NdsDNA feitas a partir de ADN de plasmídeo bacteriano. Várias cópias do molde de cadeia dupla portão ndsDNA são clonados em um plasmídeo. Os plasmídeos clonados são on transformado em E. coli e colónias na placa são seqüência verificada. Assim que a sequência é confirmada, o DNA de plasmídeo foi extraído e amplificado. Finalmente, o plasmídeo de cadeia dupla é transformada nos portões ndsDNA desejados por meio do processamento enzimático. (B) o tratamento enzimático de portões ndsDNA. A enzima de restrição PvuII é usado para libertar o portão a partir do plasmídeo. Os portões liberados serão tratados posteriormente usando enzimas nicking: Nb.BsrDI é usado para gerar nicks para Junte-AB (Painel i); Nt.BstNBI é usado para gerar cortes para forquilha BC (Panel II). Os locais de restrição e nicking são indicados como caixas com códigos de cores. (C) vista Sequência do modelo de portão de Participe AB (Painel i) e Fork BC (Painel ii). O local de restrição PvuII (em destaque na caixa roxa) é em ambas as extremidades dos portões ndsDNA. O Nb.BsrDI e NtSites nicking .BstNBI são destacados em caixas de vermelho e preto, respectivamente. Os locais de corte estão marcados com setas. Sequência N é qualquer nucleótido. (Esta figura foi modificado com a permissão de Ref 29). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3. (A) por PCR de um ADN modelo de portão. Um modelo de portão DNA contém as sequências ndsDNA portão no centro (uma região azul), e sequências separadoras em ambas as extremidades (regiões negras; estas duas sequências finais são ortogonais). Os iniciadores podem ligar-se as sequências espaçadoras de o modelo de porta, e gerar quatro fragmentos de ADN por PCR de sobreposição (sequências sobrepostas na figura são codificados por cor). Montagem (B) Gibson. Os quatro Fragmen ADN amplificadosts são então montados em um backbone plasmídeo linearizado através de Gibson método de montagem 43. (Esta figura foi modificado com a permissão de Ref 29). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4. desempenho de circuito com diferentes quantidades da enzima. (A) Uma representação simplificada do portão, reporter, fios auxiliares, e fios de sinal utilizadas para as experiências correspondentes. (B) Cinética experiências com derivados do plasmídeo Entre para o AB processadas com diferentes quantidades da enzima . Eu. 10 unidades de PvuII-HF e 45 unidades de Nb.BsrDI por 1 ug de plasmídeo; II. 10 unidades de PvuII-HF e 4 unidades de Nb.BsrDI por 1 ug de plasmídeo; iii. 4 unidades de PvuII-HF e 4 unidades de Nb.BsrDI por 1 ug de plasmídeo. Todos os fios auxiliares foram em 2x (1x = 10 nM). O complexo foi 1,5x portão, e as experiências foram realizadas a 35 ° C em 1x TAE / Mg 2+. (Esta figura foi modificado com a permissão de Ref 29). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

. Figura 5 Fluxograma de experimentos cinéticos Azul:. Materiais para adicionar à cuvette (célula de quartzo 0,875 ml sintético). Tabela de referência 14 para volumes específicos para adicionar para a experiência cinética de A + B -> B + C. Verdes: Instruções de uma espectrofluorímetro (rotulado como SPEX). Vermelho: Mistura de instruções."Target =" _ blank 53087fig5large.jpg "> Clique aqui para ver uma versão maior desta figura.

Figura 6. Enzima comportamento de dissociação e circuito. (A), uma representação simplificada do portão, reporter, fios auxiliares, e fios de sinal utilizadas para as experiências correspondentes. (B) Experiências cinética de 80 ° C de calor AB Junte-se derivados de plasmídeo usando inativação (traços verde), 0,15% dodecil sulfato de sódio (SDS) (vermelho), e um controle sem inactivação pelo calor ou a adição de SDS (azul). A concentração do padrão foi 1x = 10 nM, e todas as vertentes e auxiliares de entrada B estavam em 2x. O complexo foi 1,5x portão, e as experiências foram realizadas a 35 ° C em tampão 1 x Tris-acetato-EDTA contendo 12,5 mM de Mg2 + (1x TAE / Mg 2+ ). (Este valor foi modificado a partir Ref 29). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 7. calibração repórter. (A) Repórter C cinética. A concentração repórter foi em 3x (1x = 50 nM), e a concentração inicial de sinal C é indicado na figura. (B) Os níveis de fluorescência do sinal C na fase terminal de medição (40 min) mostra um relação linear com o concentração inicial do sinal C. Em um exemplo quantificação de Fork BC portão (linha tracejada verde), o valor de fluorescência de Fork BC foi medidad como 3 x 10 6 (au), o que corresponde a 25 Nm (0,5x) com base na curva de calibração. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 8. Cinética da reação catalítica bimolecular (A + B-> B + C). (A) Uma representação do portão, repórter, fios auxiliares, e fios de sinal utilizado para os experimentos correspondentes simplificado. As experiências foram executadas em tampão 1 x Tris-acetato-EDTA contendo 12,5 mM de Mg 2+ (1x TAE / Mg 2+). Todos os complexos foram gate 75 na concentração nM (1,5x), e fios auxiliares foram a 100 nM de concentração (2x). Cinética de dados para portões e dados derivados de plasmídeo para portões sintetizados estão apresentados em (B) e (C) , respectivamente. O sinal foi de 50 nM (1x). Diferentes quantidades de sinal (catalisador) foram introduzidos no sistema, e a reacção foi testado a 35 ° C. Portões (D) O plasmídeo derivados exibiu volume maior do que portões ADN sintetizado quando foram adicionadas quantidades reduzidas de entrada (E). A extensão de vazamento. O gráfico de barras mostra a relação entre o vazamento final para o sinal de término (C = 0 B0 / C B0 = 1) com os pontos finais (10 h). (Este valor foi modificado a partir Ref 29). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
| Portão Templates | Sequências | Comprimento (nt) |
| JoinAB | TCTAGTTCGATCAGAGCGTTATTACCAGTAGTCGATTGCTCAGCTGCTACATTGCTTCTACGAGTCATCCTTCCACCATTGCACCTTAGAGTCCGAATCCTACCATTGCTTAACCGAGTCTCACAACCAGCTGTCATTATGGACTTGACACACAGATTACACGGGAAAGTTGC | 173 |
| FORKBC | TCTAGTTCGATCAGAGCGTTATTACCAGTAGTCGATTGCTCAGCTGCCATCATAAGAGTCACCATACCCACATTGCCACATCGAGTCCCTTTTCCACCATTGCACCTTAGAGTCCGAATCCTACCATTGCTTAACCGAGTCTCACAACCAGCTGTCATTATGGACTTGACACACAGATTACACGGGAAAGTTGC | 194 |
Tabela 1. Seqüências de modelos de porta ndsDNA.
| Portão | Strand | Comprimento da cadeia inferior (nt) |
| JoinAB | JoinAB-Bottom, , , | 87 |
| ForkBC | ForkBC-Bottom, , , , | |
| 108 |
Tabela 2. Os fios que compõem Junte-se AB e BC Fork. (Este quadro foi modificado a partir Ref 29).
| Domínio | Seqüência | Comprimento (nt) |
| ta | CTGCTA | 6 |
| tuberculose | TTCCAC | 6 |
| tc | TACCCA | 6 |
| tr | TCCTAC | 6 |
| tq | AACCAG | 6 |
| uma | CATTGCTTCTACGAGTCATCC | 21 |
| b | CATTGCACCTTAGAGTCCGAA | 21 |
| c | CATTGCCACATCGAGTCCCTT | 21 |
| r | CATTGCTTAACCGAGTCTCAC | 21 |
| Eu | CTGCCATCATAAGAGTCACCA | 21 |
| Vertente Primer | Sequências | Comprimento (nt) |
| Adiante iniciador-1 | AAGAGAGACCACATGGTCCTTCTTGAGTTTGTAACAG CGTTATTACCAGTAGTCGATTGC | 60 |
| Iniciador inverso 1- | ACTACTATTTACTAATCCCATTGCGTGTTCTTATT TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Adiante iniciador 2- | AATAAGAACACGCAATGGGATTAGTAAATAGTAGT CGTTATTACCAGTAGTCGATTGC | 58 |
| Iniciador inverso 2- | GCGAAACTAGCTTGTGGTGATATTGTCTCGTGTGT TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Adiante iniciador 3- | ACACACGAGACAATATCACCACAAGCTAGTTTCGC CGTTATTACCAGTAGTCGATTGC | 58 |
| Iniciador reverso de 3- | ACATTGTACGCCTAAATCATCAAGAATAATTGTTG TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Adiante iniciador 4- | CAACAATTATTCTTGATGATTTAGGCGTACAATGT CGTTATTACCAGTAGTCGATTGC | 58 |
| Iniciador inverso 4- | GAGCGCAGCGAGTCAGTGAGCGAGGAAGCCTGCAG TAATCTGTGTGTCAAGTCCATAATG | 60 |
Tabela 4. As sequências dos iniciadores para a PCR de modelos ndsDNA porta.
| Reagente | Volume de reação 1x (ul) |
| De alta cópia plasmídeo de esqueleto (~ 300 ng / ul) | 10 |
| PvuII-HF (20000 unidades / ml) | 2 |
| PstI-HF (20000 unidades / ml) | 2 |
| Tampão inteligente de 10x Cut | 2 |
| H2O | 4 |
| Volume total | 20 d> |
Tabela 5. Protocolo para plasmídeo backbone digerir.
| Reagente | Volume de reação 1x (ul) |
| Vector de ADN (~ 50 ng / ul) | 1 |
| Fragmento amplificado por PCR-1 (~ 50 ng / ul) | 1 |
| Fragmento amplificado por PCR-2 (~ 50 ng / ul) | 1 |
| Fragmento amplificado por PCR-3 (~ 50 ng / ul) | 1 |
| Fragmento amplificado por PCR-4 (~ 50 ng / ul) | 1 |
| Assembléia Gibson 2x mestre Mix | 5 |
| Volume total | 10 |
| Reagente | Volume de reação 1x (ul) |
| O DNA de plasmídeo (~ 1 ug / mL de concentração) | 1000 |
| PvuII-HF (20000 unidades / ml) | 200 |
| Tampão inteligente de 10x Cut | 133,3 |
| Volume total | 1333,3 |
Tabela 7. Protocolo para portões ndsDNA inseridos digerir o plasmídeo com a enzima de restrição PvuII-HF.
| Reagente | Volume (uL) |
| Junte-se os portões (~ 5 mg / l de concentração) | 150 |
| Nb.BsrDI (10000 unidades / ml) | 300 |
| Tampão inteligente de 10x Cut | 50 |
| Volume total | 500 |
Tabela 8. Protocolo para juntar-se digerir com portões nicking enzima Nb.BsrDI.
| Reagente | Volume (uL) |
| Forquilha portões (~ 5 ug concentração / ul) | 150 |
| Nt.BstNBI (10000 unidades / ml) | 600 |
| Tampão NEB 10x 3.1 | 83,3 |
| Volume total | 833,3 |
Tabela 9 Protocolo para forquilha portões digerir com nicking enzima Nt.BstNBI.
. Tabela 10 sequências Strand para a implementação da reação química A + B -.> B + C (. Este quadro foi modificado a partir Ref 29)
| Reagente | Volume (uL) | A concentração final |
| ROX- em 100 mM | 10 | 10 uM (1x) |
| -rq em 100 μ; M | 13 | 13 uM (1,3x) |
| 10x TAE com Mg 2+ 125 mM | 10 | 1x TAE com 12,5 mM de Mg 2+ |
| H2O | 67 | - |
| Volume total | 100 | 10 uM (1x) |
Tabela 11. Protocolo para a montagem Repórter C.
| Reagente | Volume (uL) | A concentração final |
| H2O | 514 | - |
| 10x TAE com Mg 2+ 125 mM | 60 | 1x TAE com 12,5 mM de Mg 2+ |
| PoliT em 300 μ; M | 2 | 1 uM |
| Repórter C a 10 uM | 9 | 150 nM (3x) |
| 10% de SDS | 9 | 0,15% |
| a 5 ^ M | 6 | 50 nM (1x) |
| Volume total | 600 | - |
Tabela 12. Protocolo para a calibração do Repórter C. Os volumes fornecidos aqui é para um volume total de reacção de 600 ul (correspondentes à utilização de uma célula de quartzo ml sintético 0,875), mas pode ser modificada para trabalhar com células de diferentes tamanhos.
| Reagente | Volume (uL) | Con finalcentração |
| H2O | 493 | - |
| 10x TAE com Mg 2+ 125 mM | 60 | 1x TAE com 12,5 mM de Mg 2+ |
| poliT a 300 uM | 2 | 1 uM |
| Repórter C a 10 uM | 9 | 150 nM (3x) |
| em 100 mM | 3 | 10x |
| a 100 uM | 3 | 10x |
| em 100 mM | 3 | 10x |
| 10% de SDS | 9 | 0,15% |
| Fork BC em ~ 1 mM (concentração desconhecido) | 15 | ~ 0,5x |
| a 100 uM | 3 | 10x |
| Volume total | 600 | - |
Tabela 13. Protocolo para a calibração de forquilha aC. Os volumes fornecidos aqui é para um volume total de reacção de 600 ul, mas pode ser modificada para trabalhar com células de diferentes tamanhos.
| Reagente | Volume (uL) | A concentração final | |
| H2O | 407.2 | - | |
| 10x TAE com Mg 2+ 125 mM | 52,8 | 12,5 mM de Mg 2+ | |
| poliT a 300 uM | 2 | 1 uM | |
| Repórter C a 10 uM | 9 | 150 nM (3x) | |
| em 10 mM | 6 | 100 nM (2x) | |
| em 10 mM | 6 | 100 nM (2x) | |
| em 10 mM | 6 | 100 nM (2x) | |
| 6 | 100 nM (2x) | ||
| 10% de SDS | 9 | 0,15% | |
| Junte-se a AB em 1 PM | 45 | 75 Nm (1,5x) | |
| Garfo BC em 1 uM | 45 | 75 Nm (1,5x) | |
| em 10 mM | 3 | 50 nM (1x) | |
| em 10 mM | 3 | 50 nM (1x) | |
| Volume total | 600 | - | |
Tabela 14. Protocolo para uma reacção química A + B-> B + C. Os volumes fornecidos aqui é para um volume total de reacção de 600 ul, mas pode ser modificada para trabalhar com células de diferentes tamanhos.
| Portões sintetizados | Portões derivado de plasmídeos | ||||
| Descrição | Custo | Junte-se a portas | Portões Fork | ||
| PAGE cadeia longa purificado (100 nt; serviu como os fios de fundo de uma porta) | ~ $ 75 | Descrição </ strong> | Custo | Descrição | Custo |
| PAGE cadeia curta purificado (~ 30 nt, serviu como principais vertentes de um portão) | ~ $ 185 | Modelo de portão | ~ $ 100 | Modelo de portão | ~ $ 100 |
| Total | ~ $ 260 | Kit de extracção de plasmídeo | ~ $ 26 | Kit de extracção de plasmídeo | ~ $ 26 |
| Enzima de restrição (PvuII-HF) | ~ $ 11 | Enzima de restrição (PvuII-HF) | ~ $ 11 | ||
| Nicking enzima (Nt.BsrDI, Junte-se a portas) | ~ $ 29 | Enzima nicking (Nt.BstNBI, portões Fork) | ~ $ 62 | ||
| Total | ~ $ 166 | Total | ~ $ 199 |
fo:.. manter-com-previous.within-page = "always"> Tabela de comparação entre os portões 15 Custo derivado do plasmídeo e portões sintetizados (. Este quadro foi modificado a partir Ref 29)
| Portões sintetizados | Portões derivado de plasmídeos | ||
| Processamento | Tempo de processamento | Processamento | Tempo de processamento |
| Recozimento | 1 hr | Clonagem | 5 h |
| Purificação PAGE | 2 hr | Extração plasmídeo | 2 hr |
| Total | 3 hr | Dois passos de digestão enzimática | 0,5 hr |
| Precipitação com etanol | 1 hr | ||
| Total | 8,5 hr | ||
Comparação Tabela 16. Processamento de tempo entre portas derivado do plasmídeo e portões sintéticos. (Este quadro foi modificado a partir Ref 29).
Discussão
Este artigo descreve um método para derivar portões ndsDNA a partir de DNA de plasmídeo muito puro. Além disso, um protocolo é apresentada para caracterizar o desempenho portão utilizando um ensaio de cinética de fluorescência. Os dados experimentais mostram que o sistema de plasmídeo derivado de supera sua contraparte sintética, mesmo se o sistema sintético é montado a partir de fios purificado utilizando electroforese em gel de poliacrilamida (PAGE). Provavelmente, o melhor desempenho de portas derivado do plasmídeo é principalmente devido à muito alta pureza do DNA biológico. ADN sintético contém uma variedade de erros, em particular, deleções que resultam em oligonucleótidos de tais produtos secundários comprimento n-1, e são tipicamente não completamente removido em PAGE ou cromatograf ia líquida de alta eficiência (HPLC) procedimentos de purificação. Também foram observadas melhorias semelhantes aos aqui relatados em um estudo prévio de um amplificador hairpin catalisada que usou DNA derivado de fontes biológicas 21.
No entanto, mesmo o uso de portas derivado do plasmídeo não pode eliminar completamente os erros no desempenho portão, para as quais existem pelo menos duas razões: em primeiro lugar sobre-digestão ou falta de precisão de corte pode levar a portões com demasiados nicks ou cortes nas posições erradas. Em qualquer caso, as portas são mais susceptíveis de participar em reacções indesejadas. Tais problemas podem ser facilitado através da optimização da quantidade de enzima utilizada (ver Figura 4). Em segundo lugar, nestas experiências, a maioria das entradas e cordões eram auxiliares de ADN sintético e continha assim as supressões e mutações. Em princípio, todas as entradas de cadeia simples e fios auxiliares também poderia ser obtido a partir de ADN de fagemideo por meio de uma digestão com enzimas de nicking do genoma viral M13 pré-codificada 26. Talvez o desempenho do circuito pode ser melhorado utilizando ADNcs derivados do genoma bacteriano.
Enquanto o uso de portões derivado de plasmídeo foi encontrada para melhorar o desempenho do circuito, uma análisedos tempos de processamento e custos revelou que, enquanto a produção de portas derivado de plasmídeo é ligeiramente mais barato (Tabela 15), leva-se 2-3 vezes mais em comparação com o tempo de processamento e a purificação da montagem de portas de oligos sintetizados comercialmente (Tabela 16). Os custos primários de portões são derivados do plasmídeo síntese de genes e a utilização de enzimas de restrição. Para 300 pmol de portas (suficiente para 15 reacções em 30 nM), o custo estimado para Junte-se portas é de aproximadamente US $ 170 e US $ 200 para Fork portões, a diferença de custo ser devido ao uso de diferentes enzimas nicking. Em contraste, a síntese química dos fios para os mesmos custos de porta em torno $ 260, incluindo uma taxa de purificação PAGE. O custo do tempo primário para portas derivado do plasmídeo é no procedimento de clonagem, que, assim como a síntese de DNA, pode ser terceirizado para uma empresa de síntese de genes. No entanto, uma vez montado, portas derivado do plasmídeo tem a vantagem de que os plasmídeos hospedeiros podem ser facilmente replicadas de umaND pode ser armazenado na forma de estoques de glicerol bacterianas. Isto faz com que seja possível voltar a utilizar os portões muitas vezes.
Olhando para a frente, o melhor desempenho de portas derivado do plasmídeo poderia permitir uma gama muito maior de dinâmica de comportamentos que têm sido experimentalmente demonstrado até agora com DNA CRNs. Por exemplo, recente trabalho teórico 47,48 sugeriu que padrões espaciais auto-organizadas na macro-escala podem ser realizados com DNA CRNs através de um mecanismo de difusão reação. O método aqui apresentado fornece um caminho viável para a construção de componentes moleculares subjacentes a tais materiais de DNA auto-padronização. Apesar de desafiador, desenvolvendo morfologias macro-escala de uma forma programável teria implicações significativas em áreas que vão desde pesquisas em biomateriais para a medicina regenerativa.
Divulgações
The authors declare no competing financial interests.
Agradecimentos
As Figuras 1, 2, 3, 4, 6, 8 e quadros 2, 3, 10, 15, 16 são modificados a partir de 29 Ref. Este trabalho foi financiado pela National Science Foundation (NSF conceder-CCF 1117143 e NSF-CCF 1.162.141 para GS). Y.-JC foi apoiado por bolsas de estudo governamentais de Taiwan. SDR foi apoiada pela Fundação Nacional de Ciência Programa de Pós-Graduação Research Fellowship (GRFP).
Materiais
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity PCR Master Mix with HF Buffer | NEB | M0531S | |
| PvuII-HF | NEB | R3151L | |
| PstI-HF | NEB | R3140S | |
| Gibson Assembly Master Mix | NEB | E2611S | |
| Terrific Broth, Modified | SIGMA-ALDRICH | T0918-250G | |
| QIAprep Spin Miniprep Kit (250) | QIAGEN | 27106 | |
| QIAGEN Hispeed Maxi-prep Kit | QIAGEN | 12662 | |
| Nb.BsrDI | NEB | R0648L | |
| Nt.BstNBI | NEB | R0607L | |
| NanoDrop 2000c | Thermo Scientific | ||
| Double-stranded Genomic Blocks | IDT | ||
| Horiba Jobin-Yvon Spex Fluorolog-3 Fluorimeter | Horiba/Jobin Yvon | ||
| Synthetic Quartz Cells | Starna | 23-5.45-S0G-5 | |
| QIAGEN Gel Extraction Kit | QIAGEN | 28706 | |
| Plasmid Backbones | BioBrick | E0240-pSB1A2 | High copy number plasmid with Ampicillin resistance. Sequence can be found from http://parts.igem.org |
Referências
- Zhang, D. Y., Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 3, 103-113 (2011).
- Krishnan, Y., Simmel, F. C. Nucleic acid based molecular devices. Angew. Chem. Int. Ed. Engl. 50, 3124-3156 (2011).
- Zhang, D. Y., Winfree, E. Control of DNA strand displacement kinetics using toehold exchange. J. Am. Chem. Soc. 131, 17303-17314 (2009).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475, 368-372 (2011).
- Qian, L., Winfree, E. Scaling up digital circuit computation with DNA strand displacement cascades. Science. 332, 1196-1201 (2011).
- Zadegan, R. M., Jepsen, M. D., Hildebrandt, L. L., Birkedal, V., Kjems, J. Construction of a fuzzy and boolean logic gates based on DNA. Small. 11, 1811-1817 (2015).
- Seelig, G., Soloveichik, D., Zhang, D. Y., Winfree, E. Enzyme-free nucleic acid logic circuits. Science. 314, 1585-1588 (2006).
- Zadegan, R. M., et al. Construction of a 4 zeptoliters switchable 3D DNA box origami. ACS Nano. 6, 10050-10053 (2012).
- Andersen, E. S., et al. Self-assembly of a nanoscale DNA box with a controllable lid. Nature. 459, 73-76 (2009).
- Zhang, D. Y., Hariadi, R. F., Choi, H. M., Winfree, E. Integrating DNA strand-displacement circuitry with DNA tile self-assembly. Nat. Commun. 4, (1965).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406, 605-608 (2000).
- Green, S. J., Lubrich, D., Turberfield, A. J. DNA hairpins: fuel for autonomous DNA devices. Biophys. J. 91, 2966-2975 (2006).
- Venkataraman, S., Dirks, R. M., Rothemund, P. W., Winfree, E., Pierce, N. A. An autonomous polymerization motor powered by DNA hybridization. Nat. Nanotechnol. 2, 490-494 (2007).
- Green, S. J., Bath, J., Turberfield, A. J. Coordinated chemomechanical cycles: a mechanism for autonomous molecular motion. Phys. Rev. Lett. 101, 238101 (2008).
- Omabegho, T., Sha, R., Seeman, N. C. A bipedal DNA Brownian motor with coordinated legs. Science. 324, 67-71 (2009).
- Turberfield, A. J., et al. DNA fuel for free-running nanomachines. Phys. Rev. Lett. 90, 118102 (2003).
- Dirks, R. M., Pierce, N. A. Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. U. S. A. 101, 15275-15278 (2004).
- Seelig, G., Yurke, B., Winfree, E. Catalyzed relaxation of a metastable DNA fuel. J. Am. Chem. Soc. 128, 12211-12220 (2006).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318, 1121-1125 (2007).
- Yin, P., Choi, H. M., Calvert, C. R., Pierce, N. A. Programming biomolecular self-assembly pathways. Nature. 451, 318-322 (2008).
- Chen, X., Briggs, N., McLain, J. R., Ellington, A. D. Stacking nonenzymatic circuits for high signal gain. Proc. Natl. Acad. Sci. U. S. A. 110, 5386-5391 (2013).
- Phillips, A., Cardelli, L. A programming language for composable DNA circuits. J. R. Soc. Interface. 6, S419-S436 (2009).
- Lakin, M. R., Youssef, S., Polo, F., Emmott, S., Phillips, A. Visual DSD: a design and analysis tool for DNA strand displacement systems. Bioinformatics. 27, 3211-3213 (2011).
- Lakin, M. R., Youssef, S., Cardelli, L., Phillips, A. Abstractions for DNA circuit design. J. R. Soc. Interface. 9, 470-486 (2012).
- Zhang, D. Y., Winfree, E. Robustness and modularity properties of a non-covalent DNA catalytic reaction. Nucleic Acids Res. 38, 4182-4197 (2010).
- Ducani, C., Kaul, C., Moche, M., Shih, W. M., Hogberg, B. Enzymatic production of 'monoclonal stoichiometric' single-stranded DNA oligonucleotides. Nat. Methods. 10, 647-652 (2013).
- Lin, C., et al. In vivo cloning of artificial DNA nanostructures. Proc. Natl. Acad. Sci. U. S. A. 105, 17626-17631 (2008).
- Bhatia, D., et al. Icosahedral DNA nanocapsules by modular assembly. Angew. Chem. Int. Ed. Engl. 48, 4134-4137 (2009).
- Chen, Y. J., et al. Programmable chemical controllers made from DNA. Nat. Nanotechnol. 8, 755-762 (2013).
- Arkin, A., Ross, J. Computational functions in biochemical reaction networks. Biophys. J. 67, 560-578 (1994).
- Érdi, P., Tóth, J. . Mathematical models of chemical reactions: theory and applications of deterministic and stochastic models. , (1989).
- Magnasco, M. O. Chemical kinetics is Turing universal. Phys. Rev. Lett. 78, 1190 (1997).
- Oishi, K., Klavins, E. Biomolecular implementation of linear I/O systems. IET Syst. Biol. 5, 252-260 (2011).
- Senum, P., Riedel, M. Rate-independent constructs for chemical computation. PLoS One. 6, (2011).
- Soloveichik, D., Cook, M., Winfree, E., Bruck, J. Computation with finite stochastic chemical reaction networks. Natural Computing. 7, 615-633 (2008).
- Soloveichik, D., Seelig, G., Winfree, E. DNA as a universal substrate for chemical kinetics. Proc. Natl. Acad. Sci. U. S. A. 107, 5393-5398 (2010).
- Tyson, J. J., Chen, K. C., Novak, B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell. Biol. 15, 221-231 (2003).
- Cardelli, L. Two-domain DNA strand displacement. Math. Struct. Comput. Sci. 23, 247-271 (2013).
- Angluin, D., Aspnes, J., Eisenstat, D. A simple population protocol for fast robust approximate majority. Distrib. Comput. 21, 87-102 (2008).
- Cardelli, L., Csikasz-Nagy, A. The cell cycle switch computes approximate majority. Sci. Rep. 2, 656 (2012).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 32, 170-173 (2011).
- Lee, P. Y., Costumbrado, J., Hsu, C. Y., Kim, Y. H. Agarose gel electrophoresis for the separation of DNA fragments. J. Vis. Exp. , (2012).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 6, 343-345 (2009).
- Froger, A., Hall, J. E. Transformation of plasmid DNA into E. coli using the heat shock method. J. Vis. Exp. , e253 (2007).
- Lessard, J. C. Transformation of E. coli via electroporation. Methods Enzymol. 529, 321-327 (2013).
- Nasri, M., Thomas, D. Alteration of the specificity of PvuII restriction endonuclease. Nucleic Acids Res. 15, 7677-7687 (1987).
- Dalchau, N., Seelig, G., Phillips, A. Computational design of reaction-diffusion patterns using DNA-based chemical reaction networks. DNA Computing and Molecular Programming. , 84-99 (2014).
- Scalise, D., Schulman, R. Designing modular reaction-diffusion programs for complex pattern formation. Technology. 2, 55-66 (2014).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados