
Method Article
Deteção de tilápia Lago vírus usando convencional RT-PCR e SYBR Green RT-qPCR
Neste Artigo
Resumo
Este protocolo diagnostica vírus de Lago de tilápia (TiLV) em tecidos de tilápia, utilizando metodologias de RT-PCR. Todo o método é descrito de dissecção de tecidos para extração de RNA total, seguida pela síntese do cDNA e detecção de TiLV por PCR convencional ou PCR quantitativo usando dsDNA vinculando um corante fluorescente de vinculação.
Resumo
O objetivo desse método é facilitar a detecção rápida, sensível e específica de tilápia Lago vírus (TiLV) em tecidos de tilápia. Este protocolo pode ser usado como parte de programas de vigilância, medidas de biossegurança e nos laboratórios de pesquisa básica de TiLV. O padrão-ouro de diagnóstico de vírus normalmente envolve o isolamento do vírus seguido por técnicas complementares tais como a reação em cadeia do polymerase transcrição reversa (RT-PCR) para posterior verificação. Isso pode ser complicado, demorado e exige tipicamente pesadamente infectadas com vírus de amostras de tecido. O uso de RT-quantitativa (q) PCR na detecção de vírus é vantajoso devido à sua natureza quantitativa, alta sensibilidade, especificidade, escalabilidade e seu rápido tempo de resultar. Aqui, todo o método de PCR com base em abordagens para TiLV detecção é descrita, tilápia órgão corte, total ácido ribonucleico (RNA) extração usando uma solução de tiocianato-fenol-clorofórmio guanidium, quantificação de RNA, seguido por uma PCR em dois passos protocolo que impliquem, síntese de ácido desoxirribonucleico complementar (cDNA) e detecção de TiLV por PCR convencional ou identificação quantitativa através de qPCR usando SYBR green tingir. PCR convencional requer etapas pós-PCR e simplesmente informará sobre a presença do vírus. A última abordagem vai permitir a quantificação absoluta de TiLV para tão pouco como 2 cópias e, portanto, é extremamente útil para o diagnóstico de TiLV em casos subclínicos. Uma descrição detalhada das duas abordagens PCR, resultados representativos de dois laboratórios e uma discussão detalhada dos parâmetros críticos de ambos foram incluídos para assegurar que pesquisadores e médicos encontram o seu mais adequado e aplicável método de deteção de TiLV.
Introdução
O fornecimento de peixe per capita global atingiu um novo recorde de 20 kg em 2014, e isso foi devido ao crescimento vigoroso na aquicultura. Aquicultura continua sendo um dos setores de produzir alimentos de origem animal mais cresce em todo o mundo e é o sector de produção de alimentos só animais que está crescendo mais rápido do que a população humana1. Cichilds peixes compõem o segundo mais importante peixe de água doce cultivado em todo o mundo com uma produção global total de 6,4 milhões de toneladas (MT) e um valor estimado de 9,8 bilhões de dólares em 20152. Os produtores de top 10 da tilápia são China (1.78 MT), Indonésia (1.12 MT) e Egito (0.88 MT), seguido por Bangladesh, Vietnã, Filipinas, Brasil, Tailândia, Colômbia e Uganda2. Espera-se que a produção de tilápia global será cerca de 7,3 MT por 20303. Tilápia tornaram-se como uma fonte de alimento global importante não apenas porque eles são uma fonte barata de proteínas4 mas também porque eles são fáceis de procriar em capacidade sob uma ampla gama de água e clima condições5,6. Apenas há algumas décadas, acreditava-se que existiam poucas doenças comercialmente significativas ameaçando o cultivo de tilápia, mas isto não é mais verdade. Uma doença emergente viral chamada doença de vírus de Lago de tilápia (TiLVD) é a primeira epidemia de doença crítica já encontrados em tilápia e toda a indústria está em risco. Esta doença tem graves consequências sócio-económicas e é uma ameaça directa para a segurança alimentar milhões de pessoas na África7, Ásia e América do Sul. No início de 2018, a Organização Mundial de Saúde Animal (OIE) relatou que o agente etiológico da doença, TiLV, tinha sido detectado oficialmente em três continentes, cobrindo oito países8 e desde que este cartão de informação do patógeno foi atualizado lá foram mais de TiLV, na Tanzânia, Uganda9, Indonésia10, Formosa11 e Peru12. TiLV é um novo single-stranded RNA vírus descrito para ser um orthomyxo-como vírus porque ele contém uma variedade de características, uma reminiscência de outros orthomyoxoviruses como a gripe ou infecciosa anemia salmão vírus (ISAV)13. Ele foi identificado no rescaldo da enorme perda de caça selvagem e tilápia no lago da Galileia, Israel14. Depois disso, surtos de doenças semelhantes conhecido como mortalidade de verão e o mês síndrome de mortalidade associado com infecção TiLV foram relatados em tilápia do Nilo (Oreochromis niloticus) no Egito15 e Nilo e tilápia híbrida vermelho (Oreochromis spp.) na Tailândia16, respectivamente. Detecção de vírus de animais aquáticos é historicamente realizada pelo crescimento e isolamento de vírus em cultura de células. Várias linhas de células foram testadas para a propagação e isolamento de TiLV incluindo, E-11 células derivadas de cabeça de cobra de17,de peixe (Ophiocephalus striatus)18, OmB e TmB provenientes Oreochromis mossambicus18e OnlB e OnlL, proveniente de Nile tilapia (o. niloticus)19. Enquanto cultura de vírus tem a vantagem que fornece material para novas experiências, mas tem a desvantagem que requer pelo menos 4-7 dias para observar a formação de efeitos citopático (CPE) e crucialmente, diferentes vírus piscine, que são o mais apto para replicar podem ser propagadas e produzir CPE semelhante.
Nas últimas décadas, tem havido um movimento longe de métodos de diagnóstico tradicionais, muitas vezes demorados como cultura celular, sorologia e deteção do antígeno e substituição do ácido nucleico mais rápido e mais sensível à base de testes de deteção20, 21. Isso é evidente pelo fato de que muitos qPCR ensaios foram desenvolvidos como métodos de diagnóstico importantes para uma infinidade de doenças virais dos animais aquáticos, tais como para ISAV22,23, vírus septicemia hemorrágica viral (VHS)24 ,25, betanodavirus26,27 alphavirus salmonídeos28, peixe iridovirus29, Anguillid herpesvirus 1 (AngHV1)30e Lymphocystis doença vírus (LCDV)31 . Métodos robustos para diagnóstico e patógeno vigilância são urgentemente necessários para reduzir a propagação da TiLV. Tais métodos devem permitir a detecção precoce da infecção antes de desenvolvem sinais clínicos e detecção de vírus baixas cargas. Até à data, diferentes protocolos PCR incluindo RT-PCR14,,32, de RT-PCR aninhado18, semi aninhados de RT-PCR33e RT-qPCR32,34 foram desenvolvidos para a detecção de TiLV em tecidos de peixes. Uma comparação de isolamento de vírus e RT-qPCR em linhas de células sensíveis para a deteção de TiLV revelou que RT-qPCR era 1.000 vezes mais sensível do que o isolamento de vírus32. Embora cada protocolo PCR publicado tem relatado diferentes sensibilidades para a detecção de TiLV, a maioria dos ensaios são altamente sensíveis com os limites de detecção de cópias virais em 7,5 cópias33, 7 cópias18 ou 2 cópias32 por reação.
O objetivo deste artigo de métodos é para explicar, em detalhes, como realizar ensaios de deteção de TiLV, começando com a coleção de tecido de tilápia, para extração de RNA total, síntese do cDNA e então TiLV PCR específico com base em ensaios. Especificamente, protocolos abrangentes de RT-PCR convencional e também SYBR baseada em verde RT-qPCR têm sido descritos a apelar a uma vasta gama de cientistas com o objetivo de detectar TiLV. O primeiro é menos sensível, mas geralmente é uma opção mais barata de deteção. Este último requer uma infra-estrutura mais elaborada como uma máquina PCR quantitativa e os reagentes mais caros, mas tem as vantagens de ser quantitativa, rápido e altamente sensível, significando que ele pode ser usado para a detecção de TiLV em sub clinicamente Peixes infectados. O RT-PCR e RT-qPCR protocolos foram realizados em dois laboratórios diferentes com isolados geográficos distintos de TiLV e os resultados incluídos destaque a sensibilidade e a reprodutibilidade dos ensaios descritos aqui.
Protocolo
O protocolo de uso de animais para este estudo foi aprovado pela Comissão de ética Kasetsart University Animal sob licença número Alves 59-VET-016.
Nota: Consulte a Tabela de materiais para estendido informações relativas aos reagentes e equipamento sugerido para este protocolo.
1. coleta de amostra de tecido
- Eutanásia o peixe usando uma overdose de óleo de cravo (o volume depende do tamanho do peixe e concentração de produtos, geralmente mais de 3 mL/L). Imergir um quarto da tesoura pinça e mayo em etanol 95% (v/v), seguido pela queima o equipamento usando uma lamparina para esterilizar o equipamento.
Nota: Tricaina Metanossulfonato (MS-222) pode ser usado em vez de óleo de cravo. - Encontrar o fígado e cortou um pedaço pequeno (aproximadamente 20-100 mg) ou recolher 200 µ l de muco, usando a tampa de vidro ou lâmina cirúrgica para remover o muco do anterior posterior do peixe tilápia e colocar as amostras em um tubo de microcentrifugadora de 1,5 mL.
- Amostras do processos imediatamente, armazenam em um solução de estabilização de RNA ou movê-los a-80 ° C até utilização posterior.
Nota: A maior tarefa no trabalho com RNA é preparar as moléculas de RNA intactas e mantê-los sem danos ao longo de qualquer manuseio subsequentes do. A espinha dorsal do RNA é intrinsecamente mais sensível aos danos do DNA. Extração e isolamento de RNA total de células do tecido necessita de técnica de laboratório cuidado; Tome todas as disposições para evitar a contaminação de RNase, usando luvas, usando água livre de RNase, reagentes, equipamentos, utensílios de plástico, vidro, espaço de trabalho e usando filtro dicas para pipetar.
2. Guanidium tiocianato - fenol - clorofórmio extração de RNA
- Adicione 1 mL de solução de monofásico contendo isotiocianato de fenol e guanidina num tubo contendo a amostra de tecido da seção 1.
Atenção: Esta solução é muito tóxica e deve ser manuseada com cuidado em uma capa de fluxo laminar com equipamento protetor e usando as luvas óculos, roupas e segurança adequadas. - Triture a amostra de tecido usando um homogeneizador de pilão até homogênea.
Nota: As amostras podem também ser homogeneizadas usando um homogeneizador de poder combinado com grânulos cerâmicos. Certifique-se de que a amostra de tecido é completamente homogeneizada antes de prosseguir para o próximo passo ou parar o protocolo aqui e armazenar as amostras totalmente homogeneizadas a-80 ° C até utilização posterior. - Adicione 200 µ l de clorofórmio para separação de fases.
Cuidado clorofórmio é um potencial narcótico e é extremamente perigoso. Isso deve ser manuseado com cuidado em uma capa de fluxo laminar com equipamentos de proteção, bem como, vestindo as luvas óculos, roupas e segurança adequadas. Como um menos tóxico alternativo, 1-Bromo-3-cloropropano também pode ser usado.
Nota: Escala os volumes cima ou para baixo, se for caso disso. Por exemplo, se apenas 500 µ l de solução monofásica contendo isotiocianato de fenol e guanidina foi usado, então só Adicione 100 µ l de clorofórmio nesta etapa.- Misturar as amostras bem pela inversão por 15 s.
- Incube as amostras por 3 min à temperatura ambiente (RT).
- Centrifugar por 15 min em 12.000 × g e 4 ° C.
Nota: Deve haver uma separação clara em uma fase orgânica inferior, uma interfase branca e uma fase aquosa superior contendo RNA. Esta fase superior é normalmente incolor, mas dependendo do tipo e quantidade de tecido homogeneizado, pode ter uma aparência rosa luz. - Transferi a fase aquosa superior (aproximadamente 500 µ l) para um tubo de microcentrifugadora fresco sem perturbar a interfase.
Nota: não tente transferir toda a fase aquosa, deixar uma pequena quantidade para evitar qualquer contaminação potencial do RNA que contém a fase aquosa com o orgânico ou interfase. - Adicione 1 volume de isopropanol 100% para precipitar o RNA.
- Opcionalmente, se quantidades muito pequenas de tecido foram usadas, em seguida, adicione 1 µ l (5-10 µ g) de glicogênio RNase-livre para cada amostra a promover eficiente precipitação do RNA. Isso ajuda a identificação da pelota do RNA na etapa 2.8.
Nota: O glicogênio funciona como um portador do RNA e impedirá que pequenas quantidades de RNA de degola para o lado do tubo. - Mix tubos bem por inversão várias vezes.
- Armazenar as amostras durante 2 h durante a noite a-20 ° C.
- Opcionalmente, se quantidades muito pequenas de tecido foram usadas, em seguida, adicione 1 µ l (5-10 µ g) de glicogênio RNase-livre para cada amostra a promover eficiente precipitação do RNA. Isso ajuda a identificação da pelota do RNA na etapa 2.8.
- Amostras de centrifugar por 15 min a 12.000 x g e 4 ° C.
- Desprezar o sobrenadante, tomando cuidado para não deslocar o pellet de RNA na parte inferior do tubo de microcentrifugadora.
- Adicione 1 mL de etanol a 75% (v/v) e misturar amostras do RNA o tubo várias vezes por inversão.
- Centrifugar por 15 min em 10.000 x g e 4 ° C.
Nota: O protocolo pode ser parado aqui e as amostras compreendendo a pelota do RNA em 75% de etanol podem ser armazenadas a-20 º C até utilização posterior. - Desprezar o sobrenadante, estar atentos para não deslocar o pellet de RNA na parte inferior do tubo de microcentrifugadora.
- Opcionalmente, repita os passos 2.9-2.11 usando etanol a 70% (v/v). Completamente a pelota do RNA de lavagem minimizará qualquer sal ou reporte de contaminantes que possam interferir será sensíveis aplicações a jusante.
- Tirar o etanol restante com uma pipeta e em seguida, secar ao ar livre a pelota do RNA em temperatura ambiente por mais tempo do que 5 a 10 min.
Nota: demasiado secas pelotas será difícil para re-suspender. - Adicione 30-60 µ l de água livre de RNase, pre-aquecida a 55-60 ° C para solubilizar a pelota do RNA.
- Coloque o RNA no gelo para uso imediato ou loja a-80 ° C para uso posterior.
3. quantificar a concentração de RNA usando um Espectrofotômetro de Micro-Volume
- Mude as configurações do espectrofotómetro para RNA.
- Use 1-2 µ l de água livre de RNase como um espaço em branco.
- Use 1-2 µ l de cada amostra de RNA para avaliar a quantidade de RNA.
- Gravar as leituras em 230 nm, 260 nm e 280 nm para cada amostra.
- Diluir o RNA a 200 ng / µ l, usando a água livre de RNase.
4. síntese de DNA complementar (cDNA) usando o RNA total
- Misture 1 µ g de RNA total do protocolo 2, 2 µM oligo (dT), mistura de dNTPs de 0.5 mM e trazer o volume final de 10 µ l com água livre de nuclease. Por isso, prepare um RT-mestre-mix de acordo com o número de amostras e controles a serem testados.
Nota: Os controles são uma amostra da transcriptase reversa-sinal de menos (-RT) no qual a enzima RT é substituída com água livre de nuclease (ver passo 4.3) e um sem controle do modelo (NTC) em que água livre de nuclease é adicionada à mistura de mestre ao invés de molde de ARN.- Misture as amostras bem pipetando seguido de uma breve centrifugação.
- Incube as amostras a 65 ° C por 5 min, seguido de uma incubação de 2 min no gelo.
- Brevemente Centrifugar as amostras para coletar todo o líquido na parte inferior dos tubos.
- Juntar 1 x tampão de transcriptase reversa, 100 U reverse transcriptase e trazer o volume final de cada amostra a 20 µ l usando água livre de nuclease.
- Misture as amostras bem pipetando seguido de uma breve centrifugação.
- Incube as amostras a 42 ° C por 60 min, seguido de 85 ° C por 5 min.
- Diluir o cDNA sintetizado para uma concentração desejada adicionando um volume adequado de água livre de nuclease e colocar o cDNA no gelo para uso imediato ou armazená-lo a-20 ° C para uso posterior.
5. a TiLV PCR convencional
- Use o cDNA, amostras e controles, gerados no protocolo seção 4 como modelos para uma reação de PCR usando qualquer um dos pares de cartilha estabelecida detalhadas na tabela 1, juntamente com uma DNA polimerase de escolha.
Nota: Um controle não-modelo adicional (NTC) deve ser incluído aqui substituindo o cDNA para água nuclease livre na reação de PCR. Um controle positivo, se disponível, também deve ser incluído compreendendo previamente verificadas amostras positivas de TiLV ou o apropriado TiLV fragmento de cDNA clonado em para um plasmídeo. - Prepare uma mistura de mestre do PCR de acordo com as diretrizes do sistema DNA-polimerase em uso e o número de amostras e controles a serem testados. Esta mistura deve incluir o primer para a frente, reverso da primeira demão, dNTPs, MgCl2 e a DNA polimerase selecionado, juntamente com sua reserva.
- De acordo com as diretrizes do polymerase de ADN selecionado, combine o volume designado de mestre-mistura com a quantidade sugerida de amostras de cDNA e amostras de controle.
Nota: Preparar um 0.5 x excesso de reação é frequentemente benéfico desde que alguns do mestre-mix é perdido durante a pipetagem. - Realize PCR ciclismo condições de acordo com as diretrizes do sistema utilizado DNA polimerase e usando uma adequada temperatura do recozimento para os primers em uso (tabela 1). Geralmente, esse programa irá envolver uma desnaturação inicial a 95 ° C por 2-5 min, seguido por 30-40 ciclos de desnaturação a 95 ° C por 30 s, recozimento a temperatura recomendada para 30 s e alongamento a 72 ° C por 30 s, seguido de um alongamento final a 72 ° C, durante 5-10 min.
- Carga de 5 a 15 µ l de cada reação de PCR e uma escada de DNA apropriada em poços de um gel de agarose 1-2%, dependendo o tamanho esperado do produto do PCR. Separar o DNA amplificado por electroforese em gel e manchar o gel por brometo de etídio (EthBr) para facilitar a visualização das bandas de DNA do tamanho esperado (tabela 1) em uma máquina de documentação de gel usando luz UV.
Cuidado: EtBr é tóxico; deve ser manuseado com cuidado, vestindo a roupa de proteção apropriada e luvas de segurança.
| Segmento-alvo TiLV genoma | Cartilha para a frente 5' - 3' | Primeira demão reversa 5' - 3' | Tamanho do produto do PCR (bp) | Tm ° C | Referência original | ||||
| 1 | CCAAACGTTATCTCTTAATTACGCAC | GCAAATATTTCTCTCATTCGCCT | 1641 | 50 | Surachetpong et al, 2017 | ||||
| 1 | CCTCATTCCTCGTTGTGTAAGT | AGGAGTTGCTGTTGGGTTATAG | 1000 | 62 | Eduardo et al., 2018 | ||||
| 2 | ACTCTCTATTACCAAATACATTTACT | TTACCATATATATAGTGAAGGC | 1445 | 45 | Surachetpong et al, 2017 | ||||
| 2 | GTCCAGGGCGGTATGTATTG | CTTACGGCTGACAAGTCTCTAAG | 834 | 62 | Eduardo et al., 2018 | ||||
| 3 | GTTGGGCACAAGGCATCCTA | TATCACGTGCGTACTCGTTCAGT | 250 | 56 | Eyngor et al, 2014 | ||||
| 3 | TATGCAGTACTTTCCCTGCC | TTGCTCTGAGCAAGAGTACC | 491 | 57 | Eyngor et al, 2014 | ||||
| 3 | ACCCCTTAATCCTTAATAGACCGTTA | CCCATAATCCTCTATTAGAACGTCGT | 1352 | 50 | Surachetpong et al, 2017 | ||||
| 3 | GTCGAGGCATTCCAGAAGTAAG | GAGCTAAGGGAACGGCTATTG | 834 | 62 | Eduardo et al., 2018 | ||||
| 4 | AGCAGCAGCAGGAGAAAGAG | ACCGTCCTGTTTCTGAATGG | 358 | 60 | Nicholson et al, 2017 | ||||
| 4 | CCAAAGTTTACTCCTATTACCCAGA | GCAAATCTTTCTCCAATTACCGTCT | 1250 | 50 | Surachetpong et al, 2017 | ||||
| 4 | GCCCAATGGTTCCCATATCT | GCCCAATGGTTCCCATATCT | 524 | 62 | Eduardo et al., 2018 | ||||
| 5 | CCAAATGTTTCTCTTATCTCAGACTC | CTTTTTCTCAGTTTACCACTTTATG | 1087 | 57 | Surachetpong et al, 2017 | ||||
| 5 | CAACTCTTAGCCTCCGGAATAC | CGTTCTGCACTGGGTTACA | 696 | 62 | Eduardo et al., 2018 | ||||
| 6 | CCAAATTTTACCTCTCGCAT | TCAAGCACTTAAAACTGTACC | 1027 | 45 | Surachetpong et al, 2017 | ||||
| 6 | CCCACACGACAGGACATATAG | GAGTTGGCTTAGGGTGATAAGA | 948 | 62 | Eduardo et al., 2018 | ||||
| 7 | CTCTCTTTGCATTGCATACCGT | GACCAATTATCCCTGCTTTCA | 704 | 57 | Surachetpong et al, 2017 | ||||
| 7 | TCCTTTAGGGATTGGCACTAAC | TTCCATCGACTGCTCCTAGA | 486 | 62 | Eduardo et al., 2018 | ||||
| 8 | ACCTCATCTACACTAACATTTCCA | TCATCATTACACAAATGGAGTAGCT | 637 | 50 | Surachetpong et al, 2017 | ||||
| 8 | CTTAAGGGCCATCCTGTCATC | TGGCTCAAATCCCAACACTAA | 476 | 62 | Eduardo et al., 2018 | ||||
| 9 | TTGGTGATGTCACGATGGATA | AGTTCTATCGCCAGCCATGT | 351 | 60 | Nicholson et al, 2017 | ||||
| 9 | ACAAGTCCGATTACTTTTTCCGC | TCTTTCTCACGTCCTTAAAGTCA | 530 | 50 | Surachetpong et al, 2017 | ||||
| 9 | GATATCCTCCACATGACCCTTC | GTACGTCACTTTGTGCCATTAC | 261 | 62 | Eduardo et al., 2018 | ||||
| 10 | AACCCTACTAACACCAAATATAGCT | CTTTCCCTCTGACACCCTGT | 450 | 50 | Surachetpong et al, 2017 | ||||
| 10 | TCCTCTCTGTCCCTTCTGTT | CAGGATGAGTGTGGCAGATTAT | 276 | 62 | Eduardo et al., 2018 | ||||
Tabela 1. Cartilha publicada pares para a amplificação do cDNA de TiLV usando o ponto de extremidade do PCR. O primer conjunto mostrado em negrito foram usados para gerar os resultados representativos, mostrados na Figura 3A e 3B.
6. TiLV quantitativos Polymerase Chain Reaction (qPCR)
- Usando um plasmídeo contendo o TiLV apropriado segmento genômico 3 cDNA como um padrão, tais como pTiLV32, preparar uma série de 10 vezes de diluição serial duplicado ou triplicou.
- Preparar uma mestre-mistura de qPCR para todas as amostras, padrões e controles, tendo em conta as reações devem ser executada em duplicar ou triplicata utilizando 0,4 µ l de água livre de nuclease, 0,3 µ l de primer para a frente, 0,3 µ l de primer reverso e 5 µ l de 2 x SYBR Green Mestre-mistura de polymerase do DNA por reação.
- Use as primeiras demão em uma concentração de 10 µM e as informações da primeira demão e o pTiLV padrão, como segue:
Encaminhar a cartilha: TiLV-112F (5'-CTGAGCTAAAGAGGCAATATGGATT-3')
Reverter a primeira demão: TiLV-112R (5'-CGTGCGTACTCGTTCAGTATAAGTTCT-3')
Padrão pTiLV:10 pg / µ l
Nota: Se o número total de amostras e controles é 10 e será executado em triplica, isso equivale a um mestre de qPCR-mix composto por 12 de água livre de nuclease µ l, 9 da primeira demão µ l para a frente, primeira demão reversa de 9 µ l e 150 µ l de SYBR Green DNA polimerase mestre-mix. Adquiridos comercialmente 2 x universal SYBR Green DNA polimerase mestre-misturas contêm todos os componentes necessários para a reação de qPCR, ou seja, o SYBR Green tingir, começo quente Taq DNA polimerase, dNTPs, MgCl2 e corantes de referência passiva. Proteja o mix de SYBR Green mestre da luz.
- Use as primeiras demão em uma concentração de 10 µM e as informações da primeira demão e o pTiLV padrão, como segue:
- Dispense a 6 µ l de mistura de mestre qPCR em qPCR tira tubos ou um prato bem 96 compatível com a máquina de qPCR em uso.
- Adicione 4 µ l de modelo de cDNA, controles ou padrões de TiLV serialmente diluídas em tubos ou poços da placa de 96 bem.
- Fechar os tubos de qPCR ou selar a 96 placa bem com uma tampa da placa compatível para a máquina de qPCR em uso
- Agite suavemente os tubos de qPCR para misturar a solução e spin-down os qPCR tubos ou 96 placa bem usando uma centrífuga para coletar todo o líquido no fundo dos vasos.
- Coloca os tubos ou placa do termociclador em tempo real.
- Programar o thermocycler qPCR para executar uma desnaturação inicial a 95 ° C por 3 min, seguido de 40 ciclos de 95 ° C por 10 s e 60 ° C por 30 s para recozimento da primeira demão e alongamento, terminando com um derretimento curva passo de 65 ° C a 95 ° C com um incremento de 0,5 ° C / 5 s.
- Selecione SYBR como um corante de fluoróforo, em seguida, selecione o desconhecido como um tipo de amostra e inserir um nome em uma caixa de nome de amostra.
- Abra a tampa da máquina RT-qPCR e coloque a fita de qPCR para os poços atribuídos e, em seguida, feche a tampa.
- Realize o ensaio de RT-qPCR com as condições selecionadas. A máquina começa a funcionar depois que a tampa tenha alcançado a temperatura desejada. Recolha a fluorescência de cada amostra após cada etapa da extensão para monitorar o progresso da reação.
Nota: A máquina de qPCR e software relacionado irão automaticamente calcular todos os parâmetros do ensaio e exibir as curvas de amplificação em tempo real, enquanto a curva padrão e a curva de fusão serão gerados no final do ciclo de qPCR. - Realizar análise de dados e aquisição, assegurando-se primeiro que o derretimento curvas para cada amostra e padrão tem um pico de uniforme a temperatura esperada para o amplicon.
- Avaliar as curvas de amplificação das amostras e padrões e definir o limiar de uma região foram que a taxa de amplificação dos cDNAs é o mesmo em todas as amostras. Isto normalmente é realizado automaticamente pelo software, mas deve ser cuidadosamente verificado.
- Calcule o número de cópias de TiLV usando a curva padrão.
Resultados
Seguindo o protocolo descrito na secção 1, tilápia moribundo híbrido vermelho exibindo sinais clínicos de infecção TiLV (figura 1A) foram sacrificados por banhos em uma alta concentração de óleo de cravo, que funciona como um anestésico. Relataram sintomas clínicos são variáveis, mas os sintomas comuns parecem ser letargia, erosão da pele e descoloração, exoftalmia, escalas desanexadas, feridas aberta/lesão e comportamento anormal de16,de15,33, 35,36, alguns destes pode ser claramente visto na figura 1A. A parede abdominal foi removida para coletar órgãos internos como o fígado, baço ou cabeça rim (figura 1B). Amostras de muco foram também coletadas nesta fase raspando suavemente a pele de anterior para posterior dos peixes usando um vidro de tampa ou lâmina37.

Figura 1 . Coleção de dissecção e amostra de tilápia. A. tilápia híbrido vermelho TiLV-infectados com leisons de pele, vermelhidão ao redor da boca e opérculo, erosão da pele e opacidade da córnea. B. Sectioned vermelho híbrido de tilápia para permitir a coleta de tecido do fígado (no ponto da seta azul), baço ou órgãos rim cabeça. Clique aqui para ver uma versão maior desta figura.
{kind=link}
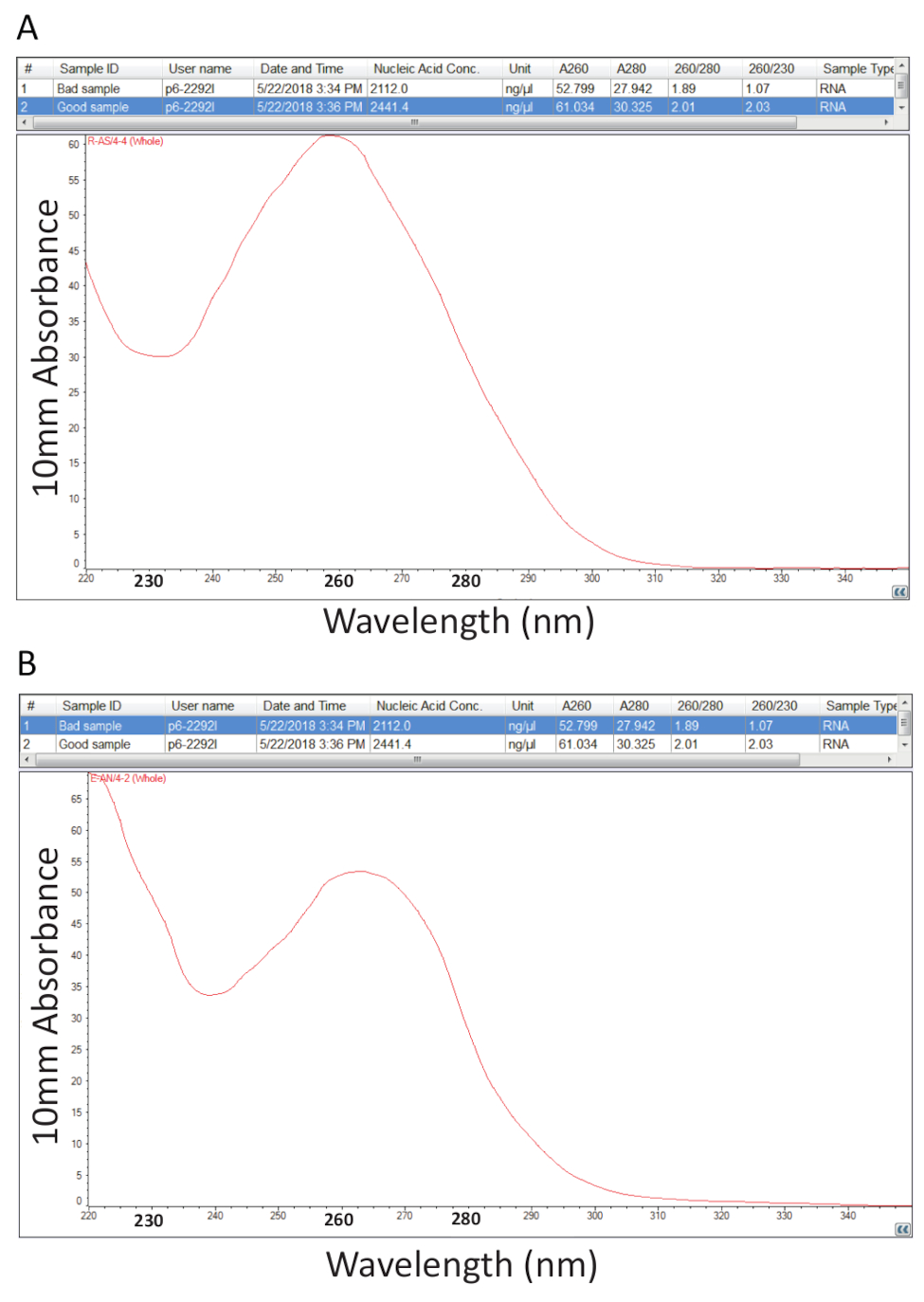
Depois, seguiu-se o protocolo detalhado na seção 2 para a extração de Guanidium tiocianato-fenol-clorofórmio do RNA total e quantificação de RNA, conforme descrito na seção 3 foi realizada para avaliar a pureza da amostra pelo cálculo das relações de pureza e análise de perfis espectrais (Figura 2). Figura 2A mostra um resultado representativo de um procedimento de extração de RNA total sucesso, enquanto Figura 2B representa uma má preparação do RNA. Ácidos nucleicos têm maxima de absorvância em 260, enquanto as proteínas têm deles em 280 nm. A relação das medições em 260 nm e 280 nm indicam a pureza de cada amostra e rácios de 1.9 a 2.1 indicam RNA puro, como é o caso para o exemplo na Figura 2A. Rácios de A260/280 baixos observados na Figura 2B indicam possível contaminação da proteína ou fenol, resto do processo de extração de RNA. Absorvância a 230 nm pode ser o resultado de contaminação da amostra e a relação de A260/230 nm é calculada também por este motivo. Esta relação deve ser no intervalo de 2.0-2.2 para preparações puras do RNA, como ilustrado por um valor de 2,03 para o exemplo na Figura 2A, enquanto Figura 2B tem uma baixa relação de A260/230 de 1,07 e o perfil espectral mostra uma mudança na calha em 230 nm para 240 nm, que é indicativo de guanidina residual ou fenol na amostra. Para o exemplo mostrado na Figura 2B, re-precipitação do RNA para remover a contaminação pode melhorar a pureza da amostra.

Figura 2 . Quantificação espectrofotométrica do RNA total extraído de tecidos doentes de tilápia. A. índices de pureza e perfis espectrais de uma preparação bem sucedida do RNA. B. como A, exceto o representante de um procedimento de extração de RNA pobre. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Para detectar TiLV por RT-PCR, amostras puras como a representada na Figura 2A foram transcrito reverso (protocolo. 4) em cDNA e usado como um modelo para o PCR do ensaio detalhado na seção 5 e resultados representativos são mostrados na Figura 3A. Primeiras demão mostradas em negrito na tabela 1 foram usados para amplificar um fragmento de bp 491 de TiLV segmento genômico 314. Os produtos PCR foram separados por eletroforese em gel e manchados com EtBr para visualização. A figura 3A mostra os resultados de uma RT-PCR em duas etapas usando 4 amostras de cDNA (S1-S4), derivadas do fígado de tilápia doente isolado na Tailândia e em cada amostra, uma limpeza única banda de aproximadamente 500 bp pode ser observado e assim, amostras de 1-4 são TiLV positivo. O mesmo produto PCR foi obtido da amostra controle positivo, compreendendo o cDNA do segmento TiLV 3 clonado em um plasmídeo32 , enquanto o não controle do modelo (NTC) não deu os produtos de PCR. O ensaio na Figura 3B foi realizado utilizando os primers mesmos como na Figura 3A , mas em um laboratório diferente, usando uma abordagem de RT-PCR de uma etapa e com 5 amostras de RNA derivadas os tecidos renais cabeça de tilápia originários da aquicultura egípcia 15. foi determinado usando este ensaio de deteção que amostras 1, 3 e 5 são TiLV positivo, enquanto amostras 2 e 4 são TiLV negativo, desde que nenhum produto PCR foi encontrado no tamanho correto. Os controles negativos, incluindo dois menos controles de transcriptase reversa e dois NTCs não gerou qualquer produtos PCR. Um ensaio de RT-PCR de uma etapa também foi realizado visando o gene ActinB de tilápia. O tamanho de amplicons de 217 bp foi gerado em cada amostra (S1-S5) como esperado38. Este ensaio serviu como um controle para a integridade das amostras do RNA, bem como permitindo uma análise semi-quantitativa das amostras positivas TiLV. Dado que o produto de tilápia ActB gerado é relativamente igual, então, as diferenças na quantidade de produto PCR TiLV específico gerado podem ser interpretadas como um verdadeiro reflexo da quantidade de TiLV em uma amostra de determinado tecido.

Figura 3 . TiLV RT-PCR. A. amostras de cDNA produzidas a partir de tecidos de fígado de tilápia doente, recolhidos da Tailândia foram tela para TiLV infecção utilizando primers específicos para o segmento 3 (mostrado em negrito na tabela 1) de TiLV usando um ensaio de RT-PCR 2-passo. M = marcador mostrado em pares de base; S1-S4 = amostras 1-4; C1 = controle positivo usando pTiLV como um modelo PCR; e C2 não = nenhum controle modelo (NTC). B. uma etapa RT-PCR utilizando primers mesmos como em A e coletadas amostras de tecidos de rim cabeça de tilápia doente do Egito15. M = marcador mostrado em pares de base; S1-S5 = amostras de 1-5. C1-C2 de controles são menos controles de transcriptase reversa e C3-C4 painel NTCs. inferior é um One-Step RT-PCR utilizando primers dirigidos contra a produção de tilápia ActinB38 (veja texto para detalhes) um produto PCR de 217 de pares de bases. Clique aqui para ver uma versão maior desta figura.
{kind=link}
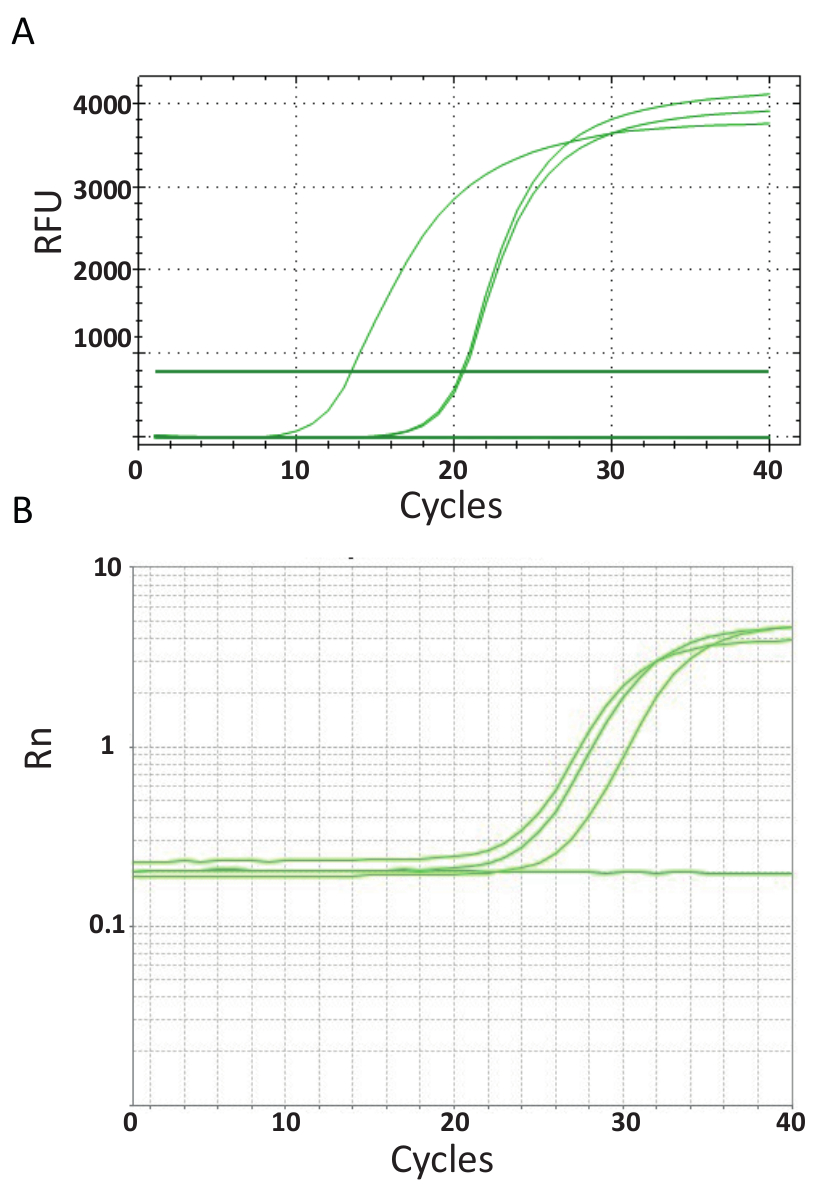
Ao contrário de ponto de extremidade de PCRs representados na Figura 3, ensaios de qPCR que são explicados no protocolo 6, medir a quantidade de produto do PCR após cada ciclo PCR. A amplificação do DNA alvo é detectada utilizando moléculas fluorescentes que interagem com o DNA gerado a partir de cada rodada de reação. Aqui, foi utilizado SYBR Green tingir, que intercala com DNA dupla-hélice. O sinal fluorescente é seguido durante a reação e sua intensidade está relacionada com a quantidade de produto formado 39,40,41,42,43. TiLV qPCR ensaios foram realizados conforme descrito no protocolo 6 em laboratórios diferentes, usando diferentes reagentes SYBR Green, qPCR máquinas e amostras de diferentes países. As curvas de amplificação resultante são mostradas na Figura 4A e 4B. Pode-se observar que para cada ensaio, o curso do experimento tem quatro fases: a fase linear terreno, primeira fase exponencial, final da fase exponencial e a fase de planalto. A fase linear terreno ocorre durante os primeiros ciclos onde a duplicação de DNA ainda não pode ser identificada devido a quantidades de DNA produzindo Rácio sinal/fundo insuficiente. Fluorescência de base é calculada durante esta fase. Daí em diante, DNA alvo começa a dupla em concentração com cada ciclo, induzindo o sinal se tornar detectável acima de fundo e aumentar exponencialmente. A eficiência de amplificação (E) de um ensaio de qPCR bem otimizado é muito alta (quase 100 por cento) no início da reação e permanece estável durante esta primeira fase exponencial a amplificação e é neste ponto que quantificação é realizada, quando eficiência da reação é ainda firme. Em ciclos posteriores o sinal começa ao platô, e a intensidade da fluorescência já não está correlacionada com o número da cópia inicial do modelo, porque os componentes da reação são exausto44. Saturação também pode ocorrer devido à concorrência de reações re-recozimento, os rácios de concentração mudança de componentes, ou a quantidade de unidades de enzima de moléculas de ADN substrato. Possivelmente, tais parâmetros representam as diferenças entre as curvas de amplificação para os ensaios, mostrados na Figura 4A e 4B. Os controles incluídos não gerou estas curvas de amplificação característico.

Figura 4 . Amplificação de parcelas para mostrar o acúmulo de produto ao longo da duração do ensaio do PCR em tempo real. A. curvas de amplificação de amostras de TiLV-positivo derivado da Tailândia, NTCs e controle positivo do plasmídeo usando um SYBR-Green qPCR 2-passo do ensaio. O gráfico foi gerado por plotagem fluorescência relativo (RFU) vs. ciclo de número. B. curvas de amplificação das amostras positivas TiLV derivado do Egito, como na Figura 3B e um NTC. A curva de amplificação é a fluorescência do repórter sinal normalizado para a fluorescência do corante ROX passivo incluído no ensaio (Rn) contra ciclo número. Clique aqui para ver uma versão maior desta figura.
{kind=link}
No final do thermocycling qPCR nas máquinas diferentes em cada laboratório, os dados foi adquiridos e analisados. Figura 5A e 5B mostram as curvas de fusão representativas de ensaios realizados em cada laboratório. Cada máquina de qPCR foi programada para realizar uma análise de curva derretimento no final. Isto foi conseguido incrementalmente, aumentando a temperatura e monitoramento a fluorescência em função da temperatura. Quando a temperatura é alta o suficiente para desnaturar dsDNA, uma grande queda na fluorescência é registrada porque a molécula de fluoróforo é liberada. O software de cada instrumento de qPCR calculado a temperatura do recozimento (Tm) a partir dos dados de curva a fusão plotando a temperatura negativa de derivativos vs primeira (Figura 5). Pode ser visto na Figura 5A e 5B , que os produtos formaram em conjuntos de amostra diferente que transição fusão uniforme a temperatura esperada de cerca de 80 ° C para o ensaio. Não há outros picos a temperaturas mais baixas foram observados. Devido ao seu pequeno tamanho, o Tm de cartilha-dímeros é tipicamente menor do que a sequência de DNA alvo. Portanto, essa diferença entre o Tmtorna mais fácil para identificar potenciais cartilha-dímeros ou outros produtos de amplificação não-específica. Os controles não gerou derreter curvas como padrões e amostras positivas de TiLV e podem ser vistos como uma linha quase horizontal na parte inferior das paradas na Figura 5A e 5B.

Figura 5 . Derreta a análise da curva para garantir a especificidade do ensaio e diferentes produtos PCR podem ser diferenciados por suas características de fusão. R. derreter a análise da curva de amostras positivas TiLV provenientes da Tailândia, controle negativo e positivo do plasmídeo controle. B. análise da curva de TiLV amostras positivas derivadas de Egipto, padrões de pTiLV e um NTC de derreter. Os gráficos em A e B ambos mostram a mudança na fluorescência dividida pela mudança de temperatura plotagem contra temperatura para produzir uma imagem clara da fusão dinâmica. Clique aqui para ver uma versão maior desta figura.
{kind=link}
A maioria das máquinas de qPCR vêm com um software, facilitando nova avaliação da qPCR executar e irá quantificar as amostras, gerando uma curva padrão traçando automaticamente o limiar de ciclo (Ct) contra o logaritmo do modelo dos padrões pTiLV Copie o número, como mostrado na figura 6A e 6B para os dois laboratórios independentes. Brevemente, o Ct é a unidade usada para avaliar resultados de qPCR. O valor det C denota o número de ciclos necessários para atingir um nível de sinal de fluorescência do limiar definido. Quanto maior a quantidade de começando modelo, quanto menos ciclos leva para atingir um nível de fluorescência detectável. Com efeito, as amostras com uma alta carga de TiLV terá Ct valores inferiores amostras com baixa carga de TiLV tais como em peixes com uma infecção sub-clínica. Para determinar os valores det C, níveis de fluorescência de fundo primeiro são deduzidos dados brutos. Em seguida, o software associado com o dispositivo de qPCR selecionará automaticamente um limiar de fluorescência pesquisando as curvas de dados para cada amostra e incorporando um Ct que representa onde a amostra cruzou o limiar. Isso é feito separadamente para cada ensaio e cada limite deve ser cuidadosamente avaliada, assegurando que o limite foi definido na parte logarítmica das curvas de amplificação e em um lugar onde todas as curvas são paralelas. Assim, o específico Ct adquirida é um valor relativo e é em relação a partida modelo cópia número45, mas também é específico para a máquina de qPCR e os reagentes, a eficiência de amplificação da PCR e a sensibilidade da deteção. Estes parâmetros contribuem para as diferenças observadas usando o mesmo ensaio na Figura 6.
Partir das curvas padrão na Figura 6, análises de regressão, incluindo cálculo de encostas de curva padrão (m) e intercepta, amplificação eficiências (100 x (101/m -1))46 e linearidade da reação foram realizados. Análises de curva padrão também foram usadas para confirmar a sensibilidade (limite de detecção), repetibilidade e reprodutibilidade do ensaio. Teoricamente, a quantidade de DNA é duplicada com a PCR cada ciclo, significando que a eficiência (E) é igual a 100%. No entanto, na prática uma eficiência tal ideal é raramente alcançada devido às condições PCR sub-ótimo tais como, inibição da DNA polimerase, contaminantes, demais erros do cDNA e pipetagem47. Normalmente, amplificação E variam de 90-110% para os ensaios de boas, na figura 6A , uma eficiência de 94,5% foi calculada usando 8 amostras de pTiLV serialmente diluída, ao mesmo tempo, a eficiência de ensaio no ensaio indicado na Figura 6B usando 7 pTiLV serialmente diluída amostras foi 101,2%. Uma eficiência de mais de 100% é geralmente devido à presença de inibidores da PCR no ensaio. Análise de regressão linear da trama padrão também permite o cálculo do número de cópias de TiLV em cada amostra41,42,45, como pode ser observado para as três amostras de TiLV mostradas em vermelho em Figura 6B que está em consonância com os resultados de amostras S1, S3 e S5, mostrado na Figura 3B.

Figura 6 . RT-qPCR curvas padrão. PCR em tempo real de 10 vezes diluições em série de pTiLV, o padrão utilizado em ambos os laboratórios. A. 8 pTiLV serialmente diluída amostras foram testadas, todas de concentração conhecida e correlacionadas com o número de cópias de TiLV / reação. A curva padrão foi gerada por plotagem registro cópia número vs ciclo limiar (Ct). A inclinação =-3.462, R2 = 0.9992 e a eficiência é 94.47%. B. como em A, exceto 7 pTiLV serialmente diluída foram testadas amostras (verde) e o gráfico exibe o ciclo limiar no eixo y e o número de cópia de TiLV (quantidade) no eixo x. A intercepção de y = 32.327, inclinação =-3.292, R2 = 0,98 e a eficiência é de 101,2%. Para ambas as curvas padrões em A e B, a inclinação, a intercepção de y e valores do coeficiente de correlação (R2) são utilizados para compreender o desempenho do ensaio. Importante, R2 valor deve ser próximo de 1, já que é uma medida da linearidade da curva padrão. A inclinação é usada para medir a eficiência do PCR, em que a eficiência de 100% corresponde a uma inclinação de-3.32, veja o texto principal para a equação e uns detalhes mais adicionais. Uma reação de qPCR bom geralmente tem uma eficiência entre 90-110%, correlacionando-se com um declive de entre-3.58 e-3.10. A curva padrão é usada para quantificação absoluta das amostras positivas de TiLV desconhecidas e determina o número exato de cópias TiLV / reação, como é o caso para as três amostras positivas de TiLV de cor vermelha em B.
Discussão
TiLV foi primeiramente relatada em 2014 em Israel14 e desde então, ele foi identificado em vários países, incluindo Egito, Colômbia, Índia, Malásia, Uganda, Tanzânia e Tailândia15,16,18, 35 , 48. conscientização global, particularmente, em países produtores de tilápia tem colocado mais atenção sobre o vírus e várias restrições e medidas de controlo das autoridades do governo têm sido implementadas a tentativa de evitar a propagação do TiLV. Aqui, um protocolo detalhado para a deteção de TiLV em tecido de tilápia, abrangendo a coleta de amostra, isolamento de RNA, síntese, PCR e qPCR ensaios de cDNA foi explicado. Há vários aspectos a estes métodos que garantem discussão específica. TiLV foi identificado em peixes, abrangendo uma variedade tamanhos9,12,14,15,49 e espécies de tilápia, até agora, incluindo a tilápia híbrida de criação (O. niloticus x o. aureus)11,14, Nile tilapia (o. niloticus)9,10,14,15,16, 33 , 35 , 36 , 49 , 50 e vermelho de tilápia (Oreochromis SP.)16,33,,48,51, como bem como selvagens Nile tilapia9,12, preto tilápia51, T. zilli14,15, S. galilaeus, o. aureus e T. simonis intermedia14 e muito recentemente TiLV foi identificado em carpa selvagem (Barbonymus schwanenfeldii)52. Amostras de tecido dos órgãos internos (gill, baço, fígado, coração, rim cabeça) ou muco37 podem ser coletadas de tilápia saudável, bem como moribunda, independentemente da idade, tamanho ou espécie e processadas para isolamento de RNA. O protocolo de extração de RNA total descrito aqui usa uma solução monofásica de fenol e guanidínio tiocianato, que é um agente de desnaturação caotrópicas. Os tecidos são homogeneizados diretamente nesta solução, seguido pela adição de clorofórmio e centrifugação para conseguir a separação de fases em que uma clara RNA que contém a fase aquosa superior, uma interfase e a fase orgânica inferior é gerado. O RNA é isolado da fase aquosa por precipitação do isopropanol, seguida de lavagem do RNA recuperado para se livrar de contaminantes. Isolamento do RNA por esta metodologia foi pioneira por Piotr Chomczynski e Nicoletta Sacchi e foi referido como guanidínio tiocianato-fenol-clorofórmio extração53,54. Este tipo de reagente utilizado para a extração de RNA pode ser comprado ou feito em laboratório comercialmente (consulte a Tabela de materiais , para mais informações). Este protocolo ligeiramente mais lento do que métodos baseados em colunas, como a purificação baseada em sílica, mas em geral, é mais custo-eficaz e produz mais de RNA.
Neste protocolo, quantificação de RNA usando valores A260 foi delineada pelo qual valores de espectrofotometria podem indicar a qualidade do RNA (A260/A280 = 1.9-2.1). Enquanto este método dará uma boa indicação de pureza da amostra, ele absolutamente não pode informar sobre a qualidade do RNA extraído. Para determinar corretamente se o RNA está intacto ou parcialmente degradadas, as amostras podem ser separação por eletroforese em gel de agarose em que manchas do EtBr manchado 18S e bandas de rRNA 28S indicam degradação de RNA. Mais verificação de qualidade de RNA pode incluir usando um instrumento de laboratório-em-um-microplaqueta. Além disso, é também importante para digerir o RNA purificado com DNase eu remover contaminantes hospedar o DNA genômico, que dependendo das aplicações a jusante pode levar a resultados falsos. Se o anfitrião gDNA ainda está a contaminar a amostra de RNA em grande medida, um tratamento adicional de DNaseI também pode ser realizado no final do processo de extração de RNA (ver Tabela de materiais).
Síntese de DNA complementar pode afetar muito os resultados globais da qPCR e é um aspecto do método que pode apresentar variação. O protocolo de cDNA defendido aqui é composto de uma configuração de componente único usando oligo (dT) e, portanto, apenas transcreve mRNAs contendo polyA caudas. Permite o controle de usuário de exatamente quais componentes para uso na reação de transcrição reversa e este modo de cDNA síntese provou bem sucedido para a deteção de TiLV32. Uma alternativa para esta montagem é um mestre-mistura comercialmente comprou contendo todos os componentes necessários para a reação de transcrição reversa e é muito rápido e simples sem o protocolo habitual de várias etapa, pipetagem e multi-temperatura. Isto é vantajoso, pois minimiza a manipulação e promove a uniformidade em todas as amostras. Tais mestre-misturas frequentemente incluem oligo (descolamento) e primers aleatórios tornando-o aplicável a diferentes modelos de RNA e gerando cópias de cDNA representativa das sequências de todo o comprimento dos RNAs em uma população (viral e tilápia hospedar mRNA) e, em teoria, todas as espécies de RNA desejada então podem ser medida por PCR convencional ou de qPCR de tal uma amostra. Esta versatilidade é a principal vantagem de uma abordagem de RT-PCR 2-passo; fornece um pool de longo prazo que pode ser usado para muitas experiências diferentes. Nos resultados, uma abordagem de RT-PCR de uma etapa tem sido representada no qual iniciadores específicos de sequência (tabela 1) foram usados e o RT e PCR foram realizadas em um tubo (veja a lista de material). Em geral, primers específicos de sequência permitem para uma maior eficiência de RT do alvo específico do RNA do que usando a escorva aleatória, mas o alvo específico do RNA é o único que pode ser quantificado em uma amostra tão cDNA que pode ser o único objetivo de determinados laboratórios (ver Tabela de materiais para sugestões de produto de síntese de cDNA).
Enquanto RT-PCR convencional parece ser comumente usado até agora na TiLV diagnóstico9,13,14,15,16,17,18, 33 , 35 , 48 , 55. RT-qPCR mostrou ser uma ferramenta mais poderosa para a detecção e quantificação de pequenas quantidades de TiLV nos tecidos de peixes ou muco32,37. Em geral, qPCR é amplamente utilizado em laboratórios de diagnóstico de virologia clínica devido a sua alta sensibilidade, especificidade, reprodutibilidade boa, ampla faixa dinâmica e velocidade21. Enquanto qPCR pode ser inicialmente mais caro de implementar do que RT-PCR convencional, ele oferece muitas vantagens importantes sobre PCR convencional; tem um mais rápido tempo de rotação da amostra para resultados e não requer quaisquer etapas de post-PCR. Este último ponto significa que há um risco mínimo para a contaminação do laboratório e pode ser mais facilmente adaptado para situações de alto rendimento como em caso de surtos. Além disso, é inerentemente mais sensível do que a RT-PCR convencional, que é de vital importância para detectar baixas cargas virais em infecções sub-clínica21. Isso exigiria uma abordagem PCR aninhada que exige a transcrição reversa, duas outras reações de PCR e depois análise de agarose electroforese do gel. Estes passos muitos levam muito tempo e aumentam as chances de erros ou de contaminação. No entanto, devido a sua alta sensibilidade, RT-qPCR exige meticuloso desenho experimental e um conhecimento aprofundado das técnicas de quantificação para gerar resultados precisos56,57.
O ligação de DNA fluoróforo, SYBR Green demonstrou-me neste protocolo. É um corante de ligação dsDNA inespecífica DNA e, assim, a especificidade do ensaio encontra-se inteiramente no conjunto de primers, que podem gerar falsos positivos58. Portanto, enquanto o dsDNA derretendo análise de curva, realizada no final de cada PCR é especialmente importante para a reação de PCR porque confirma que apenas um PCR amplicons do correto Tm é produzido (isto deve também ser alcançado por gel eletroforese quando novos ensaios estão sendo implementados). O Tm de um fragmento de DNA é dependente de uma variedade de características tais como o seu comprimento, composição de GC, sequência, vertente complementaridade, concentração, bem como no buffer componentes e potenciadores de PCR. As análises de curva derretimento nos resultados representativos mostradas de dois laboratórios não revelaram a presença de dímeros-primeira demão ou outros produtos PCR indesejados, mas se isto for observado com outras amostras e/ou experimentais set-ups, o ensaio deve ser Re-otimizado. As tecnologias mais avançadas de qPCR não requer um passo de curva tão derretendo e com efeito, desde este métodos paper foi escrito, um TaqMan baseado TiLV RT-qPCR foi desenvolvido utilizando dois primers e uma sonda, tornando-se altamente específico de TiLV34.
Sem dúvida, as primeiras demão projetadas para ensaios de RT-qPCR são fundamentais para o sucesso do ensaio e os primers aqui foram projetados com base em cima os publicamente disponíveis dados genomic da TiLV o tempo de32. No entanto, vírus de RNA são bem conhecidos para apresentam taxas de mutação alta e possíveis estirpes irão escapar os testes de diagnóstico atuais, como foi observado por ISAV59. Sempre vai ser difícil para esses tipos de vírus gerar um universal pan-TiLV RT-qPCR ensaio e desses ensaios serão apenas continuamente melhorados se dados genomic da mais TiLV de longo alcance locais e períodos de tempo se tornam disponíveis.
Finalmente, é essencial para executar duplicata ou triplicata se possível, as reacções em ambos intra e inter ensaios qPCR. Se os valores det C são muito elevados, então o uso de repetições é especialmente importante para verificar que a reação de PCR é confiável e reprodutível. Em geral, se replicam dados de reações varia mais do que 0,5 ciclos, as reações devem ser repetidos e se os valores det C consistentemente variam ciclos > 0,5 na Replica, o ensaio deve ser re-otimizado. O uso de um robô de pipetagem integrado qPCR ajuda imensamente com esta questão, mas é uma ferramenta de luxo. Como com qualquer experimento, os controles adequados e pertinentes de inclusão são de extrema importância para o desenvolvimento de ensaios moleculares robustos, especialmente em laboratórios de diagnóstico onde tais ensaios tem que ser credenciado. Controles devem incluir positivo (amostra positiva de TiLV, TiLV do plasmídeo padrão) e amostras de controles negativo (NTC e -RT), bem como a detecção de genes de tilápia endógena das tarefas domésticas. Esses controles não podem ser subestimados e devem ser incluídos em cada ensaio de entender corretamente a qualidade de cada etapa do ensaio e interpretar corretamente os resultados.
Divulgações
Os autores não têm nada para divulgar.
Agradecimentos
Estamos gratos ao Instituto de bacteriologia veterinária, faculdade Vetsuisse, Universidade de Berna, pelo seu apoio. Este trabalho foi financiado pela Comissão para a promoção da precoce carreira pesquisadores acadêmicos e gênero igualdade na faculdade Vetsuisse, Universidade de Berna, através do financiamento do modelo de 120% concedido a PN. WS e PR são suportados pelo centro de estudos avançados para agricultura e alimentação, Instituto para estudos avançados, Universidade Kasetsart, Banguecoque, Tailândia, no âmbito do ensino superior pesquisa promoção e nacional pesquisa Universidade projeto da Tailândia, escritório da Tailândia de Comissão, Ministério da educação, ensino superior. Gostaríamos de agradecer ao Dr. Kwanrawee Sirikanchana para sua narração e Piyawatchara Sikarin para edição do vídeo.
Materiais
| Name | Company | Catalog Number | Comments |
| Tissue collection | Step 1 | ||
| Tricaine methanesulfonate | Sigma-Aldrich | E10521 | An alternative to clove oil. Step 1.1 |
| RNAlater stabilization solution | Thermo Fisher Scientific | AM7020 | For storing tissues if they cannot be processed immediately Step 1.3 |
| RNA extraction | Step 2 | ||
| TRIreagent | Sigma-Aldrich | Step 2.1 | |
| TRIzol | Thermo Fisher Scientific (Invitrogen) | 15596026 | Step 2.1 |
| GENEzol | Geneaid | GZR100 | Step 2.1 |
| Trisure | Bioline | BIO-38032 | Step 2.1 |
| Homemade solution | - | - | 94.53 g/L (800 mM) guanidine thiocyanate 30.45 g/L (400 mM) ammonium thiocyanate 8.20 g/L (100 mM) sodium acetate 380 mL/L (38 % v/v) phenol 50 mL/L (5 % v/v) glycerol 1.0 g/L (0.1 % w/v) 8-quinolinol, pH 5.0 Store up to 2 years at 4oC Step 2.1 |
| MagNA Lyser Green Beads | Roche | 3358941001 | An alternative tissue homogenization method used in conjunction with tissue lysing machines detailed below Step 2.2 |
| Lysing Matrix D, 2 mL Tube | MP BIOMEDICALS | 116913050 | |
| Chloroform | Sigma-Aldrich | C2432 | Step 2.3 |
| Chloroform | RCI Labscan | AR1027E-G2.5L | Step 2.3 |
| 1-Bromo-3-chloropropane | Sigma-Aldrich | B9673 | A less toxic alternative to chloroform Step 2.3 |
| Isopropanol (GC) ≥ 99.8 % | Sigma-Aldrich | 59300 | Step 2.6 |
| Isopropanol (ACS, ISO Reag. Ph Eur) | Merck KGaA (EMSURE) | 1.09634.2500 | Step 2.6 |
| Glycogen, molecular biology grade (e.g., Sigma, cat. no. G1767) | Thermo Fisher Scientific (Thermo Scientific) | R0551 | Useful step if tissue starting material is small to maximise RNA precipitation optional |
| Ethanol (purity (GC) ≥ 99.9 % | Sigma-Aldrich (EMD Millipore) | 1.00983 | Step 2.9 |
| Ethanol (ACS, ISO Reag. Ph Eur) | Merck (EMSURE) | 1.00983.2500 | Step 2.9 |
| Nuclease-free water | Promega | P1193 | Step 2.13 |
| Nuclease-free water | Multicell | 809-115-CL | Step 2.13 |
| Ambion TURBO DNA-free kit | Thermo Fisher Scientific (Invitrogen) | AM1907 | Can be performed at the end of the RNA extraction protocol optional |
| cDNA synthesis | Step 4 | ||
| Viva cDNA Synthesis Kit | Vivantis | cDSK01 | Step 4.1 & 4.3 |
| ReverTra Ace qPCR RT MasterMix with gDNA remover | Toyobo | A1172K | An alternative option see discussion |
| ReverTra Ace qPCR RT Kit | Toyobo | FSQ-101 | An alternative option see discussion |
| AffinityScript Multiple Temperature Reverse Transcriptase | Agilent Technologies | 600107 | An alternative option |
| PCR | Step 5 | ||
| DNA polymerase systems: | Step 5.2 | ||
| - Platinum II Hot-Start Green PCR Master Mix (2X) | Thermo Fisher Scientific (Invitrogen) | 14001012a | Step 5.2 |
| - GoTaq Mastermix | Promega | M7122 | Step 5.2 |
| Separate PCR mixture components: | Step 5.2 | ||
| 10mM dNTP Mix | Vivantis | NP2409 | Step 5.2 |
| 25mM MgCl2 | Thermo Fisher Scientific | R0971 | Step 5.2 |
| 10X Taq Buffer with KCl | Thermo Fisher Scientific | 00348114 | Step 5.2 |
| Taq DNA polymerase | Vivantis | PL1202 | Step 5.2 |
| - Verso 1-step RT-PCR ReddyMix with ThermoPrime Taq | Thermo Fisher Scientific | AB1454 | One step RT-PCR exemplified in Figure 3B |
| Gel electrophoresis: | For visulation of PCR products from steps 5.1-5.4 | ||
| Ethidium Bromide solution (10 mg/mL) | Thermo Fisher Scientific | 17898 | Step 5.5 |
| Tris/Acetic/EDTA (TAE) buffer: | Step 5.5 | ||
| - Tris | Vivantis | PR0612-1KG | Step 5.5 |
| - Acetic acid (glacial) (ACS, ISO, Reag. Ph Eur) | Merck KGaA (EMSURE) | 1.00063.2500 | Step 5.5 |
| - Ethylenediaminetetraacetic acid (EDTA) | BIO-RAD | 161-0729 | Step 5.5 |
| Agarose | Vivantis | PC0701-100G | Step 5.5 |
| DNA ladders and markers | Vivantis | NL1405 | Step 5.5 |
| DNA gel loading dye (6X) | Thermo Fisher Scientific | R0611 | Step 5.5 |
| qPCR | Step 6 | ||
| PowerUP SYBR Green Master Mix | Thermo Fisher Scientific (Applied Biosystems) | A25779 | Exemplified in Figures 4-6B Step 6.2 |
| iTaq Universal SYBR Green Supermix | BIO-RAD | 1725120 | Exemplified in the video and in Figures 4-6A Step 6.2 |
| Equipment | |||
| Dounce tissue grinder pestle | Sigma-Aldrich | P1110 | Protocol 2 |
| MagNA Lyser Instrument | Roche | 3358976001 | An alternative tissue homogenizing option for protocol 2 which are used in conjunction with the lysing beads detailed above Step 2.2 |
| FastPrep-24 5G Homogenizer | MP BIOMEDICALS | 116005500 | |
| Refrigerated microcentrifuge | Eppendorf | Eppendorf 5427R | Protocol 2 Step 2.4, 2.7 & 2.10 |
| Refrigerated microcentrifuge | Eppendorf | Eppendorf 5418R | |
| Heat box | Labnet | AccuBlock Digital Dry Bath | Protocol 2 Step 2.13 |
| Microvolume spectrophotometer | Thermo Fisher Scientific (Applied Biosystems) | Nanodrop 2000 | Protocol 3 Step 3.1 - 3.4 |
| PCR machine | BIO-RAD | T100 Thermal Cycler | Protocol 5 Step 5.4 |
| Power supply | BIO-RAD | PowerPac HC | Protocol 5 Step 5.5 |
| Horizontal gel electrophoresis | BIO-RAD | Mini ReadySub-Cell GT Cell #1704487edu | Protocol 5 Step 5.5 |
| Mini microcentrifuge | Corning | LSE 6766 | Useful to quickly spin down PCR reaction tubes in protocols 4, 5 & 6 Step 6.5.1 |
| Microcentrifuge | LioFuge | LM-60 | Step 6.5.1 |
| qPCR machine and software | Thermo Fisher Scientific | 7500 Fast Real-Time PCR System with 7500 Software v2.0 | Protocol 6 Step 6.6-6.8 |
| qPCR machine and software | BIO-RAD | CFX96 Touch Real-Time PCR Detection System with CFX Manager software | |
| General Materials | |||
| Mayo scissors | Step 1.1-1.2 | ||
| Forceps | Step 1.1-1.2 | ||
| Pipette | Rainin | Pipette-Lite XLS | |
| Aerosol-barrier pipette tips | Sigma-Aldrich | Z333328, Z333336, Z333344 | |
| Nuclease-free 1.5-ml microcentrifuge tubes | Eppendorf |
Referências
- FAO. The State of World Fisheries and Aquaculture, 2014. Opportunities and Challenges. , Rome. (2014).
- FAO. The State of World Fisheries and Aquaculture, 2016. Contributing to Food Security and Nutrition for all. , Rome. (2016).
- WorldBank. FISH TO 2030: Prospects for Fisheries and Aquaculture. Agriculture and Environmental Services Discussion Paper 03. , (2013).
- Wing-Keong, N., Nicholas, R. A review of the nutrition and feeding management of farmed tilapia throughout the culture cycle. Reviews in Aquaculture. 5 (4), 220-254 (2013).
- Cleasby, N., et al. The socio-economic context for improving food security through land based aquaculture in Solomon Islands: A peri-urban case study. Marine Policy. 45, 89-97 (2014).
- Ponzoni Raul, W., et al. Genetic improvement of Nile tilapia (Oreochromis niloticus) with special reference to the work conducted by the WorldFish Center with the GIFT strain. Reviews in Aquaculture. 3 (1), 27-41 (2011).
- Hounmanou, Y. M. G., et al. Tilapia lake virus threatens tilapiines farming and food security: Socio-economic challenges and preventive measures in Sub-Saharan Africa. Aquaculture (Amsterdam, Netherlands). 493, 123-129 (2018).
- OIE. Tilapia Lake Virus (TiLV) - a novel orthomyxo-like virus. OIE technical disease cards. , http://www.oie.int/fileadmin/Home/eng/Internationa_Standard_Setting/docs/pdf/A_TiLV_disease_card.pdf (2018).
- Mugimba, K. K., et al. Detection of tilapia lake virus (TiLV) infection by PCR in farmed and wild Nile tilapia (Oreochromis niloticus) from Lake Victoria. Journal of Fish Diseases. , (2018).
- Koesharyani, I., Gardenia, L., Widowati, Z., Khumaira,, Rustianti, D. D. Studi kasus infeksi tilapia lake virus (tilv) pada ikan nila (Oreochromis niloticus). Jurnal Riset Akuakultur. 13 (1), 85-92 (2018).
- OIE. Tilapia lake virus disease (TiLV), Chinese Taipei. Immediate Notification. , http://www.oie.int/wahis_2/public/wahid.php/Reviewreport/Review?reportid=24033 (2017).
- OIE. Tilapia Lake Virus Disease (TiLV), Peru. Immediate Notification. , (2018).
- Bacharach, E., et al. Characterization of a Novel Orthomyxo-like Virus Causing Mass Die-Offs of Tilapia. MBio. 7 (2), e00431-e00416 (2016).
- Eyngor, M., et al. Identification of a novel RNA virus lethal to tilapia. Journal of Clinical Microbiology. 52 (12), 4137-4146 (2014).
- Nicholson, P., et al. Detection of Tilapia Lake Virus in Egyptian fish farms experiencing high mortalities in 2015. Journal of Fish Diseases. 40 (12), 1925-1928 (2017).
- Surachetpong, W., et al. Outbreaks of Tilapia Lake Virus Infection, Thailand, 2015-2016. Emerging Infectious Diseases. 23 (6), 1031-1033 (2017).
- Tattiyapong, P., Dachavichitlead, W., Surachetpong, W. Experimental infection of Tilapia Lake Virus (TiLV) in Nile tilapia (Oreochromis niloticus) and red tilapia (Oreochromis spp.). Veterinary Microbiology. 207, 170-177 (2017).
- Kembou Tsofack, J. E., et al. Detection of Tilapia Lake Virus in Clinical Samples by Culturing and Nested Reverse Transcription-PCR. Journal of Clinical Microbiology. 55 (3), 759-767 (2017).
- Thangaraj, R. S., et al. Derivation of two tilapia (Oreochromis niloticus) cell lines for efficient propagation of Tilapia Lake Virus (TiLV). Aquaculture (Amsterdam, Netherlands). 492, 206-214 (2018).
- Hanson, L. A., Rudis, M. R., Vasquez-Lee, M., Montgomery, R. D. A broadly applicable method to characterize large DNA viruses and adenoviruses based on the DNA polymerase gene. Virology Journal. 3, 28-28 (2006).
- Josko, D. Molecular virology in the clinical laboratory. Clinical Laboratory Science. 23 (4), 231-236 (2010).
- Munir, K., Kibenge, F. S. Detection of infectious salmon anaemia virus by real-time RT-PCR. Journal of Virological Methods. 117 (1), 37-47 (2004).
- Snow, M., et al. Developement, application and validation of a Taqman real-time RT-PCR assay for the detection of infectious salmon anaemia virus (ISAV) in Atlantic salmon (Salmo salar). Developments in Biologicals. 126, discussion 325-136 133-145 (2006).
- Matejusova, I., McKay, P., McBeath, A. J., Collet, B., Snow, M. Development of a sensitive and controlled real-time RT-PCR assay for viral haemorrhagic septicaemia virus (VHSV) in marine salmonid aquaculture. Diseases of Aquatic Organisms. 80 (2), 137-144 (2008).
- Garver, K. A., et al. Development and validation of a reverse transcription quantitative PCR for universal detection of viral hemorrhagic septicemia virus. Diseases of Aquatic Organisms. 95 (2), 97-112 (2011).
- Dalla Valle, L., et al. Development of a sensitive and quantitative diagnostic assay for fish nervous necrosis virus based on two-target real-time PCR. Veterinary Microbiology. 110 (3-4), 167-179 (2005).
- Hodneland, K., Garcia, R., Balbuena, J. A., Zarza, C., Fouz, B. Real-time RT-PCR detection of betanodavirus in naturally and experimentally infected fish from Spain. Journal of Fish Diseases. 34 (3), 189-202 (2011).
- Hodneland, K., Endresen, C. Sensitive and specific detection of Salmonid alphavirus using real-time PCR (TaqMan). Journal of Virological Methods. 131 (2), 184-192 (2006).
- Wang, X. W., Ao, J. Q., Li, Q. G., Chen, X. H. Quantitative detection of a marine fish iridovirus isolated from large yellow croaker, Pseudosciaena crocea, using a molecular beacon. Journal of Virological Methods. 133 (1), 76-81 (2006).
- van Beurden, S. J., et al. Development and validation of a real-time PCR assay for the detection of anguillid herpesvirus 1. Journal of Fish Diseases. 39 (1), 95-104 (2016).
- Ciulli, S., et al. Development and application of a real-time PCR assay for the detection and quantitation of lymphocystis disease virus. Journal of Virological Methods. 213, 164-173 (2015).
- Tattiyapong, P., Sirikanchana, K., Surachetpong, W. Development and validation of a reverse transcription quantitative polymerase chain reaction for tilapia lake virus detection in clinical samples and experimentally challenged fish. Journal of Fish Diseases. 41 (2), 255-261 (2018).
- Dong, H. T., et al. Emergence of tilapia lake virus in Thailand and an alternative semi-nested RT-PCR for detection. Aquaculture (Amsterdam, Netherlands). 476, 111-118 (2017).
- Waiyamitra, P., et al. A TaqMan RT-qPCR assay for tilapia lake virus (TiLV) detection in tilapia. Aquaculture (Amsterdam, Netherlands). 497, 184-188 (2018).
- Behera, B. K., et al. Emergence of Tilapia Lake Virus associated with mortalities of farmed Nile Tilapia Oreochromis niloticus (Linnaeus 1758) in India. Aquaculture (Amsterdam, Netherlands). 484, 168-174 (2018).
- Ferguson, H. W., et al. Syncytial hepatitis of farmed tilapia, Oreochromis niloticus (L.): a case report. Journal of Fish Diseases. 37 (6), 583-589 (2014).
- Liamnimitr, P., Thammatorn, W., U-thoomporn, S., Tattiyapong, P., Surachetpong, W. Non-lethal sampling for Tilapia Lake Virus detection by RT-qPCR and cell culture. Aquaculture (Amsterdam, Netherlands). 486, 75-80 (2018).
- Yang, C. G., et al. Evaluation of reference genes for quantitative real-time RT-PCR analysis of gene expression in Nile tilapia (Oreochromis niloticus). Gene. 527 (1), 183-192 (2013).
- Bustin, S. A. Real-time, fluorescence-based quantitative PCR: a snapshot of current procedures and preferences. Expert Review of Molecular Diagnostics. 5 (4), 493-498 (2005).
- Fleige, S., Pfaffl, M. W. RNA integrity and the effect on the real-time qRT-PCR performance. Molecular Aspects of Medicine. 27 (2-3), 126-139 (2006).
- Kubista, M., et al. The real-time polymerase chain reaction. Molecular Aspects of Medicine. 27 (2-3), 95-125 (2006).
- Mackay, I. M., Arden, K. E., Nitsche, A. Real-time PCR in virology. Nucleic Acids Research. 30 (6), 1292-1305 (2002).
- Wong, M. L., Medrano, J. F. Real-time PCR for mRNA quantitation. Biotechniques. 39 (1), 75-85 (2005).
- Bustin, S. A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. Journal of Molecular Endocrinology. 25 (2), 169-193 (2000).
- Heid, C. A., Stevens, J., Livak, K. J., Williams, P. M. Real time quantitative PCR. Genome Research. 6 (10), 986-994 (1996).
- Rutledge, R. G., Côté, C. Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Research. 31 (16), e93-e93 (2003).
- Svec, D., Tichopad, A., Novosadova, V., Pfaffl, M. W., Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomolecular Detection and Quantification. 3, 9-16 (2015).
- Amal, M. N. A., et al. A case of natural co-infection of Tilapia Lake Virus and Aeromonas veronii in a Malaysian red hybrid tilapia (Oreochromis niloticus × O. mossambicus) farm experiencing high mortality. Aquaculture (Amsterdam, Netherlands). 485, 12-16 (2018).
- Fathi, M., et al. Identification of Tilapia Lake Virus in Egypt in Nile tilapia affected by ‘summer mortality’ syndrome. Aquaculture (Amsterdam, Netherlands). 473, 430-432 (2017).
- OIE. Tilapia Lake Virus disease (TiLV), Philippines. Immediate Notification. , (2017).
- OIE. Tilapia lake virus disease (TiLV), Malaysia. Immediate Notification. , https://www.oie.int/wahis_2/public/wahid.php/Reviewreport/Review?page_refer=MapFullEventReport&reportid=24809 (2017).
- Abdullah, A., et al. First detection of tilapia lake virus (TiLV) in wild river carp (Barbonymus schwanenfeldii) at Timah Tasoh Lake, Malaysia. Journal of Fish Diseases. 41 (9), 1459-1462 (2018).
- Chomczynski, P., Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 162 (1), 156-159 (1987).
- Chomczynski, P., Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nature Protocols. 1 (2), 581-585 (2006).
- Del-Pozo, J., et al. Syncytial Hepatitis of Tilapia ( Oreochromis niloticus L.) is Associated With Orthomyxovirus-Like Virions in Hepatocytes. Veterinary Pathology. 54 (1), 164-170 (2017).
- Bustin, S. A., et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry. 55 (4), 611-622 (2009).
- Purcell, M. K., Getchell, R. G., McClure, C. A., Garver, K. A. Quantitative polymerase chain reaction (PCR) for detection of aquatic animal pathogens in a diagnostic laboratory setting. Journal of Aquatic Animal Health. 23 (3), 148-161 (2011).
- Simpson, D. A., Feeney, S., Boyle, C., Stitt, A. W. Retinal VEGF mRNA measured by SYBR green I fluorescence: A versatile approach to quantitative PCR. Molecular Vision. 6, 178-183 (2000).
- Kibenge, M. J., et al. Discovery of variant infectious salmon anaemia virus (ISAV) of European genotype in British Columbia, Canada. Virology Journal. 13, 3(2016).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados