
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Cultura e transfecção de células primárias de Zebrafish
Neste Artigo
Resumo
Apresentamos um protocolo eficiente e fácil de usar para a preparação de culturas de células primárias de zebrafish embriões para transfeccao e imagens de células vivas, bem como um protocolo para preparar as células primárias do cérebro adulto zebrafish.
Resumo
Zebrafish embriões são transparentes e desenvolvem rapidamente fora da mãe, permitindo assim excelente na vivo por imagens do processos biológicos dinâmicos em um vertebrado intacto e em desenvolvimento. No entanto, a imagem detalhada das morfologias de tipos distintos de células e estruturas subcelulares é limitada em montagens de toda. Portanto, nós estabelecemos um protocolo eficiente e fácil de usar para as células primárias ao vivo de cultura de embriões de peixe-zebra e tecido adulto.
Em breve, 2 dpf zebrafish embriões são dechorionated, deyolked, esterilizados e dissociado de células únicas com colagenase. Após uma etapa de filtração, as células primárias são banhadas em pratos de vidro inferior e cultivadas por vários dias. Culturas frescas, diferenciada de termo tanto quanto tempo os, podem ser usadas para estudos de imagiologia confocal de alta resolução. A cultura contém diferentes tipos de células, com miócitos estriados e neurônios sendo proeminente no revestimento de poli-L-lisina. Para especificamente rótulo subcellular estruturas por proteínas marcador fluorescente, também estabelecemos um protocolo de eletroporação que permite o transfeccao de Plasmideo DNA em diferentes tipos de células, incluindo neurônios. Assim, na presença do operador definido de estímulos, comportamento complexo celular e dinâmica intracelular de células primárias zebrafish pode ser avaliada com alta resolução espacial e temporal. Além disso, usando o cérebro adulto zebrafish, demonstramos que a técnica de dissociação descrita, bem como as condições básicas de cultivo, também trabalham para tecido adulto zebrafish.
Introdução
O peixe-zebra (Danio rerio, d. rerio) é um modelo popular vertebrado para inúmeros campos de pesquisa básica e biomédico1. Zebrafish embriões se desenvolvem rapidamente ex utero, são transparentes e ajuste sob um microscópio, proporcionando excelentes pré-requisitos para estudar o desenvolvimento de vertebrados em um organismo vivo. Devido a rastreabilidade genética de zebrafish2, foram criadas muitas linhas de repórter transgénicos estável com expressão de tipo específico de célula de vários marcadores fluorescentes permitindo a observação de populações de células específicas. A comunidade de zebrafish oferece uma ampla variedade de chamados linhas Gal4-motorista que carregam um transgene expressando o Kal4TA4 sintético (ou o equivalente de KalTA3-GalFF) gene com o domínio de Gal4-DNA-ligando de levedura fundido a ativação transcricional viral domínios sob o controle dos realçadores de tipo específico de célula. Estas linhas de motorista se cruzam linhas de efetor que carregam transgenes consistindo de uma definido upstream ativando sequência (UAS) fundida a um gene repórter. A proteína Kal4TA4 vincula-se ao elemento UAS, ativando assim a expressão seletiva-tipo de célula do gene repórter3,4. Esta abordagem permite estudos combinatórios altamente diversificados de quase todos os elementos disponíveis enhancer e repórter em animais transgénicos-duplo.
No entanto, imagens ao vivo em profundidade com foco em células individuais ou seus conteúdos subcellular é limitada em um embrião inteiro e constante mudança. Para abordar questões biológicas de célula específica com maior resolução, o uso de culturas de células, muitas vezes é preferível. Existem algumas linhas de célula de zebrafish, mas eles são considerados como altamente selecionado5,6,7 e sua propagação geralmente é demorada. Além disso, todas as linhas de celular disponíveis são fibroblastos derivados, limitando os experimentos utilizando cultura de células de um tipo de células. Portanto, estabelecemos um protocolo eficiente e fácil de usar para preparar as células primárias diretamente do zebrafish embriões e cérebro adulto zebrafish, juntamente com abordagens para aumentar a longevidade da cultura e ampliar a diversidade de cultivo tipos de células. Além disso, apresentamos um procedimento para transfect embrionário as células primárias com construções de expressão para marcadores fluorescentes organela. Assim, estruturas subcelulares e morfologias celulares podem ser analisadas com alta resolução espacial e temporal em tipos de células distintas que retêm suas principais características.
Protocolo
Todo trabalho animal aqui descrito está em conformidade com regulamentos legais (EU-Directiva 2010/63). Manutenção e manipulação de peixes foi aprovado pelas autoridades locais e o bem-estar dos animal representativo da Universidade Técnica de Braunschweig e Baixa Saxônia estado escritório de defesa do consumidor e da segurança alimentar (elevam, Oldenburg, Alemanha; AZ. parágrafo 4º (02.05) TSchB TU BS).
1. preparação da escola primária de células de embriões de peixe-zebra
- Preparação de 2 dias pós fertilização (dpf) embriões de zebrafish
- Dia 1: Configure vários cruzamentos da estirpe do zebrafish de escolha e de acordo com as especificações de sua facilidade de zebrafish gerente8.
- Dia 2: Acasalar peixe e coletar ovos8 diretamente após a desova em uma placa de Petri (plástico) de 10 cm. Remova ovos mortos ou contaminados com uma pipeta Pasteur (plástico). Lavagem de ovos 1 x com Danieau 30% (5,8 mM de cloreto de sódio, 0,07 mM de cloreto de potássio, sulfato de magnésio de 0,04 mM, nitrato de cálcio 0,06 mM, 5 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic ácido, pH 7,2)8 com azul de metileno de 0,0001% (p/v). Trocar a médio Danieau 30% sem azul de metileno, azul de metileno pode causar autofluorescência e incubar ovos durante a noite a 28 ° C.
Nota: Comece com um mínimo de 100 ovos por linha de peixe, a fim de obter uma quantidade suficiente de células. Não aumente mais de 150 embriões em uma placa de Petri. - Dia 3: Remover embriões mortos ou contaminados e trocar a médio e a Danieau de 30%. Determine o número de embriões por contagem. Incubar os embriões durante a noite a 28 ° C.
Nota: Ao usar uma linha transgênica expressando um repórter fluorescente, triagem no 1 dpf ou 2 dpf pode ser necessária.
Nota: Para quantidades maiores de embriões, é recomendável tomar uma imagem em preto e branco da caixa de Petri respectivos (figura 1A) e para quantificar o número de embriões com um software de imagem. - Dia 4: Embriões são agora 2 dpf. Remover chorions, adicionar 1 mL pronase com uma concentração de 1 mg/mL a 10 mL de Danieau 30%8 e incubar embriões num agitador, à temperatura ambiente até que todos os chorions são desanexados (20 – 40 min dependendo da temperatura ambiente). Lave com Danieau para remover tanto pronase e chorions e manter embriões à temperatura ambiente até o uso de mais de 30%.
- Preparação de pratos de fundo de vidro revestido de poli-L-lysine reutilizável
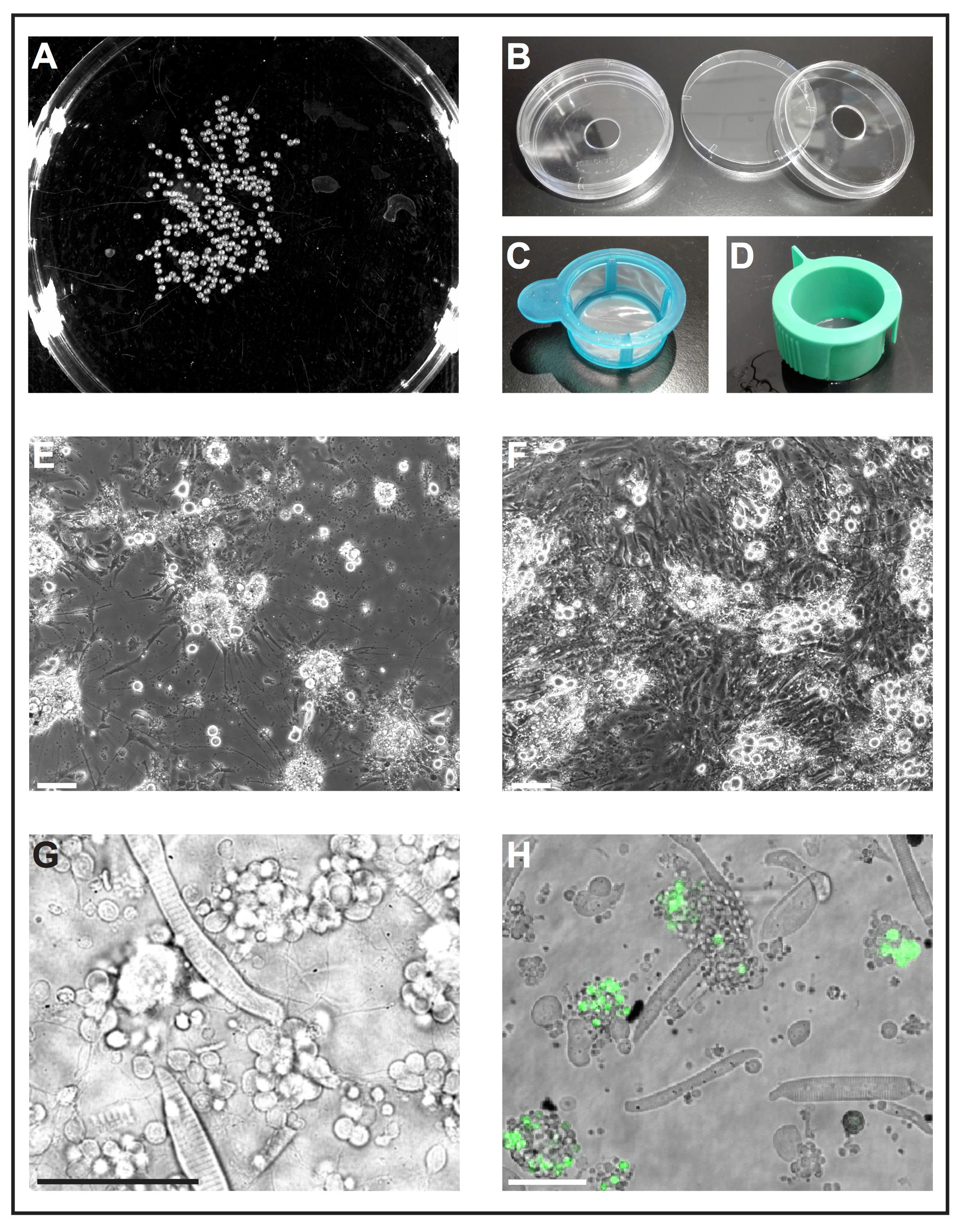
Nota: Pratos de fundo de vidro disponíveis comercialmente são projetados para uso único só e são caros. O procedimento a seguir descreve como preparar pratos de fundo reutilizável vidro self-made de materiais de laboratório padrão.- Faça um furo com um diâmetro de 10 mm na parte inferior dos pratos de cultura de célula padrão (diâmetro de 6 cm, plástico) (figura 1B). Lave o prato fundo abundantemente com água corrente para remover o pó da perfuração.
- Espalhar a graxa de silicone em torno do furo na parte inferior do fundo do prato e anexar uma lamela usando a gordura como cola. Certifique-se de que a graxa foca a lacuna entre o fundo do prato e lamela.
Nota: Certifique-se que a espessura das lamelas utilizadas é apropriada para a aplicação depois de imagem. - Lave pratos de self-made vidro inferior completamente, mas cuidadosamente, com água fria e sabão. Lave pratos de vidro fundo três vezes com água desionizada para remover o sabão. Secar ao ar livre as tampas do prato e prato fundo e armazená-los em uma caixa limpa até nova utilização.
- No dia da preparação da cultura (dia 4, ver 1.1.4): umedecer o interior das duas tampas do prato e prato fundo com etanol a 70% (v/v). Coloque as tampas do prato e prato de fundo virado para o lado interno em uma bancada estéril com fluxo laminar e luz UV. Secar ao ar livre até que o etanol é evaporado e, em seguida, aplicar UV luz por 20 min. Após este tratamento, os pratos são montados e considerados como estéril.
- Para o revestimento, pipetar 200 µ l de poli-L-lisina (0,1 mg/mL) no meio de cada prato fundo de vidro e espalhar o líquido na lamela por quebrar a tensão superficial com a ponta da pipeta. Deixe secar por 60 min, em seguida, lavar 1x com estéril 1x tampão fosfato salino (PBS). Remova o líquido. Manter pratos debaixo do banco até utilização posterior.
Nota: Outros revestimentos podem ser testados dependendo o objetivo do experimento. Encontramos o suficiente para suportar o crescimento de neurônios poli-L-lisina, Considerando que o Tratado de plástico sem qualquer revestimento adicional parecia ser favorável para o crescimento de células de fibroblastos (Figura 1E, F).
Nota: Pratos de vidro self-made inferior podem ser usados muitas vezes. Para trocar a lamela, lave com água morna da torneira, cuidadosamente desanexar a lamela e remover a gordura restante com etanol a 70% (v/v) e sabão.
- Preparação e chapeamento de células primárias
- No dia da preparação da cultura (dia 4, ver 1.1.4): transferir os embriões em um prato de cultura de células estéreis (diâmetro 6 cm), usando uma pipeta de Pasteur de plástico fresca. Remova o líquido step-wise, até que todos os embriões são reunidos em uma grande gota com um diâmetro de cerca de 2 cm ou menos.
- Coloque o prato com embriões em bancada de trabalho estéril e adicionar CO2-médio independente (suplementado com 10% (v/v) filtrado soro bovino, 1 x glutamina e 1,2% (v/v) 10.000 U penicilina-estreptomicina; médio com todos os suplementos é no seguinte conhecido como "meio de cultura celular") até que o prato está meio-cheio.
Nota: Alternativas para CO2-médio independente pode ser testado dependendo o objetivo do experimento, como por exemplo neurobasal médio, meio DMEM ou meio de Leibovitz L-15. CO2-independente médio e médio de L-15 do Leibovitz têm a vantagem de não exigir uma incubadora de CO2 . - Para remover a gema, pipetar embriões acima e para baixo usando uma ponta de 200 µ l-pipeta. Deyolking bem sucedido pode ser reconhecido pela turvação do meio.
- Encha um prato de cultura celular com etanol a 70% (v/v) e um outro prato de cultura celular com meio de cultura celular fresco. Use uma ponta de pipeta-µ l de Cut-off 1.000 para transferir os embriões em um coador de célula estéreis com punho (40 µm; A Figura 1). Pegue o filtro pelo punho e mergulhá-lo no prato com etanol, para que todos os embriões são submergidos por 5 s. imediatamente depois, mergulhe o filtro com embriões no prato com meio de cultura celular fresco.
Nota: Filtros de célula possam ser reutilizados várias vezes. Limpe com uma escova macia sob corrente de água da torneira, loja em etanol a 70% e seca e UV-tratá-los sob o trabalho estéril no banco diretamente antes de usar (ver 1.2.4). - Transferência de embriões nos tubos de reação estéril 1,5 mL (aproximadamente 100 embriões em um tubo). Adicione a colagenase (tipo 2) diluída em meio de cultura celular para uma concentração final de 4 mg/mL em um volume total de 1 mL. Incube os tubos com embriões em rotacao tubo vertical com 30 rotações por min para 45 min à temperatura ambiente.
- Dissocia os restantes aglomerados de células pipetando a mistura de embrião-colagenase acima e para baixo com uma ponta de 1.000 µ l-pipeta. Em seguida, filtrar a suspensão de células através de um filtro de célula estéril com ranhura de ventilação (40 µm; Figura 1.) em um tubo cónico de 50 mL. Lave o filtro com aproximadamente 10 mL de meio de cultura celular fresco.
- Granule células por centrifugação por 3 min a 180 x g. O sedimento pode ser quase invisível. Cuidadosamente, remover o sobrenadante e ressuspender as células em meio de cultura de células frescas µ l 200 por 30 embriões originalmente usados.
Nota: Para obter um sedimento visível, é aconselhável começar com um mínimo de 100 embriões. - Pipetar 200 µ l de suspensão de células obtida na etapa 1.3.7 diretamente sobre a área de vidro de um prato fundo de vidro self-made poli-L-lisina-revestido (ver 1.2). Incube durante 60 min à temperatura ambiente sob a bancada de trabalho estéril. Adicione 6 mL de meio de cultura celular fresco e incubar as células primárias a 28 ° C.
- Executar a aplicação de imagem desejada, usando um microscópio invertido a 28 ° C. Troca o meio de cultura celular diariamente. Culturas podem ser usadas para a imagem latente durante vários dias depois de chapeamento (dap).

Figura 1: cultura de células primárias de embriões de zebrafish. (A) imagem em preto e branco de 1 dap embriões, que podem ser processados por uma ferramenta de software para analisar o número de embriões. (B) pratos de cultura de células (diâmetro 6cm) com um orifício perfurado (diâmetro 10 mm) são usados para preparar pratos de fundo de vidro reutilizáveis de self-made. (C) filtros de célula (40 µm), com um simples identificador são usados como "pontas de camaroeiro" para mergulhar deyolked embriões em etanol e transferi-los rapidamente para o meio de cultura celular fresco. (D) célula filtros (40 µm), com ranhuras de ventilação são usados para filtrar as células após dissociação mediada por colagenase. (E) após 5 dap, as células primárias semeado no vidro revestido com poli-L-lysine principalmente formar neurônios com extensões pronunciados. Barra de escala = 100 µm. (F) depois de 5 dap em plástico tratado sem revestimento, fibroblasto, como células overgrow a cultura. Barra de escala = 100 µm. (E) e (F) foram adquiridos por um microscópio de epifluorescente. (G) transmissíveis luz imagem de células primárias derivado tipo selvagem zebrafish em 1 dap. Miócitos estriados e aglomerados de neurônios estender processos finos podem ser facilmente observados. Barra de escala = 50 µm. (H) pilhas cultivadas da linha transgénica Tg (ptf1a: eGFP) jh1, que expressa eGFP em progenitores neuronais de neurônios gabaérgica principalmente o rombencéfalo e um subconjunto de células da retina populações29, 30 , 31. barra de escala = 50 µm. (G) e (H) foram adquiridos por um laser confocal microscópio usando os pratos de fundo de vidro feitos conforme ilustrado em (B). Clique aqui para ver uma versão maior desta figura.
{kind=link}
2. transfection da escola primária de células com plasmídeo
- Ressuspender filtrado e peletizadas células obtidas no passo 1.3.7 em 1X PBS em vez de meio de cultura celular. Manche uma alíquota da suspensão celular com Trypan azul para determinar o número de células em uma contagem de câmara9.
- Misturar 0,5 milhões de células com 10 µ g ultra-pura plasmídeo em um tubo de reação de 1,5 mL e ajustar o volume total de 100 µ l com PBS 1x.
Nota: Para nossos experimentos, principalmente utilizado constrói de expressão com base no plasmídeo pCS2 +10. Expressão de frames de leitura abertos clonado no local de clonagem múltipla de pCS2 + é conduzido pelo promotor onipresente do citomegalovírus humano (promotor CMV). Outras construções de expressão e promotores podem ser testados (ver também resultados de representante e a Figura 2 e Figura 3). - Mistura de transferência do celular-DNA imediatamente para uma cubeta de eletroporação (0,4 cm), colocar a cubeta em um dispositivo de eletroporação e electroporate com as seguintes configurações: pulso único, decaimento exponencial, 280 V, 950 µF.
- Diretamente após a eletroporação, transferi a mistura de DNA de células em um tubo de reação de 1,5 mL com 300 µ l de meio de cultura celular fresco.
- 200 µ l de suspensão de células da placa conforme descrito em 1.3.8 e proceder conforme descrito em 1.3.9 dependendo da construção da expressão utilizada, expressão de proteínas fluorescentes pode ser detectável após algumas horas, ou no dia seguinte.
3. coloração de fixa as células primárias
Nota: Estruturas subcelulares também podem ser visualizadas por imunocoloração clássica em vez de usar os repórteres de proteína de fusão fluorescente. Para as células primárias de zebrafish, usamos o seguinte protocolo padrão para núcleo exemplar mancha, F-Actina e tubulina acetilada com marcadores fluorescentes.
- Células de placa em lamínulas revestidas de poli-L-lysine colocada em uma placa de prato ou multiwell de cultura celular conforme descrito anteriormente (ver 1.3.8).
- Para a fixação, remover o meio e cobrir as células com paraformaldeído 4% em 1X PBS. Incube as celulas por 10 min a 4 ° C, um agitador. Lavagem de células 3 x por 5 min com PBS 1x à temperatura ambiente. Certifique-se de que o PBS 1x cobre as células completamente e execute as etapas de lavagem em uma coqueteleira.
- Bloquear e para permeabilize as células fixas, cobrir as células com 1X PBS contendo 5% de leite desnatado e 0,3% Triton X-100. Coloque as células durante 10 minutos à temperatura ambiente em um shaker. Lave as células conforme descrito no ponto 3.2.
- Para rotular tubulina acetilada, um marcador para axônios11, diluir o 1:2,000 do anticorpo primário em 1X PBS contendo 1% de leite desnatado. Cobrir as células com esta solução e incube-os durante a noite a 4 ° C, um agitador. No dia seguinte, lave as células conforme descrito no ponto 3.2.
- Diluir o anticorpo secundário conjugado com fluorochrom verde fluoresceína isotiocianato (FITC) 1: 100 em 1X PBS contendo 1% de leite desnatado e incube as celulas com esta solução por 1h à temperatura ambiente no escuro (cobrir o prato , por exemplo, com uma caixa ou folha de alumínio) em uma coqueteleira. Lave as células conforme descrito no ponto 3.2.
- Para simultaneamente manchar o citoesqueleto de actina e núcleos, Incube as celulas em PBS 1x suplementado com faloidina12 conjugados com um fluorocromo vermelho (01:50) e 4' 6-diamidino-2-phenylindole (DAPI)13 (100 ng/mL) por 10 min no quarto temperatura no escuro em um shaker. Lave as células conforme descrito no ponto 3.2.
- Para preparar células para tratamento de imagens, colocar um portador de objeto de vidro (microscópio) sobre uma superfície limpa e coloque na gota de meio de montagem nele. Tirar uma lamela com células coradas e fixas um prato usando uma pinça e coloque-o sobre a queda com as células que enfrenta o portador de objeto. Certifique-se que esse meio de montagem se espalha sobre toda a área da lamínula. Deixar secar no escuro.
- Loja fixa e montado células no escuro a 4 ° C até que o aplicativo de imagem desejado é executado.

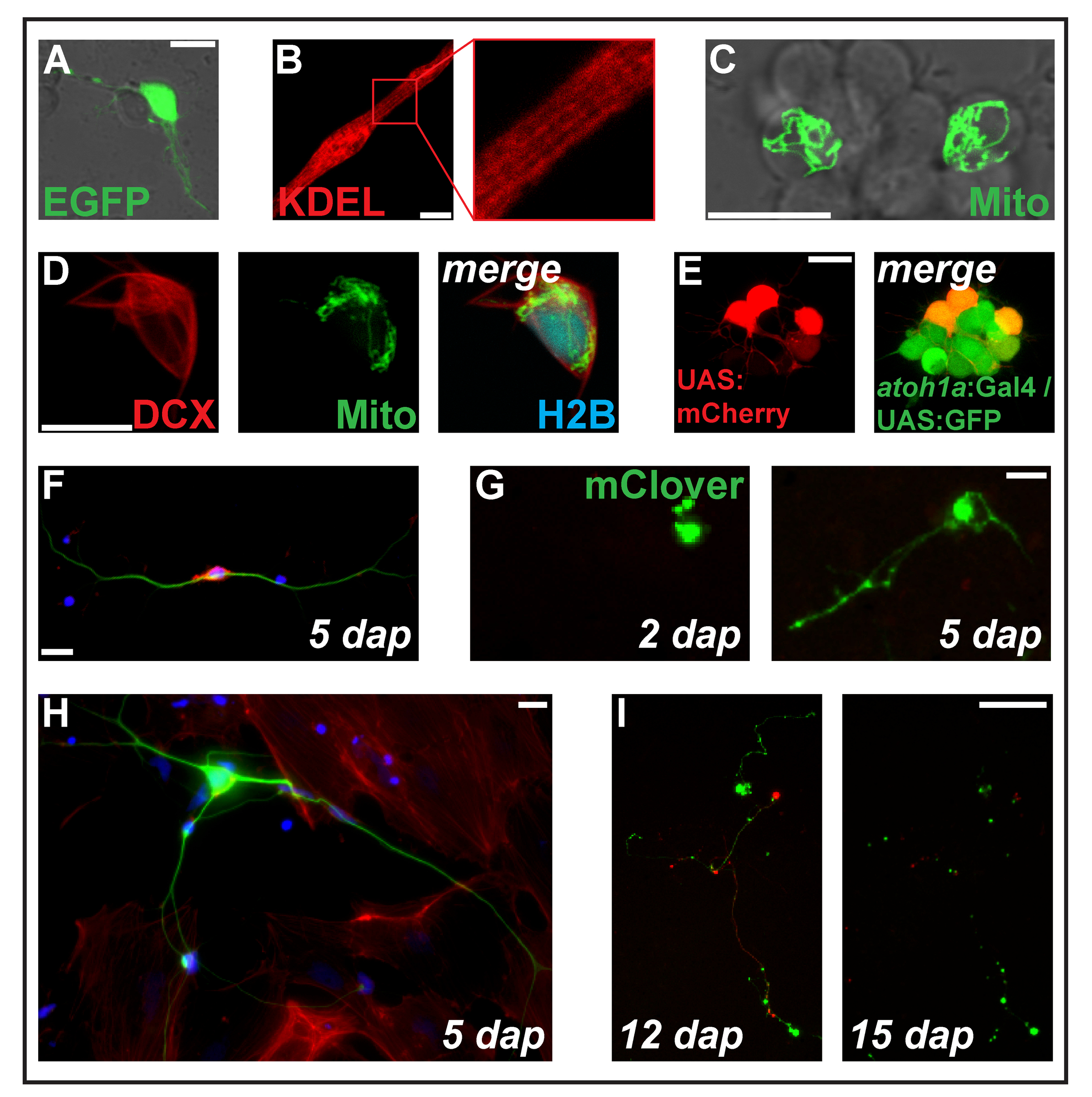
Figura 2: Transfection de expressão de construções por eletroporação. (A) Jesus Rodrigues Putative neurônio transfected com pCS-eGFP em 1. (B) estriado miócito (2 dap) expressando a proteína retículo endoplasmático-alvo ss-RFP-KDEL. (C) dois neurônios dentro de um cluster neuronal transfectadas com pCS-MitoTag-YFP em 2 dap. (D) Cell (dap 2) triplo-transfectadas com pCS-DCX-tdTomato, pCS-MitoTag-YFP e pCS-H2B-mseCFP. (E) pSK-UAS:mCherry electroporated em células primárias (1 dap) derivadas de embriões duplo-transgénicos, carregando os transgenes Tg (atoh1a: Gal4TA4) hzm222 e Tg (4xUAS:KGFPGI) hzm332 , resultando na expressão de GFP em neuronais progenitores do rombencéfalo. Barras de escala = 10 µm. (A-E) foram adquiridos por um laser confocal, microscopia usando os pratos de fundo de vidro feitos conforme ilustrado na figura 1B. (F) coloração fluorescente de neurônios primários zebrafish fixo em 5 dap. Azul: DAPI (núcleo); Vermelho: Faloidina (F-Actina); Verde: Acetilado tubulina (neurônios). Barra de escala = 10 µm. (G) neurônio-como células transfectadas com pCS-do amor. Em 2 dap, sem extensão é visível. Em 5 dap, formou-se uma estrutura de axônio. Barra de escala = 25 µm. (H) neurônio da mesma preparação como a célula em (F), rodeado por células fibroblastos. Barra de escala = 10 µm. (eu) neurônio derivado de um embrião geneticamente modificado carregando o transgene Tg (XITubb: DsRed) zf14828 transfected com pCS-do amor. Entre 12 e 15 dap, os neuritos sofrem degeneração massiva. Barra de escala = 100 µm. células mostradas na (F-eu) foram semeadas em vidro revestido de poli-L-lisina (F, H) ou plástico (G, eu), cultivado em meio de L-15, na presença de 10% de soro bovino de filtrado e o suplemento neuronal B-27 (diluídos 01:50) e fotografada com um microscópio de epifluorescente. Clique aqui para ver uma versão maior desta figura.
{kind=link}
4. preparação de células primárias do cérebro adulto de Zebrafish
-
Extração do cérebro
- Selecione um peixe adulto de pelo menos 90 dias de idade. Se trabalhar com um tipo de célula específica é necessária, escolha uma linha de transgénica na qual expressão fluorescente repórter célula específica permitirá visualizar as células desejadas.
- Coloque o peixe num copo cheio de 200 mL do anestésico tricaina8 (0,2%) em Danieau 30%. Espere até que o peixe para de se mover. Mergulhe o peixe anestesiado num copo cheio de gelo de 200 mL de água por 15 min para sacrificá-lo.
- Encha um prato de Petri (diâmetro 6cm) com etanol a 70% (v/v). Segurar o peixe pela cauda com um par de pinças e mergulhá-lo em etanol. Garantir que o peixe está completamente submersa em etanol por 5 s.
- Extrair o cérebro de acordo com o protocolo de Gupta e Mullins14 com as seguintes adaptações: use somente ferramentas autoclavadas ou estéril-embalados e dissecar a cabeça em PBS 1x estéril.
- Diretamente após a extração, colocar o cérebro em uma placa de Petri (diâmetro 3 cm) preenchido com PBS 1x estéril e mover o prato sob uma bancada de trabalho estéril para cultura de células.
-
Dissociação do cérebro e chapeamento de células primárias
- Coloque dois conjuntos de pinças estéreis (autoclavadas), uma placa de Petri estéril (diâmetro 6cm) preenchido com etanol a 70% (v/v), uma placa de Petri estéril (diâmetro 10 cm) preenchido com meio de L-15 do Leibovitz suplementado com 10% (v/v) filtrado soro bovino, B-27 (01:50) e 1,2% (v / v) 10.000 U penicilina-estreptomicina e um filtro de célula estéreis com punho (40 µm; Figura 1.) sob o banco limpo.
- Lugar o coador de célula para o prato de Petri preenchida com etanol e garantir o nível de líquido é maior que a parte inferior do filtro pelo menos 5 mm.
- Usando o primeiro conjunto de pinças, transferir o cérebro para o filtro já colocado em etanol e certifique-se de que está totalmente coberto pelo líquido. Depois de 1 s, transferência do filtro com o cérebro para a placa de Petri contendo meio de Leibovitz L-15 com o acima descrito suplementos.
- Usando o segundo conjunto de pinças, transferência o cérebro em um tubo de reação estéril 1,5 mL preenchido com média do 500 µ l Leibovitz L-15 com o acima descrito suplementos. Adicione colagenase (tipo 2) a uma concentração final de 4 mg/mL em um volume total de 1 mL.
- Incube o tubo em rotacao tubo vertical com 30 rotações por minuto para 35 min à temperatura ambiente. Mecanicamente, dissocia as touceiras de tecido restante pipetando acima e para baixo usando a ponta da pipeta 1.000 µ l para auxiliar o processo de dissociação.
- Pare a dissociação quando sem partículas visíveis permanecem em solução. Filtrar a suspensão através de um filtro de célula estéril com ranhura de ventilação (40 µm; Figura 1.) em um tubo cónico de 50 mL. Lave o filtro com aproximadamente 10 mL de meio de cultura celular fresco.
Nota: Quando a suspensão de célula única é alcançada, a etapa de filtração não é tão necessária como no caso de dissociação de embriões. O cérebro é um tecido mole e como tal é mais propenso a ser dissociado homogeneamente em suspensão de célula única. - Células de pelotas por centrifugação por 5 min a 180 x g e ressuspender as células em 1 mL de meio de L-15 do Leibovitz fresco com os suplementos descritos acima.
- Pipetar 500 µ l de suspensão de células (50% das células obtidas) em um prato fundo de vidro revestido de poli-L-lysine self-made (ver 1.2) ou em um poço de uma placa de 24. Downscale no caso de superfícies menores (ou seja, 125 solução µ l para um poço de uma placa de 96 poços). Incube durante 60 min à temperatura ambiente sob a bancada de trabalho estéril. Em seguida, adicione o volume necessário de meio fresco para encher o recipiente específico e incubar as células primárias a 28 ° C.
- Executar a aplicação de imagem desejada, usando um microscópio invertido a 28 ° C. Culturas podem ser usadas para a imagem latente durante vários dias depois do chapeamento. Substitua 50% do meio em uma base diária.
Resultados
A Figura 1 mostra uma imagem de luz transmitida de uma cultura típica derivada de embriões de tipo selvagem com miócitos estriados e aglomerados de células neurônio-como sendo o mais abundante. Para identificar determinados tipos de células mais facilmente, uma linha de transgênica com expressão de tipo específico de célula de uma proteína fluorescente pode ser usado (Figura 1 H).
Discussão
Aqui, apresentamos dois protocolos diferentes de cultura primária de células de 2 embriões de zebrafish dpf ou cérebro adulto zebrafish.
A preparação das culturas de célula primária de 2 dpf zebrafish é relativamente fácil de executar para qualquer pessoa com experiência em técnicas de cultura de célula básica. No entanto, para obter resultados reprodutíveis e boas, um número suficiente de embriões como material de partida é crucial (100 é o mínimo). Durante o levantamento ...
Divulgações
Os autores não têm nada para divulgar.
Agradecimentos
Agradecemos a T. Fritsch, A. lobo-Asseburg, I. Linde e S.-M. Tokarski para excelentes cuidados com animais e suporte técnico. Agradecemos a todos os membros do laboratório Köster discussões intensas e útil. Reconhecemos com gratidão financiamento pela Deutsche Forschungsgemeinschaft (KO 1949/5-1) e o Estado Federal da Baixa Saxónia, Niedersächsisches Vorab (VWZN2889).
Materiais
| Name | Company | Catalog Number | Comments |
| Fish lines | |||
| AB (wild-type) | established by Streisinger and colleagues, available from the Zebrafish International Resource Center (ZIRC) | ||
| Tg(ptf1a:eGFP)jh1 | stable transgenic line in which the enhancer of the zebrafish gene ptf1a drives expression of the fluorescent protein EGFP (Parsons et al., 2007) | ||
| Tg(XITubb:DsRed)zf148 | stable transgenic line in which the Xenopus neural-specific beta tubulin promoter drives expression of the fluorescent protein DsRed (Peri and Nüsslein-Volhard, 2008) | ||
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| centrifuge | Eppendorf | model 5804 R | |
| ChemiDoc MP imaging system | BioRad | model XRS+, used to acquire black-and-white images of Petri dishes containing 1 da embryos | |
| confocal laser scanning microscope | Leica microsystems | model SP8, equipped with 28 °C temperature box and a 63X objective | |
| epifluorescent microscope | Leica microsystems | model DM5500B, equipped with 28 °C temperature box and a 40X objective | |
| Gene Pulser Xcell with capacitance extender | BioRad | 1652661 | electroporation device |
| Horizontal shaker | GFL | model 3011 | |
| incubator for cell culture (28 °C) | Memmert | model incubator I | |
| incubator for embryos (28 °C) | Heraeus | type B6120 | |
| light microscope | Zeiss | model TELAVAL 31 | |
| micro pipettes | Gilson | ||
| sterile work bench | Bio Base | with laminar flow and UV light | |
| tweezers | Dumont | Style 5, Inox | |
| vertical tube rotator | Labinco B.V. | model LD-79 | |
| Name | Company | Catalog Number | Comments |
| Software | |||
| Image Lab Software | BioRad | for the ChemiDoc MP imaging system from BioRad | |
| ImageJ | National Institutes of Health | used for counting 1 dpf embryos by applying the Count particles-tool to the respective black-and-white images; Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/. (1997-2016). | |
| LAS X | Leica Microsystems | for both confocal and epifluorescent microscopes from Leica Microsystems | |
| Name | Company | Catalog Number | Comments |
| Plasmids | |||
| pCS-DCX-tdTomato | Köster Lab | # 1599 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-eGFP | Köster Lab | # 7 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-H2B-mseCFP | Köster Lab | # 2379 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-mClover | Köster Lab | # 3865 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-MitoTag-YFP | Köster Lab | # 2199 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-ss-RFP-KDEL | Köster Lab | # 4330 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-VAMP1-mCitrine | Köster Lab | # 2291 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pSK-UAS:mCherry | Köster Lab | # 1062 | based on the pBluescript-backbone of Stratagene |
| Plasmid numbers refer to the database entries of the Köster lab. Plasmids are available upon request. | |||
| Name | Company | Catalog Number | Comments |
| Plastic and glass ware | |||
| BD Falcon Cell Strainer (40 µm) | FALCON | REF 352340 | distributed by BD Bioscience, used as “landing net” to dip deyolked embryos into ethanol and to transfer them quickly to fresh cell culture medium |
| 1.5 mL reaction tubes | Sarstedt | 72690550 | |
| 24-well plate | Sarstedt | 83.3922 | |
| 50 mL falconic tube | Sarstedt | 62.547.004 | |
| 96-well plate | Sarstedt | 83.3924.005 | |
| EasyStrainer (40 µm) | Greiner Bio-One | 542 040 | with venting slots; used to filter cells after collagenase-mediated dissociation |
| electroporation cuvette (0.4 cm) | Kisker | 4905022 | |
| glass coverslips | Heinz Herenz Medizinalbedarf GmbH | 1051201 | |
| Microscope slides | Thermo Fisher Scientific (Menzel Gläser) | 631-0845 | |
| Neubauer chamber | Henneberg-Sander GmbH | 9020-01 | |
| Pasteur pipettes (plastic; 3 mL) | A. Hartenstein | PP05 | |
| Petri dishes (plastic; diameter 10 cm) | Sarstedt | 821473 | for zebrafish embryos |
| pipette tips | Sarstedt | Blue (1000 µl): 70762; Yellow (200 µl): 70760002; White (10 µL): 701116 | |
| sterile cell culture dishes (plastic; diameter 3 cm) | TPP Techno Plastic Products AG | 93040 | |
| sterile cell culture dishes (plastic; diameter 6 cm) | Sarstedt | 72690550 | |
| sterile Petri dishes (plastic; diameter 10 cm) | Sarstedt | 83.3902 | for brain dissection |
| Name | Company | Catalog Number | Comments |
| Chemicals and Reagents | |||
| sodium chloride | Roth | 0601.1 | |
| 4 % paraformaldehyde in 1x PBS | Sigma-Aldrich | 16005 | |
| 4',6-diamidino-2-phenylindole (DAPI) | Thermo Fisher Scientific | D1306 | |
| calcium nitrate tetrahydrate | Sigma-Aldrich | C1396 | |
| ethanol p.a. 100% | Sigma-Aldrich | 46139 | |
| goat α-mouse IgG (Fc specific) FITC conjugated | Thermo Fisher Scientific | 31547 | |
| HEPES | Roth | 9105.4 | |
| high vacuum grease | DOW CORNING | 3826-50 | silicon grease used for self-made glass bottom dishes |
| magnesium sulfate heptahydrate | Merck | 105886 | |
| methylene blue | Serva | 29198.01 | |
| Monoclonal Anti-Tubulin, Acetylated antibody | Sigma-Aldrich | T6793 | |
| Aqua-Poly/Mount (mounting medium) | Polyscience | 18606 | |
| poly-L-lysine | Biochrom | L 7240 | |
| potasssion chloride | Merck | 104938 | |
| Skim milk | Roth | 68514-61-4 | |
| Texas Red-X Phalloidin | Thermo Fisher Scientific | T7471 | |
| Tricaine | Sigma-Aldrich | E10521 | Synonym: Ethyl 3-aminobenzoate methanesulfonate |
| Triton X-100 | BioRad | 1610407 | |
| Trypan Blue | Gibco by Life Technologies | 15250061 | |
| Name | Company | Catalog Number | Comments |
| Enzymes | |||
| collagenase (Type 2) | Thermo Fisher Scientific | 17101015 | dissolve powder in cell culture medium (8 mg/mL) and sterile-filter the solution, store aliquots at -20 °C |
| pronase (from Streptomyces griseus) | Roche | 11459643001 | distributed by Sigma-Aldrich, dissolve in 30% Danieau (10 mg/mL) and store aliquots at -20 °C |
| Name | Company | Catalog Number | Comments |
| Medium and solutions for cell culture | |||
| 1x PBS (Dulbecco's Phosphate Buffered Saline) | Gibco by Life Technologies | 14190-169 | distributed by Thermo Fisher Scientific |
| CO2-independent medium | Gibco by Life Technologies | 18045054 | distributed by Thermo Fisher Scientific |
| filtrated bovine serum (FBS) | PAN-Biotech | individual batch | |
| glutamine 100x | Gibco by Life Technologies | 25030081 | distributed by Thermo Fisher Scientific |
| Leibovitz's L-15 medium | Gibco by Life Technologies | 11415049 | distributed by Thermo Fisher Scientific |
| PenStrep (10,000 U/mL) | Gibco by Life Technologies | 15140148 | distributed by Thermo Fisher Scientific |
Referências
- Ablain, J., Zon, L. I. Of fish and men: using zebrafish to fight human diseases. Trends in Cell Biology. 23, 584-586 (2013).
- Sassen, W. A., Köster, R. A molecular toolbox for genetic manipulation of zebrafish. Advances in Genomics and Genetics. , 151 (2015).
- Scheer, N., Campos-Ortega, J. A. Use of the Gal4-UAS technique for targeted gene expression in the zebrafish. Mechanisms of Development. 80, 153-158 (1999).
- Köster, R. W., Fraser, S. E. Tracing transgene expression in living zebrafish embryos. Developmental Biology. 233, 329-346 (2001).
- Driever, W., Rangini, Z. Characterization of a cell line derived from zebrafish (Brachydanio rerio) embryos. In Vitro Cellular & Developmental Biology - Animal. 29A, 749-754 (1993).
- Badakov, R., Jaźwińska, A. Efficient transfection of primary zebrafish fibroblasts by nucleofection. Cytotechnology. 51, 105-110 (2006).
- Senghaas, N., Köster, R. W. Culturing and transfecting zebrafish PAC2 fibroblast cells. Cold Spring Harbor Protocols. , (2009).
- Westerfield, M. . The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio). , (2007).
- Basic methods in cellular and molecular biology. Using a hemacytometer to count cells. Journal of Visualized Experiments Available from: https://www.jove.com/science-education/5048/using-a-hemacytometer-to-count-cells (2017)
- Rupp, R. A., Snider, L., Weintraub, H. Xenopus embryos regulate the nuclear localization of XMyoD. Genes & Development. 8, 1311-1323 (1994).
- Piperno, G., Fuller, M. T. Monoclonal antibodies specific for an acetylated form of alpha-tubulin recognize the antigen in cilia and flagella from a variety of organisms. Journal of Cell Biology. 101 (6), 2085-2094 (1985).
- Barden, J. A., Miki, M., Hambly, B. D., Dos Remedios, C. G. Localization of the phalloidin and nucleotide-binding sites on actin. European Journal of Biochemistry. 162 (3), 583-588 (1987).
- Kapuscinski, J. DAPI: a DNA-specific fluorescent probe. Biotechnic & Histochemistry. 70 (5), 220-233 (1995).
- Gupta, T., Mullins, M. C. Dissection of organs from the adult zebrafish. Journal of Visualized Experiments. 37, E1717 (2010).
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., Prasher, D. C. Green fluorescent protein as a marker for gene expression. Science. 263, 802-805 (1994).
- Stornaiuolo, M. KDEL and KKXX retrieval signals appended to the same reporter protein determine different trafficking between endoplasmic reticulum, intermediate compartment, and Golgi complex. Molecular Biology of the Cell. 14, 889-902 (2003).
- Lithgow, T. Targeting of proteins to mitochondria. FEBS Letters. 476, 22-26 (2000).
- Nagai, T., Ibata, K., Park, E. S., Kubota, M., Mikoshiba, K., Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nature Biotechnology. 20, 87-90 (2002).
- Sassen, W. A., Lehne, F., Russo, G., Wargenau, S., Dübel, S., Köster, R. W. Embryonic zebrafish primary cell culture for transfection and live cellular and subcellular imaging. Developmental Biology. 430, 18-31 (2017).
- Horesh, D., et al. Doublecortin, a stabilizer of microtubules. Human Molecular Genetics. 8, 1599-1610 (1999).
- Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N. G., Palmer, A. E., Tsien, R. Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nature Biotechnology. 22, 1567-1572 (2004).
- Distel, M., Hocking, J. C., Volkmann, K., Köster, R. W. The centrosome neither persistently leads migration nor determines the site of axonogenesis in migrating neurons in vivo. Journal of Cell Biology. 191, 875-890 (2010).
- Matsuda, T., Miyawaki, A., Nagai, T. Direct measurement of protein dynamics inside cells using a rationally designed photoconvertible protein. Nature Methods. 5, 339-345 (2008).
- Archer, B. T., Ozçelik, T., Jahn, R., Francke, U., Südhof, T. C. Structures and chromosomal localizations of two human genes encoding synaptobrevins 1 and 2. Journal of Biological Chemistry. 265, 17267-17273 (1990).
- Griesbeck, O., Baird, G. S., Campbell, R. E., Zacharias, D. A., Tsien, R. Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. Journal of Biological Chemistry. 276, 29188-29194 (2001).
- Shaner, N. C., et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nature Methods. 10, 407-409 (2013).
- Campbell, R. E., et al. A monomeric red fluorescent protein. Procedings of the National Academy of Sciences of the United States of America. 99, 7877-7882 (2002).
- Peri, F., Nüsslein-Volhard, C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 133, 916-927 (2008).
- Godinho, L., et al. Targeting of amacrine cell neurites to appropriate synaptic laminae in the developing zebrafish retina. Development. 132, 5069-5079 (2005).
- Jusuf, P. R., Harris, W. A. Ptf1a is expressed transiently in all types of amacrine cells in the embryonic zebrafish retina. Neural Development. 4, 34 (2009).
- Kani, S., et al. Proneural gene-linked neurogenesis in zebrafish cerebellum. Developmental Biology. 343, 1-17 (2010).
- Distel, M., Wullimann, M. F., Köster, R. W. Optimized Gal4 genetics for permanent gene expression mapping in zebrafish. Procedings of the National Academy of Sciences of the United States of America. 106, 13365-13370 (2009).
- Choorapoikayil, S., Overvoorde, J., den Hertog, J. Deriving cell lines from zebrafish embryos and tumors. Zebrafish. 10, 316-332 (2013).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados